

Abstract

Single-step retrosynthesis in organic chemistry increasingly benefits from deep learning (DL) techniques in computer-aided synthesis design. While template-free DL models are flexible and promising for retrosynthesis prediction, they often ignore vital 2D molecular information and struggle with atom alignment for node generation, resulting in lower performance compared to the template-based and semi-template-based methods. To address these issues, we introduce node-aligned graph-to-graph (NAG2G), a transformer-based template-free DL model. NAG2G combines 2D molecular graphs and 3D conformations to retain comprehensive molecular details and incorporates product-reactant atom mapping through node alignment, which determines the order of the node-by-node graph outputs process in an autoregressive manner. Through rigorous benchmarking and detailed case studies, we have demonstrated that NAG2G stands out with its remarkable predictive accuracy on the expansive data sets of USPTO-50k and USPTO-FULL. Moreover, the model’s practical utility is underscored by its successful prediction of synthesis pathways for multiple drug candidate molecules. This proves not only NAG2G’s robustness but also its potential to revolutionize the prediction of complex chemical synthesis processes for future synthetic route design tasks.
Keywords: Template-Free Retrosynthesis, Deep Learning, Chemical Reactions, Graph Generation, Single-Step Retrosynthesis Prediction
Introduction
The single-step retrosynthesis (SSR)1 is an essential operation in organic chemistry involving the reversed synthesis of a target product or intermediate in a single step. To achieve automatic multistep synthesis route design, SSR plays a critical role in building the blocks for each separated stage. Typically, the design of retrosynthesis strategies demands a thorough understanding and knowledge of organic chemistry principles, such as reaction mechanisms and reactive sites. With the emergence of computer-aided synthetic planning tools, researchers are now harnessing deep learning (DL) techniques to address this task and recognize their immense potential.
Various DL architectures have been developed and refined to suit the purpose of learning reactions for SSR tasks.2 Even though there are notable variations in their network structures and data representation formats, they mainly fall into two primary groups: template-dependent and template-independent. In the following section, we will provide a concise overview of recent DL-based methods, highlighting their model designs as well as their strengths and weaknesses.
Template or Non-template?
The chemical intuition of organic synthetic chemists is accumulated from knowledge of reaction rules. Naturally, a dictionary or so-called template of existing reactions (such as an organic synthesis textbook) will serve as a bible for SSR design. Therefore, the initial generation of retrosynthesis tools was trained to search for the most likely reaction templates in the library that could be used for generating the desired product. For instance, the program Synthia (previously named as Chematica)3 employs over 80,000 rules crafted by synthetic experts to determine the appropriate reaction step based on a huge decision tree. DL strategies, like RetroSim(4) and NeuralSym,5 use traditional molecular similarity metrics, such as fingerprints and Tanimoto similarity, to look for the templates that match well with products. Other contemporary approaches include LocalRetro,6GLN,7 and RetroComposer.8 The limitations of template-based methods are inherent in the library on which they rely on. The library may not cover all potential reactions, and there is a risk of incorrect associations between the intricate products and template structures. To avoid being overly dependent on dictionaries, semi-template approaches emerged. This method breaks down the SSR prediction process into two stages—synthons9 or intermediate detection followed by reactant generation. Both steps are critical, as the preprocessing and identification of synthon are directly associated with the reactant prediction. The advantage of the two-stage methodology includes synthon understanding, searching capabilities expansion, and reaction scheme exploration. Meantime, errors can be easily passed from the first step to the second. As the technique of semi-template approaches grows, several models include G2G,10RetroXpert,11RetroPrime,12GraphRetro,13SemiRetro,14G2Retro,15 and Graph2Edits(16) have emerged, highlighting the compatibility of graph-to-graph models with molecular topology edits, which we will discuss later.
Can a DL model learn chemistry without any prerequisite knowledge (including dictionaries, templates, synthons, intermediates, and editing strategies) given by scientists? The answer is YES. Template-free models, such as pioneering seq2seq,17SCROP,18Tied Transformer,19Augmented Transformer,20 and RetroDCVAE(21) all consider retrosynthesis as a prediction problem. The foundational idea posits that molecules can be analyzed in a manner akin to that for natural language processing (NLP) tasks. In this framework, product molecules are broken down into tokens based on their one-dimensional (1D) string representations, like the Simplified Molecular Input Line Entry System (SMILES). This tokenization allows us to treat the transformation of products into reactants, drawing parallels between chemical reactions and language translation processes. Recent studies have achieved marked improvements by applying the advanced NLP Transformer(22) model, which employs a multihead attention mechanism. This mechanism enables the model to assign varying degrees of importance to different segments of input data, enhancing its ability to manage the message-passing process between each pair of atoms within a molecule and between pairs of product and reactant.
Choices of Molecular Representation
To help computers think like chemists, it is crucial to translate reaction information, specifically, molecular-level reactants and products, into in silico “language” or so-called molecular representation. One popular approach of DL-based SSR models18−21 is to employ 1D sequences, such as SMILES. Despite its simplicity, the 1D sequence-based model exhibits several limitations: (1) The sequence disregards the extensive molecular topological information;23−25 (2) Legal SMILES follow intricate syntax rules, which magnify the difficulty of valid SMILES generation; (3) Effectively utilizing atom mapping information between products and reactants is challenging for 1D representation. Without alignment, model performance may decline due to lost atom correlations between products and reactants. (4) Due to the fact that a single molecule can have multiple SMILES representations when generating multiple candidate reactants for a product, it is possible to generate multiple reactant SMILES representing the same reaction, which may reduce the diversity of the candidates.20
To overcome the limitations of 1D sequences, models involving 2D molecular graphs, which encompass atom (node) and bond (edge) topology, have been proposed for molecular representation in SSR tasks.10,15,16,26,27 2D graphs encapsulate a wealth of information about the atomic environment, such as neighboring atoms and their connections. The graph topology offers an optimal solution for two-step tasks involving the modification of synthons and intermediates under the context of semi-template models. In order to effectively utilize the natural mapping information between atoms in products and reactants, prior approaches have employed a repeated graph edit strategy,26 wherein the input graph of the product is iteratively modified by taking edit actions (such as adding nodes, removing nodes, updating nodes or edges) until it reaches an end—the reactant. Semi-template models such as G2Retro,15MARS,27 and Graph2Edits(16) employ graph edit strategies26 for graph generation. However, the 2D graph edition requires a delicate arrangement of edit actions and editing types forehead. Despite the advanced generation processes reducing computational expenses, the iterative action–prediction cycles, which require graph input and output at each editing step, continue to add to the computational burden.
Boundaries and Pushing Boundaries
Models have improved by combining different methods with the latest machine learning designs, as we have discussed before. Even though there are still some challenges, we see great potential in the new transformer-based models that can make predictions without using templates.17,28 Despite template-dependent models taking the upper hand, we have observed that template-free methods not only demonstrate remarkable performance with neat and concise structures but are also capable of capturing the nuances of chemical reasoning themselves. Molecular representation and attention mechanism (consideration of long-range dependencies between atoms) adaption are required to introduce more imformation. For example, GET(24) merges both graphs and SMILES encoders. GTA(25) integrates topological data into attention bias. Specifically, Retroformer,200 using 1D sequence for the encoder, incorporates product-reactant atom alignments for better results.29 Graph-based template-free methods, including G2GT,30 have advanced in exploiting graph topology. Nevertheless, they have yet to leverage node-alignment strategies for enhanced performance.
To take the advantage of a template-free approach and address the above limitations, we developed NAG2G that utilizes both 2D graph and 3D coordinates, with improved efficiency of graph generation and node alignment according to proper atom mappings, as shown in Figure 1. Moreover, we implement an autoregressive approach that generates graphs node-by-node according to the aligned order, drawing inspiration from language generation techniques. NAG2G is trained using two widely recognized data sets, USPTO-50k31 and USPTO-Full7,20 with augmented data, showing great capacity compared to existing models. Additionally, our model demonstrates its proficiency in tackling real-world problems by iteratively generating step-by-step synthesis pathways for drug candidates. To gain deeper insights into the significance of each component within our methodology, we conducted ablation studies, systematically omitting certain parts of the model to evaluate their impact on performance.
Figure 1.

Overview of computer-aided SSR workflow based on template-based, semitemplate-based, and template-free design.
Method
Model Construction
Encoders are the components of a neural network that process and compress input product into a compact representation and play the critical role of learning molecular representation in the NAG2G transformer-based encoder–decoder architecture. Competent models, such as Graphormer(32) and Uni-Mol,33 have demonstrated the efficiency of encoder representation learning strategies. Thus, we adopt the encoder from Uni-Mol+,34 which incorporates both 2D graph and 3D conformation for molecular representation as shown in Figure 2. The 1D positional encoding is also taken into account, serving as the node order encoder. Formally, we can denote the process of the encoder as the following in eq 1
| 1 |
In the proposed formulation, X denotes the atom features; Penc represents the 1D positional encoding, which is supplementary to the atomic embeddings; E signifies the edge features inherent to the 2D graph structure; R corresponds to the atomic coordinates in the 3D conformation; θenc encapsulates the encoder’s learnable parameters, and Oenc is the derived molecular representation result from the encoder.
Figure 2.

Network architecture of NAG2G.
Decoders primarily operate to generate the reactant graph node-by-node through an autoregressive approach. At the i-th time step, which also corresponds to the i-th generated node (atom), the decoder receives three distinct inputs:
(1) The encoder’s output, including keys and values that help in the interaction between the encoder and decoder.
(2) The decoder outputs from prior steps (from 1 to i – 1), which is typical of autoregressive models in that the prediction of a new value is based on its preceding values. During the iterative process, 1D positional encoding is added, which is essential for NAG2G to align atom order between the encoder (product) inputs and decoder (reactant) outputs.
(3) The graph-level features of the current output graph, such as node degrees and shortest paths between nodes. Incorporating these graph-level features directly into the model presents an efficiency challenge, as the graph features vary across time steps. To address this issue, we propose an efficient method for integrating graph-level features.
Given the above inputs, the decoder generates a new node at the i-th time step autoregressively, starting from atomic type, and then associated formal charge, the number of connected hydrogen atoms, and finally its edges (types of bond) linked to prior nodes (atoms). The information for each node is produced sequentially given its above predictions. For instance, the formal charge is predicted based on the prior atomic type prediction. The process is denoted as
 |
2 |
where N1:i–1 represents the set of nodes generated from the previous i – 1 time steps, P1:idec denotes the 1D positional encoding of the current i nodes, G1:i–1 represents the graph feature extracted from previous outputs, and θdec denotes the parameters of the decoder. The atomic type, associated formal charge, and number of connected hydrogen atoms for the i-th node are represented by ti, ci, and hi, respectively. The d-th edge, denoted by ei,d = (j, b), connects the i-th node and the j-th node with the bond type b. To define an edge’s generative order, edges linked to nodes with larger 1D positions are prioritized. Generation of ci and hi is skipped if a node has zero charges or no linked hydrogen atoms to reduce the number of generative steps. The overview of NAG2G’s architecture is depicted in Figure 2.
Node Alignment and Data Augmentation
Molecular graphs, unlike sentences, lack an inherent sequence as atoms within a molecule do not have a natural order until assigned. In order to circumvent the need to consider the order of nodes in graph generation, several methods transform the graph generation task into indirect approaches, such as graph edit action prediction or SMILES prediction tasks, as discussed earlier. Alternatively, some methods utilize a one-in-all scheme, generating the entire graph output in a single step. Although this scheme avoids considering generation order, it lacks flexibility and is unsuitable for multisolution tasks such as retrosynthesis. To adopt a more flexible and autoregressive method, the output node order must be determined. A simple solution is using the canonical SMILES atom order; however, this fixed order restricts output data augmentation and limits the model’s performance. Consequently, devising a robust and flexible strategy to tackle the unordered nature of graph nodes remains a formidable challenge in graph generation tasks. The unordered nodes challenge not only the graph generation but also the encoder input data augmentation. As input graphs inherently lack sequence, graph data augmentation must rely on alternative strategies such as omitting certain node or edge information. These approaches may be unsuitable for retrosynthesis, as the omission of critical information such as different reaction sites could result in vastly different outputs. Utilizing a reactant from the training set under these circumstances may introduce inaccuracies. Therefore, we choose a more appropriate encoder input data augmentation strategy based on node order, ensuring a more accurate and reliable outcome.
To address the challenges in input and output data augmentation and enable a flexible node-by-node autoregressive generation, we propose a novel method based on product-reactant node alignment. Our method begins with the random generation of the product’s SMILES sequence by RDKit,35 as shown in Figure 3. By following the new order in the SMILES sequence, we obtain the data-augmented input graph’s node sequence order. Subsequently, the graph node order is marked by using position embedding. For the product graph with a determined order, we establish a unique and unambiguous rule that corresponds to the reactant node order for node-by-node output, as demonstrated in Figures 3 and 4. In the reactant, atoms can be classified into two types: those shared with the product and those exclusive with the reactant. The assignment of atomic order should consider both these aspects. First, in generating the order, we stipulate that the shared atoms’ order in the reactant should precede the order of nonshared atoms. For a specific ordered product input, there should exist a unique corresponding ordered reactant. This unique correlation refers to the order of shared atoms in the reactant follow the order in the product. Subsequently, the reactant SMILES is aligned with the product SMILES using RDKit to obtain the most similar SMILES. Finally, the order of nonshared atoms is extracted from the aligned reactant SMILES, ensuring the uniqueness of the nonshared atoms’ order. This approach utilizes the product-reactant alignment information by ensuring that the node generation order mirrors the input graph’s order in the training process and allows for consistent and equivariant data augmentation in both input and output. To conclude, we provide a robust and adaptable autoregressive generation procedure that can effectively handle the complexities of molecular graphs and enhance the performance of graph-to-graph generation models. NAG2G offers a concise, profound, and persuasive solution for input–output data augmentation, ensuring logical and efficient node-by-node autoregressive generation.
Figure 3.

An example to illustrate the process of data augmentation and product-reactant alignment. The red numbers indicate the atoms present in both the product and reactants, while the blue ones represent the atoms found only in the reactants.
Figure 4.

Illustration of the NAG2G generation.
Efficient Time-Varying Graph-Level Features
During the generation process through decoder, the implementation of teacher forcing during training allows for the true output from a previous time step to be used as input for the current step, rather than the model’s own prediction. This technique not only aligns the model’s learning with the correct sequence of outputs but also enables parallel processing of data at various time steps. The interaction between the current and previous time steps is addressed within the decoder’s attention layer. To avoid the inadvertent incorporation of future information, the attention matrix in a transformer model is masked with an upper triangular matrix. This ensures that a given output at a specific time step can only be influenced by preceding elements in the sequence, preserving the autoregressive property, where the prediction for each step is conditioned only on the known past information. Formally, we denote this process as
| 3 |
where 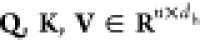 represent the query, key, and value matrices,
respectively. dh is the dimension of one
head. n is the number of time steps. M is an additive mask matrix that ensures that only the relevant information
from the current and previous time steps is considered during the
attention computation. For the sake of simplicity, we present the
calculation for only one head. The multihead attention process executes
the above single-head calculation in parallel. During the calculation
of one head, the computational complexity is
represent the query, key, and value matrices,
respectively. dh is the dimension of one
head. n is the number of time steps. M is an additive mask matrix that ensures that only the relevant information
from the current and previous time steps is considered during the
attention computation. For the sake of simplicity, we present the
calculation for only one head. The multihead attention process executes
the above single-head calculation in parallel. During the calculation
of one head, the computational complexity is 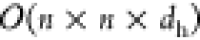 , and the peak memory consumption is
, and the peak memory consumption is  .
.
As previously mentioned, graph-level features vary across time steps, and their direct utilization poses an efficiency challenge during model training. Specifically, to maintain the time-varying graph features, a matrix with shape n × n × dh is required.a These time-varying graph features are then employed as additive positional encodings. As a result, the attention layer can be represented as
| 4 |
where  denotes the time-varying graph features
and the shape of Q, K, V is
reshaped to n × 1 × dh for broadcasting. In this process, although the computational
complexity remains unchanged, the peak memory consumption increases
to
denotes the time-varying graph features
and the shape of Q, K, V is
reshaped to n × 1 × dh for broadcasting. In this process, although the computational
complexity remains unchanged, the peak memory consumption increases
to  . Considering that dh is typically 32 or even larger, this significant increase
in peak memory consumption is considered impractical for real-world
applications.
. Considering that dh is typically 32 or even larger, this significant increase
in peak memory consumption is considered impractical for real-world
applications.
To reduce the cost, we first remove D from the V + D term. Then, the cost is bottlenecked at QDT due to the Q(K + D)T = QKT + QDT term. Furthermore, we can reduce the size of the last dimension by substituting D. Thus, the attention can be transformed to
| 5 |
where 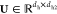 is employed to reduce the dimension of Q and
is employed to reduce the dimension of Q and 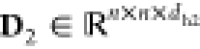 represents the time-varying graph features
with a much smaller dimension dh2. With
this configuration, the peak memory is reduced to
represents the time-varying graph features
with a much smaller dimension dh2. With
this configuration, the peak memory is reduced to 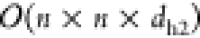 . Figure 5 illustrates the design of a self-aware layer for time-varying
graph features.
. Figure 5 illustrates the design of a self-aware layer for time-varying
graph features.
Figure 5.

Illustration of decoder attention mechanism.
Data Preparation
The NAG2G is trained and tested on two broadly acknowledged data sets, USPTO-50k31 and USPTO-Full.7,20 USPTO-50k comprises 50,016 atom-mapped reactions, categorized into 10 reaction classes. The USPTO-50K data set was split into 40,008, 5,001, and 5007 reactions for the training, validation, and test sets, respectively. We also used the filtered USPTO-Full data set with approximately 1 million atom-mapped reactions as described by Tetko et al.20 instead of the original USPTO-Full data set.7 After filtering out incorrect reactions, which leads to an approximate 4% size reduction, training, validation, and test sets contain approximately 769,000, 96,000, and 96,000 reactions. The distribution of reaction classes in the training, valid, and test sets of USPTO-50k are the same, displayed in Figure 6. Consistent with previous works, we did not benchmark the USPTO-Full results with the aid of reaction class information.
Figure 6.

Distribution of 10 types pf reactions in the USPTO-50k data set. The reaction’s legends are ranked based on their fraction from largest to smallest.
Results
NAG2G Setup
The setup of NAG2G contains a 6-layer encoder and a 6-layer decoder. The input embedding dimension was set to 768, and the number of attention heads was set to 24. We employed the Adam optimizer36 with (β1, β2) = (0.9, 0.999), and linear warmup and decay with a peak learning rate of 2.5 × 10–4. The training process took a total of 12,000 steps with a batch size of 16, requiring 6 h to finish on a single NVIDIA A100 GPU. For the training on the USPTO-Full data set, NAG2G ran 48,000 training steps with a batch size of 64, taking approximately 30 h to complete on eight NVIDIA A100 GPUs.
Results on the USPTO Data Set
To evaluate the performance of NAG2G during inference, we utilized the commonly adopted beam search method for top candidate predictions. We configure the beam size at 10, using a length penalty of 0 and a temperature of 1. Notably, data augmentation is not applied during the inference phase. Additionally, NAG2G relies on RDChiral37 to assign the atomic chirality of reactants, drawing from the product’s stereochemistry.
The definition of prediction accuracy follows the approach proposed by Liu et al.,17 which considers a prediction to be accurate only if it completely identifies all reactants for a specific chemical reaction. We measure the top-k accuracy of the predictions, defined as the proportion of test cases in which the correct answer appears among the top k candidates of the beam search results.
USTPO-50k
In Table 1, NAG2G demonstrates superior performance on the USPTO-50k data set compared to recent baseline approaches, including template-based, semi-template-based, and template-free models. (1) No supplementary techniques were implemented to aid the inference procedure in NAG2G to ensure a fair comparison. Within the template-free domain, NAG2G markedly surpasses all benchmarks across every metric. Even though certain baselines leverage extra data or methods to bolster their results (indicated by *), NAG2G continues to excel without any such enhancements. Despite the additional use of predefined rules in template-based and semitemplate-based methods, NAG2G outperforms them without prerequisite information. This marks NAG2G achieves competent results among both template-based and semi-template-based approaches, which earlier template-free benchmarks never achieved. Besides, NAG2G gives SOTA results based on MaxFrag metric proposed by Augmented Transformer,20 see the Supporting Information for details. Detailed results include testing of each reaction class on the USPTO-50k data set when trained with the class known are summarized in the Supporting Information as well.
Table 1. Top-k Accuracy for Retrosynthesis Prediction on USPTO-50ka.
| Top-k accuracy (%) |
||||||||
|---|---|---|---|---|---|---|---|---|
| USPTO-50k |
||||||||
| reaction
class known |
reaction
class unknown |
|||||||
| model | 1 | 3 | 5 | 10 | 1 | 3 | 5 | 10 |
| Template-Based | ||||||||
| RetroSim4 | 52.9 | 73.8 | 81.2 | 88.1 | 37.3 | 54.7 | 63.3 | 74.1 |
| NeuralSym5 | 55.3 | 76.0 | 81.4 | 85.1 | 44.4 | 65.3 | 72.4 | 78.9 |
| GLN7 | 64.2 | 79.1 | 85.2 | 90.0 | 52.5 | 69.0 | 75.6 | 83.7 |
| MHNreact38 | - | - | - | - | 50.5 | 73.9 | 81.0 | 87.9 |
| RetroComposer8 | 65.9 | 85.8 | 89.5 | 91.5 | 54.5 | 77.2 | 83.2 | 87.7 |
| Semi-Template-Based | ||||||||
| G2G10 | 61.0 | 81.3 | 86.0 | 88.7 | 48.9 | 67.6 | 72.5 | 75.5 |
| RetroXpert11 | 62.1 | 75.8 | 78.5 | 80.9 | 50.4 | 61.1 | 62.3 | 63.4 |
| RetroPrime12 | 64.8 | 81.6 | 85.0 | 86.9 | 51.4 | 70.8 | 74.0 | 76.1 |
| GraphRetro13 | 63.9 | 81.5 | 85.2 | 88.1 | 53.7 | 68.3 | 72.2 | 75.5 |
| SemiRetro14 | 65.8 | 85.7 | 89.8 | 92.8 | 54.9 | 75.3 | 80.4 | 84.1 |
| G2Retro15 | 63.6 | 83.6 | 88.4 | 91.5 | 54.1 | 74.1 | 81.2 | 86.7 |
| MARS27 | 66.2 | 85.8 | 90.2 | 92.9 | 54.6 | 76.4 | 83.3 | 88.5 |
| Template-Free | ||||||||
| LV-transformer39 | - | - | - | - | 40.5 | 65.1 | 72.8 | 79.4 |
| SCROP18 | 59.0 | 74.8 | 78.1 | 81.1 | 43.7 | 60.0 | 65.2 | 68.7 |
| GET24 | 57.4 | 71.3 | 74.8 | 77.4 | 44.9 | 58.8 | 62.4 | 65.9 |
| tied transformer19 | - | - | - | - | 47.1 | 67.1 | 73.1 | 76.3 |
| MEGAN26 | 60.7 | 82.0 | 87.5 | 91.6 | 48.1 | 70.7 | 78.4 | 86.1 |
| aug. transformer20 | - | - | - | - | 48.3 | - | 73.4 | 77.4 |
| aug. transformer*20 | - | - | - | - | 53.5 | 69.4 | 81 | 85.7 |
| GTA25 | - | - | - | - | 51.1 | 67.6 | 74.8 | 81.6 |
| Graph2SMILES23 | - | - | - | - | 52.9 | 66.5 | 70.0 | 72.9 |
| RetroDCVAE21 | - | - | - | - | 53.1 | 68.1 | 71.6 | 74.3 |
| DualTF40 | 65.7 | 81.9 | 84.7 | 85.9 | 53.6 | 70.7 | 74.6 | 77.0 |
| Retroformer? | 64.0 | 82.5 | 86.7 | 90.2 | 53.2 | 71.1 | 76.6 | 82.1 |
| G2GT30 | - | - | - | - | 48.0 | 57.0 | 64.0 | 64.5 |
| G2GT*30 | - | - | - | - | 54.1 | 69.9 | 74.5 | 77.7 |
| NAG2G (ours) | 67.2 | 86.4 | 90.5 | 93.8 | 55.1 | 76.9 | 83.4 | 89.9 |
The best performance is in bold, and the best results for each method type are underlined. Models denoted by an asterisk (*) employed supplementary datasets for training or incorporated techniques to enhance the accuracy during inference. In order to maintain a fair comparison, we also present their results without implementation of these additional techniques.
USTPO-FULL
Table 2 presents the performance metrics of various models evaluated on the USPTO-Full data set. As the data set size increases, the performance of all models declines due to the heightened complexity of the task. Notably, while template-based methods have shown impressive results on the USPTO-50k data set, their performance falters considerably on the larger USPTO-Full data set. This trend indicates that the reliance on template-based methods from pre-established rules becomes a limitation when confronting larger and more complex data sets. In contrast, though weakened as well, template-free methods demonstrate a more versatile and adaptive capability, particularly more suited for expansive data sets. Still, it is evident that NAG2G consistently outperforms preceding baselines across all evaluative criteria.
Table 2. Top-k Accuracy for Retrosynthesis Prediction on the USPTO-Full Data Seta.
| model |
Top-k accuracy (%) |
||||
|---|---|---|---|---|---|
| model type | methods | 1 | 3 | 5 | 10 |
| template-based | RetroSim4 | 32.8 | - | - | 56.1 |
| NeuralSym5 | 35.8 | - | - | 60.8 | |
| GLN7 | 39.3 | - | - | 63.7 | |
| semi-template-based | RetroPrime12 | 44.1 | 59.1 | 62.8 | 68.5 |
| template-free | MEGAN26 | 33.6 | - | - | 63.9 |
| aug. transformer* | 44.4 | - | - | 70.4 | |
| NAG2G (ours) | 47.7 | 62.0 | 66.6 | 71.0 | |
| aug. transformer*◦20 | 46.2 | - | - | 73.3 | |
| G2GT*◦25 | 49.3 | - | 68.9 | 72.7 | |
| NAG2G (ours)◦ | 49.7 | 64.6 | 69.3 | 74.0 | |
Models denoted by an asterisk (*) used supplementary data sets for training or incorporated techniques to improve accuracy during inference. For models denoted by a circle (◦), the invalid reactions are excluded from the test set, following the setting of the augmented transformer.20 To align our methods with the previous baselines, we adopted the approach from the augmented transformer,20 assuming that the methods failed on the removed test data, as evidenced by the results of our methods without a circle (◦).
Result Interpretation (Ablation Study)
To identify the importance of each component that has been designed for NAG2G, we performed ablation studies by studying the impact of its removal. This aids in understanding the NAG2G structure and how node alignment, data augmentation, and the incorporation of time-varying graph features can benefit the model.
Table 3 presents a comprehensive quantitative breakdown of the impact of each strategy on the model’s performance on the USPTO-50k data set, focusing specifically on Top-k accuracy percentages. When all three techniques—node alignment, data augmentation, and graph features—are utilized, the model hits a peak Top-1 accuracy of 55.1% and maintains high performance across Top-3 (76.9%), Top-5 (83.4%), and Top-10 (89.9%), emphasizing the synergistic effect of this combination. Eliminating graph features results in a marginal decrease in the accuracy of each Top-k accuracy metric. However, incorporating these graph features contributes to an approximately 1% improvement in the metrics under challenging scenarios where further progress is difficult to achieve. Omitting data augmentation leads to a more pronounced decline, with the top-1 accuracy decreasing by 5.9% and the top-10 accuracy experiencing a reduction of 9.5%, underscoring its role in enhancing model robustness and generalizability to unseen data. The performance deteriorates dramatically without node alignment, with the accuracy dropping by 50% for Top-10. This highlights the importance of node alignment in capturing the structural information of the graphs and enabling the model to make more accurate predictions. It is evident that the difference between top-1 and top-10 accuracy significantly reduces from 34.8% (when both data augmentation and node alignment are employed) to merely 3.8% (when data augmentation is used without node alignment). This illustrates that using data augmentation without node alignment leads to a lower diversity of candidate predictions compared to when both techniques are employed. However, it is worth noting that the gap remains at 22.3% when neither node alignment nor data augmentation is utilized, which is not particularly high. Consequently, this highlights that node alignment and data augmentation are complementary techniques that, when jointly employed, can enhance the performance metrics. In conclusion, our ablation study shows that node alignment, data augmentation, and graph features are all crucial components of our model, with each strategy playing a vital role in enhancing prediction accuracy. The combination of these strategies yields the best overall performance, emphasizing the importance of incorporating them in future work on graph-based reaction prediction models.
Table 3. Ablation Study on USPTO-50k with Reaction Class Unknown.
| strategies |
Top-k accuracy (%) |
|||||
|---|---|---|---|---|---|---|
| node alignment | data augmentation | graph features | 1 | 3 | 5 | 10 |
| √ | √ | √ | 55.1 | 76.9 | 83.4 | 89.9 |
| √ | √ | × | 54.1 | 75.9 | 82.6 | 88.8 |
| √ | × | √ | 49.2 | 69.2 | 75.3 | 80.4 |
| × | √ | √ | 46.1 | 47.6 | 48.5 | 49.9 |
| × | × | √ | 40.3 | 54.9 | 58.9 | 62.6 |
Table 4 illustrates the Top-k validity of various models on the USPTO-50k data set, focusing on the autoregressive node generation approach adopted by the NAG2G model. When all atomic features (type, formal charge, and number of hydrogens) are utilized, NAG2G delivers a Top-1 validity of 99.7%, with strong performances also observed in Top-3 (98.6%), Top-5 (97.1%), and Top-10 (92.9%) predictions. The exclusion of certain molecular properties during node generation reveals the importance of each feature in generating SMILES. Omitting just the formal charge results in a Top-1 validity of 89.9%, while the disregard of the hydrogens brings it to 89.6%. The most pronounced decrease is observed when both of these features are excluded, dropping the Top-1 validity to 80.8%. The results with respect to the accuracy are also studied, see the Supporting Information for details. When juxtaposed with other models such as GET, Graph2SMILES, and RetroPrime, NAG2G consistently outperforms.
Table 4. Top-k Validity of the Generated Molecules on USPTO-50k with the Reaction Class Unknown.
Furthermore, our ablation studies of the encoder reveal that the performance of our model is robust and not overly dependent on its encoder configuration. For detailed findings, please refer to the Supporting Information provided.
Case Studies
To assess the capability of designing synthetic routes for organic molecules, we picked four drug candidates as the target product and performed sequentially SSR using NAG2G trained with the USPTO-50k data set. The performance of template-free NAG2G successfully outperforms the previous model16 according to its inference results as shown in Figure 7a. All six synthetic steps documented in the original literature for Nirmatrelvir41 are accurately predicted by NAG2G, achieving all predictions within the top three ranks. The initial step involving the dehydration of the amide to form the cyano group was ranked first, while the rank-2 result predicted condensation reaction aligns with a recent advanced one-pot synthesis strategy for Nirmatrelvir.42 The exact step-2 and step-6 condensations are pulled out by NAG2G with both answers standing out among the other candidates. In the third step, the trifluoroacetylation of the amine in 6 was predicted as the third-ranked reaction by our model, an improvement over its sixth-ranked prediction in Graph2Edits. For step-4 and step-5, our model’s first- and second-ranked predictions effectively serve the protective function using different, yet common, reagents.43 The second test case, osimertinib, known by its research name AZD9291, gained approval in 2014 as a clinical treatment for patients with nonsmall cell lung cancer.44 As shown in Figure 7b, NAG2G successfully delineates a five-step synthesis, as described in the literature, tracing the synthetic pathway from commercially available pyrimidines to the final product AZD9291. The initial step of the reverse synthesis is an acylation reaction, ranking the first in order of likelihood, followed by another rank-1 reduction of the nitro group. Subsequently, it correctly identified two consecutive nucleophilic aromatic substitution steps as the top choices. In the final step, the model’s highest probability prediction was a Suzuki coupling, which was also the rank-2 reaction inferred by the Graph2Edits model.16 Although the original strategy involving a Grignard reaction was not predicted, the rank-6 result suggests the alternative pathway. For the third case, we selected salmeterol, a long-acting bronchodilator, which has been tested in the former template-based model by Coley,4LocalRetro,6 and root-aligned strategy by Zhong.29 With the presence of amine and phenol functional groups in compound 21, NAG2G recommends protecting the reactive sites, ranking these steps as first and second.45,46 The subsequent reaction is the production of compound 22 through a Williamson ether-type reaction, identified as the rank-5 choice. Moreover, rank-6 for the fourth step successfully predicts the transformation of 26 into 25 via an asymmetric Henry reaction, as described in Guo’s synthesis.46 It is noteworthy that while the original synthesis protects two hydroxyl groups in compound 27 simultaneously using 2,2-dimethoxypropane, NAG2G opts to protect only the more reactive phenol group, which aligns with the requirements of the complete synthetic scheme.
Figure 7.

Synthesis route of four drug molecules and the predicted ranks given by NAG2G.
The last example, lenalidomide, which is also involved in the work of LocalRetro(6) and Graph2Edit(16) turns out to be a more challenging task.47 NAG2G accurately predicted the first and last steps involving nitro reduction and NBS (N-bromosuccinimide) substitution as the top-1 reactions. For the cyclization step involving the formation of two C–N bonds, NAG2G proposed a precursor, compound 31, with a chlorine substituent instead of bromine. Nonetheless, NAG2G is capable of suggesting a stepwise ring closure mechanism, mirroring the results reported by LocalRetro.
Error Analysis
In the sequence-to-sequence (seq2seq) model17 context, the generation of reactants in the SMILES format can result in three outcomes: (1) Generated SMILES strings generated are invalid, corresponding to a nonchemically feasible structure. (2) The SMILES strings are chemically valid but do not represent suitable reactants capable of producing the desired product under given reaction conditions. (3) The SMILES strings represent reactive compounds that can lead to the products as common reactions, even though they may not match the ground truth reactants exactly. In evaluating the first type of error for our model on the USPTO-50k data set, we focused on validity, which gauges the percentage of valid SMILES strings generated among the top-k predictions. Our model boasts a top-1 validity of 99.7%, outperforming other advanced models.12,23,24 This superior validity is due to the autoregressive generation process, as we have discussed in the ablation study section. If the model randomly omits predictions for charge and hydrogen attachments, then the top-1 validity significantly decreases to 80.8%. The identification of the second and third types of errors involves expert evaluation by organic chemists. To impartially review the inference results without preassigned reaction classes, we selected three representatives to examine the variety of reactants that can be predicted by our model, referred to here as NAG2G. In all cases, the ground truth falls within the top 10 predictions, alongside a variety of other reaction types, showcasing the model’s extensive predictive range. The rich diversity of suggested synthetic solutions enhances the retrosynthetic route design by providing a wide array of chemical reactions to consider, thus offering alternatives for subsequent route selection and evaluation. For a detailed summary table and analysis, please refer to the Supporting Information.
Another limitation stemming from the origins of the USPTO data set is the lack of detailed reaction information, such as conditions, yields, and selectivity. With this information, the NAG2G model could provide more accurate and reliable rankings for its predictions. Additionally, single-step prediction models, including ours, might overlook interactions between consecutive steps. For example, in case 3, the selective phenolic benzylation, while an efficient protecting step, could significantly impact the subsequent asymmetric Henry reaction. This scenario illustrates that effective single-step predictions require not just accuracy but also a thorough evaluation of route complexity and yield trade-offs. In response to these challenges, we are actively developing advanced scoring methods to facilitate multistep process decisions based on the results provided by NAG2G.
Conclusions
In this study, we present the NAG2G model—a graph-based SSR method free from templates. This model employs the transformer encoder–decoder framework, generating reactant molecule graphs in an autoregressive fashion. Testing on well-established data sets, USPTO-50k and USPTO-Full, indicates that NAG2G offers competent performance against prevailing SOTA models. Ablation studies shed light on the contributions of various components, underlining the potential of our approach. The replication of case studies and error analysis highlight the promising performance of NAG2G in specific SSR tasks, suggesting that further advancements could enhance its capabilities even more.
While many SSR models, detailed in our introduction, show promise, NAG2G marks a notable stride in applying DL to single-step retrosynthesis—especially when considering a template-independent approach. Our methodology opens the door that intricate neural networks are not the sole route to achieve high quality results; careful and detailed model design, combined with precise task definitions, can yield competitive results. Our current design is tailored for single-step retrosynthesis predictions, where input and output graphs bear close resemblance. For broader graph-to-graph generation tasks, especially with considerable input–output disparities, refinements may be needed. Moving forward, our ultimate goal is to develop this method, delving into multistep synthesis planning for more intricate chemical synthesis scenarios.
Acknowledgments
L.Y. and W.G. thank the support from Tuantuan during the project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00737.
The NAG2G is available at https://github.com/dptech-corp/NAG2G. Experimental results related to NAG2G’s performance, auxiliary ablation studies, and prediction examples (PDF)
Author Contributions
∥ W.G. and Z.W. contributed equally to this work.
The authors declare no competing financial interest.
Footnotes
Here, we consider node-wise graph features, such as node degrees. Pair-wise graph features, such as the shortest path, will consume significantly more memory.
Supplementary Material
References
- Corey E. J.; Cheng X.-M.. The Logic of Chemical Synthesis; John Wiley & Sons: Nashville, TN, 1995. [Google Scholar]
- Zhong Z.; Song J.; Feng Z.; Liu T.; Jia L.; Yao S.; Hou T.; Song M. Recent advances in deep learning for retrosynthesis. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2024, 14, e1694 10.1002/wcms.1694. [DOI] [Google Scholar]
- Grzybowski B. A.; Szymkuć S.; Gajewska E. P.; Molga K.; Dittwald P.; Wołos A.; Klucznik T. Chematica: a story of computer code that started to think like a chemist. Chem 2018, 4, 390–398. 10.1016/j.chempr.2018.02.024. [DOI] [Google Scholar]
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 2017, 3, 1237–1245. 10.1021/acscentsci.7b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H.; Waller M. P. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chem.—Eur. J. 2017, 23, 5966–5971. 10.1002/chem.201605499. [DOI] [PubMed] [Google Scholar]
- Chen S.; Jung Y. Deep Retrosynthetic Reaction Prediction using Local Reactivity and Global Attention. JACS Au 2021, 1, 1612–1620. 10.1021/jacsau.1c00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H.; Li C.; Coley C.; Dai B.; Song L.. Retrosynthesis Prediction with Conditional Graph Logic Network. Advances in Neural Information Processing Systems, 2019; pp 8870–8880.
- Yan C.; Zhao P.; Lu C.; Yu Y.; Huang J. RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction. Biomolecules 2022, 12, 1325. 10.3390/biom12091325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koca J.; Kratochvil M.; Kvasnicka V.; Matyska L.; Pospichal J.. Synthon Model of Organic Chemistry and Synthesis Design; Springer Science & Business Media, 2012; Vol. 51. [Google Scholar]
- Shi C.; Xu M.; Guo H.; Zhang M.; Tang J.. A Graph to Graphs Framework for Retrosynthesis Prediction. Proceedings of the 37th International Conference on Machine Learning (ICML), 2020; pp 8818–8827.
- Yan C.; Ding Q.; Zhao P.; Zheng S.; Yang J.; Yu Y.; Huang J.. Retroxpert: Decompose retrosynthesis prediction like a chemist. Advances in Neural Information Processing Systems, 2020; Vol. 33, pp 11248–11258.
- Wang X.; Li Y.; Qiu J.; Chen G.; Liu H.; Liao B.; Hsieh C.-Y.; Yao X. RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions. Chem. Eng. J. 2021, 420, 129845. 10.1016/j.cej.2021.129845. [DOI] [Google Scholar]
- Somnath V. R.; Bunne C.; Coley C. W.; Krause A.; Barzilay R. Learning Graph Models for Template-Free Retrosynthesis. arXiv 2021, arXiv:2006.07038v2. 10.48550/arXiv.2006.07038. [DOI] [Google Scholar]
- Gao Z.; Tan C.; Wu L.; Li S. Z. SemiRetro: Semi-template framework boosts deep retrosynthesis prediction. arXiv 2022, arXiv:2202.08205v1. 10.48550/arXiv.2202.08205. [DOI] [Google Scholar]
- Chen Z.; Ayinde O. R.; Fuchs J. R.; Sun H.; Ning X. G2Retro as a two-step graph generative models for retrosynthesis prediction. Commun. Chem. 2023, 6, 102. 10.1038/s42004-023-00897-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W.; Yang Z.; Chen C. Y.-C. Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing. Nat. Commun. 2023, 14, 3009. 10.1038/s41467-023-38851-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Ramsundar B.; Kawthekar P.; Shi J.; Gomes J.; Luu Nguyen Q.; Ho S.; Sloane J.; Wender P.; Pande V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. 10.1021/acscentsci.7b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Rao J.; Zhang Z.; Xu J.; Yang Y. Predicting Retrosynthetic Reactions Using Self-Corrected Transformer Neural Networks. J. Chem. Inf. Model. 2020, 60, 47–55. 10.1021/acs.jcim.9b00949. [DOI] [PubMed] [Google Scholar]
- Kim E.; Lee D.; Kwon Y.; Park M. S.; Choi Y.-S. Valid, Plausible, and Diverse Retrosynthesis Using Tied Two-Way Transformers with Latent Variables. J. Chem. Inf. Model. 2021, 61, 123–133. 10.1021/acs.jcim.0c01074. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Karpov P.; Van Deursen R.; Godin G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. 10.1038/s41467-020-19266-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H.-R.; Wang J.; Liu Y.; Wu F.. Modeling Diverse Chemical Reactions for Single-step Retrosynthesis via Discrete Latent Variables. Proceedings of the 31st ACM International Conference on Information & Knowledge Management, 2022; pp 717–726.
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser L. u.; Polosukhin I.. Attention is All you Need. Advances in Neural Information Processing Systems, 2017.
- Tu Z.; Coley C. W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. J. Chem. Inf. Model. 2022, 62, 3503–3513. 10.1021/acs.jcim.2c00321. [DOI] [PubMed] [Google Scholar]
- Mao K.; Xiao X.; Xu T.; Rong Y.; Huang J.; Zhao P. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 2021, 457, 193–202. 10.1016/j.neucom.2021.06.037. [DOI] [Google Scholar]
- Seo S.-W.; Song Y. Y.; Yang J. Y.; Bae S.; Lee H.; Shin J.; Hwang S. J.; Yang E. GTA: Graph truncated attention for retrosynthesis. Proc. AAAI Conf. Artif. Intell. 2021, 35, 531–539. 10.1609/aaai.v35i1.16131. [DOI] [Google Scholar]
- Sacha M.; Błaż M.; Byrski P.; Dąbrowski-Tumański P.; Chromiński M.; Loska R.; Włodarczyk-Pruszyński P.; Jastrzebski S. Molecule Edit Graph Attention Network: Modeling Chemical Reactions as Sequences of Graph Edits. J. Chem. Inf. Model. 2021, 61, 3273–3284. 10.1021/acs.jcim.1c00537. [DOI] [PubMed] [Google Scholar]
- Liu J.; Yan C.; Yu Y.; Lu C.; Huang J.; Ou-Yang L.; Zhao P. MARS: A Motif-based Autoregressive Model for Retrosynthesis Prediction. arXiv 2022, arXiv:2209.13178v1. 10.48550/arXiv.2209.13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.; Xu Y.; Pei J.; Lai L. Automatic retrosynthetic route planning using template-free models. Chem. Sci. 2020, 11, 3355–3364. 10.1039/C9SC03666K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.Pushing the Limits of Interpretable End-to-End Retrosynthesis Transformer. arXiv [physics.chem-ph], 2022, http://arxiv.org/abs/2201.12475.
- Zhong Z.; Song J.; Feng Z.; Liu T.; Jia L.; Yao S.; Wu M.; Hou T.; Song M. Root-aligned SMILES: a tight representation for chemical reaction prediction. Chem. Sci. 2022, 13, 9023–9034. 10.1039/D2SC02763A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.; Yin S.; Shi L.; Zhou W.; Zhang Y. J. G2GT: Retrosynthesis Prediction with Graph-to-Graph Attention Neural Network and Self-Training. J. Chem. Inf. Model. 2023, 63, 1894–1905. 10.1021/acs.jcim.2c01302. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Lowe D. M.; Sayle R. A.; Tarselli M. A.; Landrum G. A. Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem. 2016, 59, 4385–4402. 10.1021/acs.jmedchem.6b00153. [DOI] [PubMed] [Google Scholar]
- Ying C.; Cai T.; Luo S.; Zheng S.; Ke G.; He D.; Shen Y.; Liu T.-Y.. Do Transformers Really Perform Bad for Graph Representation?. 35th Conference on Neural Information Processing Systems, 2021.
- Zhou G.; Gao Z.; Ding Q.; Zheng H.; Xu H.; Wei Z.; Zhang L.; Ke G.. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. The Eleventh International Conference on Learning Representations, 2023.
- Lu S.; Gao Z.; He D.; Zhang L.; Ke G. Highly Accurate Quantum Chemical Property Prediction with Uni-Mol+. arXiv 2023, arXiv:2303.16982v2. 10.48550/arXiv.2303.16982. [DOI] [Google Scholar]
- Landrum G.RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling, 2013; Vol. 8, p 31.
- Kingma D. P.; Ba J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:1412.6980v9. 10.48550/arXiv.1412.6980. [DOI] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application. J. Chem. Inf. Model. 2019, 59, 2529–2537. 10.1021/acs.jcim.9b00286. [DOI] [PubMed] [Google Scholar]
- Seidl P.; Renz P.; Dyubankova N.; Neves P.; Verhoeven J.; Wegner J. K.; Segler M.; Hochreiter S.; Klambauer G. Improving Few- and Zero-Shot Reaction Template Prediction Using Modern Hopfield Networks. J. Chem. Inf. Model. 2022, 62, 2111–2120. 10.1021/acs.jcim.1c01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Shen T.; Jaakkola T. S.; Barzilay R. Learning to Make Generalizable and Diverse Predictions for Retrosynthesis. arXiv 2019, arXiv:1910.09688v1. 10.48550/arXiv.1910.09688. [DOI] [Google Scholar]
- Sun R.; Dai H.; Li L.; Kearnes S.; Dai B. Energy-based View of Retrosynthesis. arXiv 2021, arXiv:2007.13437v2. 10.48550/arXiv.2007.13437. [DOI] [Google Scholar]
- Hammond J.; Leister-Tebbe H.; Gardner A.; Abreu P.; Bao W.; Wisemandle W.; Baniecki M.; Hendrick V. M.; Damle B.; Simón-Campos A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 2022, 386, 1397–1408. 10.1056/nejmoa2118542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravez J. C.; Iyer K. S.; Kavthe R. D.; Kincaid J. R.; Lipshutz B. H. A 1-pot synthesis of the SARS-CoV-2 Mpro Inhibitor Nirmatrelvir, the key ingredient in Paxlovid. Org. Lett. 2022, 24, 9049–9053. 10.1021/acs.orglett.2c03683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Finlay M. R. V.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; et al. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014, 57, 8249–8267. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Hett R.; Stare R.; Helquist P. Enantioselective synthesis of salmeterol via asymmetric borane reduction. Tetrahedron Lett. 1994, 35, 9375–9378. 10.1016/S0040-4039(00)78546-7. [DOI] [Google Scholar]
- Guo Z.-L.; Deng Y.-Q.; Zhong S.; Lu G. Enantioselective synthesis of (R)-salmeterol employing an asymmetric Henry reaction as the key step. Tetrahedron: Asymmetry 2011, 22, 1395–1399. 10.1016/j.tetasy.2011.08.008. [DOI] [Google Scholar]
- Ponomaryov Y.; Krasikova V.; Lebedev A.; Chernyak D.; Varacheva L.; Chernobroviy A. Scalable and green process for the synthesis of anticancer drug lenalidomide. Chem. Heterocycl. Compd. 2015, 51, 133–138. 10.1007/s10593-015-1670-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.