

Abstract
INTRODUCTION
Genome‐wide association studies link susceptibility to late‐onset Alzheimer's disease (LOAD) with EphA1. Sequencing identified a non‐synonymous substitution P460L as a LOAD risk variant. Other Ephs regulate vascular permeability and immune cell recruitment. We hypothesized that P460L dysregulates EphA1 receptor activity and impacts neuroinflammation.
METHODS
EphA1/P460L receptor activity was assayed in isogenic Human Embryonic Kidney (HEK) cells. Soluble EphA1/P460L (sEphA1/sP460L) reverse signaling in brain endothelial cells was assessed by T‐cell recruitment and barrier function assays.
RESULTS
EphA1 and P460L were expressed in HEK cells, but membrane and soluble P460L were significantly reduced. Ligand engagement induced Y781 phosphorylation of EphA1 but not P460L. sEphA1 primed brain endothelial cells for increased T‐cell recruitment; however, sP460L was less effective. sEphA1 decreased the integrity of the brain endothelial barrier, while sP460L had no effect.
DISCUSSION
These findings suggest that P460L alters EphA1‐dependent forward and reverse signaling, which may impact blood‐brain barrier function in LOAD.
Highlights
EphA1‐dependent reverse signaling controls recruitment of T cells by brain endothelial cells.
EphA1‐dependent reverse signaling remodels brain endothelial cell contacts.
LOAD‐associated P460L variant of EphA1 shows reduced membrane expression and reduced ligand responses.
LOAD‐associated P460L variant of EphA1 fails to reverse signal to brain endothelial cells.
Keywords: Alzheimer's disease, blood‐brain barrier, EphA1, late‐onset Alzheimer's disease, T‐cell recruitment
1. INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by inexorable cognitive decline. Neuropathological hallmarks include aggregation of cytotoxic amyloid beta (Aβ) oligomers and intracellular accumulation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein. 1 Late‐onset AD (LOAD) is the most common cause of dementia, with age considered the biggest risk factor. 2 Analysis of post mortem brains indicates that infiltration of peripheral immune cells into the brain parenchyma is also characteristic of AD neuropathology. 3 , 4 This loss of blood‐brain barrier (BBB) integrity is primarily considered to occur through age‐related processes or as a secondary consequence of Aβ deposition. 5
While age remains the biggest risk factor in the development of LOAD, heritability estimates stand at 60% to 80%. 6 Early genome‐wide association studies of Caucasians identified a member of the Eph receptor tyrosine kinase family, EphA1, as a susceptibility locus for LOAD. 7 , 8 Subsequent studies using additional datasets showed the erythropoietin EphA1 locus to be extremely complex. All single‐nucleotide polymorphisms (SNPs) with genome‐wide significance are non‐coding, lying either within intron regions of EPHA1 itself or within its neighboring antisense gene EPHA1. Further analyses of individual SNPs will be required to identify those that produce the AD‐association signal and their contribution to disease pathology.
A rare coding variant of EphA1, rs20217856, identified through targeted sequencing, segregated with LOAD in an extended Caribbean Hispanic family, supporting its significant association with the disease (p = 2.6 × 10−3). 9 This non‐synonymous variant encodes a proline to leucine substitution at position 460, with P460 being highly conserved in EphA1 across species. 10 Dissecting the structure and function of the EphA1 protein is of fundamental importance for understanding the impact of EphA1‐associated protein‐coding mutations, such as P460L.
Eph receptors and their surface‐associated ligands, ephrins, control cell–cell contacts and cell migration during developmental morphogenesis, organogenesis, pattern formation, and cell fate determination. 11 A characteristic of Eph–ephrin interactions is cell contact‐dependent, bidirectional signaling in both the Eph‐bearing (“forward signaling”) and ephrin‐bearing (“reverse signaling”) cell. While EphA1 was the first member of the transmembrane Eph family of receptor tyrosine kinases (RTKs) to be discovered, 10 it remains the most incompletely characterized Eph family member, with some properties inferred from its closest homolog, EphA2. 12 P460L is located in the second fibronectin type III repeat (FNIII‐2) of EphA1, which is outside of the conventional N‐terminal ligand binding domain required for detecting ligand expressed on opposing cells in trans. However, trans clustering of EphA2 by ephrinA5 induces a structural rearrangement in the membrane proximal FNIII‐2 domain. Moreover, the FNIII‐2 in EphA2 interfaces with cis‐expressed ephrinA ligands, 13 and cis‐expressed ephrinA5 ligand binding to the FNIII‐2 domain in EphA3 decreases its sensitivity to trans signaling. 14 These findings suggest that the P460L protein‐coding missense in the FNIII‐2 domain may impact EphA1 receptor structure and/or function.
In this study, the impact of P460L on the subcellular localization and activation of the EphA1 receptor using in silico modeling and cell‐based assays was determined. Eph–ephrin interactions have been shown to regulate tight junctions in brain endothelial cells 15 and BBB integrity, 16 as well as immune cell recruitment to inflamed tissues. 17 , 18 , 19 To determine the role of P460L in the context of neuroinflammation associated with AD, we determined the impact of P460L on EphA1‐dependent regulation of BBB integrity and its ability to recruit T cells.
RESEARCH IN CONTEXT
Systematic review: The link between the EphA1 locus and late‐onset Alzheimer's disease (LOAD) stimulated interest in understanding its role in the disease. A rare coding variant, P460L, which segregates with LOAD, provides a comparator to dissect the role of EphA1 in disease.
Interpretation: We discovered that P460L localized poorly to cell membranes and failed to internalize or undergo increased Y781 phosphorylation following ligand treatment. Soluble P460L levels released from cells are low and fail to induce reverse signaling in brain endothelial cells even at high doses. In contrast, soluble EphA1 stimulates reverse signaling in brain endothelial cells, causing T‐cell recruitment and remodeling of endothelial cell–cell contacts.
Future directions: Future studies will focus on determining the role of P460L in T‐cell biology to assess its impact on forward signaling and whether this promotes T‐cell transmigration across the BBB. This will inform whether targeting P460L activity has therapeutic potential for treating LOAD.
2. METHODS
2.1. Generation of expression constructs for EphA1, P460L and sP460L, and isogenic HEK293 Flp‐In cells
EphA1 was amplified from the pDNOR223‐EphA1 plasmid purchased from Addgene using Herculase II fusion DNA polymerase (Agilent) and the following set of polymerase chain reaction primers (Eurofins Genomics) that introduced NheI and NotI restriction sites respectively: 5′AAAATGGAGCGGCGCTGGCCCCTGGGGCTA3’ (EphA1‐Forward) and 5′AAACCAGTCCTTGAATCCCTGAATACTGCAAAG3’ (EphA1‐Reverse).
P460L was amplified using overlap extension mutagenesis in conjunction with two mutagenic primers that introduced the P to L coding sequence at position 460 into the EphA1 sequence and the EphA1‐Forward and EphA1‐Reverse primers from above. The P460L encoding primers were 5′CTGAGACTGGTGAAGAAAGAATGAGGCAACTAGAGCTGACCTGG3’ (P460L forward) 5′CCAGGTCAGCTCTAGTTGCCTCATTCTTTCTTCACCAGTCTCAG3’ (P460L reverse), with the single codon mutation underlined.
The soluble P460L (sP460L) coding sequences were amplified using P460L as the template in conjunction with EphA1‐Forward and a new reverse primer (5′TATCTCTCCTCCAGTCAGGCCCCTGGACAC3’) that introduced an XhoI restriction site and deleted the transmembrane and intracellular coding sequences of EphA1.
The coding sequences for EphA1 and P460L were cleaved with NheI and NotI prior to ligation into a pcDNA5‐V5‐His expression vector. In contrast, sP460L was cleaved with NheI and XhoI prior to ligation into a modified pcDNA5 expression vector containing the coding sequences for human Fcϒ. All expression constructs were sequenced by Eurofins Genomics, which confirmed that their coding sequences were correct and that the P460L mutation had been introduced.
The Flp‐InTMHEK293 cell line was from Invitrogen and cells were transfected with EphA1, P460L, and sP460L pcDNA5‐expression constructs in combination with the correct dose of pOG44 Flp‐recombinase plasmid using fugene 6 (Roche). Subsequent selection of transfectants in hygromycin medium was performed according to the manufacturer's instructions (Invitrogen), and expression of epitope‐tagged EphA1 and P460L was confirmed by western blotting of lysates. Mycoplasma screening was performed routinely and was negative.
2.2. Activation of EphA1 and P460L Flp‐In 293 cell lines with soluble ephrinA1
HEK transfectants were seeded at a density of 0.5 × 106 cells per well in a six‐well plate (Greiner). After 24 h, cells were treated with either 2 μg/mL ephrinA1‐Fc chimera (sEphrinA1; R&D Systems) or 2 μg/mL human IgG1 (R&D Systems) as control in DMEM/10% foetal calf serum (FCS) at 37°C with 5% CO2 for up to 3 h. In some experiments, cells were pre‐incubated with 25 μM DAPT (Enzo Life Science) or 25 μM GM6001 (Merck) or equivalent volume of DMSO (Sigma) as vehicle control prior to activation. Media were collected and stored at −80°C prior to analysis of released EphA1 and P460L by enzyme‐linked immunosorbent assay (ELISA). Cells were either stained for EphA1 protein expression and analyzed by flow cytometry or lysed and EphA1 levels analyzed by western blot.
2.3. Western blotting for total EphA1 and P460L protein
Cells were washed with ice‐cold PBS and lysed for 30 min in 35 to 50 μL of lysis buffer containing 25 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 2% glycerol, and 1% Triton‐X100 and supplemented with proteinase inhibitor (ULTRA; Roche), 4 mM 1,10‐phenanthroline (Sigma), and 1 mM sodium orthovanadate (Sigma). Lysates were centrifuged at 250 × g for 5 min at 4°C to pellet cellular debris and the supernatant retained. Protein concentrations were determined using the Pierce BCA protein assay (Thermo Fisher Scientific).
Equal protein concentrations (10 to 30 μg) were mixed with Laemmli sample buffer, heat denatured, and loaded onto 4% to 10% gradient gels (Bio‐Rad) and resolved at 100 V. Gels were transferred to polyvinylidene difluoride (PVDF) membrane (Immobilon‐PSQ) at 75 V for 1 h at room temperature (RT) or 20 V overnight (O/N) at 4°C in a Bio‐Rad transfer cell. Membranes were blocked in 5% milk (in PBS‐0.01% Tween 20; PBS‐T) for 1 h at RT prior to O/N incubation with either mouse monoclonal anti‐N‐terminal human EphA1 (MAB 638, R & D; 1 in 400) or mouse monoclonal anti‐V5 (R960‐25, Invitrogen; 1 in 1000). Blots were washed and then incubated in 5% milk with anti‐mouse horseradish peroxidase conjugated secondary antibodies (715‐035‐150‐JIR, Stratech; 1 in 7000) detected with ECL chemiluminescent western blotting reagent (Pierce) using an X‐ray film and developer. Semiquantitative densitometry analysis was performed using the Gel EZ Doc Imager software from Bio‐Rad. Antibody to GAPDH (Thermo Fisher Scientific, MA5‐15738; 1 in 1000) was used to normalize for equal protein loading across the gel.
2.4. Analysis of ephrinA1 ligand–dependent phosphorylation of Y781 in EphA1‐ and P460L‐expressing HEK293 cells
Transfected HEK293 cells were seeded at 0.5 × 105 cells per well into a six‐well plate and grown to 80% confluency. Medium was removed and cells treated with either 2 μg/mL sEphrinA1 or 2 μg/mL IgG1 control in reduced serum Opti‐MEMTM minimal essential medium. Cells were washed with ice‐cold PBS and lysed in 200 μL 50 mM HEPES, 250 mM NaCl, 2 mM EDTA, 10% glycerol,10 mM NEM, 0.5% NP‐40 supplemented with phosphatase inhibitors (PhosSTOP, 04906837001, Sigma‐Aldrich) and protease inhibitors (cOmplete Tablets EDTA‐free, 04693159001, Sigma‐Aldrich), as well as 10 mM NaF, 200 mM sodium orthovanadate, 4 mM 1,10 phenanthroline, and 1 μM benzamidine. Protein concentrations were determined using the PierceTM BCA protein assay and 25 μg total protein loaded onto 8% SDS‐PAGE gels, followed by western blotting using nitrocellulose membranes. Membranes were blocked for 1 h in 5% milk in Tris buffered saline 0.01% Tween 20 (TBST), followed by incubation with anti p‐Y781 EphA1 antibody (Thermofisher, PA5‐64783, 1 in 330) in 1% milk in TBST O/N. Membranes were washed in Tris buffered saline (TBS) and incubated with goat anti‐rabbit horseradish peroxidase (HRP) conjugate secondary antibody (Proteintech, SA00001‐2 at 1:10,000) in 1% milk in TBS for 1 h at RT prior to washing with TBS and incubation with SuperSignal Chemiluminescence substrate (Supersignal West Pico; Thermo Fisher Scientific). Blots were developed using an iBright 1500 (Invitrogen). Membranes were reprobed with mouse anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) antibody to determine equal loading and developed as described above but using goat anti‐mouse HRP conjugate secondary antibody (Proteintech SA00001‐1 at 1:10,000).
Fold changes in p‐Y781 signals for EphA1 or P460L were determined after normalizing to GAPDH for equal loading using iBright analysis software. The EphrinA1‐treated samples were compared with the relevant IgG control. Results are expressed as fold change of p‐Y781 signal.
2.5. Flow cytometric analysis of cell surface EphA1, P460L, and ephrinA1
EphA1‐expressing Flp‐In 293 and hCMEC/D3 cells were detached using Accutase (Sigma); their viability was determined by trypan blue exclusion, and cells were collected by centrifugation at 300 g for 5 min at 4°C. Cells (1 × 106) were plated per well in a round‐bottom 96‐well plate (Greiner) and incubated with 1 μg/mL LIVE/DEAD Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific) and incubated at 4°C with mouse IgG2a anti‐human EphA1 (MAB 638, R&D Systems,1 in 400), mouse IgG1 anti‐human ephrinA1 (Clone B12, Santa Cruz; 1 in 400), or isotype controls diluted in PBS plus 2% FCS (FACS buffer). After 3 washes in FACS buffer, cells were incubated in Phycoerythrin (PE) labeled goat anti‐mouse secondary antibody (Poly‐4503, R&D Systems; 1 in 400). Each incubation was performed at 4°C for 30 min, and cells were collected by centrifugation at 300 × g for 5 min. Cells were resuspended in 200 μL FACS buffer and analyzed on a FACSCanto II machine using DIVA software and data analyzed using FlowJo version 10. UltraComp eBeads (Thermo Fisher Scientific) were used following manufacturer's protocols for compensation during flow cytometry analysis.
2.6. Imaging flow cytometric analysis of membrane and intracellular EphA1 and P460L
Live EphA1 and P460L variant‐expressing cells were stained for cell surface EphA1 as described above. EphA1 stained cells were incubated with 1 μg/mL LIVE/DEAD Fixable Near infrared Dead Cell Stain (Thermo Fisher Scientific) for 30 min at 4°C. Cells were fixed and permeabilized using the True‐Nuclear Transcription Factor Buffer Set diluted as per the manufacturer's instructions (424401, BioLegend). Cells were stained with the AF647 V5‐tag antibody (411098, Thermo Fisher Scientific) for 30 min at 4°C. Nuclei were stained with NucBlue Live ReadyProbes Reagent according to the manufacturer's protocol (R37605, ThermoFisher) for 30 min at 4°C. Following staining, cells were resuspended in 30 μL of FACS buffer and placed in separate 1.5‐mL Eppendorf tubes. Data acquisition was conducted at 60× magnification using an ImageStream X Mark II imaging flow cytometer. During acquisition, singlets were exported for data analysis to limit file size. Analysis was performed using IDEAS software. Controls for ImageStream analysis were created through single stains of each antibody and relevant isotypes on live cells and following incubation at 95°C for 5 min for LIVE/DEAD stain.
Membrane and cytosolic subcellular compartment masks were created in the IDEAS analysis program to allow accurate quantification of EphA1 expression and location. To create these masks, the membrane was defined by staining EphA1‐HEK293 cells with both the N‐terminal EphA1 (PE stain) and V5‐tagged AF647 antibodies. The EphA1 negative parental cell line allowed for the accurate gating of single, in‐focus, live, and circular HEK293 cells. Gates that distinguish N‐term EphA1‐positive and V5‐tag‐positive HEK293 cells were created using negative parental cells to set background fluorescence levels. To quantitatively define the membrane and cytosol masks, 20 concentric one‐pixel (0.1 μm2) masks were created on the brightfield channel and overlaid upon both AF647 V5‐tag (top) and N‐term EphA1 (bottom) antibody channels. This enabled the extraction of the median fluorescence intensity for each antibody in individual masks. The median fluorescence intensity in the individual masks was then converted to a percentage of total fluorescence intensity for each antibody and plotted against the mask number. A two fit component Lorentzian curve analysis was performed in Graphpad Prism to derive the first component peak width, which described the membrane‐associated fluorescence for each antibody. The range of the peak widths from both the extracellular and intracellular antibody were used to quantitatively define mask numbers that covered the membrane. The membrane mask was defined as the sum of the masks that covered the membrane. The cytosol mask was defined as the remainder of the unmasked cell. This was then visually confirmed by the dataset images.
2.7. Confocal analysis of membrane and intracellular EphA1 and P460L
EphA1‐expressing cells (1 × 105) were plated on poly‐L‐lysine precoated (100 μg/mL poly‐L‐lysine for 1 h) 18‐mm square glass coverslips and cultured overnight at 37°C in DMEM/10% FCS. Cells were stimulated in triplicate with 2 μg/mL sEphrinA1 or human IgG1 as control for up to 3 h, supernatant removed, and cells washed in PBS at 37°C and fixed in 3% paraformaldehyde for 15 min. Free aldehyde groups were quenched by incubating with 50 mM NH₄Cl for 10 min, and cells were permeabilized with 0.4% saponin for 10 min. Following fixation and permeabilization, non‐specific binding sites were blocked by incubating cells in 1% bovine serum albumin (BSA) in PBS for 1 h. All antibodies were diluted in PBS containing 0.4% saponin, 2% FBS, and 2% BSA and incubated for 1 h at RT. Cells were stained for EphA1 using mouse anti‐human N‐terminal EphA1 ( MAB 638, R & D; 1 in 100) or for the V5 tag using mouse anti‐V5 (R960‐2, Invitrogen; 1 in 100) and primary antibodies detected using AF594‐conjugated goat anti‐mouse IgG (A‐11005, Molecular Probes; 1 in 400). DAPI Vectashield mounting medium was used as a nuclear counterstain and slides were imaged using a ZEISS Apotome fluorescent microscope (running ZEN software) with a 63× oil immersion objective scanning at 488 and 543 nm or the ZEISS confocal microscope, as detailed.
Region‐of‐interest (ROI) analyses were conducted using ImageJ to determine the membrane or cytosolic membrane intensity of EphA1 cells. Using the Freehand Tool, a ROI was selected (ie, the membrane or cytosolic compartment) followed by Analyze > Plot Profile with the overall pixel intensity returned. The background immediately adjacent to the ROI was selected using the same freehand‐drawn shape. This intensity was then subtracted from the cell ROI intensity to correct for any differences in background staining between slides and experiments. At least five cells were analyzed per triplicate slide.
2.8. Purification of soluble P460L protein
HEK‐293 cells (3 × 106) were seeded in TripleFlasks 500 cm2 (Thermo Fisher Scientific Nunc) in 250‐mL Serum Free Media II (SFMII) (Thermo Fisher Scientific) and grown until confluent. Conditioned medium containing the ectodomain of human P460L variant of EphA1 (Met1‐Glu547) fused to human IgG1 (Pro100‐Lys330) to generate soluble P460L (sP460L) was collected and centrifuged at 250 × g, and the cell‐free supernatant was supplemented with protease inhibitor and stored at −80°C. Protein G magnetic beads (500 μL; 5 mg) were washed twice in 10 mL of TST buffer (50 mM Tris‐HCl pH 7.6, 150 mM NaCl, 0.05% Tween 20); beads were collected using a magnetic bead stand, and the supernatant was discarded. sP460L‐containing media (400 mL) were thawed O/N at 4°C, the pH adjusted to 7.0, and sterile‐filtered through 0.22 μM nitrocellulose filter membranes (Millipore). Washed protein G magnetic beads were added and incubated O/N at 4°C on a tube roller. Protein G magnetic beads with bound sP460L were collected using the magnetic stand and the beads washed twice with Tris‐saline buffer (50 mM Tris‐HCl pH 7.6, 150 mM NaCl). The sP460L was eluted by incubating beads with 500 μL 0.1 M glycine‐HCl pH 2.7 for 5 min, supernatant was collected and neutralized with 80 μL of 1 M Tris‐HCl pH 9.0, and elution was repeated five times. A PBS buffer exchange was performed on the highest concentrations of sP460L using PD SpinTrapTM G‐25 column as per the manufacturer's instructions (GE Healthcare). Protein concentrations were determined using the BCA assay (Pierce). sP460L was concentrated using Amicon centrifugal filter units with a molecular weight (MW) cut‐off of 3 kDa and quantitated by ELISA (see next section).
2.9. Role of ectodomain shedding in release of EphA1 and P460L
EphA1‐ and P460L‐expressing HEK 293 cells were pre‐incubated with 25 μM DAPT (Enzo Life Science) or 25 μM GM6001 (Merck) or equivalent volume of DMSO (Sigma) as vehicle control prior to activation with sEphrinA1 for 3 h at 37°C. Conditioned media were removed and centrifuged at 250 × g for 5 min at 4°C to pellet cellular debris. Soluble EphA1 was quantitated by ELISA (human EphA1 ELISA, RAB1437‐1KT, Merck) according to the manufacturer's instructions and its absorbance read at 450 nm, immediately, using a Clariostar microplate reader. The detection limit for EphA1 was 0.085 ng/mL.
2.10. L‐selectin‐expressing T cells
The MOLT‐3 T‐lymphoblast cell line (henceforth referred to as MOLT‐3 T cells), derived from the peripheral blood of a 19‐year‐old male suffering from acute lymphoblastic leukemia and stably transduced with an human immunodeficiency virus–based gag T‐cell receptor, and human L‐selectin has been described in full elsewhere. 20
2.11. Human brain endothelial cell line (hCMEC/D3)
The human cerebral microvascular endothelial cell (hCMEC/D3) line was derived from a surgical excision of the temporal lobe of an adult female with epilepsy and generously provided by Pierre‐Olivier Couraud. 21 The cell line was generated through immortalization of freshly isolated endothelial cells using sequential lentiviral hTERT and SV40 large T antigen transduction. Cells were delivered cryopreserved at passage 25. Cells were cultured in Endothelial Cell Growth Basal Medium‐2 (EBM−2) supplemented with 25% strength Microvascular endothelial (MV) kit (Lonza) and grown on collagen‐coated flasks (50 μg/mL bovine tail collagen in PBS [Sigma], 2 h at 37°C) (Nunc). Cells were subcultured using ReagentPack™ Subculture Reagents (Lonza) following 15 min pre‐incubation in PBS. Cells were used up to passage 35 when endothelial cell markers and functional properties of hCMECs/D3s are maintained. 21
2.12. Impact of EphA1 on T‐cell recruitment by BBB
2.12.1. Static adhesion assays
hCMEC/D3 cells (2 × 105 in 2 mL endothelial cell medium per well) were seeded in triplicate into 24‐well plates and cultured O/N. Cells were pre‐incubated in fresh medium containing 5 μg/mL human EphA1 Met1‐Glu547 fused to human Fcγ1 (sEphA1; R&D Systems), 5 μg/mL sP460L, or 5 μg/mL human IgG1 for 18 h. MOLT‐3 T cells were labeled with 2 μM carboxyfluorescein succinimidyl ester (CFSE) in PBS for 10 min at RT. Excess dye was removed by washing twice in PBS, and cells were resuspended 2 × 106 per milliliter in DMEM 1% FCS. Pre‐incubation media were removed from hCMEC/D3 cells, and 2 × 106 CFSE‐ labeled MOLT‐3 T‐cells in 1 mL DMEM 1% FCS were added per well and incubated for 60 min at 37°C. Non‐adherent T cells were removed by three washes with 1 mL PBS at 37°C and adherent T cells were fixed to D3 cells using 2% paraformaldehyde for 30 min at RT. Cells were imaged using phase contrast to check the integrity of the hCMEC/D3 monolayer and green fluorescence for CFSE‐labeled T cells using Etaluma Lumascope LS560 Inverted Microscope. Adherent T cells were counted in three separate fields of view in each well (total area counted per well: 3.37 mm2).
2.12.2. Flow assays
MOLT‐3 T‐cell interactions with hCMEC/D3s under flow conditions were assessed using a Bioflux 200 (Fluxion Biosciences Inc., CA, USA). Channels in a 24‐well Bioflux 200 plate (0 to 20 dynes/cm2) were coated with 20 μg/mL of fibronectin (FN) (0 to 20 dynes/cm2) for 1 h prior to two 15‐min washes in endothelial cell medium. hCMEC/D3 cells (4 × 106) were pulsed through the channels and allowed to statically adhere for 2 h, then endothelial cell medium was pulsed over the cells O/N. The confluency and alignment of hCMEC/D3 cells were visually inspected prior to stimulation and flow assays. hCMEC/D3 cells were then stimulated with medium only, sEphA1 (5 μg/mL), or control hIgG (5 μg/mL) for 16 h.
CFSE‐labeled MOLT‐3 T cells at 7.5 × 106 cells/mL in DMEM 1% FCS were flowed over hCMEC/D3 cells at 0.25 dynes/cm2 and imaged using time‐lapse fluorescence microscopy taking 60 images in over 7.55 s (one image every 0.126 s). This resulted in 60 separate Tagged Image Format File (TIFF) images assembled into 60 stacked files. These stacks were converted into audio video interleaved (AVI) files, creating a video file and unstacked using ImageJ for subsequent analysis using the cell tracking analysis mode of the Fluxion BioFlux 200 software. Rolling cells were distinguished from non‐interacting cells by a rapid decrease in velocity and movement over the EC surface in the direction of flow. Rolling analyses was conducted on all rolling cells, with their interaction distance tracked until the cells detached, firmly adhered, or left the field of view. Rolling velocities were calculated by the BioFlux 200 software automatically using cell tracking mode. The total contact time of all rolling MOLT‐3 T cells was established by the number of frames in the time‐lapse sequence the cells rolled for, multiplied by the time elapsed between each frame (each frame = 0.126 s). Firmly adhered cells were identified as those that had not made forward motion for at least 40 frames (ie, 5 s) and counted.
2.13. Impact of EphA1 on BBB integrity
The electrical conductivity of hCMEC/D3 cell monolayers was measured using a real‐time cell analyzer (xCELLigence RTCA SP machine, Agilent) and expressed as cell index (CI). hCMEC/D3 cells (7 × 105) were plated in 200 μL of endothelial cell medium in quadruplicate wells of a 96‐well E‐plate (5232376001, Aglient) and incubated inside a 5% CO2 incubator at 37°C. The conductivity (CI) was recorded every 15 min; and after 70 to 75 h, when D3 cells reached confluency, CI reached a plateau. Recording was paused, media removed and 200ul μL of pre‐warmed media containing 5 μg/mL sEphA1, 5 μg/mL sP460L, 5 μg/mL human IgG as control or equivalent volume of PBS as vehicle control. Recording was restarted and continued for up to 20 h.
The CI was normalized using the following equation in the RTCA software package:
where CIti = CI at a specific time point, and CInml_time = CI at the time point prior to the addition of reagents. This removes interwell variation in cell seeding attachment and growth. CI values rise with the increasing cellular coverage of the electrode due to attachment and proliferation of D3 cells. After reaching a plateau at confluency, decreases in CI reflect reduced electrical impedance of the D3 monolayer. The normalized CI is presented over the duration of the experiment.
2.14. Statistical analysis
Experiments were repeated at least three times, and data were combined from independent repeats. Statistical analyses were conducted using GraphPad Prism 6 (GraphPad Software, CA, USA).
3. RESULTS
3.1. In silico modeling of P460L
We first investigated where the P460L mutation localized in a model of a functional EphA receptor, which might give an indication of potential structural changes in the receptor. The human EphA1 protein sequence for the FNIII type repeats (domains 3 and 4) was uploaded to SWISS‐MODEL to produce a three‐dimensional (3D) model based on structures released in the Protein Data Bank (PDB) with similar sequences. The target sequence was threaded onto the top 7 (default setting) 3D models with the best matching sequences. Only three structures had enough sequence match with the target sequence with just over 40% identity. These were PDB entries 3FL7, 2X10, and 2X11 and represent structures for EphA2 crystallized under different conditions: 3FL7 was crystallized at pH 5.5, 2X10 was crystallized at pH 8, and 2X11 was co‐crystallized with a ligand at pH 5. Gross features of the three models showed a significant difference among them. The molecule comprised four domains in a long chain, with a hinge angle between domains 3 and 4 that was more acute in 3FL7 and 2X11, making a pronounced bend at that point. These two crystal structures were from crystals grown at low pH, 5 to 5.5, while the pH 8 structure, 2X10, was a much straighter molecule. None of the crystal structures showed dimerization.
The common FNIII type domains 3 and 4 in EphA2 match a similar FNIII domain in irisin, PDB entry 4LSD. The latter comprises four copies of a homodimer of FNIII domains, where the dimer of chains A and B was used to construct a dimer of EphA1 (Figure 1). Domain 4 of 2X10 (436 to 529) was matched with 4LSD chain A or irisin, while domain 4 of a copy of 2X10 was matched with 4LSD chain B or irisin. A different dimer could be constructed by matching domain 3 of 2X10 and its copy with 4LSD A/B, but it was discounted as it put the C‐termini of the dimer too far apart. The transmembrane helix homodimer structure of EphA1 was solved with nuclear magnetic resonance (NMR) spectroscopy, showing their N‐termini relatively close together (PDB entry 2K1K), favoring a close separation between the C‐termini of the EphA1 ectodomain dimer, as would be obtained by a dimer based on matching domains 4 of irisin, leading us to propose an EphA1 model shown in Figure 1.
FIGURE 1.
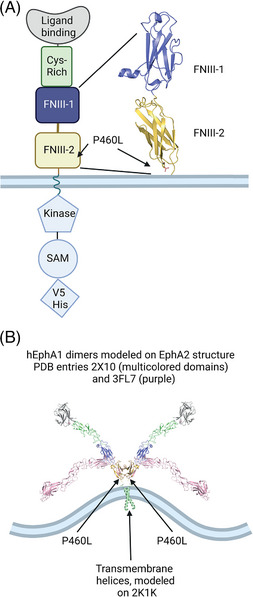
Domain structure and in silico modeling of EphA1. (A) Domain structure of epitope‐tagged EphA1 and location of P460L adjacent to outer plasma membrane. (B) In silico model of EphA1 dimer based on homologous structures of EphA2 (pH 8.0) and FN III domains in irisin as well as EphA1 transmembrane helix homodimer. Colors are shared with domain structure shown in panel (A) and location of P460L shown. The EphA1 structure in pink represents the bend between Domains 3 and 4 detected in other members of EphA family crystallized at low pH (5 to 5.5). Created with Biorender.
The position of EphA1 P460 is at the C‐terminus of second FNIII type immunoglobulin domain adjacent to the cell surface (Figure 1B). On the scale of the whole molecule, the size difference between Pro and Leu is not significant. Both residues are relatively small, and they have a neutral charge. Proline residues break helices, so the substitution by leucine may impact the secondary structure of the EphA1 ectodomain, its flexibility, dimerization, or the signaling capability of the full‐length receptor. 13 Furthermore, P460L may interact with the lipid bilayer, 22 which may cause ligand‐independent activation. Therefore, the impact of P460L on EphA1 receptor expression and responses to ligand engagement were determined by generating isogenic cell lines using the Flp‐In expression system, which enables direct comparisons between variants of EphA1 due to site‐specific integration of the coding sequences into the cellular genome.
3.2. P460L reduces expression of EphA1 at the cell surface
Full‐length EphA1 and the P460L variant were tagged at the carboxy terminus with a V5/His tag for imaging and biochemical analyses of receptor function (Figure 1A). Flp‐In HEK293 cells expressing EphA1 and P460L were analyzed by western blot, flow cytometry, and immunocytochemistry (ICC) for V5 signals to determine expression levels and cellular localization. To determine whether EphA1 was membrane localized, immunolabeling of the V5 tag was performed (Figure 2A, top panel). These data indicate that full‐length EphA1 is predominantly membrane localized with little intracellular staining evident (Figure 2A, top panels). In comparison with full‐length EphA1, the P460L variant was predominantly localized intracellularly (Figure 2A, bottom panels). Western blot analysis indicated that EphA1 and P460L were of the predicted molecular mass at around 100 kD (Figure 2B), although expression levels of P460L appeared to be reduced (Figure 2A,B).
FIGURE 2.
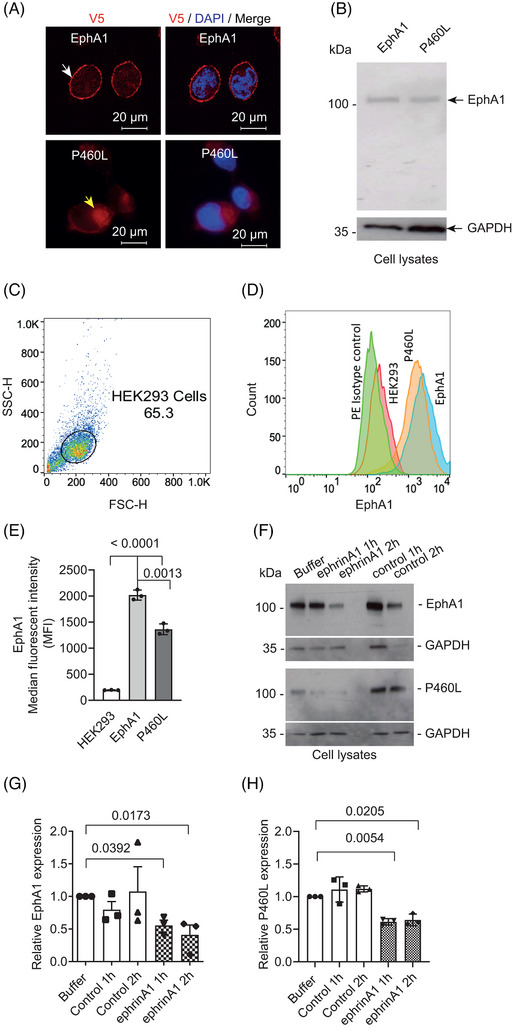
Impact of P460L on expression, subcellular localization, and ligand‐induced downregulation of EphA1 receptor. (A) Confocal images of EphA1‐ and P460L‐expressing isogenic HEK293 cell lines stained for cytotail (V5) and nucleus (DAPI). (B) Full‐length EphA1 and P460L variant detected by western blotting of solubilized HEK cells. (C–E) Flow cytometric analysis of cell surface EphA1 and P460L showing (C) gating of HEK cells; (D) representative overlay histograms of EphA1 (blue), P460L (orange), parental HEK cells (pink), and isotype control stained cells (green); and (E) bar charts showing mean fluorescence intensity ± SD, n = 3. (F–H) Impact of ligand engagement on receptor levels detected by western blotting showing (F) representative western blots of full‐length proteins in solubilized EphA1 and P460L cells with molecular weights in kDa, (G) total levels of EphA1, and (H) P460L variant following incubation with sEphrinA1, control (IgG1), or buffer alone for 1 and 2 h. Bands were normalized to the loading control GAPDH. Bar charts show mean levels of full‐length proteins compared to buffer alone ± SEM, n = 3. Statistical analysis used Student t test (E) and one‐way ANOVA (G, H).
To compare the expression levels of EphA1 and P460L at the cell surface, EphA1 transfectants and parental HEK293 live cells were stained using an EphA1 ectodomain‐specific antibody and analyzed by flow cytometry (Figure 2C). HEK293 cells do not express endogenous EphA1, whereas EphA1 protein was detected at the surface of both EphA1 and P460L transfectants (Figure 2D). However, a comparison of mean fluorescent intensities showed that P460L cells expressed significantly lower cell surface receptor than EphA1 cells (Figure 2E).
3.3. Impact of P460L on receptor‐mediated degradation following ligand engagement
To mimic cross‐linking by membrane‐expressed ligand, EphA1‐ and P460L‐expressing cells were incubated with its cognate ligand, sEphrinA1, presented as a homodimer in the form of ephrinA1‐Fc. HEK293 cells did not express endogenous ephrinA1 (Figure S1), thereby excluding potential inhibitory cis interactions with expressed EphA1 and P460L receptors. Human IgG1 was used as a control to ensure that the effects seen were not due to the Fc portion of the ligand fusion protein. Incubation of cells with human IgG (control) for up to 2 h had no effect on full‐length EphA1 or P460L, in comparison with buffer alone as assessed by quantitative immunoblot analysis of lysates for V5‐tagged C‐terminus (Figure 2F–H). EphrinA1 cross‐linking of cell surface EphA1 indicated that ligand engagement caused a decrease in full‐length EphA1 expression by ∼50% compared to buffer at 1 and 2 h (Figure 2G). P460L expression was also reduced by ∼40% following incubation with ephrinA1 compared to buffer (Figure 2H), indicating that the P460L variant does not prevent ligand‐induced degradation of full‐length EphA1 2 h following ligand activation.
The impact of P460L on the subcellular distribution of EphA1 was investigated further using imaging flow cytometry. Cells were stained for V5 cytotail, and single cells were analyzed for membrane and cytosolic signals as described in Figure S2. Levels of total EphA1 (Figure 3A), membrane EphA1 (Figure 3B), and cytosolic EphA1 (Figure 3C) were measured in both the EphA1 and P460L cells using the parental HEK293 cell line to control for non‐specific staining. Representative images for parental, EphA1, and P460L variant expressing HEK293 cells are shown for C‐terminal V5 tag (Figure 3D). EphA1 localized to the plasma membrane (Figure 3D, EphA1 panel), whereas the P460L variant mainly localized to the cytosol (Figure 3D, P460L panel). Total receptor expression as determined by V5 antibody did not differ in EphA1 and P460L cells (Figure 3E). However, the ratio of membrane to cytosolic V5 signal was much lower for P460L‐expressing HEK cells at 0.5 compared to 2.0 for EphA1 (Figure 3F). Staining of HEK293 cells using an ectodomain antibody for EphA1 also showed that membrane expression was higher in EphA1 than in P460L‐expressing cells (Figure 3G).
FIGURE 3.
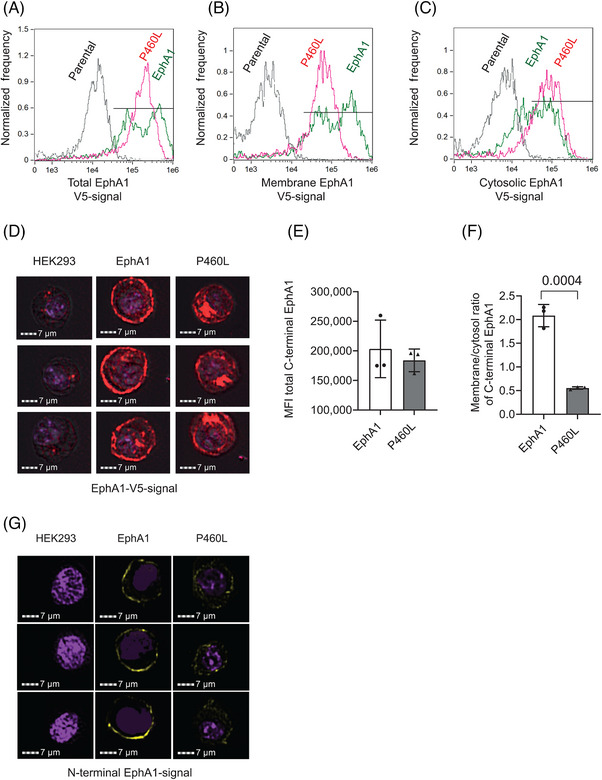
Subcellular localization of EphA1 receptor within EphA1‐ and P460L‐expressing HEK293 cells. EphA1, P460L, and parental HEK293 cell lines were stained for EphA1 receptor expression by staining for V5‐tagged cytotail or for N‐terminal ectodomain and subcellular localization of receptors analyzed by imaging flow cytometry. See Figure S3 for creation of membrane and cytosolic subcellular masks of live, single HEK293 cells. (A) Representative overlay histograms showing total level of receptor expression in parental (black), EphA1 (green), and P460L (pink) HEK293 cells. (B) Representative overlay histograms showing EphA1 receptor expression present within membrane of parental (black), EphA1 (green), and P460L (pink) HEK293 cells. (C) Representative overlay histograms showing EphA1 receptor expression present within cytosol of parental (black), EphA1 (green), and P460L (pink) HEK293 cells. (D) Representative images generated by ImageStream showing distribution of EphA1 receptor expression within parental (left), EphA1 (middle) and P460L (right) HEK293 cells. (E) Median fluorescence intensity of total V5‐tagged EphA1 receptor in EphA1 and P460L HEK293 cells. (F) Ratio of receptor expression in the membrane and cytosolic subcellular compartments of EphA1 and P460L HEK293 cells. (G) Representative confocal microscope images of N‐terminal EphA1 receptor expression in parental (left), EphA1 (middle), and P460L (right) HEK293 cells. Bars represents mean ± SD, n = 3. Statistical analysis used Student t test.
To study ligand‐mediated endocytosis of the receptor, HEK cells expressing full‐length EphA1 or P460L were incubated with sEphrinA1 for up to 3 h. Fixed and permeabilized cells were stained for V5, and membrane localization was analyzed by confocal microscopy. In the absence of ligand, EphA1 localized largely at the plasma membrane in EphA1‐expressing HEK cells (Figures 1, 4). Ligation by ephrinA1 stimulated a transient increase in membrane levels of EphA1 at 1 h followed by a marked reduction to 20% of the starting level in untreated cells after 2 and 3 h (Figure 4B). Loss of fluorescent signal for membrane EphA1 was accompanied by increased cytosolic V5 staining close to the nuclei of the EphA1‐expressing cells at the 2‐ and 3‐h time points (Figure 4A), although changes in cytosolic staining were not significant (Figure 4B). The decrease in total fluorescence for EphA1‐V5 signal at 2 and 3 h indicates that the receptor is degraded in response to ligand engagement (Figure 4C) and agrees with findings from the Western blot analysis at 2 h (Figure 2G, EphA1 panel treated with ephrinA1). The preferential reduction in membrane over cytosolic EphA1‐V5 led to a marked decrease in the membrane:cytosol ratio to < 0.2 at 3 h (Figure 4B).
FIGURE 4.
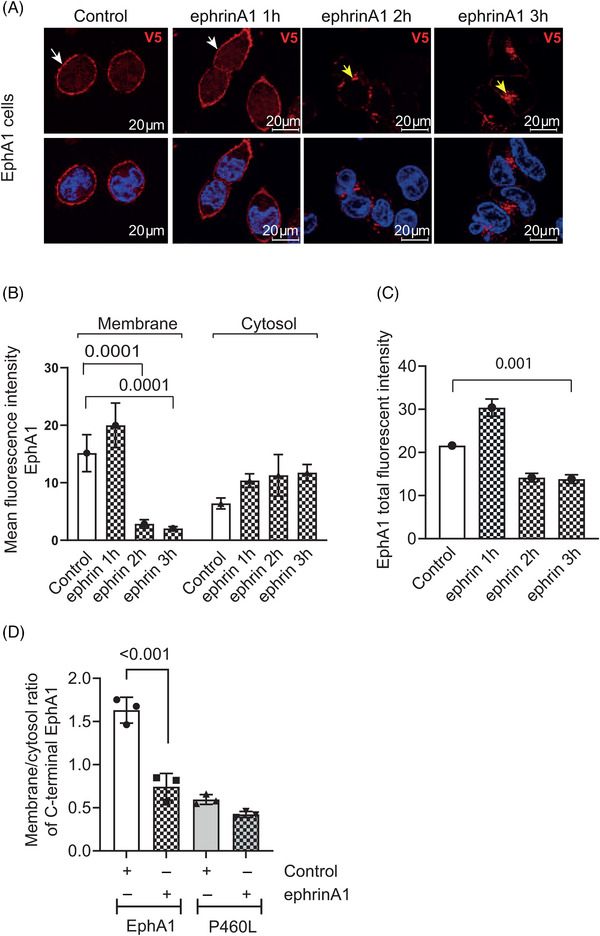
Receptor engagement stimulates internalization and degradation of EphA1 and P460L. EphA1‐ and P460L‐expressing HEK293 cells were incubated with sEphrinA1 or human IgG1 (control) and subcellular distribution of EphA1 receptor determined by immunohistochemical localization of C‐terminal V5 tag at 1 to 3 h (A–C) or by imaging flow cytometry at 3 h (D). Representative confocal microscope images of control and ephrin A1‐treated EphA1 (A) cells stained for V5 (red) and nucleus (blue). White arrows indicate membrane localization; yellow arrows indicate internalization of EphA1. Bar charts show (B) membrane and cytosolic staining in EphA1 and (C) total staining in EphA1‐expressing HEK cells. The ratio of membrane:cytosol EphA1 was determined by V5 staining and analysis by imaging flow cytometry. Bar charts show mean fluorescence intensity ± SEM in (B, C) and mean ratio of membrane:cytosol staining ± SD in (D) (n = 3). Statistical analysis used one‐way ANOVA in (B, C) and Student t test in (D). ***p < 0.001.
In contrast to clear membrane localization of EphA1, P460L staining using the V5 tag was less well defined by confocal microscopy (Figures 1A, 2A). Therefore, we used ImageStream analysis to compare ligand‐induced endocytosis of EphA1 and P460L receptors. As found by confocal microscopy, the ratio of membrane:cytosol V5 tag in EphA1‐expressing HEK293 cells was reduced from 1.63 ± 0.15 to 0.74 ± 0.15 (mean ± SD, n = 3) following incubation with sEphrinA1 for 3 h (Figure 4D). The membrane:cytosol ratio in P460L‐expressing HEK cells was slightly but not significantly reduced from 0.59 ± 0.06 to 0.42 ± 0.03 (mean ± SD, n = 3) following incubation with ligand for 3 h (Figure 4D). Evidence for ephrinA1‐induced degradation of full‐length P460L was evident by Western blotting at 2 h (Figure 2H). The fact that the membrane:cytosol ratio of P460L‐V5 was not significantly altered by ligand suggests that levels of P460L‐V5 at the membrane and in the cytosol were reduced to similar extents.
3.4. The role of proteases in EphA1 and P460L variant turnover at cell surface and inside cell
When EphA1‐ and P460L‐expressing HEK cells were compared directly by confocal microscopy and flow cytometry, the levels of membrane‐inserted P460L were consistently lower than levels of EphA1 in the absence of ligand (Figures 2, 3, 4). To determine whether the lower level of membrane P460L was due to accelerated release into the medium, sEphA1 and sP460L were quantitated by ELISA. In addition, we wanted to determine whether ligand‐induced loss of membrane expression in EphA1 HEK cells was due to proteolytic shedding.
Media were collected from EphA1‐ and P460L‐expressing cells incubated for 3 h in the absence or presence of sEphrinA1 and analyzed by ELISA. First, we ensured that exogenously added soluble ephrinA1 would not impede detection of sEphA1 released into the media. Standard curves generated in the absence and presence of sEphrinA1 did not interfere with sEphA1 detection (Figure S3). In the absence of ligand, sEphA1 and sP460L were detectable in the media, but the levels of sP460L were significantly lower (Figure 5A). After correcting for the reduced cell surface expression, the release of P460L was 50% lower than that of EphA1 (Figure 5B), indicating that under homeostatic conditions, the release of soluble P460L is impaired. Incubation with sEphrinA1 for 3 h did not stimulate the release of sEphA1 or sP460L (Figure 5C). Inclusion of a broad‐spectrum metalloprotease inhibitor GM6001 that targets matrix metalloproteinases and ADAMs reduced sEphA1 levels by around 25% in the absence or presence of ligand (Figure 5D), suggesting that the release of EphA1 under basal and ligand‐activating conditions is partially metalloproteinase‐dependent, although the data were not statistically significant. Soluble P460L levels were not influenced by GM6001 treatment and remained identical across treatment conditions. The potential involvement of γ‐secretase was studied, since it is involved in proteolytic processing of AD‐associated proteins, and it can cleave cell surface proteins without prior shedding of the ectodomain. 23 Inclusion of the γ‐secretase inhibitor DAPT had no effect on the release of receptor from EphA1 or P460L cells in the absence or presence of ligand (Figure 5D).
FIGURE 5.
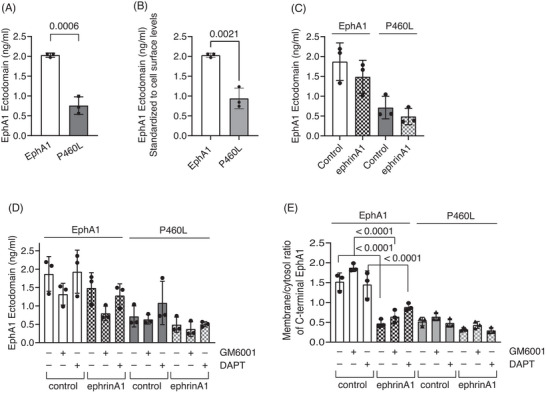
Role of proteolysis in membrane localization and release of soluble receptor by EphA1 and P460L HEK cells under basal and activating conditions. The levels of soluble EphA1 released from unactivated and activated EphA1‐ and P460L‐expressing HEK cells were determined using a human EphA1 ELISA kit that detected the N‐terminal ectodomain (ECD). (A) Total EphA1 ECD released under basal conditions by EphA1 and P460L HEK cells (p = 0.006). (B) Normalized EphA1 ECD released under basal conditions by EphA1 and P460L HEK cells (p =0 .0021). (C) EphA1 ECD released from EphA1‐ and P460L‐expressing HEK cells following incubation with 2 μg/mL sEphrinA1 (ephrinA1) or human IgG (control) for 2 h. (D) EphA1 ECD released from EphA1‐ and P460L‐expressing HEK cells following incubation with ephrinA1 or control in presence of 25 μM GM6001, 25 μM DAPT, or DMSO equivalent for 2 h. (E) Subcellular localization of receptor in EphA1 and P460L HEK cells following incubation with ephrinA1 or control in presence of 25 μM GM6001, 25 μM DAPT, or DMSO equivalent for 2 h by imaging flow cytometry (p = < .0001). Bar charts show mean and error bars indicate ± SD, n = 3.
Finally, we determined whether metalloproteinase or γ‐secretase activity regulated ligand‐induced downregulation of cell‐associated EphA1 and P460L seen by Western blotting and imaging (Figures 2, 3, 4). We used imaging flow cytometry to determine the subcellular localization of the V5 carboxy terminus in EphA1‐ and P460L‐expressing HEK cells following incubation with ephrinA1 ligand for 3 h in the absence and presence of the protease inhibitors. Inclusion of GM6001 increased the membrane:cytosol ratio of EphA1‐V5 tag from 1.63 ± 0.15 to 1.96 ± 0.11 in the absence of ligand, which is commensurate with the reduction in sEphA1 detected by ELISA (Figure 5D vs 5E). Incubation with ephrinA1 reduced the membrane:cytosol ratio of EphA1‐V5 tag to 0.74 ± 0.15 in the absence and 1.11 ± 0.13 in the presence of metalloproteinase inhibitor. Inclusion of γ‐secretase inhibitor had no effect on the membrane:cytosol EphA1 ratio in unstimulated or ephrinA1‐stimulated cells. Interestingly, the membrane:cytosol ratio for P460L in the absence of ligand at 0.59 ± 0.06 was similar to that in ligand‐activated EphA1 cells at 0.74 ± 0.15. Addition of ligand reduced the ratio slightly, but not significantly, to 0.42 ± 0.03. Inclusion of GM6001 or DAPT had no significant effect on membrane:cytosol ratios in P460L cells in the absence or presence of ligand (Figure 5E).
Together, these results suggest that soluble receptor is constitutively released from EphA1 and P460L cells. However, the release of P460L is significantly lower, which is due in part to reduced cell surface levels in P460L cells. Ligand‐induced downregulation of full‐length receptor from EphA1 and P460L cells seen by western blotting and confocal microscopy is not due to increased release of soluble receptor from the cell surface. Interestingly, evidence of constitutive metalloproteinase‐dependent shedding of EphA1 is presented; however, this was not the dominant pathway of EphA1 release. There was no evidence for metalloproteinases or γ‐secretase regulating changes in the subcellular distribution of EphA1 or P460L under either basal or ligand activating conditions.
3.5. Impaired ligand‐dependent tyrosine phosphorylation in P460L cells
We hypothesized that the reduced membrane and increased intracellular localization of P460L might be due to constitutive activation of receptor. The activation levels of both the EphA1 and P460L receptors were analyzed via quantitation of phosphotyrosine‐EphA1 levels under control and ligand‐stimulated conditions.
EphA1‐ and P460L‐expressing HEK293 cells were stimulated with sEphrinA1 for 5 min to activate the EphA1 receptor. Cell lysates were analyzed by SDS‐PAGE and western blotting, probing for total EphA1 receptor expression via V5‐tag staining and phosphorylation of Y781 in EphA1 and P460L (Figure 6). Control cell lysates from each time point were used to measure fold changes in phospho‐Y781 signals upon ephrinA1 stimulation.
FIGURE 6.
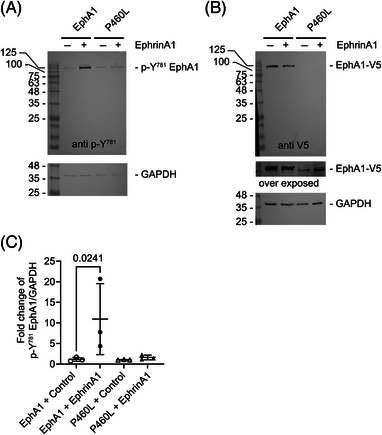
EphrinA1 treatment causes phosphorylation of Y781 on EphA1. EphA1 and P460L HEK transfectants were incubated with 2 μg/mL sEphrinA1 (EphrinA1) or human IgG (control). Cell lysates were run on the same gel to compare EphA1 and P460L side by side and the blot cut into two for staining. (A) Western blot analysis of cell lysates for p‐Y781 phosphorylation comparing EphA1 and P460L. (B) Western blot analysis of the same lysate samples shown in (A) for V5‐tagged EphA1 and P460L. Blots were overexposed to detect reduced levels of membrane P460‐V5 expressed in HEK cells. (C) Comparison of p‐Y781/GAPDH levels in response to control IgG or EphrinA1 treatment in EphA1‐ or P460L‐expressing cells. Statistical analysis was performed in GraphPad Prism using ANOVA and Sidak's multiple‐comparisons test.
Staining for phospho‐Y781 revealed a single band at 120 kD, detectable in both EphA1 and P460L HEK transfectants (Figure 6A) in the absence of ephrinA1 stimulation, which corresponded to the single species of full‐length EphA1 and P460L receptors detected by staining for V5 tag (Figure 6B). The reduced V5 signal for P460L compared to EphA1 may relate to reduced cell surface expression reported in Section 3.2. EphrinA1 stimulation for 5 min significantly increased phospho‐Y781 EphA1 signal when compared to the 5 min IgG control treatment in EphA1 cells (Figure 6C). This increase was not seen for the P460L receptor–expressing cells, where no significant changes in phospho‐Y781 signal were seen at 5 min of ephrinA1 stimulation (Figure 6C).
3.6. Impact of P460L variant on T‐cell recruitment by BBB
Interactions between EphA1 and ephrinA1 have been shown to regulate T‐cell adhesive interactions with specialized vascular endothelia outside the central nervous system (CNS). 17 We hypothesized that EphA1‐ephrinA1 regulated T‐cell interactions with specialized vascular endothelial cells that control the BBB. Using human cerebral microvascular endothelial cells (hCMEC/D3) as an in vitro model, 24 we first determined cell surface expression of EphA1 and ephrinA1 and found that hCMEC/D3 cells expressed ephrinA1 but not EphA1 (data not shown). Since hCMEC/D3 cells constitutively express a ligand for EphA1, we used soluble EphA1 to engage ephrinA1 on hCMEC/D3 cells prior to determining the impact on the binding of T cells. MOLT‐3 T cells co‐expressing the adhesion receptor LFA‐1 25 were transduced with a rolling receptor (human L‐selectin 20 ) and used in these studies. Using a previously described static adhesion assay, 26 pretreatment of hCMEC/D3 cells with sEphA1 for 18 h increased adhesion of T cells by 2.4‐fold from 197 ±38 to 470 ± 68 cells/mm2 (mean ± SE n = 5, Figure 7A). Interestingly, sP460L stimulated T‐cell adhesion to hCMEC/D3 cells by 2.0‐fold to 404 ± 6 cells/mm2 (mean ± SE, n = 3), although the increase was not statistically significant. These findings suggest that both sEphA1 and sP460L stimulate BBB endothelial cells to increase T‐cell recruitment under static conditions.
FIGURE 7.
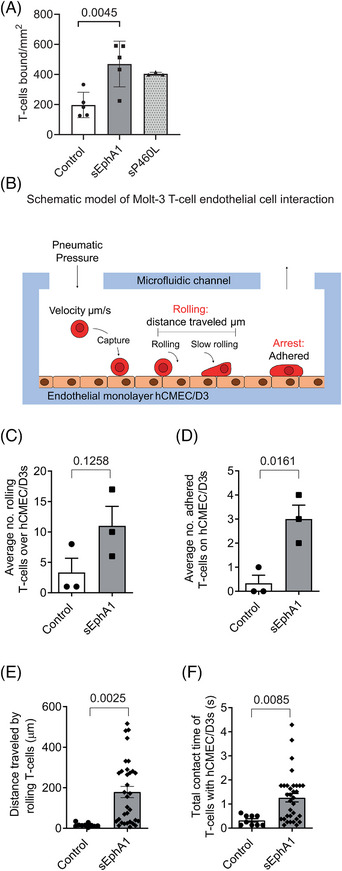
Impact of soluble EphA1 and P460L on T‐cell recruitment by blood‐brain barrier (BBB) endothelial cells. (A) hCMEC/D3s were pretreated with 5 μg/mL EphA1‐Fc (sEphA1), 5 μg/mL P460L‐Fc (sP460L), or 5 μg/mL human IgG (control) for 18 h, incubated with CFSE‐labeled MOLT‐3 T cells for 1 h under non‐flow conditions and non‐interacting T cells removed by washing. Numbers of adherent T cells were enumerated by fluorescence microscopy. (B) Schematic of Bioflux 200 system used to measure T‐cell recruitment to human brain endothelial cells under flow conditions. Pneumatic pressure pushes T cells through the inlet well at a defined shear stress across endothelial cells attached to the bottom of the microfluidic channel, and images are collected. Rolling and different stages of T‐cell adhesion are assessed: rolling cells by number, velocity (μm/s), distance traveled (μm), and total interaction time of all rolling cells (seconds); adhered cells by number. (C–F) hCMEC/D3s were pretreated with 5 μg/mL EphA1‐Fc (sEphA1) or 5 μg/mL human IgG (control) for 18 h at 1 dyne/cm2. 7.5X105 CFSE‐labeled MOLT‐3 T cells were passed over hCMEC/D3s at 0.25 dynes/cm2. Rolling velocity (μm/s) and distance traveled (μm) were calculated by the Bioflux 200 software following manual identification of rolling cells using the cell tracking analysis mode. (C) Numbers of T cells rolling on sEphA1‐primed endothelial cells (p = 0.1258). (D) Numbers of T cells adhered to sEphA1‐primed endothelial cells (p = 0.0161). (E) Total distance traveled by rolling T cells (data points show individual cells; bars show average distance; p ≤ 0.0025). (F) Total contact time of rolling T cells (data points show individual cells; bars show average time; p ≤ 0.0085). Bar charts show means ± SD, n = 3.
To investigate this possibility further, an assay that more closely recapitulates blood flow through the vasculature was used to investigate T‐cell recruitment by sEphA1‐primed hCMEC/D3 cells. Using the Bioflux 200 system (Fluxion), microfluidic flow assays were used to assess the impact of sEphA1 priming of vascular endothelial cells on T‐cell binding to the BBB using hCMEC/D3 cells and MOLT‐3 T‐cells (see Figure 7B for diagram of Bioflux system). MOLT‐3 T cells were labeled with CFSE and flowed over sEphA1‐primed hCMEC/D3 cells under shear force conditions and interactions between T cells, and the underlying endothelial cells were recorded by time‐lapse photography. Individual fluorescently labeled T cells were categorized as rolling and adhered T cells (see Figure 7B for a schematic detailing the adhesion cascade steps assessed and Figure S4 for definitions of non‐interacting, rolling, and adhered T cells).
sEphA1 pretreatment of hCMEC/D3 cells increased the number of T cells that interacted with the endothelial cell layer either through rolling or firm adhesion. Not all rolling T cells became firmly adhered. By tracking the movement of individual T cells that interacted with endothelial cells in sequential time frames, we calculated the duration of interaction with endothelial cells and defined adherent cells not moving forward for at least 5 s. The number of rolling T cells increased threefold in comparison with control IgG–treated cells, although this was not statistically significant (Figure 7C). sEphA1 pretreatment had a greater effect on the number of firmly adhered cells resulting in a sixfold increase (Figure 7D). For T cells defined as rolling, they interacted over a longer distance or traveled further between transient interactions with the endothelial cells following EphA1 pretreatment (Figure 7E), resulting in an increased total contact time between T cells and BBB endothelial cells (Figure 7F). Representative videos showing the behavior of T cells interacting with control and sEphA1‐activated hCMEC/D3s are shown in Videos S1 and S2, respectively.
3.7. Impact of P460L variant on BBB integrity
Having found that exogenously added sEphA1 and sP460L proteins increased T‐cell recruitment by BBB endothelial cells, we next determined their impact on the integrity of BBB endothelial cells. The electrical impedance of endothelial cells grown on gold‐film electrodes in an xCelligence RTCA machine was measured and impedance before and after treatment with sEphA or sP460L was monitored. hCMEC/D3 cells were plated and grown in E‐plates until impedance stabilized at confluency (70 to 75 h, data not shown). Increasing concentrations of sEphA1 or vehicle control were added to hCMEC/D3 cells, and impedance was recorded every 15 min. The vehicle control showed a transient increase in impedance in the first 15 min, which reverted to pretreatment levels by 30 min and was maintained for up to 5 h. Doses of sEphA1 from 1 to 500 ng/mL showed transient increases in impedance similar to vehicle controls, likely due to buffer changes in the E‐plates. However, higher doses of sEphA1 showed a significant reduction in impedance (Figure 8A). Doses of 1 to 5 μg/mL sEphA1 induced a rapid reduction in impedance, which was detectable within 15 min. From 15 to 45 min, impedance increased but stabilized at significantly lower levels than those in unstimulated cells or cells treated with low doses of sEphA1, as shown by the comparison of impedance in all treated groups at 2.5 h (Figure 8B). Control IgG at 5 μg/mL gave similar results as vehicle control, showing that reduction in electrical impedance by EphA1 was not due to the Fc tail (Figure 8C). Having shown that exogenously added sEphA1 reduced electrical impedance of hCMEC/D3 monolayers, we compared the effect of sP460L (Figure 8C). Strikingly, sP460L did not reduce electrical impedance in hCMEC/D3 cells during the 5‐h period of study (Figure 8C,D).
FIGURE 8.
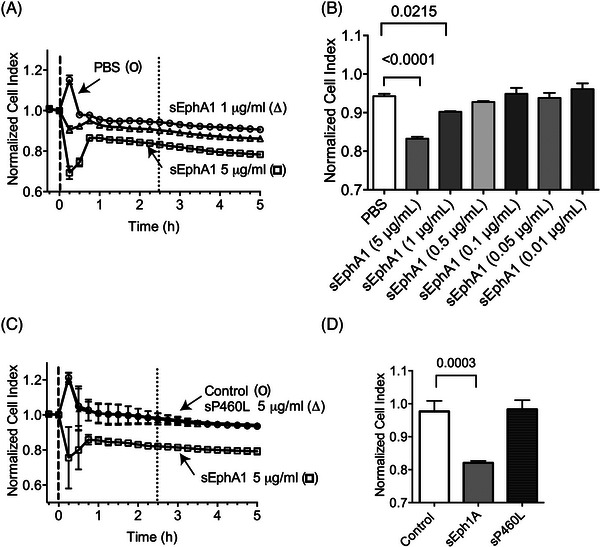
Impact of soluble EphA1 and P460L on integrity of blood‐brain barrier (BBB) endothelial cells. hCMEC/D3 cells were cultured in 96‐well E‐plates for 70 to 75 h until electrical impedance reached a plateau at cell confluency. Media were replaced with EphA1‐Fc (sEphA1), P460L‐Fc (sP460L), or human IgG (control) and electrical impedance recorded every 15 min for up to 5 h. Data were normalized to impedance values immediately prior to addition of stimuli and expressed as normalized cell index. (A, B) Normalized cell indices of endothelial cells treated with increasing doses of sEphA1 from 0.001 to 5 μg/mL or PBS vehicle (A) and for all treatments compared at 2.5 h (B). (C, D) Normalized cell indices for endothelial treated with single dose of 5 μg/mL sEphA1, sP460L, and control (C) and for all treatments compared at 2.5 h (D). Data show means ± SD, n = 3.
4. DISCUSSION
The link between the EphA1 locus and LOAD has stimulated interest in understanding its role in adult brain, as well as in the development and progression of AD. A rare coding variant of EphA1, P460L, allows comparison with its wildtype equivalent to understand P460L signaling and function.
In silico modeling of transmembrane EphA1 showed that P460L localized at the end of the first β sheet of FNIII‐2 domain, adjacent to the cell membrane. The substitution in FNIII‐2 does not impact charge and unlikely disrupts secondary structure, but membrane proximity of the mutation might impact receptor activation status. Our model agrees with a model of the FNIII‐2 domain proposed by Kim et al. 2021. 22 Isogenic cell lines expressing either EphA1 or the P460L variant were generated in HEK cells lacking endogenous EphA1 or ephrinA1 ligand, so differences seen are the result of the specific EphA1 variant that we overexpressed.
This is the first report showing a striking impact of P460L on the subcellular distribution of EphA1 with a 50% reduction in receptor levels at the cell surface under homeostatic conditions. Although we cannot exclude the possibility that P460L alters the extracellular conformation of EphA1, as proposed by our in silico modeling, the fact that antibodies against both the ectodomain and C‐terminal V5 tag detected similar reductions in cell surface levels of P460L suggests that this is not due to restricted antibody access. Total cell levels of P460L measured via the V5 tag reached EphA1 levels, suggesting that reduced membrane expression was unlikely due to accelerated intracellular degradation. Reduced cell surface expression of P460L was independent of membrane ectodomain shedding. Both EphA1 and P460L were released, but the levels of sP460L were lower than sEphA1, even after normalizing to cell surface receptor expression levels.
Following ligand engagement with soluble ephrinA1, EphA1 undergoes receptor internalization, causing increased intracellular, vesicular V5‐tag staining, and degradation, as was reported for other EphAs. 27 , 28 In contrast, P460L cell surface changes were less pronounced and not significant in response to ligand engagement, with signal intensities significantly lower than for EphA1. We excluded that receptor loss from the membrane was due to accelerated release, as sEphA1 and sP460L levels were stable in the presence of ligand and independent of metalloproteinase or γ‐secretase activity. Our cells lack endogenous ephrinA1, thereby excluding potential inhibitory cis interactions with expressed EphA1 and P460L receptors regulating responses to exogenous ephrinA1.
We next determined ligand‐dependent EphA1 and P460L Y781 phosphorylation. Phosphoproteomics has shown that Y781 in the EphA1 kinase domain is the main target of phosphorylation. 29 , 30 Therefore, we used a phospo‐Y781 EphA1 antibody to quantitate ligand‐dependent phosphorylation in EphA1 and P460L cells. EphrinA1 treatment caused a significant increase in phosphorylation of Y781 in EphA1 but not in P460L cells. These data indicate that ligand responses in P460L cells were delayed or impaired. This may be related to the markedly reduced levels of P460L receptor compared to EphA1 that we report here. Previous studies by Kim et al. showed that EphA1‐expressing HEK cells responded to ephrinA1 by increased tyrosine phosphorylation. 22 Tyrosine‐phosphorylation levels were significantly increased in their P460L cells under basal conditions and further increased in response to ligand stimulation, detected using an immunoprecipitation approach. 22 Our approach cannot detect tyrosine phosphorylation of EphA1 and P460L outside Y781.
Interestingly, Kim et al. 22 showed EphA1 cells spread on FN, with ephrinA1 treatment causing rounding. Their P460L cells rounded and failed to respond to ephrinA1, indicating that P460L was constitutively active. A kinase‐dead P460L variant restored cell spreading on FN to EphA1 levels, thereby rescuing the P460L phenotype and demonstrating that P460L is constitutively active. EphA1 receptor activation caused RhoA activation and Rac1 downregulation. 31 A relative increase in RhoA over Rac1 activity was linked to the inhibition of cell motility and inhibition of cell spreading. 32 , 33 , 34 However, in CD4 and CD8 T cells, EphA1 activation by ephrinA1 stimulated cell migration. 17 , 35 The outcome for cells expressing the P460L variant of EphA1 on cytoskeletal dynamics, cell–cell contacts, and cell migration are, therefore, likely to vary depending on cell type. It is difficult to predict how T cells in P460L carriers will behave due to a lack of knowledge of how forward signaling in T cells contributes to infiltration and neuroinflammation in LOAD compared to EphA1 T cells. In AD, it will be important to determine which cells express EphA1 and its cognate ligands and their roles in neuro‐glial cell interactions, as EphA1 knockout mice have normal CNS. 36
Soluble receptor is constitutively released from both EphA1 and P460L transfected cells in the absence of ligand engagement and is not blocked by a broad‐spectrum metalloproteinase inhibitor. However, further investigation is warranted, as ADAM12 binds EphA1 37 and is not efficiently inhibited by the metalloproteinase inhibitor. EphA1 does not undergo regulated intramembrane proteolysis, which is supported by previous results. 38 Serine proteases may be involved in EphA1 receptor processing as shown for stress‐induced neuroplasticity due to EphB2 cleavage by neurospin. 39
The demonstration that P460L affects forward signaling stimulated by ligand engagement in EphA1‐expressing HEK cells led us to speculate that P460L may impact reverse signaling in ephrinA1‐expressing cells. Eph receptors directly regulate T‐cell chemotaxis and interactions of monocytes with blood endothelial cells, 17 , 18 , 19 but the potential role of EphA1 in regulating peripheral immune cell interactions with the BBB in AD was unknown. A human brain microvascular endothelial cell line model of human BBB function was treated with recombinant sEphA1 and sP460L to stimulate reverse signaling. First, we determined the impact on T‐cell adhesion and found that pretreatment of brain endothelial cells with sEphA1 increased T‐cell adhesion. sP460L pretreatment also increased T‐cell adhesion; however, the effect was not significant. To further dissect EphA1 reverse signaling on T‐cell recruitment by BBB, additional assays were performed under flow conditions. EphA1 activation increased the number of rolling T cells and reduced their rolling velocity. Consequently, both the number of contacts and contact time between T cells and brain endothelial cells were increased. As found in static adhesion assays, the number of stably adherent T cells increased under flow. Further studies will be required to dissect the underlying mechanisms, but these data demonstrate, for the first time, reverse signaling by EphA1 in brain endothelial cells and its potential impact on T‐cell recruitment.
Next, we determined the impact of EphA1 reverse signaling on the integrity of the BBB using electrical impedance, which measures changes in cell–cell contacts in real time. EphA1 stimulated rapid and dose‐dependent transient reduction in basal levels of electrical impedance of confluent, growth‐arrested cerebral microvascular endothelial cells, which was detectable within 15 min and stabilized after 45 min at a lower level of electrical impedance. This suggested non‐reversible effects of sEphA1 on endothelial cell contacts. In marked contrast, sP460L had no effect on basal levels of electrical impedance of confluent endothelial cells, suggesting a reverse signaling defect of P460L. Interestingly, a recent analysis of EphA1, P460L, and their endogenous ligand, ephrin, in Drosophila demonstrated that mis‐ or overexpression of EphA1 or P460L variant receptors caused hyper arousal, reduced sleep, stimulated a stronger circadian rhythm, and increased neuron activity and excitability in the absence of memory loss. 40 These findings indicate that it is not necessarily forward signaling by mutant receptors that is of concern and that alterations in reverse signaling may be causal and altered by the P460L variant, as seen here.
We demonstrated such a scenario using an impedance assay in our BBB model. The fact that EphA1 induces changes to cell–cell contacts in the electrical impedance assay and increases T‐cell adhesion supports the hypothesis that EphA1 stimulates reverse signaling in BBB endothelium. The lack of effect of the LOAD‐associated P460L variant on BBB integrity is striking. The pathophysiological relevance of EphA1‐dependent changes in cell–cell contacts measured in the electrical impedance assay to BBB breakdown in LOAD and whether these are linked to T‐cell recruitment will require further studies. Models that express endogenous levels of P460L on T cells will be required to evaluate its impact on T‐cell transmigration through the BBB.
The recruitment of immune cells, particularly T cells, and loss of BBB integrity are normally considered in the context of pathology such as in multiple sclerosis or CAR T‐cell neurotoxicity. 41 The presence of T cells in post mortem AD brains has been linked to breakdown of the BBB. The mechanisms controlling T‐cell recruitment across the BBB and the contribution of T cells to the progression of AD are poorly understood. Our study, using cell‐based models, shows for the first time the potential role of EphA1 reverse signaling in controlling T‐cell recruitment as well as the integrity of the BBB and the differential impact of P460L. Further investigation using preclinical in vivo models will be required to fully understand the role of LOAD‐associated P460L variant of EphA1 in disease progression. This will require determining the impact of P460L forward signaling on T‐cell biology, including adhesion, homing, and activation, which we could not address here, and how this may be related to BBB breakdown.
AUTHOR CONTRIBUTIONS
Ann Ager and Vera Knäuper conceived the study. Helen A. Owens, Lauren E. Thorburn, Elisabeth Walsby, Owen R. Moon, Subuhi Sherwani, Caroline L. Tinsley, Louise Rogers, and Vera Knäuper performed experiments with assistance from Camilla Cerutti. Pierre Rizkallah performed in silico analyses. Ann Ager and Vera Knäuper supervised the study with critical input from Julie Williams and Anne J. Ridley. Helen A. Owens, Lauren E. Thorburn, Vera Knäuper, and Ann Ager wrote the manuscript with contributions from all authors.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Author disclosures are available in the Supporting information.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Professor Chris Pepper for use of the Biolfux 200, Maria Stack and Joanne Jones for technical assistance, and Dr. Catherine Hogan and Professor Ed Rainger for critical appraisal of the study.
H.A.O. was funded by Neuroscience and Mental Health Research Institute (NMHRI), Cardiff University and Sir Geraint Evans Cardiovascular Fund, Cardiff University (grant to A.A. and A.J.R.), L.E.T. by UK Dementia Research Institute, Cardiff University, O.R.M. by School of Medicine, Cardiff University, S.S. and C.L.T. by Eli Lilly (grant to J.W. and A.A.), and C.C. by School of Cellular and Molecular Medicine, University of Bristol. The Bioflux 200 was funded by the Leukemia Research Appeal for Wales, and iBright 1500 was funded by the Ser Cymru II programme (grant CU209 to V.K. and Manon Pritch, which was partially funded by Cardiff University and the European Regional Development Fund through the Welsh Government (Grant CU209 to V.K. and Manon Pritchard).
Owens HA, Thorburn LE, Walsby E, et al. Alzheimer's disease‐associated P460L variant of EphA1 dysregulates receptor activity and blood‐brain barrier function. Alzheimer's Dement. 2024;20:2016–2033. 10.1002/alz.13603
Helen Owens and Lauren Thorburn are first authors.
Vera Knäuper and Ann Ager are senior authors.
REFERENCES
- 1. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sloane PD, Zimmerman S, Suchindran C, et al. The public health impact of Alzheimer's disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health. 2002;23:213‐231. doi: 10.1146/annurev.publhealth.23.100901.140525 [DOI] [PubMed] [Google Scholar]
- 3. Togo T, Akiyama H, Iseki E, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. 2002;124:83‐92. [DOI] [PubMed] [Google Scholar]
- 4. Zenaro E, Pietronigro E, Della Bianca V, et al. Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med. 2015;21:880‐886. doi: 10.1038/nm.3913 [DOI] [PubMed] [Google Scholar]
- 5. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388‐405. doi: 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leonenko G, Baker E, Stevenson‐Hoare J, et al. Identifying individuals with high risk of Alzheimer's disease using polygenic risk scores. Nat Commun. 2021;12:4506. doi: 10.1038/s41467-021-24082-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429‐435. doi: 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43:436‐441. doi: 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol. 2015;78:487‐498. doi: 10.1002/ana.24466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717‐1720. doi: 10.1126/science.2825356 [DOI] [PubMed] [Google Scholar]
- 11. Klein R. Eph/ephrin signaling during development. Development. 2012;139:4105‐4109. doi: 10.1242/dev.074997 [DOI] [PubMed] [Google Scholar]
- 12. Miao H, Li DQ, Mukherjee A, et al. EphA2 mediates ligand‐dependent inhibition and ligand‐independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9‐20. doi: 10.1016/j.ccr.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph‐ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398‐402. doi: 10.1038/nsmb.1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carvalho RF, Beutler M, Marler KJ, et al. Silencing of EphA3 through a cis interaction with ephrinA5. Nat Neurosci. 2006;9:322‐330. doi: 10.1038/nn1655 [DOI] [PubMed] [Google Scholar]
- 15. Zhou N, Zhao WD, Liu DX, et al. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc Res. 2011;82:113‐121. doi: 10.1016/j.mvr.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 16. Malik VA, Di Benedetto B. The Blood‐brain barrier and the ephr/ephrin system: perspectives on a link between neurovascular and neuropsychiatric disorders. Front Mol Neurosci. 2018;11:127. doi: 10.3389/fnmol.2018.00127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aasheim HC, Delabie J, Finne EF. Ephrin‐A1 binding to CD4+ T lymphocytes stimulates migration and induces tyrosine phosphorylation of PYK2. Blood. 2005;105:2869‐2876. doi: 10.1182/blood-2004-08-2981 [DOI] [PubMed] [Google Scholar]
- 18. Jellinghaus S, Poitz DM, Ende G, et al. Ephrin‐A1/EphA4‐mediated adhesion of monocytes to endothelial cells. Biochim Biophys Acta. 2013;1833:2201‐2211. doi: 10.1016/j.bbamcr.2013.05.017 [DOI] [PubMed] [Google Scholar]
- 19. Pfaff D, Heroult M, Riedel M, et al. Involvement of endothelial ephrin‐B2 in adhesion and transmigration of EphB‐receptor‐expressing monocytes. J Cell Sci. 2008;121:3842‐3850. doi: 10.1242/jcs.030627 [DOI] [PubMed] [Google Scholar]
- 20. Mohammed RN, Wehenkel SC, Galkina EV, et al. ADAM17‐dependent proteolysis of L‐selectin promotes early clonal expansion of cytotoxic T cells. Sci Rep. 2019;9:5487. doi: 10.1038/s41598-019-41811-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weksler BB, Subileau EA, Perriere N, et al. Blood‐brain barrier‐specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872‐1874. doi: 10.1096/fj.04-3458fje [DOI] [PubMed] [Google Scholar]
- 22. Kim Y, Lasso G, Patel H, Vardarajan B, Santa‐Maria I, Lefort R†. Alzheimer's disease‐associated P460L mutation in ephrin receptor type A1 (EphA1) leads to dysregulated Rho‐GTPase signaling. bioRxiv. Published online June 18, 2021. doi: 10.1101/2021.06.17.448790 [DOI] [Google Scholar]
- 23. Laurent SA, Hoffmann FS, Kuhn PH, et al. Gamma‐secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6:7333. doi: 10.1038/ncomms8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood‐brain barrier. Fluids Barriers CNS. 2013;10:16. doi: 10.1186/2045-8118-10-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phongpradist R, Chittasupho C, Okonogi S, et al. LFA‐1 on leukemic cells as a target for therapy or drug delivery. Curr Pharm Des. 2010;16:2321‐2330. doi: 10.2174/138161210791920450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faveeuw C, Di Mauro ME, Price AA, Ager A. Roles of alpha(4) integrins/VCAM‐1 and LFA‐1/ICAM‐1 in the binding and transendothelial migration of T lymphocytes and T lymphoblasts across high endothelial venules. Int Immunol. 2000;12:241‐251. [DOI] [PubMed] [Google Scholar]
- 27. Beauchamp A, Lively MO, Mintz A, Gibo D, Wykosky J, Debinski W. EphrinA1 is released in three forms from cancer cells by matrix metalloproteases. Mol Cell Biol. 2012;32:3253‐3264. doi: 10.1128/MCB.06791-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walker‐Daniels J, Riese DJ 2nd, Kinch MS. c‐Cbl‐dependent EphA2 protein degradation is induced by ligand binding. Mol Cancer Res. 2002;1:79‐87. [PubMed] [Google Scholar]
- 29. Organ SL, Tong J, Taylor P, et al. Quantitative phospho‐proteomic profiling of hepatocyte growth factor (HGF)‐MET signaling in colorectal cancer. J Proteome Res. 2011;10:3200‐3211. doi: 10.1021/pr200238t [DOI] [PubMed] [Google Scholar]
- 30. Steen NV, Potze L, Giovannetti E, et al. Molecular mechanism underlying the pharmacological interactions of the protein kinase C‐beta inhibitor enzastaurin and erlotinib in non‐small cell lung cancer cells. Am J Cancer Res. 2017;7:816‐830. [PMC free article] [PubMed] [Google Scholar]
- 31. Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y. EphA1 interacts with integrin‐linked kinase and regulates cell morphology and motility. J Cell Sci. 2009;122:243‐255. doi: 10.1242/jcs.036467 [DOI] [PubMed] [Google Scholar]
- 32. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235‐1244. doi: 10.1083/jcb.144.6.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Etienne‐Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629‐635. doi: 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- 34. Taylor H, Campbell J, Nobes CD. Ephs and ephrins. Curr Biol. 2017;27:R90‐R95. doi: 10.1016/j.cub.2017.01.003 [DOI] [PubMed] [Google Scholar]
- 35. Hjorthaug HS, Aasheim HC. Ephrin‐A1 stimulates migration of CD8+CCR7+ T lymphocytes. Eur J Immunol. 2007;37:2326‐2336. doi: 10.1002/eji.200737111 [DOI] [PubMed] [Google Scholar]
- 36. Duffy SL, Coulthard MG, Spanevello MD, et al. Generation and characterization of EphA1 receptor tyrosine kinase reporter knockout mice. Genesis. 2008;46:553‐561. 10.1002/dvg.20434 [DOI] [PubMed] [Google Scholar]
- 37. Ieguchi K, Tomita T, Omori T, et al. ADAM12‐cleaved ephrin‐A1 contributes to lung metastasis. Oncogene. 2014;33:2179‐2190. doi: 10.1038/onc.2013.180 [DOI] [PubMed] [Google Scholar]
- 38. Merilahti JAM, Ojala VK, Knittle AM, Pulliainen AT, Elenius K. Genome‐wide screen of gamma‐secretase‐mediated intramembrane cleavage of receptor tyrosine kinases. Mol Biol Cell. 2017;28:3123‐3131. doi: 10.1091/mbc.E17-04-0261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Attwood BK, Bourgognon JM, Patel S, et al. Neuropsin cleaves EphB2 in the amygdala to control anxiety. Nature. 2011;473:372‐375. doi: 10.1038/nature09938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buhl E, Kim YA, Parsons T, et al. Effects of Eph/ephrin signalling and human Alzheimer's disease‐associated EphA1 on Drosophila behaviour and neurophysiology. Neurobiol Dis. 2022;170:105752. doi: 10.1016/j.nbd.2022.105752 [DOI] [PubMed] [Google Scholar]
- 41. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7:1404‐1419. doi: 10.1158/2159-8290.CD-17-0698 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information