

Abstract
Human age estimation from trace samples may give important leads early in a police investigation by contributing to the description of the perpetrator. Several molecular biomarkers are available for the estimation of chronological age, and currently, DNA methylation patterns are the most promising. In this study, a QIAGEN age protocol for age estimation was tested by five forensic genetic laboratories. The assay comprised bisulfite treatment of the extracted DNA, amplification of five CpG loci (in the genes of ELOVL2, C1orf132, TRIM59, KLF14, and FHL2), and sequencing of the amplicons using the PyroMark Q48 platform. Blood samples from 49 individuals with ages ranging from 18 to 64 years as well as negative and methylation controls were analyzed. An existing age estimation model was applied to display a mean absolute deviation of 3.62 years within the reference data set.
Key points
Age determination as an intelligence tool during investigations can be a powerful tool in forensic genetics.
In this study, five laboratories ran 49 samples and obtained a mean absolute deviation of 3.62 years.
Five markers were analyzed on a PyroMark Q48 platform.
Keywords: forensic genetics, age estimation, DNA methylation, pyrosequencing, trace sample
Introduction
The estimate of the chronological age of an unknown person who left a trace at the scene of a crime can display an important lead for ongoing police investigations. Alongside, externally visible traits, such as gender, pigmentary traits, and biogeographic ancestry, information on the age of an individual are useful to describe an alleged perpetrator and thus narrowing down the pool of suspects.
Several methods for molecular age estimation have been tested, and currently, the investigation of the methylation pattern at specific age-dependent methylated CpG loci (cytosine followed by guanine in a DNA sequence) is the most promising method [1–20]. Common techniques for methylation analysis rely on the use of sodium bisulfite (NaHSO3), which chemically converts un-methylated cytosines (C) into uracils, while methylated cytosines remain unaffected. In the subsequent PCR reaction, uracils are amplified as thymine (T) nucleotides, which results in a final sequence of methylated cytosines (displayed as cytosines) and unmethylated cytosines (displayed as thymines). Consequently, the methylation level of each CpG locus is represented by the C/T ratio at the particular position [21]. Several methods for quantitative measurements of these sites have been employed, including single-base extension analyzed by capillary electrophoresis [22, 23], microarrays [3, 6–9, 24], mass determination of transcribed PCR products using mass spectrometry [5, 10], and DNA sequencing, e.g. pyrosequencing [1, 2].
One of the most frequently used instrument for pyrosequencing, especially in the context of molecular age estimation, is the PyroMark Q48 (QIAGEN). The technique relies on a PCR amplification of the regions of interest using one biotinylated PCR primer. The biotin labels allow the immobilization of the amplicons to streptavidin-coated beads for subsequent pyrosequencing. Pyrosequencing is based on a cascade of enzymatic reactions [25]. First, the DNA polymerase incorporates one or more complementary nucleotides into the growing DNA strand, releasing a proportional number of pyrophosphate (PPi) molecules. ATP is generated from PPi by the ATP sulfurylase enzyme and activates the luciferase-mediated conversion of luciferin to oxyluciferin, which emits light captured in real time by the instrument. The C/T ratio at the CpG positions gets automatically calculated by the PyroMark Q48 Autoprep Software (QIAGEN) and reported as percent methylation [26–28].
In this work, we tested a CpG-based age estimation assay that has been converted into an assay suitable for commercial use. The age assay consists of five age-associated CpG sites (located in the genes ELOVL2, C1orf132, TRIM59, KLF14, and FHL2) individually analyzed and sequenced using the PyroMark Q48. The assay was originally developed by testing 41 CpG loci in blood samples from 300 individuals and selecting the five most informative CpG loci. Based on the reference data, the age-dependent methylation changes were modelled by applying multivariate linear regression (MLR). The prediction model was tested in an independent sample set of 120 individuals displaying a mean absolute deviation (MAD) between the predicted and chronological age of 3.9 years [1].
In this study, five forensic genetic laboratories conducted a collaborative study of the feasibility of the method and comparability of results among the laboratories by testing blood samples from a limited number of individuals (49) with ages ranging from 18 to 64 years.
Materials and methods
Samples and reagents
The work was performed as a collaboration between Palacky University, the Czech Republic; Laboratoire d’Hématologie Médico-Légale, France; Florida International University, USA; University of Cologne, Germany; and University of Copenhagen, Denmark. QIAGEN kindly provided each laboratory with reagents for bisulfite conversion, PCR amplification, and sequencing reagents together with methylated and unmethylated controls to each of the laboratories. The amount of reagents made available to the laboratories was limited so that ~1 000 tests could be performed in total. In all laboratories, peripheral blood samples from individuals with known age (18–64 years) were collected with written informed consent, and all samples were anonymized. The Human EpiTect Control DNA was either “high methylated” or “low methylated”. A “medium methylated” control was generated by mixing the two controls in equal amounts. Altogether, 49 individuals and three control samples were analyzed in triplicates, which resulted in a total of 960 sequencing runs and 192 CpG analyses. Each laboratory sequenced and analyzed between five and 15 samples in addition to the three methylation controls.
DNA extraction and Bisulfite conversion
DNA was extracted from the blood samples using either the DNeasy Blood & Tissue kit (QIAGEN) or the EZ1 DNA Investigator Kit (QIAGEN) according to the manufacturer’s instructions in each of the laboratories. DNA concentrations were measured using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). The EpiTect Fast DNA Bisulfite Kit (QIAGEN) was used for bisulfite conversion following the manufacturer’s protocol: Bisulfite Conversion of Unmethylated Cytosines in DNA [21].
Amplification and sequencing
Five different amplicons containing a CpG site of interest were amplified individually using 10 ng of bisulfite-converted DNA in 25-μL reactions in each of the five laboratories. The reaction mix contained 12.5 μL 2× PyroMark PCR Master Mix, 2.5 μL 10× of CoralLoad Concentrate together with variable volumes of both primers, RNase-free water, and sample. The concentrations of each set of PCR primers are shown in Supplementary Table S1. The PCR cycling programme consisted of an initial PCR activation step at 95°C for 15 min, followed by 50 cycles of 30 s at 95°C, 30 s at 48°C, and 30 s for 72°C. The annealing step at 48°C was ramped up to 52°C during cycle 1–10 and kept at 52°C from cycle 11. A final extension of 10 min at 72°C and a hold at 4°C was used. PCR products from selected samples (n = 22) were analyzed using a Qubit 2.0 (Thermo Fisher Scientific) and a 2100 Bioanalyzer (Agilent Technologies).
Sequencing on the PyroMark Q48 was performed according to the manufacturer’s instructions using predesigned assays (QIAGEN) in each of the five laboratories. Briefly, 10 μL of the amplicons, 3 μL of the PyroMark Q48 Magnetic Beads, and 2 μL sequencing primer were mixed and loaded onto the PyroMark Q48 disc before sequencing. The primers used for amplification and sequencing were kindly provided by (QIAGEN) (Supplementary Table S1). All samples were amplified and sequenced in triplicates.
Pyrosequencing analysis
The PyroMark Q48 software assessed the quality of each sequencing result and categorized individual sequencing runs as either (i) “Passed” for a result without complications, (ii) “Check” for a result with possible complications, which may include declining signal strength, a signal in a nucleotide cycle where no signal was expected, or a cytosine signal in the bisulfite control (an unmethylated cytosine nucleotide followed by an adenine or thymine nucleotide), or (iii) “Failed” for a result with likely errors, which may be too little signal or imbalanced signals. Each run was also visually inspected, and the result was either accepted or rejected. Furthermore, the reason for the PyroMark Q48 software categorization was documented. The results were accepted (i) if an unexpected peak was less than half the height of the expected peaks, (ii) if the bisulfite conversion control peaks (cytosines not followed by a guanine) were less than 5% of a single nucleotide signal intensity, and (iii) if the expected nucleotide sequences were identified despite low peak heights or declining signal strength.
The median value of the three replicates was used to estimate the age of the individual. For three samples, only two reliable results were available, and the averages of the two C/T ratios were used. The following age model was used [11]: Predicted age (years) = 3.268 477 847 518 17 + 0.465 445 549 010 653 × methC7-ELOVL2–0.355 450 171 437 202 × methC1-C1orf132 + 0.306 488 541 137 007 × methC7-TRIM59 + 0.832 684 435 238 792 × methC1-KLF14 + 0.237 081 243 617 191 × methC2-FHL2.
Statistical analysis
Calculations of standard deviation and coefficient of variance, as well as linear regression analyses, were performed in Excel.
Results
Analysis of the PCR products
Each of the five CpG loci were amplified in singleplex PCR reactions. Electropherograms from the 2100 Bioanalyzer revealed that multiple PCR products were generated by amplification of two of the five CpG loci. Four and two products with different sizes were observed for ELOVL2 and FHL2, respectively (data not shown). Nevertheless, as the nucleotide sequences obtained from the pyrosequencing analysis corresponded to the expected sequences, we concluded that the artificial amplification products did not interfere with the sequencing reaction.
Pyrosequencing analysis
Table 1 shows how the PyroMark Q48 software evaluated the 960 sequencing runs from samples and controls. The number of sequencing runs without complications (Passed category) ranged from 48% to 94% for the different loci. Most warnings for poor quality were observed for C1orf132 (52%) and FHL2 (34%). However, the coefficients of variance (CV) for the triplicate C/T ratios were relatively low (6% and 8%, respectively) for these loci, indicating that the assays performed reproducibly even though the software indicated poor sequencing quality. Overall, there was little to no correlation between the CV for the triplicate C/T ratios and the PyroMark Q48 software categorization (Table 1).
Table 1.
Assay quality.
| Categorya | ELOVL2 | C1orf132 | TRIM59 | KLF14 | FHL2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of observations | CV for triplicates | Number of observations | CV for triplicates | Number of observations | CV for triplicates | Number of observations | CV for triplicates | Number of observations | CV for triplicates | |
| Passed | 147 | 4% | 87 | 7% | 166 | 8% | 171 | 24% | 117 | 6% |
| Check | 17 | 8% | 66 | 4% | 9 | 28% | 4 | 17% | 33 | 12% |
| Failed | 18 | 8% | 29 | 7% | 7 | 29% | 7 | 46% | 28 | 11% |
Evaluation by the PyroMark Q48 software. CV: Coefficient of variance.
Each pyrogramme was analyzed by a trained analyst in each of the five laboratories, and the analyst either accepted or rejected the result. Despite the many sequencing runs with complications somewhere in the sequence, only three C/T ratios were not accepted. They were all observed in the KLF14 locus and were rejected because the peaks in the pyrogrammes were too low, and the sequences could not be recognized as a KLF14 sequence. For these three samples, average C/T ratios were calculated from the two approved replicates and used in the downstream analyses. For all other samples, the median C/T ratios of triplicates were used to estimate the age of the individual.
The “high”, “low”, and “medium” methylated Human EpiTect Control DNA were also analyzed with the age assay. Table 2 shows the median C/T ratios for the five CpG loci observed in the five laboratories. There were some differences between the laboratories.
Table 2.
C/T ratios for methylation control samples.
| Sample | Laboratory | ELOVL2 | C1orf132 | TRIM59 | KLF14 | FHL2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Median | SD | Median | SD | Median | SD | Median | SD | Median | SD | ||
| High methylated | 1 | 72 | 71 | 98 | 78 | 55 | |||||
| 2 | 80 | 78 | 88 | 76 | 75 | ||||||
| 3 | 80 | 85 | 90 | 80 | 56 | ||||||
| 4 | 85 | 43 | 91 | 82 | 67 | ||||||
| 5 | 73 | 4.86 | 73 | 14.34 | 88 | 3.69 | 77 | 2.15 | 38 | 12.51 | |
| Low methylated | 1 | 11 | 6 | 4 | 3 | 12 | |||||
| 2 | 6 | 5 | 4 | 3 | 4 | ||||||
| 3 | 10 | 1 | 6 | 2 | 5 | ||||||
| 4 | 9 | 5 | 4 | 2 | 3 | ||||||
| 5 | 12 | 2.06 | 4 | 1.72 | 5 | 0.80 | 3 | 0.49 | 7 | 3.19 | |
| 1:1 of high and low methylated | 1 | 38 | 26 | 49 | 49 | 33 | |||||
| 2 | 34 | 40 | 36 | 43 | 25 | ||||||
| 3 | 34 | 42 | 36 | 41 | 22 | ||||||
| 4 | 38 | 40 | 40 | 43 | 35 | ||||||
| 5 | 37 | 1.83 | 36 | 5.74 | 42 | 4.80 | 41 | 2.94 | 18 | 6.47 | |
SD: Standard deviation for all laboratories collected.
Age estimation
Figure 1 depicts the methylation levels of each of the five loci compared with the chronological age of the individuals. The best linear correlation between methylation levels and age was observed for ELOVL2, KLF14, and FHL2, while the C1orf132 locus did not appear to be correlated with age in this study (R2 = 0.1314).
Figure 1.
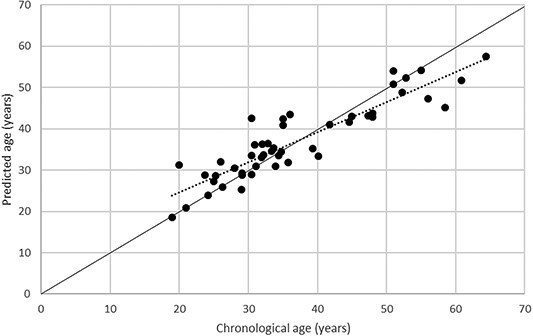
Chronological age of the participants plotted against the predicted age estimated using the age assay. Solid line: the perfect correlation between estimated and chronological age according to the MLR model [11]; dotted line: the line with the best fit (R2 =0.8234) between the predicted and the chronological age in the dataset.
The solid line indicates the perfect correlation between estimated and chronological age according to the MLR model [11]. The dotted line indicates the line with the best fit (R2 = 0.8234) between the predicted and the chronological age in our dataset.
Figure 2 shows the chronological age of the participants compared with the predicted age. The MAD was 3.62 years for the 49 individuals and lowest for the individuals in the 20–29 years age group (Table 3).
Figure 2.
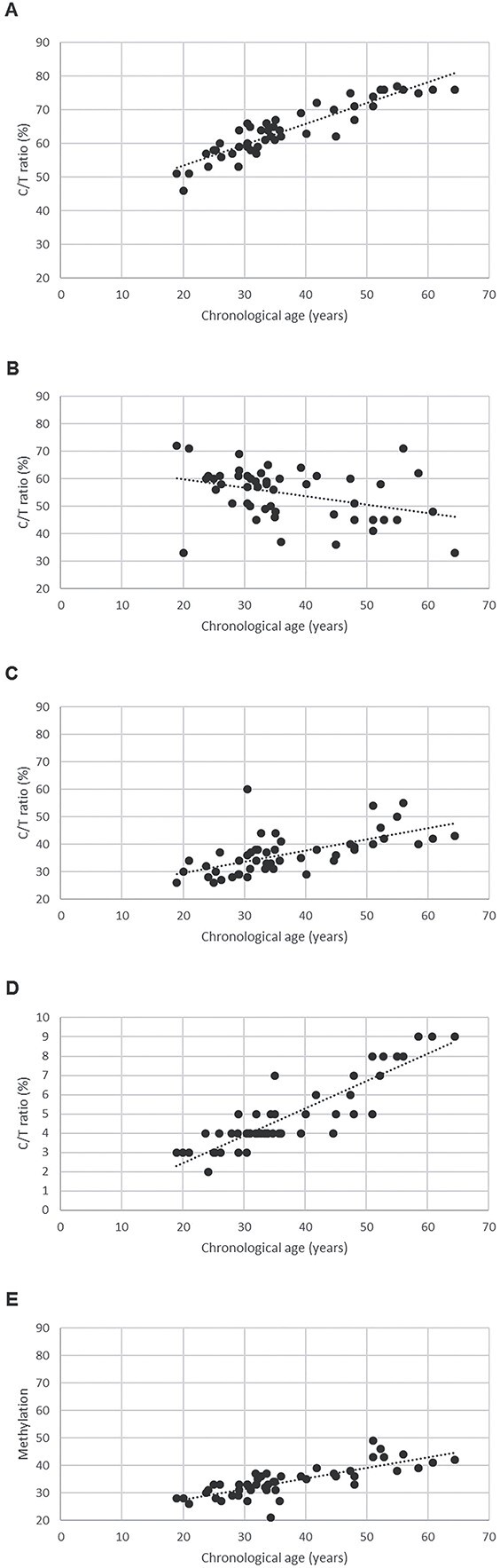
Chronological age plotted against C/T ratio. (A) ELOVL2 locus (R2 = 0.8230), (B) C1orf132 locus (R2 = 0.1314), (C) TRIM59 locus (R2 = 0.3237), (D) KLF14 locus (R2 = 0.7920), and (E) FHL2 locus (R2 = 0.6120). For comparison, all vertical axes are of the same size with the exception of KLF14 given the very low C/T ratios for this locus.
Table 3.
Mean absolute deviation (MAD) for the age groups included in this study.
| Age group (years) | Number of individualsa | MAD |
|---|---|---|
| 20–29 | 12 | 2.94 |
| 30–39 | 20 | 3.47 |
| 40–49 | 7 | 3.73 |
| 50–59 | 7 | 4.27 |
Three samples (age 19, 61, and 64) were not included in this table.
Discussion
Five forensic genetic laboratories tested the age assay designed for the PyroMark Q48 platform. Only a limited amount of reagents was made available to the laboratories. The main purpose of the collaboration was to study the feasibility of the method and comparability of results among the laboratories. The age assay and prediction model was based on the original work by Zbiec-Piekarska and co-workers [11], who selected five CpG loci and developed an age prediction model based on 300 reference samples that was tested on 120 individuals in the age range of 2–75 years. The MAD was 3.9 years for the independent test set [11]. Here, we tested 49 individuals in the age range of 18–64 years and observed an MAD of 3.62 years. This was comparable to other age prediction models with MAD values of 3.07 [5], 3.16 [8], 3.34 [14], and 5.2 [3] years .
We tested the age assay using the same reagents for bisulfite conversion, PCR amplification, and pyrosequencing. Nevertheless, we observed differences in performance between the laboratories. Laboratory 1 had the largest number of sequencing runs that were not immediately accepted by the PyroMark Q48 software (Passed category), and high standard deviations between the laboratories were observed for the CpG loci typed in the Human EpiTect Control DNA samples. This indicates that the methylation controls might not be suited for standardization as it was difficult to identify the reason(s) for the differences. However, it underlines the importance of proficiency tests and continuous collaborations between forensic genetic laboratories that wish to apply age estimation in casework.
The age assay is based on five singleplex PCR and pyrosequencing assays, which require more sample material and more laboratory work than commonly used STR profiling multiplex assays applied in standard forensic genetic workflows. Fast reporting of information such as age or phenotype estimations is important for ongoing police investigations. Ultimately, it would be preferable to utilize a multiplex PCR assay with a more standardized analysis tool that could calculate the estimated age automatically. Such a protocol could be based on a recently published multiplex strategy, where pyrosequencing is performed on a single aliquot of DNA sample subjected to multiplex amplification of several CpG markers [29].
In the present work, we essentially ignored the quality assessment of the PyroMark Q48 software and only used it as an initial screening before manual analysis. Only three of the 960 sequencing runs were rejected by the analyst even though 218 runs were categorized as Check or Failed by the PyroMark Q48 software. This raised the question of whether the quality assessment of the PyroMark Q48 software is adequate. The PyroMark Q48 software uses the entire sequence to evaluate the quality of the run, which may not be relevant for the analysis of the C/T ratio of a single nucleotide. Further validation of each pyrosequencing assay is needed to establish more suitable criteria for the data analysis. It will also be necessary to investigate whether the chosen C1orf132 CpG locus is associated with age or not. In our study of 49 individuals, the C1orf132 locus does not seem to be highly correlated with age even though the gene C1orf132 was associated with age in other studies [11, 30, 31]. The reason for this discrepancy may be that the C1orf132 CpG locus (position chr1:207823681) analyzed with this age assay is different from the CpG loci analyzed in other studies (position chr1:207823715 [5] and chr1:207823702/05 [32]). Furthermore, multiple amplification products were observed for the ELOVL2 and FHL2 loci. During sequencing, only one sequencing product was observed, and therefore, the designed sequencing primers were specific and only annealed to the PCR product of interest. Nevertheless, it would be advantageous to design amplification primers with only one binding site to optimize the PCR efficiency and avoid the risk of noise in the pyrosequencing reaction.
In this work, an age prediction tool was evaluated and showed promising results. An MAD of 3.62 years and a maximum deviation of 13 years based on only five CpG markers are noteworthy. Thus, while further improvements in PCR design and software would be helpful, along with additional validation involving more samples and individuals with other biogeographic backgrounds, this age assay presents a useful new addition to the analytical toolkits utilized in forensic genetic casework even in the current form.
Supplementary Material
Acknowledgements
The authors would like to thank laboratory technicians Anja Ladegaard Jørgensen, Barbora Blumova, and Veronika Holinkova for help with the laboratory work.
Contributor Information
Marie-Louise Kampmann, Section of Forensic Genetics, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Jan Fleckhaus, Institute of Legal Medicine, Faculty of Medicine and University Clinic, University of Cologne, Cologne, Germany.
Claus Børsting, Section of Forensic Genetics, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Helena Jurtikova, Institute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacky University Olomouc and the University Hospital Olomouc, Olomouc, the Czech Republic.
Alice Piters, Laboratoire d’Hématologie Médico-Légale, Bordeaux Cedex, France.
Julien Papin, Laboratoire d’Hématologie Médico-Légale, Bordeaux Cedex, France.
Quentin Gauthier, Department of Chemistry and Biochemistry, Florida International University, Miami, FL, USA.
Mirna Ghemrawi, Department of Chemistry and Biochemistry, Florida International University, Miami, FL, USA.
Christian Doutremepuich, Laboratoire d’Hématologie Médico-Légale, Bordeaux Cedex, France.
Bruce McCord, Department of Chemistry and Biochemistry, Florida International University, Miami, FL, USA.
Peter M Schneider, Institute of Legal Medicine, Faculty of Medicine and University Clinic, University of Cologne, Cologne, Germany.
Jiri Drabek, Institute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacky University Olomouc and the University Hospital Olomouc, Olomouc, the Czech Republic.
Niels Morling, Section of Forensic Genetics, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Authors’ contributions
Marie-Louise Kampmann performed the sample analysis for samples running at the University of Copenhagen and further collected all samples, analyzed the collected data, and wrote the manuscript. Jan Fleckhaus analyzed the samples from the University of Cologne together with participated in the analysis of the collected data. Claus Børsting helped to interpret the results. Helena Jurtikova analyzed the results from Palacky University. Alice Piters and Julien Papin analyzed the data from Laboratoire d’Hématologie Médico-Légale. Quentin Gauthier and Mirna Ghemrawi analyzed the results from Florida International University. Christian Doutremepuich, Bruce McCord, Peter M. Schneider, Jiri Drabek, and Niels Morling coordinated the project together with reviewed the manuscript. All authors contributed to the review work and approved the final manuscript.
Compliance with ethical standards
In all laboratories, peripheral blood samples from individuals with known age were collected with informed consent, and all samples were anonymized. Samples were collected in the individual laboratories according to the regulations in the respective countries.
Disclosure statement
The bisulfite conversion reagents, amplification reagents, and sequencing reagents were kindly provided by QIAGEN with no costs. QIAGEN had no influence on the design of the study, the laboratory analyses, and analyzing the results of this project.
Funding
This work was supported by LM2018125 for the work performed in Palacky University, Czech Republic; by the National Institute of Justice, Department of Justice, USA under grant 2017-NE-BX-0001 for work performed at the Department of Chemistry and Biochemistry, Florida International University, Miami, FL, USA. Points of view in the document are those of the authors and do not necessarily represent the official view of the US Department of Justice.
References
- 1. Aliferi A, Gallidabino MD, Thurtle H, et al. . DNA methylation-based age prediction using massively parallel sequencing data and multiple machine learning models. Forensic Sci Int Genet. 2018;37:215–226. [DOI] [PubMed] [Google Scholar]
- 2. Vidaki A, Aliferi A, Miller TH, et al. . DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci Int Genet. 2017;28:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bocklandt S, Lin W, Sehl ME, et al. . Epigenetic predictor of age. PLoS One. 2011;6:e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eipel M, Mayer F, Arent T, et al. . Epigenetic age predictions based on buccal swabs are more precise in combination with cell type-specific DNA methylation signatures. Aging (Albany NY). 2016;8:1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freire-Aradas A, Phillips C, Mosquera-Miguel A, et al. . Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena bioscience EpiTYPER system. Forensic Sci Int Genet. 2016;24:65–74. [DOI] [PubMed] [Google Scholar]
- 6. Garagnani P, Bacalini MG, Pirazzini C, et al. . Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012;11:1132–1134. [DOI] [PubMed] [Google Scholar]
- 7. Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY). 2011;3:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park JL, Kim JH, Seo E, et al. . Identification and evaluation of age-correlated DNA methylation markers for forensic use. Forensic Sci Int Genet. 2016;23:64–70. [DOI] [PubMed] [Google Scholar]
- 9. Xu C, Qu H, Wang G, et al. . A novel strategy for forensic age prediction by DNA methylation and support vector regression model. Sci Rep. 2015;5:17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yi SH, Jia YS, Mei K, et al. . Age-related DNA methylation changes for forensic age-prediction. Int J Leg Med. 2015;129:237–244. [DOI] [PubMed] [Google Scholar]
- 11. Zbiec-Piekarska R, Spolnicka M, Kupiec T, et al. . Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci Int Genet. 2015;17:173–179. [DOI] [PubMed] [Google Scholar]
- 12. Bram Bekaert AK, Zapico SC, Van de Voorde, et al. . Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics. 2015;10:922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weidner CI, Lin Q, Koch CM, et al. . Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia-Donas JG, Bonicelli A, Scholl AR, et al. . Rib histomorphometry: a reliability and validation study with a critical review of histological techniques for forensic age estimation. Leg Med (Tokyo). 2021;49:101827. [DOI] [PubMed] [Google Scholar]
- 16. Ubelaker DH, Khosrowshahi H. Estimation of age in forensic anthropology: historical perspective and recent methodological advances. Forensic Sci Res. 2019;4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valsecchi A, Irurita Olivares J, Mesejo P. Age estimation in forensic anthropology: methodological considerations about the validation studies of prediction models. Int J Leg Med. 2019;133:1915–1924. [DOI] [PubMed] [Google Scholar]
- 18. Ruengdit S, Troy Case D, Mahakkanukrauh P. Cranial suture closure as an age indicator: a review. Forensic Sci Int. 2020;307:110111. [DOI] [PubMed] [Google Scholar]
- 19. Hermetet C, Saint-Martin P, Gambier A, et al. . Forensic age estimation using computed tomography of the medial clavicular epiphysis: a systematic review. Int J Leg Med. 2018;132:1415–1425. [DOI] [PubMed] [Google Scholar]
- 20. Verma M, Verma N, Sharma R, et al. . Dental age estimation methods in adult dentitions: an overview. J Forensic Dent Sci. 2019;11:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frommer M, Mcdonald LE, Millar DS, et al. . A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. P Natl Acad Sci USA. 1992;89:1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyd VL, Zon G. Capillary electrophoretic analysis of methylation status in CpG-rich regions by single-base extension of primers modified with N6-methoxy-2,6-diaminopurine. Anal Biochem. 2008;380:13–20. [DOI] [PubMed] [Google Scholar]
- 23. Hong SR, Jung SE, Lee EH, et al. . DNA methylation-based age prediction from saliva: high age predictability by combination of 7 CpG markers. Forensic Sci Int-Gen. 2017;29:118–125. [DOI] [PubMed] [Google Scholar]
- 24. Hannum G, Guinney J, Zhang L, et al. . Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nyren P. Enzymatic method for continuous monitoring of DNA polymerase activity. Anal Biochem. 1987;167:235–238. [DOI] [PubMed] [Google Scholar]
- 26. Nilsson M, Styrman H, Andréasson H, et al. . Sensitive forensic analysis using the pyrosequencing technology. Int Congr Ser 2006;1288:625–627. [Google Scholar]
- 27. Allen MAH, Andréasson H. Mitochondrial D-loop and coding sequence analysis using pyrosequencing. Methods Mol Biol. 2005;297:179–196. [DOI] [PubMed] [Google Scholar]
- 28. Andreasson H, Asp A, Alderborn A, et al. . Mitochondrial sequence analysis for forensic identification using pyrosequencing technology. Biotechniques. 2002;32:124–133. [DOI] [PubMed] [Google Scholar]
- 29. Fleckhaus J, Schneider PM. Novel multiplex strategy for DNA methylation-based age prediction from small amounts of DNA via pyrosequencing. Forensic Sci Int Genet. 2020;44:102189. [DOI] [PubMed] [Google Scholar]
- 30. Spolnicka M, Pospiech E, Peplonska B, et al. . DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int J Leg Med. 2018;132:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dias HC, Cordeiro C, Pereira J, et al. . DNA methylation age estimation in blood samples of living and deceased individuals using a multiplex SNaPshot assay. Forensic Sci Int. 2020;311:110267. [DOI] [PubMed] [Google Scholar]
- 32. Freire-Aradas A, Phillips C, Giron-Santamaria L, et al. . Tracking age-correlated DNA methylation markers in the young. Forensic Sci Int Genet. 2018;36:50–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.