

Abstract
Cerebral cavernous malformations (CCMs) are vascular lesions that can cause severe neurological complications due to intracranial hemorrhage. Although the CCM disease genes, CCM1, CCM2, and CCM3, have been known for more than 15 years now, our understanding of CCM pathogenesis is still incomplete. CCM research currently focuses on three main disease mechanisms: (1) clonal expansion of endothelial cells with biallelic inactivation of CCM1, CCM2, or CCM3, (2) recruitment of cells with preserved CCM protein expression into the growing lesion, and (3) disruption of endothelial cell–cell junctions in CCMs. We here describe novel CRISPR/Cas9-based in vitro models of CCM and discuss their strengths and limitations in the context of high-throughput drug screening and repurposing approaches.
Keywords: cerebral cavernous malformations, CRISPR/Cas9 genome editing, human endothelial cells, cell junctions, spheroid sprouting
Introduction
Cerebral cavernous malformations (CCMs) are mulberry-like lesions in the microvasculature of the central nervous system (Fig. 1A,B) which are found with a prevalence of 0.5 % in the general population. They consist of densely packed, thin-walled, and leaky endothelial channels. Depending on their location and size, they can lead to a diverse spectrum of clinical signs and symptoms. While many of these vascular malformations are asymptomatic, some cause focal neurological deficits, epileptic seizures, and stroke-like symptoms due to intracranial hemorrhage. Especially brainstem lesions may lead to significant neurological complications [1]. However, CCMs can not only manifest with symptomatic hemorrhage but also with nonhemorrhagic focal neurological deficits.
Figure 1.
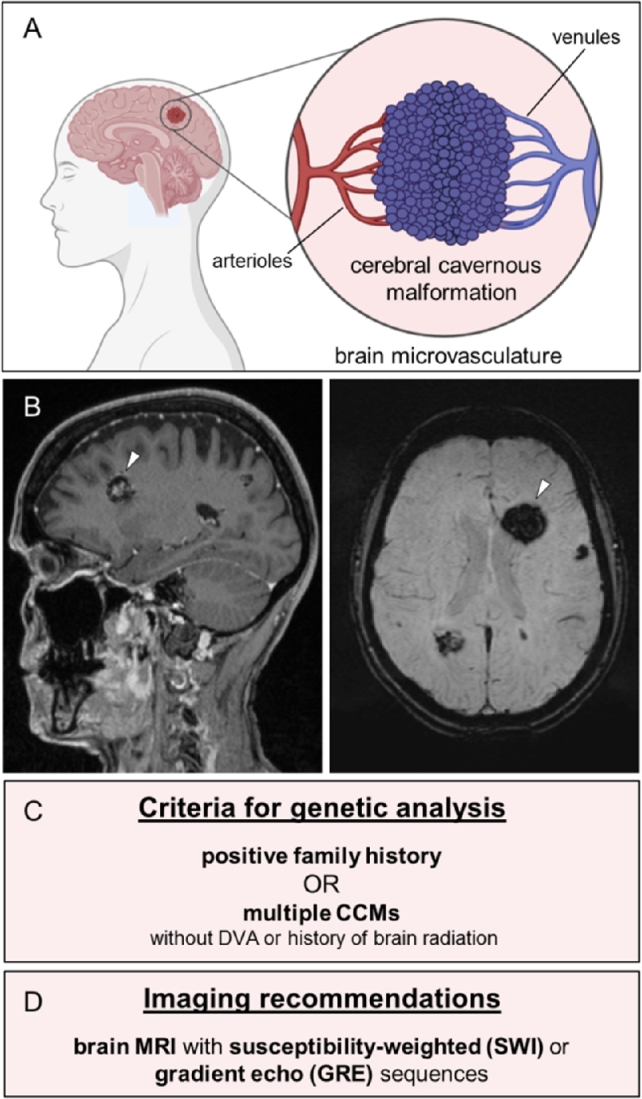
Clinical features of CCMs. (A) CCMs are mulberry-like vascular lesions in the microvascular bed of the central nervous system. (B) Sagittal T1-weighted (left) and axial susceptibility-weighted magnetic resonance images (right) show a large CCM (white arrowhead) and multiple smaller CCMs in both hemispheres of a patient with a pathogenic CCM1 germline variant. (C) Criteria for genetic testing of patients with CCMs (adapted from [2]). DVA = developmental venous anomaly. (D) Recommended imaging techniques for diagnosis or follow-up of CCMs (adapted from [2]).
Besides sporadic cases, about 6–7 % of CCMs occur in a familial form that is inherited in an autosomal dominant manner and caused by loss-of-function germline variants in CCM1 (KRIT1; OMIM: *604214), CCM2 (OMIM: *607929), or CCM3 (PDCD10; OMIM: *609118) [3], [4]. Familial cases usually become symptomatic in the fourth to fifth decade of life [5]. In this context, CCMs can be a significant cause of neurologic morbidity in middle-aged adults. Genetic counseling and testing should be offered to patients with a positive family history or multiple CCMs (Fig. 1C). For patients without a positive family history, however, current best practice guidelines only recommend genetic testing if there is no associated developmental venous anomaly (DVA) and no history of brain radiation, as these features usually indicate a sporadic case [2]. Magnetic resonance imaging (MRI) with susceptibility-weighted (SWI) or gradient echo (GRE) sequences is essential for making the correct diagnosis [2], [6] (Fig. 1D). Especially small CCMs can often only be detected with these special imaging techniques. Predictive genetic testing in children is possible because the results may guide the decision to perform an MRI examination, which may require sedation in young children [2].
Although more than 20 years have passed since the first disease gene, known as CCM1 or KRIT1 [7], [8], was identified, there is still no specific or targeted therapy for CCM patients. While symptomatic and easily accessible lesions may be treated with neurosurgical resection, conservative management is often the only option for patients with cavernous malformations in eloquent areas. Therefore, finding new pharmaceutical targets is a primary goal of CCM research. CCM studies in mice have recently added the mTOR inhibitor rapamycin and the third-generation tyrosine kinase inhibitor ponatinib to the short list of potential novel therapies [9], [10]. Unfortunately, in vivo studies are time consuming, expensive, and complex. Simplified in vitro systems are not perfect disease models either, but they can be used to study specific aspects of CCM pathobiology in more detail. Since they are less complex, less expensive, and compatible with the 3R principle (namely replacement, reduction, and refinement of animal experiments), they also qualify as first-line approach in high-throughput drug discovery studies. In the second or third line, in vivo models can then be used to validate novel drug candidates. It is important to realize that patients with sporadic CCMs could also benefit from new pharmacological treatments identified in those combined in vitro/in vivo screening assays because a substantial number of sporadic cases is caused by biallelic somatic CCM1, CCM2, or CCM3 mutations [11], [12].
This article reviews the currently available CCM cell culture models and illustrates their strengths and limitations. In particular, we focus on our recent efforts to establish new CRISPR/Cas9-based in vitro models of CCM disease.
Modeling the clonal expansion of mutant endothelial cells in CCMs
In efforts to find a treatment that can block disease progression, hope rests on a better understanding of the molecular mechanisms that trigger CCM formation. In reminiscence of Knudson’s two-hit model for retinoblastoma [13], DNA sequencing and immunohistochemical analyses of human CCMs demonstrated that CCM1, CCM2, or CCM3 gene expression is completely inactivated by a germline and a second somatic mutation or by two somatic mutations in many cavernous malformations [10], [12], [14], [15], [16], [17]. However, the vascular lesions do not only consist of mutant endothelial cells. Instead, a mosaic pattern of mutant endothelial cells and heterozygous or wild-type cells is found in CCM mouse models and human CCM tissue samples of familial and sporadic cases, respectively [17], [18], [19]. How these cells interact and whether the mosaic state is necessary for the survival of mutant cells in vivo is not yet understood.
Using CRISPR/Cas9 genome editing, we were recently able to study the effects of the second hit in blood outgrowth endothelial cells (BOECs) of a CCM patient with a pathogenic CCM1 germline mutation. Signs of endothelial dysfunction, namely the disruption of intercellular junctions, the formation of actin stress fibers, and the upregulation of the transcription factor KLF2, were only observed after inactivation of the second CCM1 allele [20], [21]. Interestingly, we were able to model a phenomenon in vitro that has recently been observed in CCM mouse models: clonal expansion of mutant endothelial cells [18], [19]. BOECs and immortalized human umbilical vein endothelial cells (CI-huVECs) demonstrated a striking survival advantage when co-cultured with BOECs or CI-huVECs (Fig. 2A–C), respectively [21], [22]. We also noticed resistance of BOECs and CI-huVECs to apoptosis, a feature reminiscent of malignant tumors. Even treatment with the broad-spectrum protein kinase inhibitor staurosporine, a potent inducer of apoptosis, only led to minimal activation of caspase-3 in CCM1- and CCM3-deficient cells [21], [22]. In a proof-of-principle approach, CRISPR/Cas9 genome editing also enabled us to study the feasibility of a targeted gene repair. While we were able to correct the CCM1 germline mutation in a significant number of BOECs in vitro, corrected BOECs were replaced by highly proliferative BOECs in co-culture [21]. In human CCM, where CRISPR/Cas9-mediated gene repair would not eradicate all mutant endothelial cells, the therapeutic benefit of such a genome editing approach would therefore be limited.
Figure 2.

gene disruption promotes clonal expansion of endothelial cells. (A) In patients with a CCM3 germline mutation (), a second somatic CCM3 mutation in an endothelial cell () initiates CCM formation. A mutant endothelial cell undergoes clonal expansion and forms a CCM that is characterized by endothelial mosaicism of and endothelial cells. The impaired endothelial barrier function can lead to bleeding into the surrounding brain tissue. (B) CCM3 knockout endothelial cells were generated with CRISPR/Cas9 genome editing. Biallelic loss-of-function variants were introduced into the first coding exon of CCM3 (knockout [KO] clones 1 and 2: c.[87_88insAG];[87_88insAG] [p.[Phe30Serfs*5];[Phe30Serfs*5]]; KO3: c.[90dupT];[87_88insAGTTGGATAAACATGTTTATCCAACT] [p.[Asn31*];[Phe30Serfs*13]]). (C) CI-huVECs demonstrated significant expansion in co-culture with CI-huVECs. Knockout and wild-type (WT) allele frequencies were determined by amplicon deep sequencing after six days of co-culture.
The new hypothesis that the tumor-like behavior of mutant endothelial cells represents a suitable therapeutic target has also been supported by the detection of PIK3CA mutations in CCMs [10]. The identification of somatic variants in this well-known oncogene suggests a three-hit mechanism in CCM pathogenesis. Following this intriguing model, only the combination of inactivating mutations in CCM genes acting as vascular “suppressor genes” and activating variants in vascular “oncogenes” can provoke a severe or aggressive course of CCM disease [10].
Cell culture models of the endothelial barrier dysfunction in CCMs
Apart from blocking CCM formation, restoration of an intact endothelial barrier is another primary objective in CCM therapy. As part of the blood–brain barrier (BBB), vascular endothelial cells participate in the tightly regulated exchange of ions, molecules, and cells between the blood and the brain [23]. However, no targeted therapies have yet been approved to prevent CCM bleeding and hemorrhage-associated neurological complications.
Endothelial tight and adherence junctions are indispensable to maintain BBB integrity but are highly dysfunctional in CCMs. Claudins, occludin, and junctional adhesion molecules (JAMs) are major components of endothelial tight junctions. These transmembrane proteins are linked to the actin cytoskeleton by scaffold proteins like zonula occludens protein 1 (ZO-1) (Fig. 3A). Destabilization of tight junctions and reduced expression of claudin-5, occludin, and ZO-1 have been observed in human CCM tissues [24], [25] and CCM mouse models [26], [27], [28]. These features of CCM disease can be perfectly reproduced in vitro [25], [28]. However, not only tight junctions but also adherens junctions are disorganized in CCMs. Using CRISPR/Cas9 genome editing, we could mimic disruption of adherens junctions in CI-huVECs (Fig. 3B) and BOECs [20]. In line with previous literature reports [21], [22], [29], [30], [31], the dysfunction of endothelial cell–cell junctions was accompanied by an increased formation of actin stress fibers (Fig. 3B). These phenotypes are useful surrogate markers for the hyperpermeability and increased bleeding risk of CCMs.
Figure 3.

Disorganized cell junctions and impaired function in 3D models of angiogenesis upon CCM3 gene inactivation. (A) Scheme of endothelial tight and adherens junctions. ICS = intracellular space. ECS = extracellular space. ZO-1 = zonula occludens protein 1. JAM = junctional adhesion molecule. (B) In contrast to wild-type CI-huVECs, CI-huVECs demonstrated numerous small gaps (white arrowheads) and a less homogeneous pattern in VE-cadherin staining (red). Scale bar 20 µm. They also displayed significant actin stress fiber formation (green, phalloidin staining). Scale bar 25 µm. DAPI (blue) was used to stain cell nuclei. (C) CI-huVECs demonstrated impaired spheroid formation and VEGF-induced sprouting. The number and length of sprouts formed by spheroids upon stimulation with 25 ng/ml VEGF-A were significantly reduced. Scale bar 100 µm. (D) CCM3 gene inactivation had cell type-specific effects on endothelial tube formation on Matrigel. While tubes formed by CI-huVECs were unstable and had fallen apart 17 h after seeding on Matrigel, hCMEC/D3 cells formed more stable meshes than hCMEC/D3 wild-type controls. Scale bar 500 µm.
Three-dimensional cell culture models in CCM research
The impaired interaction of mutant endothelial cells can also be visualized in three-dimensional (3D) cell culture models. More than 20 years ago, Thomas Korff and Hellmut G. Augustin developed a 3D spheroid formation and sprouting assay to analyze endothelial cell differentiation, cell–cell and cell–matrix interactions, and capillary sprouting [32], [33]. Using this assay, we demonstrated that CI-huVECs could only form irregular and barely demarcated spheroids upon CRISPR/Cas9-induced CCM1, CCM2, or CCM3 gene disruption [22], [29]. Furthermore, CCM3 gene inactivation in CI-huVECs and the immortalized human brain microvascular endothelial cell line D3 (hCMEC/D3) significantly impaired sprouting (Fig. 3C) [22]. Since transient CCM3 knockdown and genetic CCM3 knockout modulate this fundamental process differently [22], [26], compensatory mechanisms likely influence the angiogenic behavior of endothelial cells.
Disruption of endothelial junctions after CCM inactivation has also been found in transwell permeability assays. Upon CCM1, CCM2, or CCM3 depletion, the permeability of HUVEC monolayers was significantly increased [31], [34]. These results demonstrate that not only an altered 3D organization and angiogenic behavior of mutant cells, but also the leaky phenotype seen in CCMs can be modeled in in vitro systems. However, it can be sometimes challenging to directly compare the results of different in vitro models. An example is endothelial tube formation of mutant endothelial cells on Matrigel, which is another widely used in vitro angiogenesis assay. CI-huVECs form endothelial tubes that rapidly disintegrate (Fig. 3D) [22], a phenomenon that has also been reported for primary HUVECs after short hairpin RNA-mediated knockdown of CCM1 and CCM2 [35]. In contrast, hCMEC/D3 cells were able to form stable tubes on Matrigel (Fig. 3D). The different behavior on Matrigel might be a cell type-specific effect related to the fact that CI-huVECs and hCMEC/D3 cells are derived from endothelial cells from different vascular beds. However, an effect of different culture media and supplement concentrations cannot be excluded either. A combination of different assays is therefore the best way to obtain valid results.
High-content screening in CCM drug discovery
Since drug discovery and development studies are time consuming and cost-intensive, drug repurposing approaches have become popular in recent years. In the context of CCM, endothelial barrier function and cell proliferation assays have already been used successfully in drug repurposing screens. Gibson and colleagues defined the reversion of VE-cadherin disassembly and actin stress fiber formation in CCM2-silenced human dermal microvascular endothelial cells as primary read-out parameters [36]. Using in vitro transcellular resistance analyses, dermal permeability assays in inducible endothelial-specific Ccm2 knockout mice, and magnetic resonance imaging as secondary, tertiary, and quaternary screens, they identified tempol and cholecalciferol as promising candidates for CCM therapy [36]. Nishimura and colleagues also used a multi-step screening approach [37]. Drugs that could inhibit the proliferation of CCM3-deficient mouse astrocytes were validated in an RNAi-based Drosophila model and two mouse models of CCM disease. With this screening strategy, the authors identified the combination of fluvastatin and zoledronate to be effective in vivo and in vitro [37]. Finally, Otten and colleagues used ccm2 mutant zebrafish embryos, kri-1 (CCM1), and ccm-3 ablated C. elegans, as well as CCM2 knockdown HUVECs in a multi-organism-based screening approach. In downstream analyses, they validated that indirubin-3-monoxime treatment rescued VE-cadherin and actin phenotypes in CCM1-, CCM2-, and CCM3-silenced HUVECs [38].
Another positive example of drug repurposing in the context of CCM disease is propranolol. This pleiotropic β-blocker has recently been shown to reduce lesion burden in CCM mouse and zebrafish models [39]. A further study demonstrated that propranolol treatment also increased pericyte coverage and prevented vascular leakage in inducible endothelial-specific Ccm3 knockout (CCM3iECKO) mice [40]. After encouraging case reports on propranolol treatment in CCM patients, its effectiveness is now assessed in the Treat_CCM study, a multicenter, open-label, randomized trial [41].
Drug repurposing and discovery studies have relied on Ccm knockout mouse models or RNAi-based in vitro gene knockdown models in human endothelial cells so far. Both have strengths and limitations. In particular, discrepancies between transient gene knockdowns and genetic knockouts, as well as the limited predictive value of some mouse studies for humans are inherent weaknesses of these studies. Because they are easy-to-handle, cost-effective, and 3R-compliant, the use of novel human CRISPR/Cas9-based in vitro models in primary screens may help to accelerate the process of finding effective drugs for CCM patients.
Co-culture and iPSC-based CCM models
Notably, several studies have disclosed that CCM formation and disease progression are caused by more than just endothelial dysfunction. Pericytes which interact with the abluminal side of endothelial cells and astrocytic endfeet which enclose blood vessels are also major components of the BBB and participate in CCM pathogenesis [42], [43], [44]. Wang and colleagues, for example, demonstrated that specific Ccm3 deletion in mural cells induces a CCM phenotype in mice [44]. In particular, they found reduced cell spreading and migration of CCM3-deficient pericytes which caused impaired association with endothelial cells [44]. Additionally, a recent publication highlighted the crosstalk between endothelial cells and astrocytes in CCM lesion development. Increased endothelial NO synthase (eNOS)/nitric oxide (NO)-dependent signaling in dysfunctional endothelial cells leads to elevated levels of the astrocyte-derived angiogenesis factor VEGF, which contributes to endothelial cell junction disassembly linked to an increased risk of hemorrhage [43]. These observations provide a first explanation of why CCMs only arise in the central nervous system, although the CCM proteins are ubiquitously expressed. They also suggest that endothelial monocultures may not adequately illustrate CCM disease. In vitro co-culture models of endothelial cells, astrocytes, and pericytes may be more suitable. Patient-specific induced pluripotent stem cells (iPSCs) and their direct differentiation into all three cell types may allow the development of new co-culture models in the future. Co-culturing these cells in transwell or microfluidic models can improve the barrier properties and allows studying BBB dysfunction in an isogenic system [45]. Measuring the transendothelial electrical resistance (TEER) of knockout BBB co-cultures in a compound library screen may help to find a drug that can reduce the bleeding risk of CCMs. A fascinating direction of CCM disease modeling might also be the combination with a CCM xenograft model [46]. Implantation of human spheroid co-cultures into murine models might reconstruct the in vivo cellular environment of CCM by keeping the human origin of the affected cells.
Outlook
Although there will be no “one-stop shopping” in CCM drug discovery in the near future, we now have a broad toolkit of in vitro models to study CCM pathogenesis and search for new CCM therapies. In particular, CRISPR/Cas9 genome editing has become an invaluable tool to model fundamental cellular and molecular processes of CCM formation, disease progression, and endothelial barrier disruption. Targeted gene inactivations in endothelial cells, pericytes, and astrocytes will facilitate more complex 3D co-culture models of CCM. Combined with live-cell imaging and new CRISPR tools, e. g., CRISPR activation (CRISPRa) or CRISPR interference (CRISPRi), these models will help to understand the dynamics of BBB dysfunction in CCMs better. However, higher complexity is usually accompanied by lower compatibility with high-throughput drug screening assays. Therefore, future efforts to model CCM disease in vitro will likely go into two directions: (1) simple but high-throughput-compatible in vitro assays and (2) complex cell culture models to closely mimic the in vivo situation.
Acknowledgment
We thank the CCM patient for granting permission to publish her MRI images. Dr. rer. nat. Stefanie Spiegler is thanked for providing confocal microscopy images of VE-cadherin staining. Figures 1A, 2A, and 3A were created with BioRender.com. We apologize to all colleagues whose work has not been cited due to space limitations.
Funding Statement
This work was supported by the German Research Foundation (DFG RA2876/2-2), the German Federal Ministry of Education and Research (BMBF; grant number 161L0276), and the Research Association Molecular Medicine of the University Medicine Greifswald (FVMM, grant numbers FOVB-2021-07, FOVB-2020-01, FOVB-2019-01, and FOVB-2018-06). MR is supported by a scholarship from the Gerhard Domagk program of the University Medicine Greifswald.
Footnotes
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
Conflict of interest: The authors state no conflict of interest.
Informed consent: Written informed consent was obtained from the CCM patient to publish the medical information and MRI images presented here.
Ethical approval: This article does not contain any studies with human or animal subjects.
References
- [1].Horne MA, Flemming KD, Su IC, Stapf C, Jeon JP, Li D, Maxwell SS, White P, Christianson TJ, Agid R, Cho W-S, Oh CW, Wu Z, Zhang J-T, Kim JE, ter Brugge K, Willinsky R, Brown RD, Murray GD, Al-Shahi Salman R. Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol. 2016;15:166–73. doi: 10.1016/S1474-4422(15)00303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Akers A, Al-Shahi Salman R, Awad IA, Dahlem K, Flemming K, Hart B, Kim H, Jusue-Torres I, Kondziolka D, Lee C, Morrison L, Rigamonti D, Rebeiz T, Tournier-Lasserve E, Waggoner D, Whitehead K. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the Angioma Alliance Scientific Advisory Board Clinical Experts Panel. Neurosurgery. 2017;80:665–80. doi: 10.1093/neuros/nyx091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral Cavernous Malformations: An Update on Prevalence, Molecular Genetic Analyses, and Genetic Counselling. Mol Syndromol. 2018;9:60–9. doi: 10.1159/000486292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Batra S, Lin D, Recinos PF, Zhang J, Rigamonti D. Cavernous malformations: natural history, diagnosis and treatment. Nat Rev Neurol. 2009;5:659–70. doi: 10.1038/nrneurol.2009.177. [DOI] [PubMed] [Google Scholar]
- [5].Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier-Lasserve E. Société Française de Neurochirurgie. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550–6. doi: 10.1002/ana.20947. [DOI] [PubMed] [Google Scholar]
- [6].Flemming KD, Lanzino G. Cerebral cavernous malformation: What a practicing clinician should know. Mayo Clin Proc. 2020;95:2005–20. doi: 10.1016/j.mayocp.2019.11.005. [DOI] [PubMed] [Google Scholar]
- [7].Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- [8].Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil-Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED, Marchuk DA. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325–33. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- [9].Choi JP, Wang R, Yang X, Wang X, Wang L, Ting KK, Foley M, Cogger V, Yang Z, Liu F, Han Z, Liu R, Baell J, Zheng X. Ponatinib (AP24534) inhibits MEKK3-KLF signaling and prevents formation and progression of cerebral cavernous malformations. Sci Adv. 2018;4:eaau0731. doi: 10.1126/sciadv.aau0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ren AA, Snellings DA, Su YS, Hong CC, Castro M, Tang AT, Detter MR, Hobson N, Girard R, Romanos S, Lightle R, Moore T, Shenkar R, Benavides C, Beaman MM, Müller-Fielitz H, Chen M, Mericko P, Yang J, Sung DC, Lawton MT, Ruppert JM, Schwaninger M, Körbelin J, Potente M, Awad IA, Marchuk DA, Kahn ML. PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature. 2021;594:271–6. doi: 10.1038/s41586-021-03562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Awad IA, Polster SP. Cavernous angiomas: deconstructing a neurosurgical disease. J Neurosurg. 2019;131:1–13. doi: 10.3171/2019.3.JNS181724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McDonald DA, Shi C, Shenkar R, Gallione CJ, Akers AL, Li S, De Castro N, Berg MJ, Corcoran DL, Awad IA, Marchuk DA. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet. 2014;23:4357–70. doi: 10.1093/hmg/ddu153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911–8. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gault J, Shenkar R, Recksiek P, Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005;36:872–4. doi: 10.1161/01.STR.0000157586.20479.fd. [DOI] [PubMed] [Google Scholar]
- [16].Gault J, Awad IA, Recksiek P, Shenkar R, Breeze R, Handler M, Kleinschmidt-DeMasters BK. Cerebral cavernous malformations: somatic mutations in vascular endothelial cells. Neurosurgery. 2009;65:138–45. doi: 10.1227/01.NEU.0000348049.81121.C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rath M, Pagenstecher A, Hoischen A, Felbor U. Postzygotic mosaicism in cerebral cavernous malformation. J Med Genet. 2020;57:212–6. doi: 10.1136/jmedgenet-2019-106182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Detter MR, Snellings DA, Marchuk DA. Cerebral cavernous malformations develop through clonal expansion of mutant endothelial cells. Circ Res. 2018;123:1143–51. doi: 10.1161/CIRCRESAHA.118.313970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Malinverno M, Maderna C, Abu Taha A, Corada M, Orsenigo F, Valentino M, Pisati F, Fusco C, Graziano P, Giannotta M, Yu QC, Zeng YA, Lampugnani MG, Magnusson PU, Dejana E. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun. 2019;10:2761. doi: 10.1038/s41467-019-10707-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Much CD, Sendtner BS, Schwefel K, Freund E, Bekeschus S, Otto O, Pagenstecher A, Felbor U, Rath M, Spiegler S. Inactivation of Cerebral Cavernous Malformation Genes Results in Accumulation of von Willebrand Factor and Redistribution of Weibel-Palade Bodies in Endothelial Cells. Front Mol Biosci. 2021;8:622547. doi: 10.3389/fmolb.2021.622547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spiegler S, Rath M, Much CD, Sendtner BS, Felbor U. Precise CCM1 gene correction and inactivation in patient-derived endothelial cells: Modeling Knudson’s two-hit hypothesis in vitro. Mol Genet Genomic Med. 2019;7:e00755. doi: 10.1002/mgg3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schwefel K, Spiegler S, Ameling S, Much CD, Pilz RA, Otto O, Völker U, Felbor U, Rath M. Biallelic CCM3 mutations cause a clonogenic survival advantage and endothelial cell stiffening. J Cell Mol Med. 2019;23:1771–83. doi: 10.1111/jcmm.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schneider H, Errede M, Ulrich NH, Virgintino D, Frei K, Bertalanffy H. Impairment of tight junctions and glucose transport in endothelial cells of human cerebral cavernous malformations. J Neuropathol Exp Neurol. 2011;70:417–29. doi: 10.1097/NEN.0b013e31821bc40e. [DOI] [PubMed] [Google Scholar]
- [25].Stamatovic SM, Sladojevic N, Keep RF, Andjelkovic AV. PDCD10 (CCM3) regulates brain endothelial barrier integrity in cerebral cavernous malformation type 3: role of CCM3-ERK1/2-cortactin cross-talk. Acta Neuropathol. 2015;130:731–50. doi: 10.1007/s00401-015-1479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou JH, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A, Toomre D, Fuh G, Yan M, Kluger MS, Wu D, Min W. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med. 2016;22:1033–42. doi: 10.1038/nm.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zeineddine HA, Girard R, Saadat L, Shen L, Lightle R, Moore T, Cao Y, Hobson N, Shenkar R, Avner K, Chaudager K, Koskimäki J, Polster SP, Fam MD, Shi C, Lopez-Ramirez MA, Tang AT, Gallione C, Kahn ML, Ginsberg M, Marchuk DA, Awad IA. Phenotypic characterization of murine models of cerebral cavernous malformations. Lab Invest. 2019;99:319–30. doi: 10.1038/s41374-018-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lopez-Ramirez MA, Fonseca G, Zeineddine HA, Girard R, Moore T, Pham A, Cao Y, Shenkar R, de Kreuk BJ, Lagarrigue F, Lawler J, Glass CK, Awad IA, Ginsberg MH. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J Exp Med. 2017;214:3331–46. doi: 10.1084/jem.20171178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schwefel K, Spiegler S, Kirchmaier BC, Dellweg PKE, Much CD, Pané-Farré J, Strom TM, Riedel K, Felbor U, Rath M. Fibronectin rescues aberrant phenotype of endothelial cells lacking either CCM1, CCM2 or CCM3. FASEB J. 2020;34:9018–33. doi: 10.1096/fj.201902888R. [DOI] [PubMed] [Google Scholar]
- [30].Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–54. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, Zhang L, Austin C, Akers AL, Gallione CJ, Rorrer A, Gunel M, Min W, De Souza JM, Lee C, Marchuk DA, Awad IA. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med. 2015;17:188–96. doi: 10.1038/gim.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol. 1998;143:1341–52. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Korff T, Augustin HG. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J Cell Sci. 1999;112:3249–58. doi: 10.1242/jcs.112.19.3249. [DOI] [PubMed] [Google Scholar]
- [34].Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207:881–96. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chernaya O, Zhurikhina A, Hladyshau S, Pilcher W, Young KM, Ortner J, Andra V, Sulchek TA, Tsygankov D. Biomechanics of endothelial tubule formation differentially modulated by cerebral cavernous malformation proteins. iScience. 2018;9:347–58. doi: 10.1016/j.isci.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gibson CC, Zhu W, Davis CT, Bowman-Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, Shiu YT, Grossmann AH, Thomas KR, Donato AJ, Lesniewski LA, Whitehead KJ, Li DY. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation. 2015;131:289–99. doi: 10.1161/CIRCULATIONAHA.114.010403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nishimura S, Mishra-Gorur K, Park J, Surovtseva YV, Sebti SM, Levchenko A, Louvi A, Gunel M. Combined HMG-COA reductase and prenylation inhibition in treatment of CCM. Proc Natl Acad Sci USA. 2017;114:5503–8. doi: 10.1073/pnas.1702942114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Otten C, Knox J, Boulday G, Eymery M, Haniszewski M, Neuenschwander M, Radetzki S, Vogt I, Hähn K, De Luca C, Cardoso C, Hamad S, Igual Gil C, Roy P, Albiges-Rizo C, Faurobert E, von Kries JP, Campillos M, Tournier-Lasserve E, Derry WB, Abdelilah-Seyfried S. Systematic pharmacological screens uncover novel pathways involved in cerebral cavernous malformations. EMBO Mol Med. 2018;10:e9155. doi: 10.15252/emmm.201809155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li W, Shenkar R, Detter MR, Moore T, Benavides C, Lightle R, Girard R, Hobson N, Cao Y, Li Y, Griffin E, Gallione C, Zabramski JM, Ginsberg MH, Marchuk DA, Awad IA. Propranolol inhibits cavernous vascular malformations by β1 adrenergic receptor antagonism in animal models. J Clin Invest. 2021;131:e144893. doi: 10.1172/JCI154909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oldenburg J, Malinverno M, Globisch MA, Maderna C, Corada M, Orsenigo F, Conze LL, Rorsman C, Sundell V, Arce M, Smith RO, Yau ACY, Billström GH, Mägi CÖ, Beznoussenko GV, Mironov AA, Fernando D, Daniel G, Olivari D, Fumagalli F, Lampugnani MG, Dejana E, Magnusson PU. Propranolol reduces the development of lesions and rescues barrier function in cerebral cavernous malformations: A preclinical study. Stroke. 2021;52:1418–27. doi: 10.1161/STROKEAHA.120.029676. [DOI] [PubMed] [Google Scholar]
- [41].Lanfranconi S, Scola E, Bertani GA, Zarino B, Pallini R, d’Alessandris G, Mazzon E, Marino S, Carriero MR, Scelzo E, Faragò G, Castori M, Fusco C, Petracca A, d’Agruma L, Tassi L, d’Orio P, Lampugnani MG, Nicolis EB, Vasamì A, Novelli D, Torri V, Meessen J, Al-Shahi Salman R, Dejana E, Latini R. Treat-CCM Investigators. Propranolol for familial cerebral cavernous malformation (Treat_CCM): study protocol for a randomized controlled pilot trial. Trials. 2020;21:401. doi: 10.1186/s13063-020-4202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schulz GB, Wieland E, Wüstehube-Lausch J, Boulday G, Moll I, Tournier-Lasserve E, Fischer A. Cerebral cavernous malformation-1 protein controls DLL4-Notch3 signaling between the endothelium and pericytes. Stroke. 2015;46:1337–43. doi: 10.1161/STROKEAHA.114.007512. [DOI] [PubMed] [Google Scholar]
- [43].Lopez-Ramirez MA, Lai CC, Soliman SI, Hale P, Pham A, Estrada EJ, McCurdy S, Girard R, Verma R, Moore T, Lightle R, Hobson N, Shenkar R, Poulsen O, Haddad GG, Daneman R, Gongol B, Sun H, Lagarrigue F, Awad IA, Ginsberg MH. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J Clin Invest. 2021;131:e139570. doi: 10.1172/JCI139570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang K, Zhang H, He Y, Jiang Q, Tanaka Y, Park IH, Pober JS, Min W, Zhou HJ. Mural cell-specific deletion of cerebral cavernous malformation 3 in the brain induces cerebral cavernous malformations. Arterioscler Thromb Vasc Biol. 2020;40:2171–86. doi: 10.1161/ATVBAHA.120.314586. [DOI] [PubMed] [Google Scholar]
- [45].Appelt-Menzel A, Oerter S, Mathew S, Haferkamp U, Hartmann C, Jung M, Neuhaus W, Pless O. Human iPSC-Derived Blood-Brain Barrier Models: Valuable Tools for Preclinical Drug Discovery and Development? Curr Protoc Stem Cell Biol. 2020;55:e122. doi: 10.1002/cpsc.122. [DOI] [PubMed] [Google Scholar]
- [46].Wüstehube J, Bartol A, Liebler SS, Brütsch R, Zhu Y, Felbor U, Sure U, Augustin HG, Fischer A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci USA. 2010;107:12640–5. doi: 10.1073/pnas.1000132107. [DOI] [PMC free article] [PubMed] [Google Scholar]