

Summary
Targeting cancer metabolism to limit cellular energy and metabolite production is an attractive therapeutic approach. Here, we developed analogs of the bisbiguanide, alexidine, to target lung cancer cell metabolism and assess a structure-activity relationship (SAR). The SAR led to the identification of two analogs, AX-4 and AX-7, that limit cell growth via G1/G0 cell-cycle arrest and are tolerated in vivo with favorable pharmacokinetics. Mechanistic evaluation revealed that AX-4 and AX-7 induce potent mitochondrial defects; mitochondrial cristae were deformed and the mitochondrial membrane potential was depolarized. Additionally, cell metabolism was rewired, as indicated by reduced oxygen consumption and mitochondrial ATP production, with an increase in extracellular lactate. Importantly, AX-4 and AX-7 impacted overall cell behavior, as these compounds reduced collective cell invasion. Taken together, our study establishes a class of bisbiguanides as effective mitochondria and cell invasion disrupters, and proposes bisbiguanides as promising approaches to limiting cancer metastasis.
Subject areas: Cellular physiology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
Bisbiguanides with long linkers and branched side chains reduce cancer cell growth
-
•
AX-4 and AX-7 bisbiguanides preferentially disrupt mitochondria cristae structure
-
•
AX-4 and AX-7 induce mitochondrial depolarization and bioenergetic stress
-
•
AX-4 and AX-7 reduce lung cancer cell collective invasion
Cellular physiology; Cell biology; Cancer
Introduction
Our understanding of cancer metabolism has rapidly grown in recent decades, leading to enhanced interest in identifying therapeutic metabolic vulnerabilities.1,2,3 Cancers adopt varied metabolic alterations to enable proliferation and survival throughout different stages of tumor progression from primary tumor formation to the metastatic cascade.1,2,4 Efforts to develop inhibitors of cancer metabolism have expanded to target various nodes, including those within mitochondrial metabolism.3,5,6 One class of small-molecules that alters metabolism and mitochondrial function are the biguanides.7,8,9,10,11 Several biguanide compounds are in experimental and clinical use with varying degrees of efficacy. The most well-known biguanide is metformin, which has been extensively studied for its effects on type 2 diabetes and cancer.12,13,14 Other members include phenformin and buformin; however, clinical trials in diabetes ended early due to toxicity from lactic acidosis.15,16,17 Therefore, there is a continued interest in studying biguanides as cancer therapies and improving tolerability.18,19,20,21,22
One biguanide currently being adapted as a cancer therapeutic is the bisbiguanide, alexidine.23,24,25 Alexidine and its family member, chlorhexidine, share structural similarity to metformin, but have two biguanide units connected by a hexamethylene linker and different side chains. Traditionally, alexidine and chlorhexidine are cationic antibiotics used in dentistry against plaque and gingivitis, likely due to their affinity for the negatively charged cell wall and phospholipids on the bacterial cell membrane.26,27,28,29,30,31,32 Mitochondria likely evolved from ancestrally engulfed bacteria33,34,35; therefore, bactericides, such as alexidine, are promising metabolic-targeting agents. While a study was conducted using different analogs of chlorhexidine and alexidine on their effects on common bacteria in the oral microbiome,36 the effects of alexidine analogs on human cells, particularly cancer cells, are understudied.
Through a chemical biology screen of over 3,000 compounds, we discovered that alexidine causes lung cancer cell growth arrest, reduced invasion through a 3D recombinant basement membrane matrix, and alterations in metabolism.37 We therefore sought to develop analogs of alexidine and establish a structure-activity relationship (SAR) in lung cancer cells. The current study details the development and assessment of bisbiguanides designed from the alexidine parent molecule, capable of disrupting mitochondrial activity while maintaining in vivo tolerability. We report that from a screen of seven analogs of alexidine, AX-4 and AX-7 reduce lung cancer cell growth by cell-cycle arrest. While alexidine, AX-4, and AX-7 are all tolerated in mice, AX-7 has a longer terminal half-life and higher concentration in blood plasma, suggesting longer duration of action and establishing it as a promising in vivo compound. Furthermore, we determine that AX-4 and AX-7 cause dramatic structural defects to the mitochondrial cristae, loss of mitochondrial membrane potential, and reduced oxidative phosphorylation, whereby disruption in mitochondrial metabolism corresponds with reduced collective cell invasion. Taken together, our findings support AX-4 and AX-7 as promising mitochondrial disrupters and that lung cancer cells require mitochondrial polarization for collective invasion. Our work proposes the benefit of utilizing modified biguanides for targeting metabolic vulnerabilities to reduce cancer metastasis.
Results
Development of alexidine analogs to inhibit cell proliferation
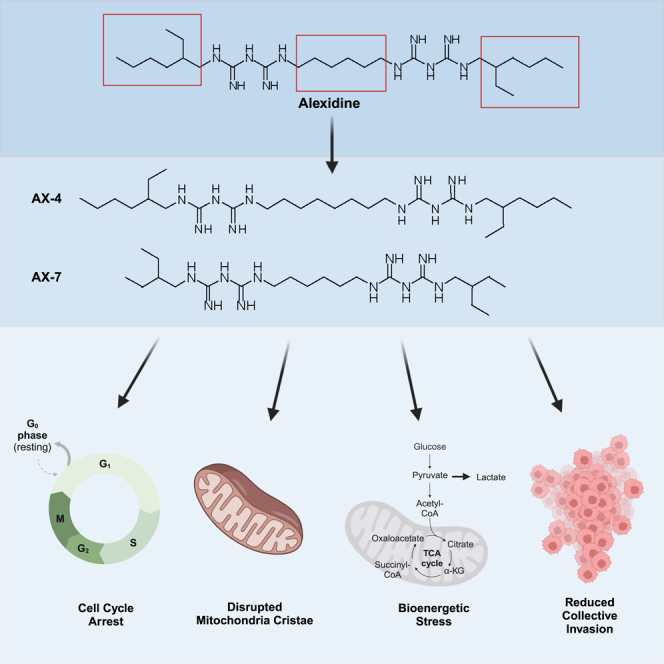
To determine whether we could improve the potency of alexidine with structural modifications and derive a SAR among the derivatives, we systematically altered the linker length connecting the two biguanidine moieties in alexidine or altered the side chains on the ends of the compounds (Figure 1A), as these structures were found to be important in bisbiguanide activity against bacteria.36,38 We were able to synthesize seven analogs, termed AX-1, AX-2, AX-3, AX-4, AX-5, AX-7, and AX-10 (Figures 1B and S1, Data S1), for further characterization. AX-6, AX-8/9, and AX-11 were unable to be synthesized.
Figure 1.

Development of alexidine analogs to inhibit cell proliferation
(A) Schematic of alexidine (AD) with the side chain, biguanide, and linker regions annotated where alterations were made to develop the analogs.
(B) Schematic of the seven resulting analogs (AX-). See also STAR methods and Figure S1.
(C) Primary proliferation screen on H1299 lung cancer cells to identify candidate analogs at the indicated concentrations for 72 h using the SRB assay.
(D) A complimentary proliferation screen using the WST-8 assay with the top candidates, AX-4 and AX-7, and negative control analog, AX-2, for 72 h. See also Table 1. For (C) and (D), three biological replicates with three technical replicates each were performed. Mean ± SEM is shown.
(E) Cell cycle analysis of H1299 cells using flow cytometry after 48 h of treatment with the analogs at the indicated doses. Mean ± SD is shown. Significance was assessed using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test and is indicated above the respective phase of the cell cycle (i.e., G1/G0 and S). ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
(F) Protein levels of CDK1, CDK4, CDK6, cyclin D1, and phosphorylated histone H3 at serine 10 (pHH3 (S10)) in H1299 cells treated for 48 h and detected by western blot. Actin was used as a loading control. See also Figure S2 for quantification of western blots. For (E) and (F), three biological replicates were performed.
To select top analog candidates, we conducted a dose response of the alexidine derivatives on H1299 non-small cell lung cancer cell proliferation for 72 h. The initial screen was performed with a sulforhodamine B (SRB) assay (Figure 1C), which assesses only substrate adherent cells; using the top candidates from the SRB screen, a complimentary screen was performed with an add-and-read water-soluble tetrazolium (WST)-8 assay to assess both adherent and non-adherent cells (Figure 1D). The IC50 values and 95% confidence intervals (CI) were determined to compare the analogs (Table 1). Alexidine inhibited H1299 cell proliferation with an IC50 of 1.0 μM in the initial SRB screen and with an IC50 of 2.5 μM using the WST assay. The difference in IC50 values corresponds with the differences in experimental procedures, in which the SRB assay removes non-adherent and loosely adherent cells, thereby resulting in a lower IC50 value. A truncated analog that contains a single biguanide core, AX-1, lost potency by 10-fold (IC50 = 10.1 μM). AX-2, which has the same linker between the guanidine units as alexidine, but the side chain 2-ethylhexylmine units are replaced by simple methyl hydrocarbon units, also caused loss of potency by greater than 10-fold, as an IC50 could not be achieved. Likewise, AX-3, with two fewer carbons in the linker (four carbons in AX-3 vs. six carbons in alexidine) that connects both the guanidine units, achieved better potency than AX-1 or AX-2; however, the IC50 was still above that of alexidine (2.0 vs. 1.0 μM). Moreover, increasing the linker length to eight carbons in AX-4 maintained similar potency as the parent compound in the SRB assay, (IC50 = 0.8 vs. 1.0 μM); however, AX-4 was less effective when assessed via the WST assay (3.7 vs. 2.5 μM). These results suggest that 4–7 carbon linkers can maintain the IC50 in the low micromolar range.
Table 1.
Cell proliferation screen of alexidine analogs
| Compound | SRB IC50 (μM) | Fit (R2) | 95% CI | WST IC50 (μM) | Fit (R2) | 95% CI |
|---|---|---|---|---|---|---|
| AX-1 | 10.1 | 0.9461 | 5.9–773.4 | – | – | – |
| AX-2 | N/A | N/A | N/A | N/A | 0.2487 | N/A |
| AX-3 | 2.0 | 0.9835 | 1.8–2.2 | – | – | – |
| AX-4 | 0.8 | 0.9497 | 0.6–0.9 | 3.7 | 0.9553 | 3.2–4.4 |
| AX-5 | 3.4 | 0.8944 | N/A | – | – | – |
| AX-7 | 0.6 | 0.7184 | 0.4–1.5 | 3.0 | 0.9520 | 2.5–3.8 |
| AX-10 | N/A | 0.1939 | 0.01-N/A | – | – | – |
| AD | 1.0 | 0.8977 | 0.7–1.3 | 2.5 | 0.9500 | 2.1–3.0 |
IC50 concentrations, goodness of fit (Fit R2), and 95% confidence intervals (CI) for the SRB and WST-8 proliferation assays in Figure 1 were determined using nonlinear regression. N/A indicates values that were unable to be accurately computed.
We next explored other side chain hydrocarbons replacing the 2-ethylhexylmine moiety in alexidine. AX-5, in which the 2-ethylhexylmine unit was replaced with 2-octylamine, reduced the potency by approximately 3-fold compared to alexidine (3.4 vs. 1.0 μM). Interestingly, compound AX-7, with 2-ethylbutyl units in place of 2-ethylhexyl moieties, trended toward improved potency in comparison to parent alexidine in the SRB assay (0.6 vs. 1.0 μM), but had similar potency as alexidine in the WST assay (3.0 vs. 2.5 μM). Lastly, we tested a derivative, AX-10, with straight chain methyl-butyl ether side chains. This led to a loss of potency by greater than 10-fold, as the IC50 could not be achieved. Therefore, this suggests that branched terminal side chains are important to maintain activity, as AX-2 (lacking the 2-ethylhexylmine units) was also ineffective. Overall, with a set of seven derivatives of alexidine, we found AX-7 as more potent than the other derivatives and similarly as potent as alexidine.
Based upon the proliferation screens, AX-4 and AX-7 emerged as promising candidates for further study. AX-2 was chosen as a negative control analog, as it was ineffective at reducing H1299 cell growth. We next tested the ability of these analogs to alter cell cycle, as observed with alexidine in our previously published work.37 After 48 h of treatment to H1299 cells, AX-4 and AX-7 induced a G1/G0 cell-cycle arrest to similar levels as alexidine and in a dose-dependent manner (Figure 1E). Concordantly, there was a reduction in the percentage of cells in S phase. While there was not a significant decrease in the percentage of cells in G2/M phase, AX-4, AX-7, and alexidine did markedly reduce the amount of phosphorylated histone H3 at serine 10 and cyclin-dependent kinase 1 (CDK1) from protein isolated in analog-treated cells (Figures 1F and S2), indicating reduced proliferation. Additionally, protein levels of CDK4, CDK6, and cyclin D1, key mediators of progression from G0/G1 to S phase, were reduced in a dose-dependent manner in AX-4, AX-7, and alexidine-treated cells, indicating a lack of signaling to transition cells into S phase and supporting the flow cytometry results. There was not a significant increase in the sub-G1 population (Figure 1E), further suggesting the reduction in cell growth is due to G1/G0 cell-cycle arrest rather than apoptosis. AX-2 did not alter the proportion of cells in each phase compared to the DMSO control (Figures 1E and 1F), consistent with the viability results in Figures 1A and 1B. Taken together, these studies identified two analog candidates, AX-4 and AX-7, capable of inhibiting cell proliferation with similar capacity as alexidine.
In vivo applicability of alexidine analogs
We next assessed the applicability of our lead analogs, AX-4 and AX-7, for in vivo use. To determine the relative permeability to approximate analog absorption in the intestines, we used standard in vitro absorption, distribution, metabolism, and excretion (ADME) approaches. Caco-2 cells are a human colon cancer cell line that have been historically used to study drug permeability and efflux prior to use in animals or humans.39 Monolayers of Caco-2 cells were treated with the analogs and the apparent permeability was determined (Table S1). Propranolol and Digoxin were used as controls for highly absorbed and highly effluxed compounds, respectively. Interestingly, AX-2 had the most favorable efflux ratio among the analogs (0.60), despite its lack of biological activity in other assays. AX-7 had a more favorable efflux ratio than AX-1, AX-4, and alexidine (1.95 vs. >236.12 vs. >6.88 vs. >5.37). Caco-2 cell monolayer integrity was intact throughout the process, as indicated by negligible Lucifer Yellow leakage (Table S2).
Drugs are commonly metabolized and cleared in the liver, so typical in vitro ADME approaches use isolated hepatocytes and liver microsomes to assess the liver metabolism of test compounds prior to use in vivo.40,41,42 Because AX-7 had a more favorable efflux ratio over AX-4 and alexidine, AX-7 was tested for its metabolic stability in liver microsomes (Table S3) and hepatocytes (Table S4). Verapamil and 7-OH coumarin were used as positive controls, which are known to be metabolized by the liver. Microsomes and hepatocytes from different species were used to predict metabolic stability in humans, monkeys, dogs, rats, and mice. Intrinsic clearance (Clint) was scaled-up based on each species’ liver weight and microsome/hepatocyte concentration. In liver microsomes, AX-7 had longer half-life (T1/2) than Verapamil and 7-OH coumarin across all species tested (Table S3). For example, in mouse microsomes, AX-7 T1/2 was 64.63 min, while T1/2 was 10.32 min for Verapamil. Similarly, the Clint was markedly lower with AX-7 treatment than with Verapamil or 7-OH coumarin. In mouse samples, the scale-up Clint was 94.35 mL/min/kg with AX-7 treatment and 590.70 mL/min/kg with Verapamil treatment. This trend was consistent when tested in hepatocytes (Table S4). In mouse hepatocytes, the T1/2 was 129.48 min with AX-7 treatment and only 16.43 min with Verapamil treatment. The scale-up Clint in mouse hepatocytes was 126.45 mL/min/kg with AX-7 treatment and 996.52 mL/min/kg with Verapamil. Taken together, AX-7 has acceptable Caco-2 permeability and is likely not rapidly metabolized by the liver, suggesting AX-7 could have reasonable duration of action in an in vivo setting.
We next tested the pharmacokinetics (PK) of AX-4, AX-7, and alexidine to determine the suitability of the analogs in vivo. Female BALB/c mice were treated with 5 mg/kg of the compounds by intraperitoneal injection and the blood plasma was collected starting at 15 min post-injection and sampled over 24 h (Figure 2A). The terminal elimination half-life (T1/2) of AX-7 was approximately three times longer than for AX-4 (17.0 vs. 5.15 h; Figure 2B), indicating AX-7 may have longer-lasting biological effects than AX-4. AX-7 trended toward an increased T1/2 than alexidine (17.0 vs. 9.5 h). In addition to a longer T1/2, AX-7 also had the highest maximum plasma concentration (Cmax) among the three compounds tested (Figure 2C). The Cmax was defined as the highest concentration of each individual compound when sampled throughout the 24-h period, regardless of timepoint. The Cmax for AX-7 was nearly four times higher than AX-4 (540 vs. 140 ng/mL) and twice as high as alexidine (540 vs. 268 ng/mL).
Figure 2.

In vivo applicability of alexidine analogs
(A–C) Female BALB/c mice were intraperitoneally (IP) injected with 5 mg/kg of the indicated compounds. Blood was sampled at the indicated times after injection and the concentrations of the compounds in the plasma were determined. The half-life (B) represents the terminal elimination half-life of each compound.
(D) Male BALB/c mice were IP injected with 2 mg/kg of AX-7, once per day for five days. Plasma, lungs, and kidneys were harvested at 0.25, 1, and 4 h after the last dose for determination of AX-7 concentration (ng/mL for plasma, ng/g for kidney and lung tissues). For (A–D), n = 3 mice per group. Mean ± SD is shown. An ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to determine significance, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
Based upon the PK data above, AX-7 emerged as the most promising in vivo candidate; therefore, we next tested the tissue distribution of AX-7 in the plasma, lungs, and kidneys by treating male mice once a day for five days with 2 mg/kg AX-7. Tissues and blood were collected at 0.25, 1, and 4 h after the last injection. While the AX-7 concentration in the blood plasma decreased over time, the amount in the lungs and kidneys persisted and at higher concentrations (Figure 2D). By 4 h post-treatment, the concentration of AX-7 in the lungs was about 22 times higher than in the plasma (418 ng/g vs. 18.3 ng/mL) and approximately 370 times higher in the kidneys than the plasma (6783 ng/g vs. 18.3 ng/mL). Urine was collected within 4 h of the last treatment to test for AX-7 excretion. The amount of AX-7 detected in mouse urine 4 h post-treatment was only 0.017% of the final dose (Table S5), suggesting that while AX-7 may be excreted by the kidneys, it may be negligible after 4 h. Importantly, no abnormal clinical symptoms were observed in the mice throughout the duration of the experiment, suggesting the accumulation of AX-7 in these tissues was tolerable.
Alexidine analogs disrupt mitochondrial cristae structure
After having established AX-4 and AX-7 as anti-proliferative in vitro and AX-7 with favorable absorption, distribution, metabolism, and excretion (ADME)-PK properties for in vivo use, we sought to determine the mode of action. We previously reported that alexidine alters cellular metabolism37 and hypothesized that the analog-induced reduction in cell proliferation was a result of energy imbalance. Furthermore, we performed a pilot proteomics study and analyzed proteins from H1299 cells treated with 1 μM alexidine or DMSO control using Gene Ontology (GO) and PANTHER. Analyses using GO Biological Process and GO Cellular Component revealed enrichment of processes and components involved with an active cell cycle (ex: positive regulation of chromosome condensation, positive regulation of mitotic metaphase/anaphase transition, ribonucleoside-diphosphate reductase complex) in DMSO-treated cells rather than alexidine-treated cells (Tables S6, S7, and S8), which corroborates the cell cycle data in Figure 1. Interestingly, there was also enrichment in DMSO-treated cells of mitochondria-related processes and components (ex: mitochondrial translation, mitochondrial small ribosomal subunit, mitochondrial inner membrane) (Tables S6, S7, and S8). Additionally, others reported that alexidine disrupts mitochondria structure, particularly by inducing damage to the cristae.23,43 Therefore, we investigated the mitochondria ultrastructure of the analog-treated cells, as our pilot proteomics study and previous data suggested mitochondria may be affected by bisbiguanides. After 48 h of treatment, AX-4, AX-7, and alexidine disrupted mitochondrial structure as observed using electron microscopy (Figure 3A). In DMSO- and AX-2-treated cells, most mitochondria had a typical morphological structure with intact cristae. At 1 μM of AX-4, AX-7, and alexidine, there were still several identifiable mitochondria. However, with 2 μM treatment, it was difficult to identify normal mitochondria structures; these cells contained mitochondria with seemingly intact outer membranes, but the cristae were disordered or absent. To further characterize mitochondria with and without intact cristae, we used confocal microscopy to probe for the mitochondrial import receptor subunit TOM20 homolog (TOM20), as a marker of the outer mitochondrial membrane, and cytochrome c oxidase assembly factor 7 (COA7), which is involved with the formation of electron transport chain complexes in the cristae and has been shown to be depleted upon alexidine treatment43,44 (Figure 3B). In DMSO and AX-2 treated cells, both COA7 and TOM20 were present and exhibited mitochondrial localization. With AX-4, AX-7, and alexidine treatment, the TOM20 signal remained persistent, while there was a dose-dependent decrease in COA7 signal compared to DMSO. This corresponded with a smaller Pearson’s correlation coefficient (Pearson’s R value) for AX-4, AX-7, and alexidine treatment than DMSO and AX-2 treatment (Figure 3C), indicating that the analogs differentially affected COA7 and TOM20 levels. These findings were confirmed using western blot, which showed a dramatic dose-dependent decrease in COA7 protein with AX-4, AX-7, and alexidine treatment, while TOM20 remained present (Figures 3D and S3). Taken together, these alexidine analogs preferentially disrupt the mitochondrial cristae, while leaving the outer membrane largely intact.
Figure 3.

Alexidine analogs disrupt mitochondrial cristae structure
(A) Representative images of transmission electron microscopy micrographs of H1299 cells treated with the indicated analogs at 1 or 2 μM for 48 h. Regions outlined in white are shown below as a zoomed image focusing on the mitochondria. Scale bar = 1 μm for upper panels; zoomed scale bar = 250 nm.
(B) Representative confocal immunofluorescence images of H1299 cells treated as in (A) and probed for COA7 (magenta) and TOM20 (green) to mark the outer mitochondrial membrane. Merged images include DAPI to stain the nuclei and TOM20/COA7 overlapping signal appears white. Scale bar = 10 μm.
(C) The Pearson’s correlation coefficient (R) comparing COA7 and TOM20.
(D) COA7 and TOM20 were detected by western blot of whole cell lysates of cells treated with the analogs as in (A). Actin was used as a loading control. See Figure S3 for quantification of western blots. For all experiments, three biological replicates were performed. For (C), Mean ± SD is shown of three biological replicates with at least 60 cells analyzed for each treatment per replicate. Significance was assessed using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
Alexidine analogs induce mitochondrial bioenergetic stress
Since mitochondrial structure was drastically damaged with AX-4, AX-7, and alexidine treatment, we next assessed if mitochondrial bioenergetics was also altered. When cells are actively using mitochondria for oxidative phosphorylation, a proton gradient and mitochondrial membrane potential (ΔΨm) are generated between the intermembrane space and matrix components of the mitochondria.45,46 The relative ΔΨm can be assessed using a cationic fluorescent dye, such as tetramethylrhodamine methyl ester (TMRM).47 We assessed the TMRM levels by flow cytometry in cells treated for 48 h with 1 μM of alexidine or the analogs. As observed with the other assays, AX-2 did not influence ΔΨm (Figure 4A). However, AX-4 and AX-7 depolarized the mitochondria to similar levels as alexidine, in congruence with the observed structural mitochondrial defects (Figure 3A). Interestingly, when probed with MitoTracker Green, which localizes to the mitochondria independent of the ΔΨm,48 the MitoTracker signal remained largely unchanged after AX-4, AX-7, and alexidine treatment, in contrast to the marked reduction in TMRM signal (Figure 4A). Therefore, the reduction in ΔΨm is not wholly due to a decrease in mitochondrial load, but rather, by depolarizing events. We also co-treated cells with the analogs and carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), an uncoupler of mitochondrial oxidative phosphorylation and a depolarizing agent.49 Cells treated with AX-2 were only depolarized if co-treated with FCCP (Figure 4B). AX-4, AX-7, and alexidine all depolarized the cells to near maximum, as the addition of FCCP only marginally depolarized cells further. To quantitatively assess changes in ΔΨm with analog treatment, cells were treated with the analogs for 48 h and subjected to short treatment with oligomycin, a known inhibitor of the mitochondrial ATPase. The addition of oligomycin forces mitochondria into a hyperpolarized state due to the lack of proton movement to generate ATP.50,51,52 Under DMSO or AX-2 conditions, approximately 1% of the cells were unable to hyperpolarize with the addition of oligomycin (Figure 4C). In contrast, AX-4, AX-7, and alexidine induced an increase in the percentage of cells unable to hyperpolarize (5.25, 12.0, and 18.1%, respectively), indicative of impaired electron transport chain and/or ATPase activity. AX-7 was more detrimental to ΔΨm than AX-4, as it produced over 2-fold more cells with the inability to hyperpolarize (12.0 vs. 5.25%). To confirm the analogs were depolarizing mitochondria, lysates from treated cells were assessed for the levels of phosphorylated ubiquitin at serine 65 (pUb (S65)) and sequestosome-1 (SQSTM1). Serine/threonine-protein kinase PINK1 (PINK1) accumulates on depolarized mitochondria and specifically phosphorylates ubiquitin on mitochondrial outer membrane proteins to mark the mitochondria as damaged and initiate mitophagy responses, such as the recruitment of adapters like SQSTM1.53,54,55,56,57,58 AX-4, AX-7, and alexidine induced drastic increases in pUb (S65) compared to the DMSO control, while total ubiquitin levels remained relatively unchanged (Figure 4D). Concordantly, SQSTM1 levels also increased with AX-4, AX-7, and alexidine treatment (Figures 4D and S4). Treatment with AX-2 resulted in pUb (S65) and SQSTM1 levels comparable to the DMSO control, consistent with the maintenance of ΔΨm.
Figure 4.

Alexidine analogs induce mitochondrial bioenergetic stress
(A) H1299 cells were treated with 1 μM of the indicated analogs for 48 h. Mitochondrial membrane potential was detected by flow cytometry using TMRM and mitochondrial amount was detected in parallel using MitoTracker Green. Representative histograms normalized to the mode are shown.
(B) Cells were treated as in (A). At the time of TMRM staining, FCCP was added to the samples and cells were analyzed by flow cytometry. Representative histograms normalized to the mode are shown.
(C) Cells were treated as in (A). At the time of TMRM staining, oligomycin was added to the samples and cells were analyzed by flow cytometry. The quantification of the TMRM-low population of three biological replicates is shown in the graph to the right. An ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to determine significance.
(D) Cells were treated for 48 h with the analogs as indicated. Lysates were separated by western blot and probed for phosphorylated ubiquitin (pUb (S65)), total ubiquitin (Ub), SQSTM1, and actin. See Figure S4 for quantification of SQSTM1.
(E) Cells were treated with 1 μM of the indicated analogs for 48 h. The oxygen consumption rate (OCR) was detected over time by the Agilent Seahorse XF Analyzer with the addition of oligomycin (oligo), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and a rotenone/antimycin mixture at the indicated timepoints. A representative curve is shown.
(F) The basal respiration, maximal respiration, spare respiratory capacity, and ATP-linked respiration were calculated from the OCR in (E).
(G) Cells were treated with the indicated analogs for 48 h. The media was collected and lactate amount detected. Lactate concentration was normalized relative to cell count and DMSO control. Significance was assessed using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test. For all experiments, three biological replicates were performed. Mean ± SD is shown. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
We next evaluated whether the mitochondrial damage induced by the analogs and alexidine impacted mitochondrial metabolism. Oxygen consumption rate (OCR) was measured after 48 h of treatment with 1 μM of the compounds. Sequential treatment with oligomycin, FCCP, and a rotenone/antimycin mixture to affect processes of mitochondrial respiration allowed for comprehensive analysis of mitochondrial bioenergetics.59 AX-2 did not alter OCR in comparison to the DMSO control (Figures 4E and 4F). Conversely, AX-4 and AX-7 drastically depleted the overall OCR to levels similar to alexidine. Basal OCR and ATP-linked respiration were dramatically reduced, indicating that cells treated with AX-4, AX-7, or alexidine were generating less ATP via the mitochondrial ATPase than control- or AX-2- treated cells. Additionally, the maximal respiration and spare respiratory capacity were also reduced in AX-4-, AX-7-, and alexidine-treated cells compared to DMSO- or AX-2-treated cells, supporting the observations of impaired cristae, where oxidative phosphorylation occurs.
Through glycolysis, glucose is converted to pyruvate, which can be converted to lactate by lactate dehydrogenase A/C (LDHA/C) or converted to acetyl-CoA by the pyruvate dehydrogenase (PDH) complex and utilized for the TCA cycle and oxidative phosphorylation. If mitochondrial metabolism is disrupted, cells may rely more on glycolysis alone for energy production and thus produce more lactate. Therefore, since analog treatment reduced mitochondrial respiration, we asked whether cells conversely increased lactate production. AX-2 had no effect on extracellular lactate compared to control (Figure 4G). AX-4, AX-7, and alexidine treatment increased lactate concentration with both 1 μM and 2 μM treatments, suggesting a switch to glycolysis. Interestingly, 2 μM AX-4 treatment produced higher lactate levels than 2 μM AX-7 and was equally as effective as alexidine. Additionally, we probed for protein levels of key metabolic enzymes after treatment with the analogs. Pyruvate kinase M2 (PKM2) and LDHA/C protein levels remained unchanged (Figures S5A and S5B), indicating that the increase in extracellular lactate was likely due to an increase in pyruvate availability for and activity of LDHA/C, not an increase of the protein level itself. We also probed for pyruvate dehydrogenase (PDHA1) and phosphorylated PDH at serine 293 (pPDH S293), an inhibitory phosphorylation. We observed a modest decrease in the levels of PDHA1 with AX-4, AX-7, and alexidine treatment (Figure S5C), likely due to the disruption of mitochondrial structure, as PDHA1 localizes to the mitochondria. Additionally, the ratio of pPDH S293:PDHA increased with AX-4, AX-7, and alexidine treatment (Figure S5D), indicative of decreased activity of the PDH complex, which likely increased the available pyruvate to be processed by LDHA/C to produce lactate. Taken together, the analogs AX-4 and AX-7 are inducers of mitochondrial depolarization and bioenergetic stress.
Alexidine analogs inhibit cell invasion
Since AX-4 and AX-7 dramatically reduced mitochondria-derived energy, we hypothesized that the mitochondrial defects would impact cell behavior. Others have shown that mitochondria localized to the leading edge of cells are important to provide energy during cell invasion60 and we previously determined that alexidine inhibits H1299 collective cell invasion.37 Therefore, we tested whether AX-4 and AX-7 maintain that ability to reduce invasion. Cells embedded as spheroids in recombinant basement membrane matrix were treated with the analogs for 48 h. AX-4 and AX-7 inhibited invasion to the extent of alexidine with 2 μM treatment, reducing the invasive area to 24.7%, 34.8%, and 36.8% of the control, respectively (Figures 5A and 5B). To assess canonical signaling pathways involved in cell motility, cells treated with the analogs in 2D cell growth conditions were harvested for protein extraction. Phosphorylated focal adhesion kinase (pFAK (Y397)) trended toward a significant decrease with AX-4, AX-7, and alexidine treatment in a dose-dependent manner (Figures 5C and S6), corroborating the reduction in invasion. There was no observable difference in the levels of pFAK (Y397) with AX-2 treatment. Therefore, AX-4 and AX-7 not only induce bioenergetic stress, but also impact cell phenotype. Taken together, we have created analogs of alexidine, AX-4 and AX-7, capable of inducing drastic mitochondrial damage, reductions in cell growth and invasion, and applicable for use in vivo.
Figure 5.

Alexidine analogs inhibit cell invasion
(A) H1299 cells were embedded as spheroids in recombinant basement membrane matrix and treated with the indicated analogs for 48 h. Representative images of cell invasion are shown. The borders of cell invasion are outlined in white. Scale bar = 250 μm.
(B) Quantification of invasive area of cells treated as in (A) and normalized to DMSO control.
(C) Cells were treated for 48 h with the analogs, as indicated, in 2D cell culture. Lysates were separated by western blot and probed for phosphorylated FAK (pFAK (Y397)) and total FAK. Actin was used as a loading control. See Figure S6 for quantification of the western blots. For all experiments, three biological replicates were performed. For (A) and (B), at least four spheroids per treatment were assessed for each biological replicate. Mean ± SD is shown. Significance was assessed using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test, ∗p ≤ 0.05, ∗∗p ≤ 0.01.
Discussion
Targeting cancer metabolism has emerged as a promising therapeutic approach to cancer.3,5 In this study, we designed analogs of the bisbiguanide small molecule, alexidine, and found AX-4 and AX-7 to be potent inducers of mitochondrial structural and bioenergetic stress in non-small cell lung cancer. The disruption in cellular metabolism corresponded with G1/G0 cell-cycle arrest and reduced invasive capacity, consistent with our previous study using alexidine,37 as well as reports for other biguanides such as metformin.22 Our initial screen included seven analogs of the parent compound, alexidine, each with differing carbon linker length or side chains (Figures 1A and 1B). We found that decreasing the linker length to four carbons from six (AX-3) did not confer better efficacy than alexidine and the analog with a single biguanide unit and subsequently no linker (AX-1) lost activity (Figure 1C). Increasing the linker to eight carbons (AX-4) had better efficacy than those with short or no linker (Figures 1C and 1D). These findings are consistent with a previous report of chlorhexidine (a bisbiguanide with similar structure to alexidine) analogs, where reducing the linker to four carbons resulted in minimal activity against panels of bacteria strains, while longer linkers had increased activity.36 This was potentially due to physical strain on the bacterial membrane components with increased analog linker length, causing “leakage” of contents.38 The terminal side chain structure also may influence leakage, as analogs with short, three carbon long linkers did not cause leakage.38 This could provide rationale as to why AX-2 was ineffective in our study, as it contained only one-carbon methyl hydrocarbon side chains (Figure 1B). Finally, it was suggested that branching of the side chains improves efficacy against bacteria compared to straight chain groups of the same length.36 This may suggest why AX-10, with straight chain methyl-butyl ether groups, was also ineffective (Figure 1C). Our lead compounds, AX-4 and AX-7, both contained branched side chains of differing carbon lengths; however, there may be a steric limitation on branch length, as AX-5 with longer branched terminal groups was not as effective. Overall, a combination of long linker length with branched carbon side chains resulted in the most effective analogs in our viability screens, possibly due to physical strain on the mitochondrial membranes.
Alexidine reduces tumor growth in several cancers, including head and neck, liver, and leukemia,23,24,25 and we, therefore, propose that AX-7 will be useful in the control of cancer progression across many cancer types. Notably, we established in vivo tolerability of AX-4 and AX-7 and identified AX-7 as superior to AX-4 and alexidine in length of terminal half-life and maximum plasma concentration (Figure 2). AX-7 was retained in lung and kidney tissues, suggesting AX-7 could have a long duration of action in these organs. Further studies on dosing schedule will be useful for future in vivo applications, as it may be possible to reduce dosing to only two or three times a week and still retain accumulation in organs of interest. Therefore, further testing to establish on- or off-target toxicities and therapeutic range will be necessary to ensure the safety of these analogs.
After establishing the feasibility of using AX-4 and AX-7 in vivo, we continued in vitro characterization of the analogs by determining their mode of action. As mitochondria are thought to be derived from bacteria,33,34,35 it is possible the mechanism of action of our analogs on mitochondrial membranes is similar to the mechanisms of physical membrane disruption proposed in the bacterial screens described above. While some reports suggest that alexidine is a phosphatidylglycerophosphatase and protein-tyrosine phosphatase 1 (PTPMT1) inhibitor,61 others recently reported a broader structural effect on mitochondria, underscored by alexidine’s preferential binding to inner mitochondrial membrane phospholipids and disruption of the mitochondrial cristae.31,43 Therefore, it is likely that AX-4 and AX-7 have a similar preferential affinity to the phospholipids of the cristae rather than the outer mitochondrial membrane. This is evidenced by the observation that cells treated with AX-4 and AX-7 have damaged mitochondrial cristae and loss of the respiratory chain assembly factor, COA7, while the outer membrane remained intact, marked by TOM20 (Figure 3). These mitochondrial structural changes are in accordance with historical observations that chlorhexidine affects bacteria permeability and renders the bacteria as “empty shells”.62,63 After treatment with AX-4, AX-7, or alexidine, several mitochondria have a similar “empty shell” phenotype, as observed using electron microscopy (Figure 3A). Additionally, many mitochondria, particularly in the AX-4-treated samples, exhibited disordered cristae, whereby the cristae membranes created concentric layers within each other, similar to the “onion-like” effect Houston et al. observed.43 Taken together, we propose that AX-4 and AX-7 interact with phospholipids in the mitochondrial cristae and cause physical strain to the membrane resulting in deformed and dysfunctional cristae.
Most targeted therapies are aimed at affecting a singular protein or family of proteins. By creating alexidine analogs, we leveraged the broader impact on mitochondrial metabolism via disruption of the physical mitochondrial structure rather than a single node in the metabolic signaling cascade. This may create a larger bioenergetic impact among cancers with varying modes of heightened mitochondrial respiration. Our analogs reduce overall oxygen consumption rate and render the cells less capable of responding to changes in energy demands (Figures 4E and 4F). Additionally, as disruption of the cristae not only affects the electron transport chain, but also the TCA cycle, AX-4 and AX-7 may have detrimental effects on other biosynthetic pathways beyond limiting mitochondrial ATP production.5 Therefore, we created small molecules with a multimodal approach to targeting cancer metabolism. Our analogs contribute to the field of anti-cancer strategies aimed at targeting the mitochondria and other critical organelles. For example, there are promising approaches that utilize reactive oxygen species (ROS) produced in mitochondria to release and amplify mitochondria-targeted drugs and induce mitochondrial stress in cancer cells.64 Photodynamic therapy can be improved by the use of mitochondria-targeted photosensitizers.65 Additionally, approaches to not only target the mitochondria, but dually-target the mitochondria and the endoplasmic reticulum (ER) allow for enhanced organelle dysfunction and cytotoxic effects.66 These examples show promising cancer cell growth inhibition both in vitro and in vivo. Taken together, the analogs, AX-4 and AX-7, are part of a growing field of mitochondrial disruptors.
Furthermore, we demonstrated that AX-4 and AX-7 cause depolarization of the ΔΨm and initiation of mitophagy (Figure 4) in concert with reduced oxidative phosphorylation. As alterations in mitochondrial metabolism and autophagy underly several diseases, further applications of AX-4 and AX-7 could be extended as tools to better understand neurodegenerative disorders, such as Parkinson’s,67,68 Huntington’s,69 and Alzheimer’s70 diseases. Additionally, as AX-4 and AX-7 caused metabolic rewiring, these analogs could be useful to combat metabolic disorders, such as diabetes71 and obesity.72
In summary, we developed bisbiguanide compounds with strong efficacy in inducing mitochondrial dysfunction, metabolic stress, and reduced cell invasion. Further, we established the feasibility of using AX-7 for in vivo studies, as it is retained in mouse tissue and daily dosing is tolerated. Overall, the alexidine analogs will further our understanding of the importance of mitochondrial metabolism as a therapeutic target for metastatic cancers.
Limitations of the study
In this study, the alexidine analogs we developed leverage the structural components necessary for effective bacterial targeting, thus producing potent anti-mitochondrial effects in human cells. While we established that the analogs, AX-4 and AX-7, induce mitochondrial cristae damage and overall metabolic rewiring, further studies are needed to elucidate the exact mechanism the analogs use to interact with the mitochondria. Future experiments to determine binding partners of the analogs and analog localization within the mitochondria will be useful to determine how the analogs induce structural damage to the mitochondria. Additionally, we established the tolerability of AX-4 and AX-7 in vivo; however, further pre-clinical studies are needed to determine the effects of the analogs in in vivo cancer models. These studies will be important to establish the anti-proliferative and/or anti-metastatic effects of the analogs, and will support the continued development and study of bisbiguanides for use in human disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Histone H3 (Ser10) XP rabbit mAb | Cell Signaling | Cat# 53348; RRID: AB_2799431 |
| Cdc2/CDK1 (POH1) mouse mAb | Cell Signaling | Cat# 9116; RRID:AB_2074795 |
| CDK4 (D9G3E) rabbit mAb | Cell Signaling | Cat# 12790; RRID:AB_2631166 |
| CDK6 (DCS83) mouse mAb | Cell Signaling | Cat# 3136; RRID:AB_2229289 |
| Cyclin D1 (E3P5S) XP rabbit mAb | Cell Signaling | Cat# 55506; RRID:AB_2827374 |
| Actin, rabbit polyclonal | MilliporeSigma | Cat# A2066; RRID:AB_476693 |
| COA7, rabbit polyclonal | Proteintech | Cat# 25361-1-AP; RRID:AB_2880043 |
| TOM20 (D8T4N) rabbit mAb | Cell Signaling | Cat# 42406; RRID:AB_2687663 |
| TOM20 mouse mAb | Proteintech | Cat# 66777-1-Ig; RRID:AB_2882123 |
| Phospho-Ubiquitin (Ser65) (E2J6T) rabbit mAb | Cell Signaling | Cat# 62802; RRID:AB_2799632 |
| Total Ubiquitin (EPR8830) rabbit mAb | Abcam | Cat# ab134953; RRID:AB_2801561 |
| SQSTM1/p62 (D5E2) rabbit mAb | Cell Signaling | Cat# 8025; RRID:AB_10859911 |
| PKM2 (D78A4) XP rabbit mAb | Cell Signaling | Cat# 4053; RRID:AB_1904096 |
| LDHA/LDHC (C28H7) rabbit mAb | Cell Signaling | Cat# 3558; RRID:AB_2135109 |
| PhosphoDetect Anti-PDH-E1α (pSer293) rabbit pAb | Millipore Sigma | Cat# AP1062; RRID:AB_10616069 |
| PDHA1 monoclonal antibody (9H9AF5) | ThermoFisher | Cat# 459400; RRID:AB_2532238 |
| Phospho-FAK (Tyr397) rabbit Polyclonal | ThermoFisher | Cat# 44-624G; RRID:AB_2533701 |
| Total FAK mouse mAb | BD Biosciences | Cat# 610087; RRID:AB_397494 |
| Chemicals, peptides, and recombinant proteins | ||
| Alexidine dihydrochloride | MilliporeSigma | A8986 |
| AX-1, AX-2, AX-3, AX-4, AX-5, AX-7, AX-10 | This paper | N/A |
| propidium iodide | ThermoFisher | P1304MP |
| RNase A | Qiagen | 19101 |
| tetramethylrhodamine methyl ester (TMRM) | ThermoFisher | I34361 |
| MitoTracker Green | ThermoFisher | M7514 |
| FCCP | MilliporeSigma | C2920 |
| oligomycin | MilliporeSigma | 495455 |
| Matrigel Basement Membrane Matrix | Corning | 356237 |
| Critical commercial assays | ||
| WST-8 | Dojindo | CK04 |
| Pierce BCA assay | ThermoFisher | 23227 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | 103015–100 |
| Lactate Assay Kit-WST | Dojindo | L256-20 |
| Experimental models: Cell lines | ||
| NCI-H1299 | ATCC | CRL-5803; RRID:CVCL_0060 |
| Caco-2 | ATCC | HTB-37; RRID:CVCL_0025 |
| Human Microsomes | Corning | 452117 |
| Monkey Microsomes | XENOTECH | P2000 |
| Dog Microsomes | XENOTECH | D1000 |
| Rat Microsomes | XENOTECH | R1000 |
| Mouse Microsomes | BioreclamationIVT | M00501 |
| Human Hepatocytes | BioreclamationIVT | X008001 |
| Monkey Hepatocytes | RILD | HP-SXH-02M |
| Dog Hepatocytes | BioreclamationIVT | M00205 |
| Rat Hepatocytes | BioreclamationIVT | M00005 |
| Mouse Hepatocytes | BioreclamationIVT | M005052 |
| Experimental models: Organisms/strains | ||
| Mice: BALB/c | Pharmaron (CRO); SPF (Beijing) Biotechnology Co. Ltd | B201 |
| Software and algorithms | ||
| GraphPad Prism 9.3.1 (471) | GraphPad Software | https://www.graphpad.com/features |
| FlowJo (v10.8.0) | BD Life Sciences | https://www.flowjo.com/ |
| ImageJ/FIJI | Schindelin et al.73 | https://fiji.sc/ |
| Gene Ontology (version 2023-11-15) powered by PANTHER (version 18.0) | Ashburner et al.74; The Gene Ontology Consortium75; Thomas et al.76 | https://geneontology.org/ |
| Other | ||
| μ-Slide 4 Well Glass Bottom | ibidi | 80427 |
| Seahorse XF Cell Culture Microplate | Agilent | 102342–100 |
| Amicon Ultra-0.5 mL filter (3K cutoff) | MilliporeSigma | UFC500396 |
| Bio-Rad ChemiDoc Imaging System | Bio-Rad | 12003153 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Adam Marcus (aimarcu@emory.edu).
Materials availability
The bisbiguanide analogs (patent pending) generated in this study can be requested through the Emory University Office of Technology Transfer.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell culture
H1299 non-small cell lung cancer cells derived from a male patient were purchased from ATCC (Manassas, VA) and grown in RPMI media supplemented with 10% FBS and 1% pen/strep. Cells were maintained in a humidified incubator at 37°C and 5% CO2 and passaged every two or three days.
Mouse studies
All mouse studies were performed by Pharmaron (Contract Research Organization, Beijing, China) in accordance with their approved protocols. Adult male and female BALB/c mice aged 8–14 weeks were used for the studies described below.
Method details
Synthesis methods of the bisbiguanide analogs
The bisbiguanide analogs based on the parent structure, alexidine, were synthesized in consultation with Anthem Biosciences (Contract Research and Innovation Service Provider (CRISP), Bangalore, India). The synthesized analogs were dissolved in DMSO to make a 20 mM stock for in vitro studies. A general scheme is shown in Figure S1. Analytical (1H NMR and HPLC) spectra of the alexidine analogs are shown in Data S1. The steps of the synthesis are described below and structures are provided in Figure S1.
Synthesis of alexidine analog (AX-1)
Typical method for preparation of HCl starting materials 1, 4, 5, 7, 7a-c, 8, and 10 from their free bases
To a solution of n-propyl amine (free base) (20 mmol, 1 equiv.) in methyl-tertiary-butyl ether (MTBE) (20 mL) at 0°C, was added a solution of HCl in 1,4-dioxane (4.5 M, 1.2 equiv.) dropwise and slowly warmed up to room temperature and stirred for 2 h. The reaction mixture was concentrated to get crude off-white gummy solid (i.e., 1). These starting materials have shown appropriate matching NMR signals and used for the next reaction.
Synthesis of precursor 3
A suspension of 4 (9 mmol, 1 equiv.) and 2 (9 mmol, 1 equiv.) in n-BuOH (10 mL) was heated up to 120°C for 16 h. The reaction was monitored by LCMS. At the conclusion of consumption of 4, the reaction mixture was cooled to 25°C–30°C and stirred for 30 min and filtered the suspension to get pure compound 3 as an off-white solid. Yield: 1.1 g (62%); 1H NMR (400 MHz, DMSO-d6): δ 7.25 (br s, 1H), 6.75 (br s, 2H), 2.95–3.00 (m, 2H), 1.35–1.20 (m, 9H), 0.90–0.70 (m, 6H). LCMS (M + H): m/z calcd. for C10H21N4 197.30, found 197.60.
Synthesis of AX-1
A suspension of precursor 3 (5.6 mmol, 1 equiv.) and amine 1 (6.7 mmol, 1.2 equiv.) in n-BuOH (5.5 mL) was heated up to 120°C for 30 h. The reaction was monitored by LCMS. When 93% of the precursor was converted, the reaction mixture was concentrated under reduced pressure to get crude. The crude was purified by preparative-HPLC to get AX-1 as off-white semisolid. Yield: 800 mg (56%); 1H NMR (400 MHz, DMSO-d6): δ 7.79–7.78 (br s, 2H), 6.95 (br s, 3H), 3.03 (br s, 4H), 1.47–1.40 (m, 3H), 1.32–1.24 (m, 8H), 0.87–0.84 (m, 9H). LCMS (M + H): m/z calcd. for C13H29N5 255.41, found 256.50. Purity by HPLC: 99.8%
Synthesis of alexidine analog AX-2
Synthesis of precursor 6
To a suspension of 5 (4.5 mmol, 1 equiv.) and 2 (9 mmol, 2 equiv.) in n-BuOH (5 mL) was heated up to 120°C for 16 h. The reaction was monitored by TLC. The reaction mixture was cooled to 25°C–30°C and stirred for 30 min and filtered the suspension. The collected solid was dried to get off-white solid 6. Yield: 1.2 g (94%); 1H NMR (400 MHz, DMSO-d6): δ 7.10 (br s, 2H), 6.69 (br s, 4H), 3.10–2.85 (m, 4H), 1.50–1.10 (m, 8H). LCMS (M + H): m/z calcd. for C10H19N8 251.31, found 251.30.
Synthesis of AX-2
A suspension of 6 (4. mmol, 1 equiv.), methylamine hydrochloride 7 (8. mmol, 2 equiv.) in n-BuOH (5 mL) was heated up to 120°C for 16 h. The reaction was monitored by TLC. The reaction mixture was cooled to 25°C–30°C and dissolved in MeOH and filtered. The filtrate was concentrated to get crude compound as a yellow gummy-solid. Which was purified by prep-HPLC to provide AX-2. Yield: 175 mg (10% as TFA salt); 1H NMR (400 MHz, DMSO-d6): δ 8.23 (br s, 4H), 7.35–6.75 (br s, 6H), 3.10–3.05 (m, 4H), 2.71 (s, 6H), 1.46 (s, 4H), 1.27 (s, 4H). LCMS (M + H): m/z calcd. for C12H28N10 312.42, found 313.60. Purity by HPLC: 99.8%.
Synthesis of alexidine analog(AX-3)
Synthesis of precursor 9
To a suspension of 8 (1.2 mmol, 1 equiv.) and 2 (2.4 mmol, 2 equiv.) in n-BuOH (10 mL) was heated up to 120°C for 16 h. The reaction was monitored by TLC. TLC indicated complete consumption of 8. The reaction mixture was cooled to 25°C–30°C and stirred for 30 min and filtered the suspension and resulting solid was dried to get 9. Yield: 2.1 g (77%): 1H NMR (400 MHz, DMSO-d6): δ 7.24 (br s, 2H), 6.75 (br s, 4H), 3.15–2.95 (m, 4H), 1.50–1.30 (m, 4H).
Synthesis of AX-3
To a suspension of 9 (4.5 mmol, 1 equiv.) and 4 (9 mmol, 2 equiv.) in n-BuOH (5 mL) was heated up to 155°C for 20 h. The reaction was monitored by LCMS for 20 h. The reaction mixture was cooled to rt and dissolved in MeOH and filtered, the filtrate was concentrated to get crude which was purified through column chromatography using basic alumina (20% MeOH in DCM) to afford the solid which was further triturated with MTBE: isopropanol (IPA) (9:1) to get pure compound AX-3 as off-white solid. Yield: 150 mg (7%); 1H NMR (400 MHz, DMSO-d6): δ 7.68 (br s, 4H), 6.95 (br s,6H), 3.05–3.00 (m, 8H), 1.46 (br s, 6H), 1.30–1.22 (m, 16H), 0.85–0.80 (m, 12H). LCMS (M + H): m/z calcd. for C24H52N10 480.75, found 481.90. Purity by HPLC: 99.2%.
Synthesis of alexidine analog(AX-4)
Synthesis of precursor 11
To a suspension of 10 (4.5 mmol, 1 equiv.) and 2 (9 mmol, 2 equiv.) in n-BuOH (5 mL) was heated up to 120°C for 16 h. The reaction was monitored by TLC. The reaction mixture was cooled to 25°C–30°C and stirred for 30 min and filtered the suspension and dried to get off-white solid 11. Yield: 1.2 g (94%); 1H NMR (400 MHz, DMSO-d6): δ 7.00–6.30 (m, 6H), 3.15–2.90 (m, 4H), 1.50–1.10 (m, 12H).
Synthesis of AX-4
To a suspension of 11 (3.5 mmol, 1 equiv.) and 4 (7.2 mmol, 2.2 equiv.) in n-BuOH (5 mL) was heated up to 120°C for 20 h. The reaction was monitored by LCMS for 20 h. The RM was concentrated under reduced pressure to get crude as a gummy solid, which was purified by preparative-HPLC to provide 11. Yield: 80 mg (6%). 1H NMR (400 MHz, DMSO-d6): δ 7.78 (br s, 4H), 6.95 (br s, 6H), 3.10–3.00 (m, 8H), 1.42 (br s, 6H), 1.27–1.22 (m, 24H), 0.90–0.70 (m, 12H). LCMS (M + H): m/z calcd. For C28H60N10 536.86, found 537.90. Purity by HPLC: 98.5%.
Synthesis of alexidine analogs AX-5, AX-7 and AX-10
Synthesis of AX-5
A suspension of 6 (0.8 mmol, 0.4 equiv.) and 7a (2.0 mmol, 2.5 equiv.) in n-BuOH (5mL) was heated up to 120°C for 16 h. After 16 h by LCMS showed only 50% desired bis-coupled product and 20% of mono-coupled product. The RM was concentrated to get crude semisolid, which was purified by prep HPLC to provide AX-5. Yield: 150 mg (16%); 1H NMR (400 MHz, DMSO-d6): δ 7.27 (br s, 4H), 6.73 (br s, 5H), 3.08–3.03 (m, 8H), 1.44 (br s, 5H), 1.25–1.15 (m, 30H), 0.90–0.85 (m, 12H). LCMS (M + H): m/z calcd. for C30H64N10 564.91, found 566.0. Purity by HPLC: 97.6%.
Synthesis of AX-7
A suspension of 6 (4 mmol) and 7b (8.42 mmol, 2.1 equi.) in n-BuOH (10 mL) was heated up to 120°C for 32 h. After 32 h, LCMS showed 64% of desired bis-coupled product and 21% of mono-coupled. The RM was concentrated to get crude semisolid, which was purified by prep-HPLC. Yield: 175 mg (8%) (formate salt). 1H NMR (400 MHz, DMSO-d6): δ 8.41 (s, 2H), 8.12 (br s, 3H), 7.05 (br s, 7H), 3.05–3.00 (m, 8H), 1.43–1.27 (m, 4H), 1.27–1.20 (m, 10H), 1.05–0.90 (m, 2H), 0.90–0.70 (m, 12H). LCMS (M + H): m/z calcd. for C22H48N10 452.41, found 453.90. Purity by HPLC: 99.4%.
Synthesis of AX-10
A suspension of 6 (160 mg, 1.11 mmol) and 7c (143 mg, 0.54 mmol) in n-BuOH (5 mL) was heated up to 130°C for 20 h. After 20 h, LCMS showed 70% of desired product and 21% of mono-coupled product. The RM was concentrated to get crude, which was purified by prep-HPLC. Yield: 25 mg (10%). 1H NMR (400 MHz, CD3OD): δ 8.50 (br s, 2H), 3.43–3.40 (m, 4H), 3.33–3.30 (m, 6H), 3.22–3.21 (m, 8H), 1.61–1.57 (m, 14H), 1.39–1.35 (m, 5H), NH protons are not detected. LCMS (M + H): m/z calcd. for C20H44N10O2 456.64, found 457.80. Purity by HPLC: 92.2%.
Cell growth assays
For cell growth assays, 1 × 103 cells/well were seeded in a 96-well plate and grown overnight. The next day, cells were treated with serial half dilutions of the specified analogs, starting at 20 μM and diluting down to 0.078 μM. Cells were incubated with the drug for 72 h. For detection using the sulforhodamine B (SRB) assay, cells were fixed with cold 10% TCA for 1 h, washed with water, and dried. Cells were then stained using 0.4% SRB solution for 15 min, washed with water and 1% acetic acid, and dried. 10 mM Tris (pH 10.5) was added to each well. The optical density (OD) was measured at 560 nm. For detection using the WST-8 reagent (Dojindo, CK04), the WST-8 reagent was added directly to the cell culture media at a 1:10 dilution. After 2 h of incubation at 37°C, the optical density was measured at 450 nm.
For both assays, three biological replicates were performed, each with three technical replicates. The OD was normalized to the DMSO control. Data were fit with a nonlinear regression model in GraphPad Prism to determine the IC50 and 95% confidence intervals.
Cell cycle analysis
For measurement of the proportion of cells in each stage of the cell cycle, 1.75 × 105 cells were seeded in 10 cm plates and grown overnight. The next day, cells were treated with 0.5, 1, or 2 μM of the analogs or DMSO as the control. After 48 h, attached and floating cells were collected as a pellet and resuspended in 0.5 mL DPBS. 4.5 mL of cold 70% ethanol was added dropwise with light agitation for approximately 1 min. Cells were stored at 4°C overnight. After washes with PBS and pelleting, cells were resuspended with 40 μg/mL propidium iodide (ThermoFisher, P1304MP) and 100 μg/mL RNase A (Qiagen, 19101) and incubated at 37°C for 30 min with gentle shaking. Stained cells were analyzed by flow cytometry using the BD FACSymphony A3. Cell cycle frequency was modeled using FlowJo. Three biological replicates were performed.
Western blot
For analysis of protein levels in treated cells, 1.75 × 105 cells were seeded in 10 cm plates and grown overnight. The next day, cells were treated with 1 or 2 μM of the analogs or DMSO as the control. After 48 h, cell media was collected to harvest any floating cells. Lysis buffer (2% SDS, 50 mM Tris pH 8.0, 100 mM NaCl, Halt protease/phosphatase inhibitors (ThermoFisher, 78442)) was added to the plates and cells were scraped, collected, and sonicated. Protein concentration was determined by Pierce BCA assay (ThermoFisher, 23227). Proteins were separated using SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and detected using the Bio-Rad ChemiDoc Imaging System (Bio-Rad, 12003153). The following primary antibodies were used: pHistone H3 (S10) (Cell Signaling, 53348); Cdc2/CDK1 (Cell Signaling, 9116); CDK4 (Cell Signaling, 12790); CDK6 (Cell Signaling, 3136); cyclin D1 (Cell Signaling, 55506); actin (MilliporeSigma, A2066); COA7 (Proteintech, 25361-1-AP); TOM20 (Cell Signaling, 42406); pUb (Ser65) (Cell Signaling, 62802); Total Ubiquitin (Abcam, ab134953); SQSTM1/p62 (Cell Signaling, 8025); PKM2 (Cell Signaling, 4053); LDHA/LDHC (Cell Signaling, 3558); pPDH (S293) (MilliporeSigma, AP1062); PDHA1 (ThermoFisher, 459400); pFAK (Y397) (ThermoFisher, 44624G); Total FAK (BD Biosciences, 610087). Three biological replicates were performed. Quantification of the integrated density of western blot bands was performed using ImageJ/FIJI.73 Integrated density was normalized to actin. For pPDH (S293) and pFAK (Y397), integrated density was normalized to their corresponding total protein levels, PDHA1 and FAK, respectively. Normalized values were then compared to the DMSO control and analyzed using GraphPad.
Caco-2 permeability assay
Permeability of the analogs was performed by Pharmaron (Beijing, China). Briefly, Caco-2 cells were grown to a confluent monolayer on transwell plates and assessed by measuring the transepithelial electrical resistance (TEER) across the monolayer. 5 μM of the test compounds were added to either the apical or the basolateral compartment to determine directionality of drug transport. After 2 h, samples from the donor and receiver sides were assessed by LC-MS/MS. Lucifer Yellow was used to determine any leakage.
Hepatic metabolic stability
Metabolic stability of AX-7 was performed by Pharmaron (Beijing, China) in different species of liver microsomes and hepatocytes.40,41,77 Briefly, 2 μM of AX-7 was added to liver microsomes and incubated at 37°C. Aliquots of the reaction solution were taken at 0, 15, 30, 45, and 60 min and assessed by LC-MS/MS. For assessment in hepatocytes, 1 μM of AX-7 was added to the cells and aliquots were taken at 0, 15, 30, 60, 90, and 120 min for assessment by LC-MS/MS.
In vivo pharmacokinetic studies
In vivo pharmacokinetics of AX-4, AX-7, and alexidine were performed by Pharmaron (Beijing, China). Two studies were conducted. For the first study, 5 mg/kg AX-4, AX-7, or alexidine (in 1% DMSO, 99% water solution) was administered via intraperitoneal (IP) injection into female BALB/c mice. Animals were observed to determine any abnormal behavior. Blood plasma was collected at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h. Calibration standards, quality control samples, and test samples were prepared and injected into a liquid chromatography with tandem mass spectrometry (LC-MS/MS) system to quantitatively determine the terminal elimination half-life (T1/2) and maximum concentration. n = 3 mice per treatment.
A second study was conducted in which 2 mg/kg AX-7 (in 1% DMSO, 99% water solution) was administered via IP injection into male BALB/c mice, once a day for five days. Animals were observed to determine any abnormal behavior. Blood plasma, lung, and kidneys were collected at 0.25, 1, and 4 h after the last dose for LC-MS/MS analysis to determine tissue concentration, as described above. Urine samples were collected 0–4 h after the last dose. Lung and kidney samples were homogenized in water at a 1:4 tissue to water ratio. The concentration calculated by LC-MS/MS (in ng/mL) was multiplied by 5 to achieve the actual tissue concentration in ng/g. For urine samples, the percentage of AX-7 excreted was calculated based on the amount detected via LC-MS/MS 0–4 h after the last dose and the initial amount of the final dose administered. For plasma, lung, and kidney samples, n = 3 mice per timepoint. For urine samples, n = 3 mice at the final timepoint.
Proteomics
To conduct a pilot proteomics study of treated cells, 1.75 × 105 H1299 cells were seeded in 10 cm plates and grown overnight. The next day, cells were treated with 1 μM of alexidine or DMSO as the control. After 48 h, cells were washed in DPBS, trypsinized, and collected as a pellet. The cell pellets were washed twice with DPBS, pelleted, and frozen on dry ice and stored at −80°C until further processing. Pellets were then lysed in 8M urea buffer and protein concentration was quantified by BCA assay. 100 μg protein were treated with 5 mM dithiothreitol (DTT) at room temperature for 30 min, followed by treatment with 10 mM iodoacetimide (IAA) for 30 min in the dark. Protein digestion was adapted from a previously published protocol.78 Proteins were then digested with 4 μg of lysyl endopeptidase (Wako) at room temperature overnight and were further digested overnight with 4 μg trypsin (Promega) at room temperature. The resulting peptides were desalted with HLB column (Waters) and were dried under vacuum.
Peptides were analyzed by LC-MS/MS, adapting a published protocol.79 Derived peptides were resuspended in the loading buffer (0.1% trifluoroacetic acid, TFA) and were separated on a Water’s Charged Surface Hybrid (CSH) column (150 μm internal diameter (ID) x 15 cm; particle size: 1.7 μm). The samples were run on an EVOSEP liquid chromatography system and were monitored on a Q-Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer (ThermoFisher Scientific). The mass spectrometer cycle was programmed to collect one full MS scan followed by 20 data dependent MS/MS scans.
Label-free quantification (LFQ) analysis was performed using MaxQuant.79 Spectra were searched in Andromeda and quantification of proteins was performed using summed peptide intensities given by MaxQuant. The quantification method only considered razor plus unique peptides for protein level quantification. LFQ intensities were then compared between DMSO-treated (control) and alexidine-treated cells. Proteins with intensities that were at least 2-fold higher in control cells than alexidine-treated cells were analyzed using Gene Ontology (GO) Enrichment Analysis74,75 (version 2023-11-15) and PANTHER76 (version 18.0) using GO Biological Process and GO Cellular Component annotation datasets. One biological replicate was used for this pilot study.
Electron microscopy
To visualize mitochondrial structure by electron microscopy, 1.75 × 105 cells were seeded in 10 cm plates and grown overnight. The next day, cells were treated with 1 or 2 μM of the analogs or DMSO as the control. After 48 h, cells were washed in DPBS, scraped, and collected as a pellet. The cell pellets were fixed for 4 h at room temperature in a mixture of 2.0% paraformaldehyde and 2.5% glutaraldehyde in 0.1M cacodylate buffer at pH 7.2. Fixed cell pellets were then washed in 0.1 M cacodylate buffer and stored at 4°C until further processing. The samples were then post fixed in 1.0% Osmium tetroxide for 1 h, washed with water, and then stained en bloc with 2% aqueous uranyl acetate for 20 min at 60°C. The samples were then dehydrated in a series of increasing ethanol concentrations up to three changes in 100% ethanol, followed by two changes in 100% propylene oxide. Samples were embedded in Eponate 12 resin and polymerized for 24 h at 60°C. Ultrathin sections were cut with a Leica EM UC6 ultramicrotome, stained with uranyl acetate and lead citrate, and imaged in a Hitachi HT7700 TEM, or a Jeol JEM 1400 TEM both operated at 80KV. Three biological replicates were performed.
Immunofluorescence
To identify in situ COA7 and TOM20 localization, 5 × 103 or 1 × 104 cells were seeded in glass-bottom slides (ibidi, 80427) and grown overnight. The next day, cells were treated with 1 or 2 μM of the analogs or DMSO as the control. After 48 h, cells were fixed in 4% paraformaldehyde for 20 min at room temperature. Cells were then washed (0.2% Triton X-100 in PBS with Ca2+ and Mg2+) and blocked in 3% BSA (in wash buffer) for 1 h. Cells were incubated with primary antibody overnight at 4°C. Primary antibodies used were TOM20 (Proteintech, 66777-1-Ig) and COA7 (Proteintech, 25361-1-AP). The next day, cells were washed and incubated with secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) for 50 min at room temperature. Washed cells were then imaged using a Leica SP8 confocal microscope with a 63× objective. At least 60 cells were imaged per treatment and per three biological replicates. Images were analyzed, processed, and the Pearson’s correlation coefficient determined using ImageJ/FIJI.73
Mitochondrial flow cytometry assays
Mitochondrial membrane potential (ΔΨm) was detected using tetramethylrhodamine methyl ester (TMRM) (ThermoFisher, I34361). 1.75 × 105 cells were seeded in 10 cm plates and grown overnight. The next day, cells were treated with 1 μM of the analogs or DMSO as the control. After 48 h, attached and floating cells were collected as a pellet, washed, and resuspended in flow buffer (RPMI, no phenol red, 2% dialyzed FBS) with or without 2 nM TMRM. Cells were incubated for 30 min at 37°C with modest agitation, then pelleted and resuspended in flow buffer for analysis via flow cytometry using the BD FACSymphony A3.
For the detection of mitochondrial load, cells were prepared as described above and stained with 20 nM MitoTracker Green (ThermoFisher/Invitrogen, M7514) instead of TMRM. Flow buffer with MitoTracker Green did not contain FBS. Cells were incubated for 30 min at 37°C with modest agitation. Samples were then processed as above.
For experiments with the addition of carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) or oligomycin, cells were prepared as described above. During the incubation with TMRM, 200 nM FCCP (MilliporeSigma, C2920) or 1 μM oligomycin (MilliporeSigma, 495455) was added to the cells. Samples were then processed as above.
All data were further analyzed using FlowJo. Three biological replicates were performed for each experiment.
Metabolic assays
To determine the effects of the analogs on oxygen consumption rate (OCR), cells were analyzed using the Agilent Seahorse Cell Mito Stress Test (Agilent Technologies, 103015-100) and a Seahorse XFe24 analyzer, according to the manufacturer’s protocols.59 Briefly, 7.5 × 103 cells were seeded per well in a 24-well Seahorse XF Cell Culture Microplate (Agilent Technologies, 102342-100). The next day, cells were treated with 1 μM of the analogs or DMSO as the control. Four technical replicates were performed for each drug treatment. After 48 h, the cells were prepared for OCR assessment. The day prior, the Seahorse cartridge was hydrated in the provided calibrant. On the day of the assay, cells were incubated in Seahorse XF RPMI media, supplemented with 10 mM glucose, 1 mM sodium pyruvate, and 2 mM glutamine in a non-CO2 incubator for approximately 1 h. During that time, 2.5 μM oligomycin, 2 μM carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and a 0.5 μM rotenone/antimycin mixture were prepared, added to the sensor cartridge, and the cartridge calibrated. Fresh Seahorse media was added to the cells just prior to starting the assay. OCR was detected before and after sequential treatment with the oligomycin, FCCP, and rotenone/antimycin. Cells were trypsinized and counted at the end of the experiment. OCR was normalized per relative cell count. Data was analyzed using the Wave software. Three biological replicates were performed.
For assessment of extracellular lactate amount, 3.75 × 104 or 7.5 × 104 cells were seeded per well in a 6-well plate and grown overnight. The next day, cells were treated with 1 or 2 μM of the analogs or DMSO as the control. After 48 h, media was collected and added to an Amicon Ultra-0.5 mL filter (3K cutoff) (MilliporeSigma, UFC500396) and centrifuged at 14,000 × g for 15 min at 4°C. Cells were trypsinized, collected, and counted to use for normalization. Extracellular lactate was determined using the Lactate Assay Kit-WST (Dojindo, L256-20) using the manufacturer’s protocol. Media was diluted 1:20 and assessed in triplicate. Lactate levels were determined using a lactate standard curve and normalized to the cell count and DMSO control. Three biological replicates were performed.
3D spheroid invasion assay
Spheroids were formed, as previously described.80 Briefly, 3 × 103 cells/well were seeded in low-attachment 96-well plates (Corning) and incubated for three days to form spheroids. Spheroids were then embedded in Matrigel containing 0.5, 1, or 2 μM of the indicated analogs and cultured in media with the indicated analogs in a 35 mm glass-bottom dish. At least four spheroids were embedded per treatment, per three biological replicates. Spheroids were incubated for 48 h. Images were captured at Day 0 and Day 2 using an Olympus IX51 microscope with the 4× objective and Infinity2 CCD camera. Invasion area was measured by outlining the perimeter of the spheroid using ImageJ/FIJI73 and calculated by subtracting the Day 0 spheroid area from the Day 2 area.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism. Experiments in which multiple doses of multiple analogs were compared, significance was assessed using an ordinary two-way ANOVA with a Tukey’s multiple comparisons test. For experiments in which there was a single variable and three or more comparisons made, an ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to determine significance. Data is represented as mean ± SD of three biological replicates, unless specified in the figure legend. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
Acknowledgments
The work presented here was supported by R01 funding from the National Institutes of Health/National Cancer Institute (NIH/NCI) to AIM (R01 CA250422 and R01 CA236369), R01 CA247367 to AIM and MS, P01 funding to AIM and HF (P01 CA257906), F32 CA257436 to JLA, and R50 CA265345 to JKM. This study was also supported by the Robert P. Apkarian Integrated Electron Microscopy Core (IEMC) (RRID:SCR_023537) at Emory University, which is subsidized by the School of Medicine and Emory College of Arts and Sciences. Additional support to the IEMC was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under award number UL1TR000454. Transmission electron micrographs that were collected on the JEOL JEM-1400, 120kV TEM were supported by the NIH Grant S10 RR025679. Research reported in this publication was also supported in part by the Pediatrics/Winship Flow Cytometry Core of Winship Cancer Institute of Emory University, Children's Healthcare of Atlanta (RRID:SCR_022324), the Emory University Integrated Cellular Imaging Core (RRID:SCR_023534) of the Winship Cancer Institute of Emory University, the Emory Integrated Proteomics shared resource (RRID:SCR_023530), and NIH/NCI under award number, 2P30CA138292-04. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. We also thank Dr. Vance Lyon of Davos Pharma for assistance with the synthesis of the analogs and Dr. Yuan Liu of the Emory Biostatistics Shared Resource for assistance with data analysis. The graphical abstract was created with BioRender.com.
Author contributions
Conceptualization, C.M.K., J.L.A., J.K.M., H.F., T.G., and A.I.M.; Methodology, C.M.K., J.L.A., R.D.H., J.K.M.,T.G., and A.I.M.; Formal analysis, C.M.K., J.L.A., I.E.R., and J.K.M.; Investigation, C.M.K, J.L.A., I.E.R., V.M., T.O.K., and J.K.M.; Writing- Original Draft, C.M.K., T.G., and A.I.M.; Writing- Review & Editing, C.M.K., J.L.A., I.E.R., T.O.K., M.S., J.K.M., H.F., T.G., and A.I.M.; Visualization, C.M.K., I.E.R., and J.K.M.; Supervision, R.D.H., M.S., and A.I.M.; Funding Acquisition, J.L.A., M.S., J.K.M., H.F., and A.I.M.
Declaration of interests
The authors declare no competing interests.
Published: March 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109591.
Supplemental information
References
- 1.Pavlova N.N., Thompson C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metabol. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlova N.N., Zhu J., Thompson C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metabol. 2022;34:355–377. doi: 10.1016/j.cmet.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemberg K.M., Gori S.S., Tsukamoto T., Rais R., Slusher B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Invest. 2022;132 doi: 10.1172/JCI148550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elia I., Doglioni G., Fendt S.M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018;28:673–684. doi: 10.1016/j.tcb.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Vasan K., Werner M., Chandel N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metabol. 2020;32:341–352. doi: 10.1016/j.cmet.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair R., Gupta P., Shanmugam M. Mitochondrial metabolic determinants of multiple myeloma growth, survival, and therapy efficacy. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bridges H.R., Jones A.J.Y., Pollak M.N., Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drahota Z., Palenickova E., Endlicher R., Milerova M., Brejchova J., Vosahlikova M., Svoboda P., Kazdova L., Kalous M., Cervinkova Z., Cahova M. Biguanides inhibit complex I, II and IV of rat liver mitochondria and modify their functional properties. Physiol. Res. 2014;63:1–11. doi: 10.33549/physiolres.932600. [DOI] [PubMed] [Google Scholar]
- 9.Dykens J.A., Jamieson J., Marroquin L., Nadanaciva S., Billis P.A., Will Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008;233:203–210. doi: 10.1016/j.taap.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 10.El-Mir M.Y., Nogueira V., Fontaine E., Avéret N., Rigoulet M., Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 11.Owen M.R., Doran E., Halestrap A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 12.LaMoia T.E., Shulman G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021;42:77–96. doi: 10.1210/endrev/bnaa023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pernicova I., Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 14.Pollak M. Overcoming Drug Development Bottlenecks With Repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat. Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 15.McKENDRY J.B., Kuwayti K., Rado P.P. Clinical experience with DBI (phenformin) in the management of diabetes. Can. Med. Assoc. J. 1959;80:773–778. [PMC free article] [PubMed] [Google Scholar]
- 16.Ungar G., Freedman L., Shapiro S.L. Pharmacological studies of a new oral hypoglycemic drug. Proc. Soc. Exp. Biol. Med. 1957;95:190–192. doi: 10.3181/00379727-95-23163. [DOI] [PubMed] [Google Scholar]
- 17.Luft D., Schmülling R.M., Eggstein M. Lactic acidosis in biguanide-treated diabetics: a review of 330 cases. Diabetologia. 1978;14:75–87. doi: 10.1007/BF01263444. [DOI] [PubMed] [Google Scholar]
- 18.Lee J.O., Kang M.J., Byun W.S., Kim S.A., Seo I.H., Han J.A., Moon J.W., Kim J.H., Kim S.J., Lee E.J., et al. Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer Res. 2019;21:115. doi: 10.1186/s13058-019-1204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S.H., Jeon Y., Kang J.H., Jang H., Lee H., Kim S.Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers. 2020;12 doi: 10.3390/cancers12061382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park J.H., Kim Y.H., Park E.H., Lee S.J., Kim H., Kim A., Lee S.B., Shim S., Jang H., Myung J.K., et al. Effects of metformin and phenformin on apoptosis and epithelial-mesenchymal transition in chemoresistant rectal cancer. Cancer Sci. 2019;110:2834–2845. doi: 10.1111/cas.14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park S.Y., Kim D., Kee S.H. Metformin-activated AMPK regulates beta-catenin to reduce cell proliferation in colon carcinoma RKO cells. Oncol. Lett. 2019;17:2695–2702. doi: 10.3892/ol.2019.9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Xu W., Yan Z., Zhao W., Mi J., Li J., Yan H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018;37:63. doi: 10.1186/s13046-018-0731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao M.H.R., Yang C., Tse A.P.W., Wei L., Lee D., Zhang M.S., Goh C.C., Chiu D.K.C., Yuen V.W.H., Law C.T., et al. Genome-wide CRISPR-Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2020.108676. [DOI] [PubMed] [Google Scholar]
- 24.Muselli F., Mourgues L., Rochet N., Nebout M., Guerci A., Verhoeyen E., Krug A., Legros L., Peyron J.F., Mary D. Repurposing the Bis-Biguanide Alexidine in Combination with Tyrosine Kinase Inhibitors to Eliminate Leukemic Stem/Progenitor Cells in Chronic Myeloid Leukemia. Cancers. 2023;15 doi: 10.3390/cancers15030995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yip K.W., Ito E., Mao X., Au P.Y.B., Hedley D.W., Mocanu J.D., Bastianutto C., Schimmer A., Liu F.F. Potential use of alexidine dihydrochloride as an apoptosis-promoting anticancer agent. Mol. Cancer Therapeut. 2006;5:2234–2240. doi: 10.1158/1535-7163.MCT-06-0134. [DOI] [PubMed] [Google Scholar]
- 26.Gjermo P., Eriksen H.M. Unchanged plaque inhibiting effect of chlorhexidine in human subjects after two years of continuous use. Arch. Oral Biol. 1974;19:317–319. doi: 10.1016/0003-9969(74)90193-9. [DOI] [PubMed] [Google Scholar]
- 27.Joyston-Bechal S., Smales F.C., Duckworth R. The use of a chlorhexidine-containing gel in a plaque control programme. Br. Dent. J. 1979;146:105–111. doi: 10.1038/sj.bdj.4804204. [DOI] [PubMed] [Google Scholar]
- 28.Lobene R.R., Soparkar P.M. The effect of an alexidine mouthwash on human plaque and gingivitis. J. Am. Dent. Assoc. 1973;87:848–851. doi: 10.14219/jada.archive.1973.0512. [DOI] [PubMed] [Google Scholar]
- 29.Löe H., Schiott C.R. The effect of mouthrinses and topical application of chlorhexidine on the development of dental plaque and gingivitis in man. J. Periodontal. Res. 1970;5:79–83. doi: 10.1111/j.1600-0765.1970.tb00696.x. [DOI] [PubMed] [Google Scholar]
- 30.Spolsky V.W., Forsythe A.B. Effects of alexidine.2HCL mouthwash on plaque and gingivitis after six months. J. Dent. Res. 1977;56:1349–1358. doi: 10.1177/00220345770560111101. [DOI] [PubMed] [Google Scholar]
- 31.Chawner J.A., Gilbert P. Interaction of the bisbiguanides chlorhexidine and alexidine with phospholipid vesicles: evidence for separate modes of action. J. Appl. Bacteriol. 1989;66:253–258. doi: 10.1111/j.1365-2672.1989.tb02476.x. [DOI] [PubMed] [Google Scholar]
- 32.McDonnell G., Russell A.D. Antiseptics and disinfectants: activity, action, and resistance. Clin. Microbiol. Rev. 1999;12:147–179. doi: 10.1128/CMR.12.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.John P., Whatley F.R. Paracoccus denitrificans and the evolutionary origin of the mitochondrion. Nature. 1975;254:495–498. doi: 10.1038/254495a0. [DOI] [PubMed] [Google Scholar]
- 34.John P., Whatley F.R. Paracoccus denitrificans: a present-day bacterium resembling the hypothetical free-living ancestor of the mitochondrion. Symp. Soc. Exp. Biol. 1975:39–40. [PubMed] [Google Scholar]
- 35.Margulis L. Yale University Press; 1970. Origin of Eukaryotic Cells; Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth. [Google Scholar]
- 36.Baker P.J., Coburn R.A., Genco R.J., Evans R.T. Structural determinants of activity of chlorhexidine and alkyl bisbiguanides against the human oral flora. J. Dent. Res. 1987;66:1099–1106. doi: 10.1177/00220345870660060301. [DOI] [PubMed] [Google Scholar]
- 37.Commander R., Wei C., Sharma A., Mouw J.K., Burton L.J., Summerbell E., Mahboubi D., Peterson R.J., Konen J., Zhou W., et al. Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion. Nat. Commun. 2020;11:1533. doi: 10.1038/s41467-020-15219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher R.G., Quintana R.P. Surface-chemical studies on chlorhexidine and related compounds: II. Interactions with monomolecular-film systems. J. Dent. Res. 1975;54:25–31. doi: 10.1177/00220345750540013001. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z., Hop C.E., Leung K.H., Pang J. Determination of in vitro permeability of drug candidates through a caco-2 cell monolayer by liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2000;35:71–76. doi: 10.1002/(SICI)1096-9888(200001)35:1<71::AID-JMS915>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Barter Z.E., Bayliss M.K., Beaune P.H., Boobis A.R., Carlile D.J., Edwards R.J., Houston J.B., Lake B.G., Lipscomb J.C., Pelkonen O.R., et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metabol. 2007;8:33–45. doi: 10.2174/138920007779315053. [DOI] [PubMed] [Google Scholar]
- 41.Iwatsubo T., Suzuki H., Sugiyama Y. Prediction of species differences (rats, dogs, humans) in the in vivo metabolic clearance of YM796 by the liver from in vitro data. J. Pharmacol. Exp. Therapeut. 1997;283:462–469. [PubMed] [Google Scholar]
- 42.Jia L., Liu X. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr. Drug Metabol. 2007;8:822–829. doi: 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Houston R., Sekine Y., Larsen M.B., Murakami K., Mullett S.J., Wendell S.G., Narendra D.P., Chen B.B., Sekine S. Discovery of bactericides as an acute mitochondrial membrane damage inducer. Mol. Biol. Cell. 2021;32:ar32. doi: 10.1091/mbc.E21-04-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozjak-Pavlovic V., Prell F., Thiede B., Götz M., Wosiek D., Ott C., Rudel T. C1orf163/RESA1 is a novel mitochondrial intermembrane space protein connected to respiratory chain assembly. J. Mol. Biol. 2014;426:908–920. doi: 10.1016/j.jmb.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Phil. Soc. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 46.Zorova L.D., Popkov V.A., Plotnikov E.Y., Silachev D.N., Pevzner I.B., Jankauskas S.S., Babenko V.A., Zorov S.D., Balakireva A.V., Juhaszova M., et al. Mitochondrial membrane potential. Anal. Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perry S.W., Norman J.P., Barbieri J., Brown E.B., Gelbard H.A. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50:98–115. doi: 10.2144/000113610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pendergrass W., Wolf N., Poot M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A. 2004;61:162–169. doi: 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- 49.Terada H. Uncouplers of oxidative phosphorylation. Environ. Health Perspect. 1990;87:213–218. doi: 10.1289/ehp.9087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rego A.C., Vesce S., Nicholls D.G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1-7 neural cells. Cell Death Differ. 2001;8:995–1003. doi: 10.1038/sj.cdd.4400916. [DOI] [PubMed] [Google Scholar]
- 52.Ward M.W., Rego A.C., Frenguelli B.G., Nicholls D.G. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kane L.A., Lazarou M., Fogel A.I., Li Y., Yamano K., Sarraf S.A., Banerjee S., Youle R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D.G., Ritorto M.S., Hofmann K., Alessi D.R., Knebel A., Trost M., Muqit M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 56.Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., Sou Y.S., Saiki S., Kawajiri S., Sato F., et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 59.Divakaruni A.S., Paradyse A., Ferrick D.A., Murphy A.N., Jastroch M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014;547:309–354. doi: 10.1016/B978-0-12-801415-8.00016-3. [DOI] [PubMed] [Google Scholar]
- 60.Cunniff B., McKenzie A.J., Heintz N.H., Howe A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell. 2016;27:2662–2674. doi: 10.1091/mbc.E16-05-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doughty-Shenton D., Joseph J.D., Zhang J., Pagliarini D.J., Kim Y., Lu D., Dixon J.E., Casey P.J. Pharmacological targeting of the mitochondrial phosphatase PTPMT1. J. Pharmacol. Exp. Therapeut. 2010;333:584–592. doi: 10.1124/jpet.109.163329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hugo W.B., Longworth A.R. Some Aspects of the Mode of Action of Chlorhexidine. J. Pharm. Pharmacol. 1964;16:655–662. doi: 10.1111/j.2042-7158.1964.tb07384.x. [DOI] [PubMed] [Google Scholar]
- 63.Hugo W.B., Longworth A.R. Cytological Aspects of the Mode of Action of Chlorhexidine Diacetate. J. Pharm. Pharmacol. 1965;17:28–32. doi: 10.1111/j.2042-7158.1965.tb07562.x. [DOI] [PubMed] [Google Scholar]
- 64.Zhang W., Hu X., Shen Q., Xing D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019;10:1704. doi: 10.1038/s41467-019-09566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng Z., Liu H., Zhai S., Zhang H., Shan G., Kwok R.T.K., Ma C., Sung H.H.Y., Williams I.D., Lam J.W.Y., et al. Highly efficient singlet oxygen generation, two-photon photodynamic therapy and melanoma ablation by rationally designed mitochondria-specific near-infrared AIEgens. Chem. Sci. 2020;11:2494–2503. doi: 10.1039/c9sc06441a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang W., Zhang Y., Wang Z., Liu X., Lu S., Hu X. A Native Drug-Free Macromolecular Therapeutic to Trigger Mutual Reinforcing of Endoplasmic Reticulum Stress and Mitochondrial Dysfunction for Cancer Treatment. ACS Nano. 2023;17:11023–11038. doi: 10.1021/acsnano.3c03450. [DOI] [PubMed] [Google Scholar]
- 67.Clark E.H., Vázquez de la Torre A., Hoshikawa T., Briston T. Targeting mitophagy in Parkinson's disease. J. Biol. Chem. 2021;296 doi: 10.1074/jbc.REV120.014294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malpartida A.B., Williamson M., Narendra D.P., Wade-Martins R., Ryan B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson's Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021;46:329–343. doi: 10.1016/j.tibs.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 69.Ma T.C., Buescher J.L., Oatis B., Funk J.A., Nash A.J., Carrier R.L., Hoyt K.R. Metformin therapy in a transgenic mouse model of Huntington's disease. Neurosci. Lett. 2007;411:98–103. doi: 10.1016/j.neulet.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 70.Xu X., Sun Y., Cen X., Shan B., Zhao Q., Xie T., Wang Z., Hou T., Xue Y., Zhang M., et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell. 2021;12:769–787. doi: 10.1007/s13238-021-00858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanchez-Rangel E., Inzucchi S.E. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60:1586–1593. doi: 10.1007/s00125-017-4336-x. [DOI] [PubMed] [Google Scholar]
- 72.Wang C., Niederstrasser H., Douglas P.M., Lin R., Jaramillo J., Li Y., Oswald N.W., Zhou A., McMillan E.A., Mendiratta S., et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 2017;8:2270. doi: 10.1038/s41467-017-02332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gene Ontology Consortium. Aleksander S.A., Balhoff J., Carbon S., Cherry J.M., Drabkin H.J., Ebert D., Feuermann M., Gaudet P., Harris N.L., et al. The Gene Ontology knowledgebase in 2023. Genetics. 2023;224 doi: 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thomas P.D., Ebert D., Muruganujan A., Mushayahama T., Albou L.P., Mi H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022;31:8–22. doi: 10.1002/pro.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davies B., Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. (N. Y.) 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 78.Soucek S., Zeng Y., Bellur D.L., Bergkessel M., Morris K.J., Deng Q., Duong D., Seyfried N.T., Guthrie C., Staley J.P., et al. The Evolutionarily-conserved Polyadenosine RNA Binding Protein, Nab2, Cooperates with Splicing Machinery to Regulate the Fate of pre-mRNA. Mol. Cell Biol. 2016;36:2697–2714. doi: 10.1128/MCB.00402-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seyfried N.T., Dammer E.B., Swarup V., Nandakumar D., Duong D.M., Yin L., Deng Q., Nguyen T., Hales C.M., Wingo T., et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer's Disease. Cell Syst. 2017;4:60–72.e4. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konen J., Summerbell E., Dwivedi B., Galior K., Hou Y., Rusnak L., Chen A., Saltz J., Zhou W., Boise L.H., et al. Image-guided genomics of phenotypically heterogeneous populations reveals vascular signalling during symbiotic collective cancer invasion. Nat. Commun. 2017;8 doi: 10.1038/ncomms15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.