

Abstract
Post-exercise recovery is essential to resolve metabolic perturbations and promote long-term cellular remodeling in response to exercise. Here, we report that muscle-generated brain-derived neurotrophic factor (BDNF) elicits post-exercise recovery and metabolic reprogramming in skeletal muscle. BDNF increased the post-exercise expression of the gene encoding PPARδ (peroxisome proliferator-activated receptor δ), a transcription factor that is a master regulator of lipid metabolism. After exercise, mice with muscle-specific Bdnf knockout (MBKO) exhibited impairments in PPARδ-regulated metabolic gene expression, decreased intramuscular lipid content, reduced β-oxidation, and dysregulated mitochondrial dynamics. Moreover, MBKO mice required a longer period to recover from a bout of exercise and did not show increases in exercise-induced endurance capacity. Feeding naïve mice with the bioavailable BDNF mimetic 7,8-dihydroxyflavone resulted in effects that mimicked exercise-induced adaptations, including improved exercise capacity. Together, our findings reveal that BDNF is an essential myokine for exercise-induced metabolic recovery and remodeling in skeletal muscle.
INTRODUCTION
Exercise recovery refers to the time period between the end of a bout of exercise and the subsequent return to a resting state, which is divided into immediate (<1 h after exercise), early (1 – 4 h after exercise), and late recovery periods (> 4 h after exercise) (1, 2). It is an essential physiological process to replenish cellular materials depleted by exercise, increase protein synthesis for repair and adaptation, and eliminate reactive oxygen species (ROS) (3). Therefore, insufficient recovery might impede tissue metabolism and performance (4). Although glycogen content is an energy source that sustains muscle function, intramyocellular triacylglycerol (IMTG) is equally important for muscle contraction during exercise. Indeed, IMTG might contribute up to half of the energy supply for ATP production during exercise (5). Hence, high IMTG without alterations in insulin sensitivity might benefit muscle performance, which is commonly seen in elite endurance atheletes (6). In addition to serving as an energy source, muscle TG also functions as building blocks for membrane phospholipids, which is particularly important for the regeneration of damaged organelles after exercise (7). Furthermore, increases in fatty acid (FA) uptake and lipogenesis after exercise is an important process to spare glucose for glycogen re-synthesis (8). Nevertheless, studies on the molecular mechanisms that govern post-exercise lipid metabolism are limited when compared with the many reports on the metabolic changes in muscle during exercise.
After exhaustive exercise, the expression of genes involved in glucose utilization, glycogen synthesis, FA oxidation (FAO), and fuel selection control is increased in skeletal muscle, indicating a critical role for transcriptional regulators in post-exercise recovery (9). Because FA uptake is increased during the recovery period (10), the FA-responsive peroxisome proliferator-activated receptor (PPAR) family of nuclear hormone factors has been proposed to be a crucial player in the process (2). PPARs control the transcription of key genes in lipid transport, β-oxidation, glucose sparing, and uncoupling reactions (1). PPARδ is the most abundant PPAR isoform in skeletal muscle (11) and Ppard expression in muscle induced by endurance exercise (12, 13) is responsible for shifting fuel utilization from glucose to lipids (14, 15). In mice expressing a form of PPARδ with increased activity due to fusion with the VP16 activation domain, running capacity is increased (16). Hence, PPARδ is considered to be an essential metabolic sensor and regulator of exercise-induced metabolic changes (1). However, it remains unknown how PPARδ abundance increases in exercised muscle.
In addition to facilitating physical movement, skeletal muscle is recognized to be an endocrine organ that produces hundreds of secretory proteins (such as myokines) that act locally on the tissue itself or distally to coordinate systemic metabolism during exercise (17, 18). Brain-derived neurotrophic factor (BDNF) is an exercise-induced myokine (19, 20). BDNF induces the dimerization and autophosphorylation of its receptor tropomyosin receptor kinase B (TrkB), which leads to the initiation of three distinct signaling cascades: phosphoinositide 3-kinase (PI3K)/protein kinase B (also known as Akt), mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK), and phospholipase C γ (PLCγ)/cAMP response element-binding protein (CREB) pathways in neurons, which underlies neuronal functions such as pro-survival gene expression, dendritogenesis, and long-term potentiation initiation (21). Although the functions of BDNF in the central nervous system (CNS) are well documented (21), the physiological roles of muscle-derived BDNF remain incompletely understood, although Bdnf expression increases in muscles and CNS after exercise (22). Experiments performed in cultured L6 cells have led to the proposal that muscle-derived BDNF is responsible for β-oxidation by activating the AMP-activated protein kinase (AMPK) during exercise (19). We also showed that BDNF signaling in muscles is essential for FA homeostasis during fasting and high-fat diet feeding (23–26). Furthermore, BDNF from muscle is an insulin secretagogue that stabilizes blood glucose concentrations (27), which might explain the stimulatory effect of BDNF on muscle glucose uptake (28). Together, these findings suggest that muscle-derived BDNF might play an autocrine role in remodeling the metabolic phenotype of skeletal muscles, but its functional role in exercise has not been verified. We report here that muscle-derived BDNF is indispensable for metabolic recovery after exercise and long-term metabolic adaptation to exercise.
RESULTS
Exercise enhances Bdnf expression in glycolytic muscle, liver, and white adipose tissue
BDNF expression increases in the skeletal muscle of rats and humans after acute exercise (19, 35), but whether this also occurs in mice has not been explored. Moreover, it remains unknown if BDNF production in other peripheral tissues is responsive to running exercise. Therefore, we measured Bdnf expression in various tissues of female mice without [sedentary (Sed)], during (0 h), and after (1–24 h) a mid-intensity (~60% VO2 max) treadmill running (30). Immediate, early, and late recovery periods were defined as 1 hour, 2 hours, and 6–24 hours after exercise, respectively. Only female mice were examined because we have found a sex-dimorphic effect of BDNF, such that male mice do not activate BDNF signaling in muscle in response to fasting (25). Compared with the Sed group, we did not detect changes in Bdnf expression in skeletal muscles during exercise (0 h). However, Bdnf expression significantly increased in the glycolytic myofiber-enriched gastrocnemius and extensor digitorum longus (EDL) muscles at early and late recovery periods (Fig 1A). Bdnf expression did not significantly change in oxidative myofiber-rich soleus muscle after exercise, implying that exercise preferentially elicits BDNF production in glycolytic myofibers (Fig 1A). In mouse liver and gonadal white adipose tissues (gWAT), Bdnf expression was significantly increased throughout the immediate, early, and late recovery periods but not during exercise (Fig 1A). The effect of acute exercise on Ntrk2 (which encodes TrkB) expression in muscle has not been examined, and increased BDNF synthesis might act as an autocrine signal to change Ntrk2 expression as observed in hippocampal neurons (36). Ntrk2 expression increased in EDL during the late recovery phase but not in gastrocnemius muscle, soleus muscle, or liver (Fig 1B). Ntrk2 expression in gWAT was significantly increased during exercise, remained elevated throughout the early recovery periods, and returned to basal level in the late recovery period (Fig 1B). Consistent with the elevated Bdnf expression in various tissues, serum BDNF content significantly increased during the late recovery phase (Fig 1C). Indeed, muscle-secreted BDNF partly contributes to the pool of BDNF in blood because the circulating BDNF concentration was lower in muscle-specific Bdnf knockout (MBKO) mice (16) after exercise (Fig 1D). Hence, our data suggest that increased expression of Bdnf in peripheral tissues and elevated circulating BDNF content are post-exercise responses in mice.
Fig. 1. Post-exercise induction of Bdnf expression in skeletal muscle.
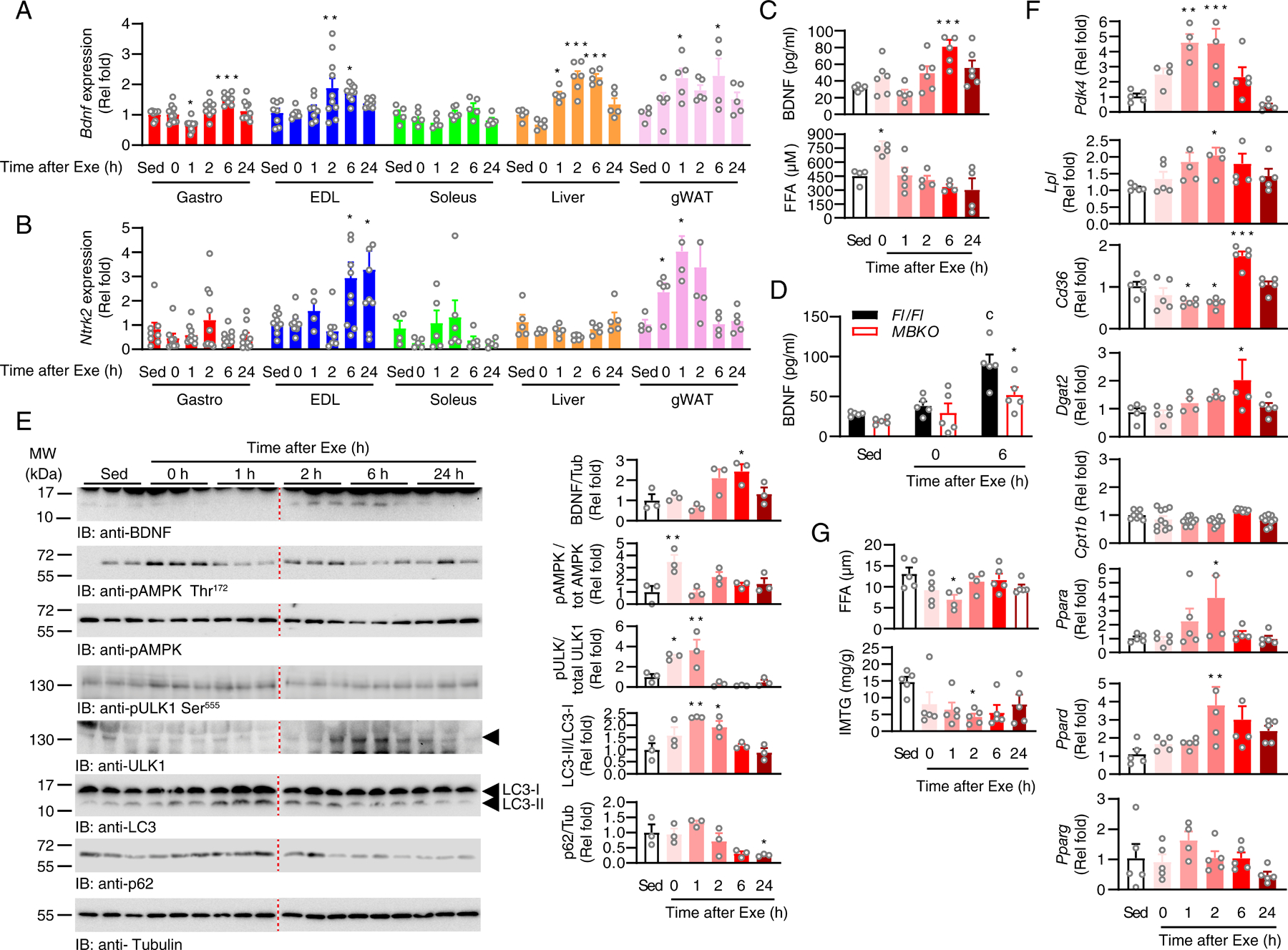
(A) Expression of Bdnf in different tissues of sedentary (Sed) mice or mice that performed a single bout of endurance exercise (Exe) (*: P<0.05, **: P<0.01, ***: P<0.001 compared to Sed, one-way ANOVA, n=5–10 mice/group). (B) Expression of Ntrk2 in different tissues of sedentary (Sed) mice or mice that performed a single bout of endurance exercise (Exe) (*: P<0.05 compared to Sed, one-way ANOVA, n=5–10 mice/group). (C) Circulating BDNF and free fatty acid (FFA) content in sedentary (Sed) mice or mice that performed after a single bout of endurance exercise (*: P<0.05, ***: P<0.001 compared to Sed, one-way ANOVA, n=4–6 mice/group). (D) Circulating BDNF content in Fl/Fl and MBKO mice after a single bout of endurance exercise (c: P<0.001 compared to Sed, *: P<0.05 compared to Fl/Fl for the same treatment, two-way ANOVA, n=5 mice/group). (E) Immunoblotting analysis of signaling molecules in the gastrocnemius muscle of sedentary (Sed) mice or mice that performed a single bout of endurance exercise (Exe). The arrowhead indicates the band with the correct molecular mass. Bar graphs show quantification of immunoblot signals (*: P<0.05, **: P<0.01 compared to Sed, one-way ANOVA, n=3 mice/group). (F) Expression of metabolic genes in the muscle of sedentary (Sed) mice or mice that performed a single bout of endurance exercise (Exe) (*: P<0.05, **: P<0.01, ***: P<0.001 compared to Sed, one-way ANOVA, n=4–10 mice/group). (G) The concentration of FFA and intramyocellular triacylglycerides (IMTG) in the gastrocnemius muscle of sedentary (Sed) mice or mice that performed a single bout of endurance exercise (Exe) (*: P<0.05 compared to Sed, one-way ANOVA, n=5 mice/group).
We also analyzed the amount of BDNF protein in the mouse gastrocnemius muscle after exercise. Aligned with the gene expression data, intramyocellular BDNF content was only increased during the early-to-late recovery period after a single bout of exercise, which returned to basal levels after 24 h (Fig 1E). Because BDNF has been proposed to be an upstream activator of AMPK during exercise (19), we measured the phosphorylation pattern of AMPK in the mouse muscle. BDNF production was not synchronized with AMPK phosphorylation because the phosphorylation of Thr172 in AMPK peaked during exercise and returned to basal levels in the immediate recovery phase (Fig 1E). The AMPK-mediated phosphorylation of Ser555 in Unc-51 like autophagy activating kinase (ULK1), which is important for exercise-induced mitophagy (37), also transiently increased during exercise and the immediate recovery period (Fig 1E). Consistent with the sequence of events in autophagosome formation, LC3 lipidation increased after ULK1 activation during the early recovery period (Fig 1E), which was followed by a reduction in p62 content, which remained low even after 24 h (Fig 1E).
The elevated production of BDNF in muscle during recovery prompted us to hypothesize that BNDF might not be responsible for the metabolic changes during exercise but regulates post-exercise remodeling. We have shown that BDNF is important for maintaining FA utilization during fasting (25), which is also the major fuel source to replenish IMTG after exercise (8). Hence, we assessed correlation between the expression of genes involved in FA metabolism and Bdnf expression in exercised muscle. Pdk4 encodes pyruvate dehydrogenase kinase 4, which controls the shift in cellular fuel preference from glycolysis to FAO by inhibiting the pyruvate dehydrogenase complex (38), and its expression increased during the early recovery phase (Fig 1F), implying that a shift from glucose to FA utilization occurs during this period. However, FA in the circulation was transiently increased during exercise (Fig 1C), suggesting the exogenous source of FA contributes only to the energy demands of skeletal muscle during exercise (39). Indeed, exercise promotes FA import from lipoproteins as an additional fuel source (40), which is consistent with the increase in the expression of Lpl (which encodes lipoprotein lipase) after exercise (Fig 1F). However, the expression of Cd36 (which encodes FA translocase) slightly decreased during exercise, but significantly increased in the late recovery period, which returned to basal status after 24 h (Fig 1F). This increase in Cd36 expression may be a compensatory response to replenish depleted intramyocellular FAs after exercise (Fig 1G). Endurance exercise also exhausted the IMTG (Fig 1G), which could be restored by the increased expression of Dgat2 (which encodes diacylglycerol acyltransferase 2) during the late recovery period (Fig 1F). Expression of Cpt1b, which encodes the rate-determining enzyme in FAO, carnitine palmitoyltransferase I, remained unchanged (Fig 1F). We also analyzed the expression of genes encoding PPARs, which are key transcription factors for lipid metabolism, and the PPAR co-activator peroxisome proliferator-activated receptor γ co-activator 1-α (PGC-1α; which is encoded by Ppargc1a). Ppargc1a expression increased during the early recovery period, and the expression of Ppara (which encodes PPARα) and Ppard (which encodes PPARδ), but not that of Pparg (which encodes PPARγ), was significantly increased 2 h after exercise (Fig 1F). Correlation analysis confirmed that Bdnf expression in mouse muscle was directly proportional to that of Cd36, Cpt1, and Ppard (Fig S1).
Deficiency of BDNF in muscle impairs exercise performance and post-exercise lipid metabolism
The temporal dissociation of AMPK phosphorylation and Bdnf expression in the skeletal muscle suggested that BDNF might be dispensable for exercise-induced AMPK activation. Indeed, AMPK activity was comparable in the gastrocnemius of female Fl/Fl and MBKO mice during and after running exercise (Fig 2A). However, during an exhaustive running test, MBKO mice reached a fatigue state more frequently as shown by the greater number of electrical shocks compared to Fl/Fl mice (Fig 2B). MBKO mice also had poorer exercise endurance than Fl/Fl mice in running distance, time, and speed (Fig 2C). Moreover, MBKO mice had a shorter wire suspension time than Fl/Fl mice during the four-limb hanging test (Fig 2D), further confirming their fatigue-prone phenotype. To further support the importance of BDNF in muscle recovery after exercise, mice were subjected to two consecutive exhaustive tests with a 6 h recovery in between, which is sufficient to replenish the IMTG in normal mice (41). Although Fl/Fl mice displayed no reduction in running endurance after the first test, the running capability of MBKO mice was significantly reduced in the second test (Fig 2E), suggesting that BDNF in muscle is crucial to recovery of muscle performance after exercise.
Fig. 2. Deficiency of BDNF production in muscle impairs exercise performance and post-exercise lipid metabolism.
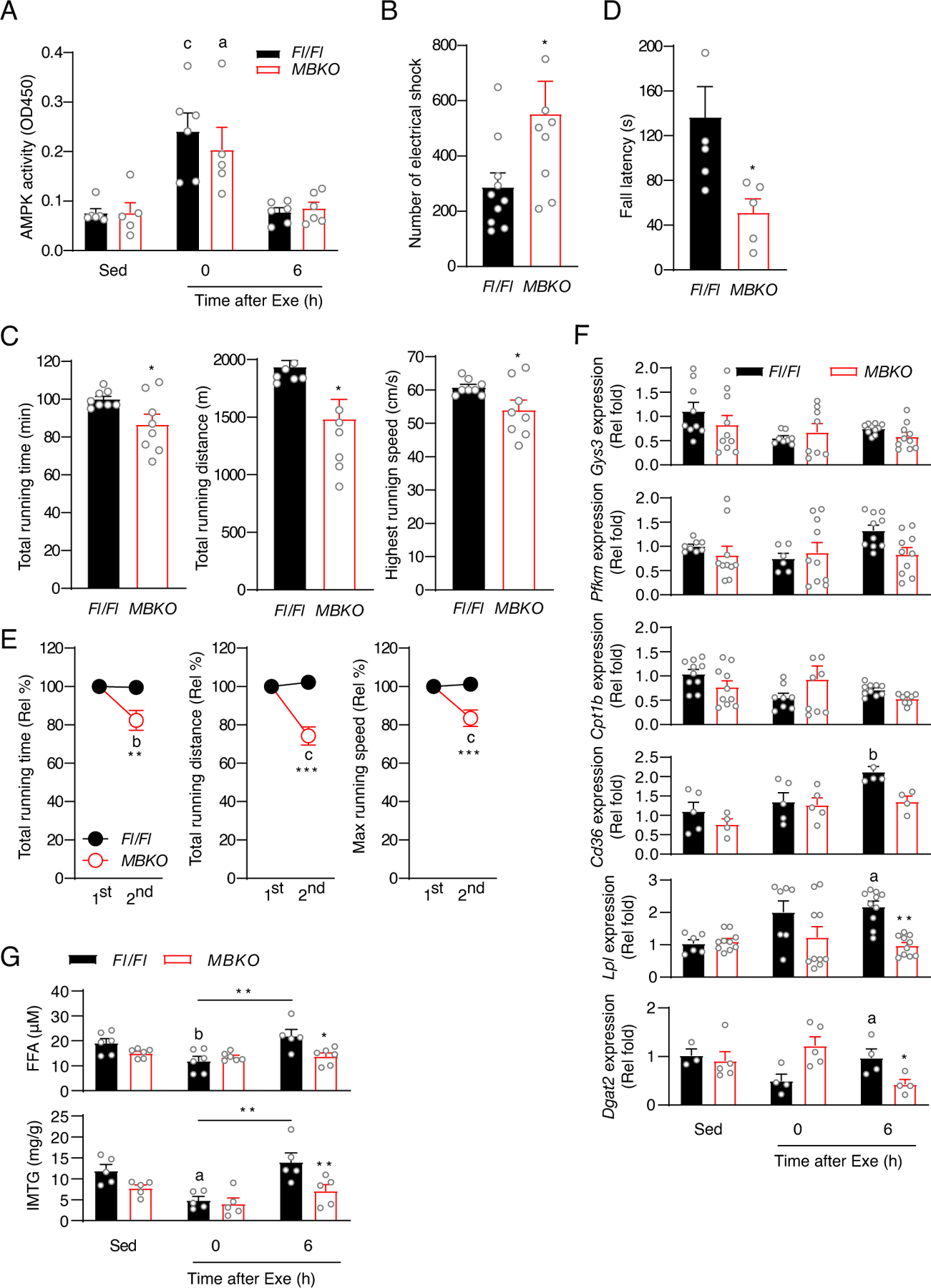
(A) AMPK activity as determined by ELISA in the gastrocnemius muscles of Fl/Fl and MBKO mice that performed a single bout of endurance exercise (n=5–6 mice/group). (B) The number of electrical shocks received by Fl/Fl and MBKO mice during the exhaustive running test (*: P<0.05, Student’s t-test, n=8–10 mice/group). (C) Running capacity of Fl/Fl and MBKO mice during the exhaustive running test (*: P<0.05, Student’s t-test, n=8 mice/group). (D) Duration of wire hanging of Fl/Fl and MBKO mice during the 4-limb hanging test (*: P<0.05, Student’s t-test, n=5 mice/group). (E) Exercise performance of Fl/Fl and MBKO mice in two consecutive exhaustive running tests spaced 6 h apart (b: P<0.01, c: P<0.001 compared to the same genotype in the first test, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice in the same test, two-way ANOVA, n=7 mice/group). (F) Expression of metabolic genes in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed a single bout of endurance exercise (a: P<0.05, b: P<0.01 compared to the Sed group of the same genotype; *: P<0.05, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice in the time interval, two-way ANOVA, n=5–10 mice/group). (G) The concentration of FFA and IMTG in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed a single bout of endurance exercise (b: P<0.01 compared to the Sed group of the same genotype; *: P<0.05, ***: P<0.001 compared to Fl/Fl mice in the time interval, **:P<0.01, two-way ANOVA, n=5–6 mice/group).
Although MBKO muscle had a small amount of centrally nucleated and swelled myofibers (Fig S2A), the total lean mass of MBKO mice was comparable to that of Fl/Fl mice (Fig S2B). Moreover, MBKO and Fl/Fl gastrocnemius muscle had similar oxidative and glycolytic myofiber composition (Figs S2C and S2D). Hence, the functional impairment in the muscle of MBKO mice could be due to metabolic defects. To test this hypothesis, we compared the expression of metabolic genes in the muscle of Fl/Fl and MBKO mice after a single bout of exhaustive exercise. This single bout of exercise did not alter the expression of key enzymes in glucose metabolism [Gys3 (which encodes glycogen synthase) and Pfkm (which encodes 6-phosphofructokinase)] and FAO (Cpt1b) in either genotype (Fig 2F). In contrast, acute exercise promoted the expression of Cd36, Lpl, and Dgat2 in Fl/Fl muscle but not MBKO muscle during the late-recovery period (Fig 2F). In association with the impaired expression of these metabolic genes, MBKO muscle had lower TG and FFA content than Fl/Fl muscle after exercise (Fig 2G). Together, these data suggest that FFA transport across the sarcolemma and intramyocellular lipogenesis during recovery may be impaired in the absence of BDNF in muscle.
BDNF modulates lipid metabolism in skeletal muscle through PPARδ
To verify that the defective metabolic reprogramming in the muscle of exercised MBKO mice is caused by the ablation of the autocrine activity of BDNF, we first tested if BDNF stimulation induced the expression of metabolic genes in cultured myotubes. BDNF stimulation upregulated the expressions of Cd36, Lpl, Ppargc1a, and Prkn [which encodes Parkin, a critical mitochondrial ubiquitin ligase for mitophagy induction (42)] in C2C12 myotubes (Fig 3A). In contrast, infection of C2C12 myotubes with adenovirus carrying shRNA directed against Bdnf (Ad-shBDNF) (26) significantly reduced the expression of Lpl, Acadl (which encodes acyl-CoA dehydrogenase), Cpt1b, Cpt2, and Prkn (Fig 3B), providing support for an autocrine role of BDNF in stimulating FA uptake, β-oxidation, and mitophagy through inducing changes in gene expression. Consistent with the downregulation of the expression of genes involved in FA uptake, Bdnf knockdown reduced lipid accumulation (Fig 3C), reminiscent of the lower FA content in the gastrocnemius muscle of exercised MBKO mice (Fig 2G). We also found that Ppard expression was decreased in BDNF-depleted myotubes (Fig 3B) and in the muscle of exercised MBKO mice (Fig 3D) but was increased in C2C12 myotubes stimulated with BDNF (Fig 3E), further supporting the notion that BDNF stimulates PPARδ in muscle. Neither Bdnf depletion (Fig 3B) nor BDNF stimulation (Fig 3E) changed the expression of the genes encoding PPARα or PPARγ, indicating an isoform-specific effect for BDNF. We have previously shown that BDNF activates AMPK in skeletal muscle through Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) (26). Knockdown of Camkk2 (Figs 3F and G) or pharmacologically inhibiting CaMKK2 activity with STO-609 (Fig 3H) significantly blocked the BDNF-induced increase in Ppard expression in C2C12 myotubes. This effect was not seen with the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin (Fig 3H), suggesting CaMKK2 is a critical signaling molecule in the BDNF pathway that controls Ppard expression.
Fig. 3. BDNF controls the transcription of metabolic genes by inducing Ppard expression.
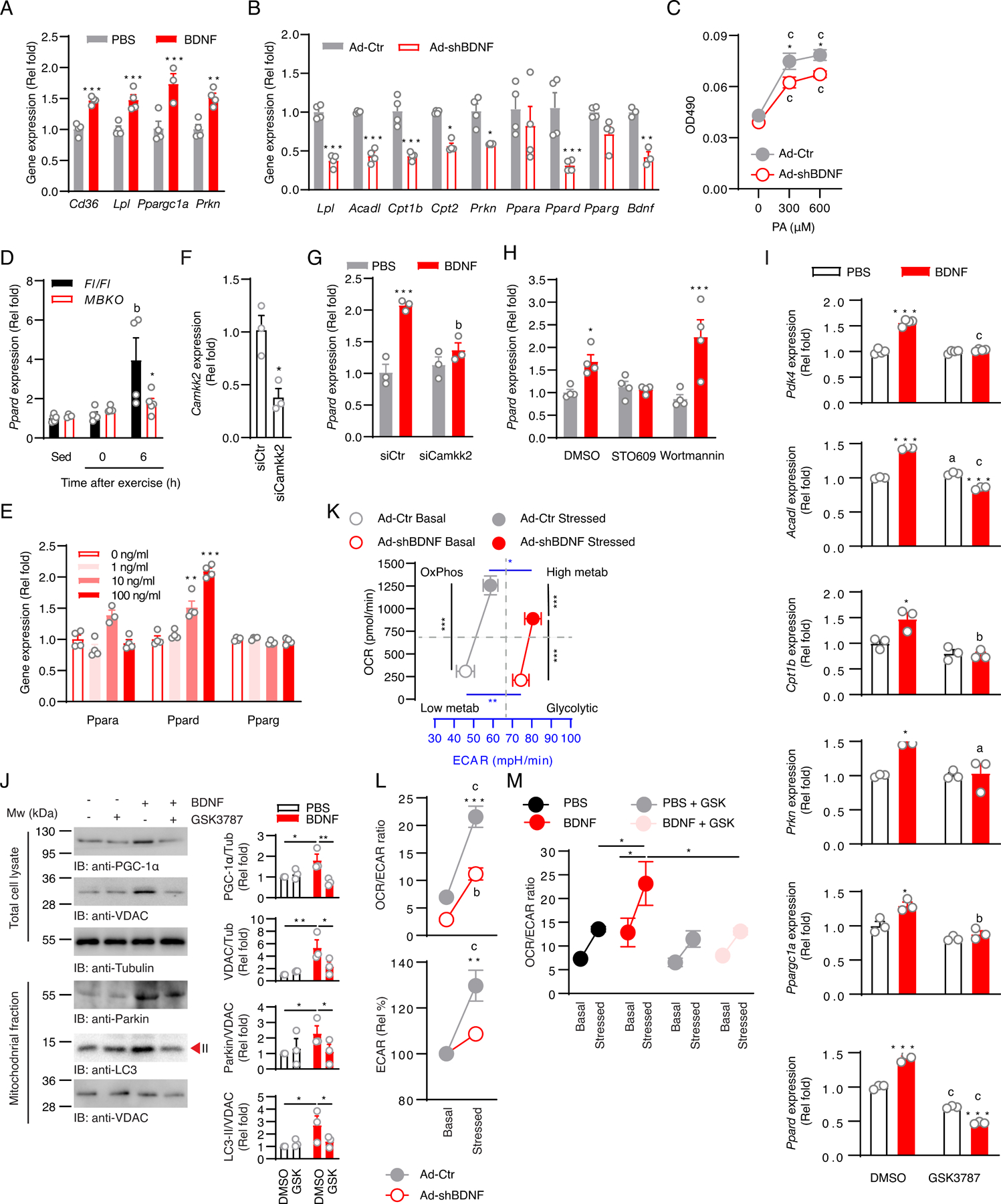
(A) Expression of metabolic genes in C2C12 myotubes stimulated with PBS or BDNF (100 ng/ml, 24 h) (**: P<0.01, ***: P<0.001 compared to PBS, Student’s t-test, n=3–4 biological replicates/group). (B) Expression of metabolic genes in C2C12 myotubes infected with control adenovirus (Ad-Ctr) or adenovirus expressing shRNA against Bdnf (Ad-shBDNF) (*: P<0.05, **: P<0.01, ***: P<0.001 compared to Ad-Ctr, Student’s t-test, n=3–4 biological replicates/group). (C) Fatty acid accumulation as determined by Oil Red O staining in Ad-Ctr- or Ad-shBDNF-infected myotubes stimulated with palmitic acid (PA) (c: P<0.001 compared to 0 µM, *: P<0.05, compared to different Ad-shBDNF-infected cells under the same PA treatment, Student’s t-test, n=5–6 biological replicates/group). (D) Expression of Ppard in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed a single bout of endurance exercise (b: P<0.01 compared to the Sed group of the same genotype; *: P<0.05 compared to Fl/Fl mice in the same time period, two-way ANOVA, n=4–6 mice/group). (E) Expression of Ppar isoforms in C2C12 myotubes stimulated with BDNF (100 ng/ml) for the indicated times (**: P<0.01, ***: P<0.001 compared to 0 ng/ml, Student’s t-test, n=4 biological replicates/group). (F) Camkk2 knockdown as assessed by real-time PCR in C2C12 myotubes transfected with siCtr (Ctr) or siCamkk2 (si) for 24 h (*: P<0.05, Student’s t-test, n=3 biological replicates/group). (G) C2C12 myotubes were transfected with control siRNA (siCtr) or siRNA against Camkk2 (siCamkk2). Twenty four hours after transfection, myotubes were stimulated with BDNF (100 ng/ml) for 24 h. Ppard expression was assessed by real-time PCR (***: P<0.001 compared to PBS, b: P<0.01 compared to BDNF, two-way ANOVA, n=3 biological replicates/group). (H) C2C12 myotubes were pre-treated with DMSO, STO609 (10 μg/ml), or Wortmannin (1 μM) for 2 h, then with PBS or BDNF (100 ng/ml) for 24 h. Ppard expression was examined by real-time PCR (*: P<0.01, ***: P<0.001 compared to PBS, two-way ANOVA, n=4 biological replicates/group). (I) Expression of PPARδ-regulated genes in C2C12 myotubes pre-treated with DMSO or the PPARδ inhibitor GSK3787 (1µM) for 1 h before being stimulated with DMSO or BDNF (100 ng/ml, 24 h) (a: P<0.05, b: P<0.01, c: P<0.001 compared to DMSO pre-treatment; *: P<0.05, **: P<0.01, ***: P<0.001 compared to PBS control, two-way ANOVA, n=3 biological replicates/group). (J) C2C12 myotubes were treated with different combinations of GSK3787 (1µM) and BDNF (100 ng/ml) for 24 h and the amounts of various proteins involved in mitochondrial biogenesis and mitophagy were measured by Western blotting. Representative blots are shown and the bars graphs show quantification of band intensities (*: P<0.05, **: P<0.01, two-way ANOVA; n=3 biological replicates/group). (K) Metabolic phenotyping of Ad-Ctr- or Ad-shBDNF-infected myotubes was assessed by extracellular flux analysis (*: P<0.05, blue **: P<0.01, ***: P<0.001, two-way ANOVA, n=4 biological replicates/group). (L) OCR/ECAR ratio and change of ECAR of Ad-Ctr- or Ad-shBDNF-infected myotubes (b: P<0.01, c: P<0.001 compared to the basal group within the same infection group; **: P<0.01, ***: P<0.001 compared to stressed Ad-shBDNF, two-way ANOVA, n=4 biological replicates/group). (M) C2C12 myotubes were treated with different combinations of GSK3787 (1µM) and BDNF (100 ng/ml) and the OCR/ECAR ratio was assessed by extracellular flux analysis (*: P<0.05, two-way ANOVA, n=5 biological replicates/group).
Because the expression of some PPARδ target genes such as Cpt1b, Lpl, and Acadl (43) was reduced in BDNF-depleted C2C12 myotubes (Fig 3B), we suspected that BDNF controlled the expression of these metabolic genes by activating PPARδ. Indeed, treating C2C12 myotubes with the selective PPARδ inhibitor GSK3787 (44) abolished the stimulatory effect of BDNF on the expression of PPARδ target genes, including Pdk4, Acadl, Cpt1b, and Ppargc1a (Fig 3I). GSK3787 also diminished basal and BDNF-induced Ppard expression (Fig 3I), which corroborated the auto-regulatory activity of PPARδ in muscle (45). In addition, PPARδ inhibition by GSK3787 in C2C12 myotubes also attenuated the BDNF-induced increases in the abundance of proteins involved in mitochondrial biogenesis and mitophagy, including PGC-1α, the mitochondrial voltage-dependent anion-selective channel (VDAC), and mitochondrially-tethered parkin and LC3-II (Fig 3J).
Because PPARδ promotes a metabolic switch from glycolysis to FAO by enhancing Pdk4 expression (38, 46), the low Pdk4 expression in Ad-shBDNF-infected myotubes might alter lipid and glucose utilization. As anticipated, metabolic phenotyping analysis revealed that, compared to control cells, Bdnf-depleted myotubes had a higher extracellular acidification rate (ECAR, an indicator of glycolysis), as well as a statistically insignificant decrease in oxygen consumption rate [OCR, an indicator of oxidative phosphorylation] (Fig 3K). The lower OCR/ECAR ratio of Bdnf-depleted myotubes further demonstrated their metabolic reliance on glycolysis (Fig 3L). The Bdnf-depleted myotubes also had impaired mitochondrial capacity as demonstrated by the smaller OCR increase when stressed with the ATPase inhibitors oligomycin and protonophore 2-[2-[4-(trifluoromethoxy)phenyl]hydrazinylidene]-propanedinitrile (FCCP) (Fig 3L). In contrast to control myotubes, Bdnf-depleted myotubes did not significantly increase ECAR upon inhibition of mitochondrial ATP synthesis by oligomycin and FCCP (Fig 3L), demonstrating an impairment in cellular oxidative-to-glycolytic transition in the absence of BDNF. However, BDNF stimulation resulted in a slightly higher basal OCR/ECAR ratio in Bdnf-depleted myotubes, which became significant when the cells were stressed by oligomycin and FCCP (Fig 3M). This increase in OCR/ECAR ratio was abolished in the presence of GSK3787, further supporting a role of PPARδ in the BDNF-mediated glycolytic-to-oxidative transition (Fig 3M). Together, our data indicate that BDNF promotes the expression of Ppard to reprogram gene expression for lipid metabolism, fuel selection, and mitochondrial activity in muscle.
BDNF production in muscle is required for exercise-improved muscle endurance
Chronic training upregulates Ppard expression in glycolytic myofiber-enriched muscle (12) and increases performance in mice with a muscle-specific deletion of Ppard to only 50% of that in control mice (14). We speculated that the compromised PPARδ signaling in MBKO mice might suppress their responses to chronic exercise training. To test this hypothesis, mice were subjected to a previously reported 4-week training protocol that is effective in improving muscle endurance in mice (30). Although Fl/Fl mice gradually received fewer electrical shocks with each training session, MBKO mice required more electrical shocks to finish the workout (Fig 4A), suggesting exercise training successfully promoted the endurance capacity of Fl/Fl mice but not that of MBKO mice. In support of this notion, only Fl/Fl mice showed improvements in total running time, total running distance, and maximal running speed (Fig 4B). Moreover, exercise training improved hanging time in the four-limb hanging test in Fl/Fl mice, but not in MBKO mice (Fig 4C), further confirming the lack of exercise-induced functional improvement in MBKO muscle. The increased exercise capacity in the trained Fl/Fl mice was not a result of myofiber composition remodeling because the expression of markers for oxidative (Myh7) and glycolytic (Myh4 and Myh1) myofibers were not altered (Figs S2C and S2D). Collectively, our data suggest that the presence of BDNF in muscles is essential for exercise-improved endurance performance.
Fig. 4. Endurance exercise-promoted metabolic reprogramming in skeletal muscle requires the presence of BDNF.
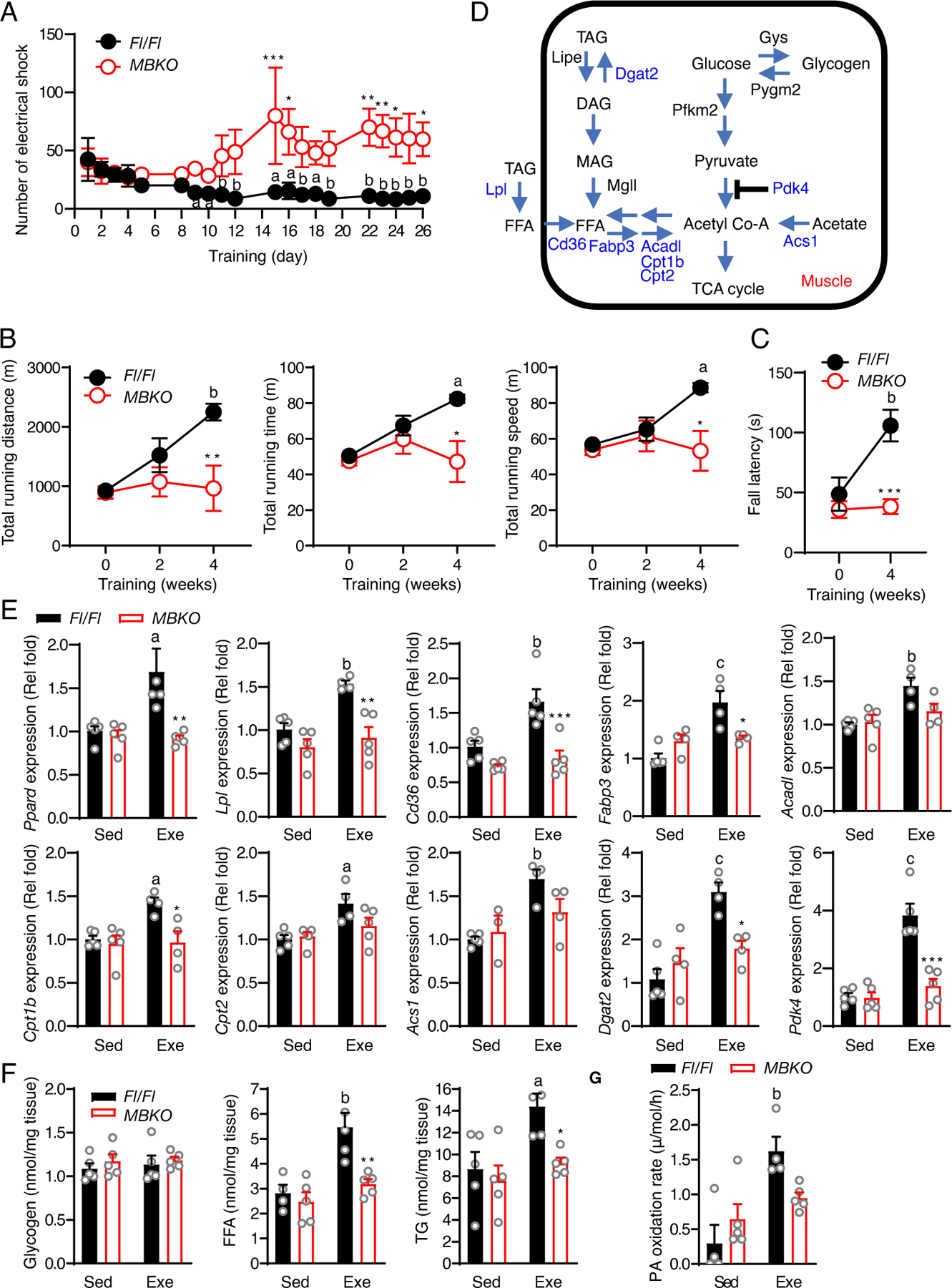
(A) The number of electrical shocks received by Fl/Fl and MBKO mice during exercise training (a: P<0.05, b: P<0.01 compared to day 0 of the same genotype; *: P<0.05, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice in the time interval, two-way ANOVA, n=4–5 mice/group). (B) The running capacity of Fl/Fl and MBKO mice after exercise training (a: P<0.05, b: P<0.01 compared to week 0 of the same genotype; *: P<0.05, **: P<0.01 compared to Fl/Fl mice in the time interval, two-way ANOVA, n=5–7 mice/group). (C) Duration of wire hanging of Fl/Fl and MBKO mice after 4 weeks of exercise training (b: P<0.01 compared to week 0 of the same genotype; ***: P<0.001 compared to Fl/Fl mice in the time interval, two-way ANOVA, n=5 mice/group). (D) Schematic of genes involved in the glucose and lipid metabolism of skeletal muscle. (E) Expression of metabolic genes in the gastrocnemius muscle of Fl/Fl and MBKO mice after 4 weeks of exercise training (Exe) (a: P<0.05, b: P<0.01, c: P<0.001 compared to the Sed group of the same genotype; *: P<0.05, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice of the same training group, two-way ANOVA, n=4–5 mice/group). (F) The concentration of glycogen, free fatty acids (FFA), and intramolecular triacylglycerides (IMTG) in the gastrocnemius muscle of Fl/Fl and MBKO mice after 4 weeks of endurance exercise training (Exe) (b: P<0.01 compared to the Sed group of the same genotype; *: P<0.05, **: P<0.01 compared to Fl/Fl mice of the same training group, two-way ANOVA, n=4–5 mice/group). (G) Palmitic acid (PA) oxidation rate of cell lysates prepared from the gastrocnemius muscle of Fl/Fl and MBKO mice after 4 weeks of exercise training (Exe) (b: P<0.01 compared to the Sed group of the same genotype, two-way ANOVA, n=4-mice/group).
Chronic endurance training does not promote metabolic reprogramming in the muscle of MBKO mice
To verify if muscle-generated BDNF is required in chronic exercise-induced metabolic remodeling, we examined the expression of key genes in lipid and glucose metabolism (Fig 4D). BDNF was dispensable to basal Ppard expression in vivo because Ppard expression was similar in MBKO and Fl/Fl muscle (Fig 4E). However, chronic exercise failed to induce Ppard expression in the skeletal muscle of MBKO mice, implying that BDNF is only important for exercise-induced Ppard expression (Fig 4E). The expression of genes encoding enzymes involved in glycogenolysis [Pygm2, which encodes glycogen phosphorylase], glycogenesis (Gys), and glycolysis (Pfkm) was not affected by aerobic exercise training or the absence of Bdnf in muscle (Fig S3). Exercise training did not alter intramyocellular glycogen content in either genotype (Fig 4F). In contrast, we found that the expression of PPARδ-regulated genes encoding enzymes for FA uptake (Lpl and Cd36) and intracellular FA transportation [Fabp3 (which encodes fatty acid binding protein 3)] was increased in exercise-trained Fl/Fl mice, but not in MBKO mice (Fig 4E). In association with the elevated FA uptake and utilization, exercise training resulted in a higher intramyocellular concentration of FFA in the muscle of Fl/Fl mice but not in that of MBKO mice (Fig 4F), suggesting BDNF in muscle is essential to exercise-induced FA accumulation. Chronic exercise also promoted the expression of Acadl, Cpt1b, and Cpt2 in the muscle of Fl/Fl mice, but not in that of MBKO mice (Fig 4E). This finding implied that exercise training failed to improve FAO capacity in the muscle of MBKO mice, a notion that was further supported by a reduced ability to oxidize 14C-labelled palmitic acid (PA) (Fig 4G). Moreover, chronic exercise increased the expression of lipogenesis-promoting genes (Acs1, which encodes acyl-CoA synthase) and Dgat2 (Fig 4E). However, the exercise-induced increases in lipogenic gene expression were not seen in the muscle of MBKO mice, which was consistent with their lower IMTG content (Fig 4F). The expression of Pdk4 was also significantly enhanced only in the muscle of Fl/Fl mice after exercise training (Fig 4E), implying that fuel selection in MBKO muscle might be disrupted. Together, our data indicates that BDNF in muscle is an important factor in promoting chronic exercise-induced functional improvement and metabolic changes in skeletal muscle.
Exercise-induced mitochondrial remodeling is impaired in the muscle of MBKO mice
Consistent with the ability of endurance exercise training to increase mitochondrial content in skeletal muscle (47) and the requirement for PPARδ to chronic exercise-induced mitochondrial biogenesis (16), the muscle of exercise-trained Fl/Fl mice had increased abundance of the mitochondrial proteins succinate dehydrogenase (SDHA) and VDAC (Fig 5A), which was associated with greater expression of various genes important for mitochondrial biogenesis, including Ppargc1a (Fig 5, A and B), Tfam (which encodes mitochondrial transcription factor A) and Nrf1 (which encodes nuclear respiratory factor 1) (Fig 5B). In contrast, exercise training did not alter the protein abundance or the expression of genes encoding mitochondrial biogenesis regulators in the muscle of MBKO mice (Fig 5B). Exercise training also increased PPARδ protein amounts in Fl/Fl muscle, but not MBKO muscle (Fig 5A).
Fig. 5. Exercise-induced mitochondrial remodeling is impaired in the muscle of MBKO mice.
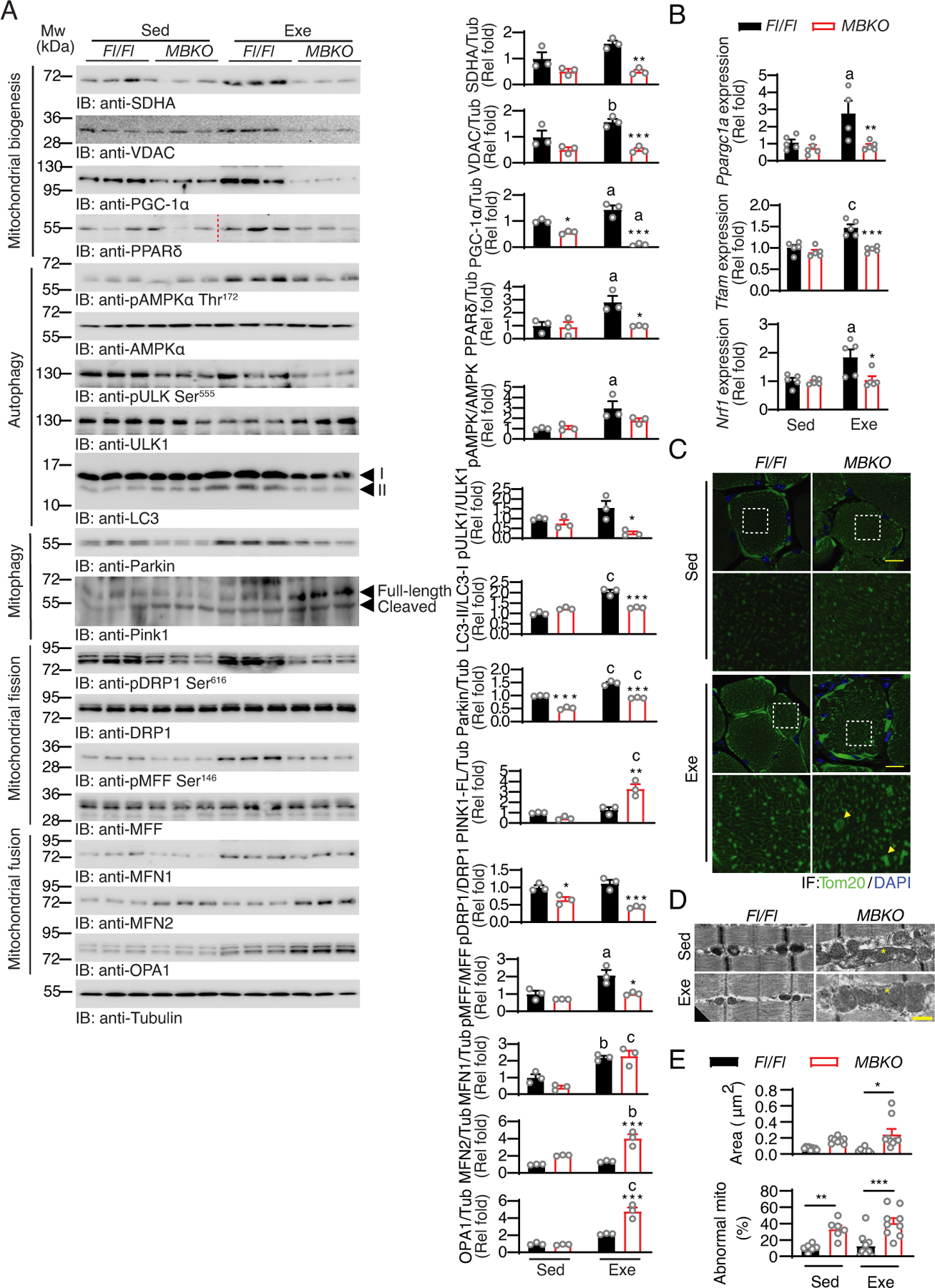
(A) Immunoblotting analysis of signaling proteins in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed 4 weeks of endurance exercise training (Exe). Bar graphs show quantification of immunoblot signals (a: P<0.05, b: P<0.01, c: P<0.001 compared to the Sed group of the same genotype; *: P<−0.05, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice of the same training group, two-way ANOVA, n=3 mice per group). (B) Expression of genes involved in mitochondrial biogenesis in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed 4 weeks of endurance exercise training (Exe) (a: p<0.05, c: P<0.001 compared to the Sed group of the same genotype; *: P<0.05, **: P<0.01, ***: P<0.001 compared to Fl/Fl mice of the same training group, two-way ANOVA, n=5 mice per group). (C) Representative immunofluorescence staining of Tom20 and 4’,6-diamidino-2-phenylindole (DAPI) in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed 4 weeks of exercise training (Exe). Magnified views of the white dashed boxes are shown in the lower panels. The yellow arrows indicate examples of enlarged mitochondria. Scale bar: 20 μm. N=1 section from 3 mice per group. (D) Representative transmission electron microscopy images showing the mitochondria morphology in the gastrocnemius muscle of sedentary (Sed) Fl/Fl and MBKO mice or mice that performed 4 weeks of endurance exercise training (Exe). The asterisk indicates enlarged mitochondria. Scale bar: 500 nm. (E) Quantification of the average area of the mitochondria and the number of enlarged mitochondrial per image are also shown (c: P<0.001 compared to the Sed group of the same genotype; **: P<0.01, ***: P<0.001 compared to Fl/Fl mice of the same training group, two-way ANOVA, N=3–6 sections from 2 mice per group).
In addition to the augmented mitochondrial protein content, exercise training also switched mitochondrial morphology from discrete globules to interconnected reticulum in the skeletal muscle of Fl/Fl mice (Fig 5C). In contrast, mitochondria in the muscle of exercise-trained MBKO mice retained a punctate morphology as evidenced by the large clusters of Tom20-positive puncta (Fig 5C). Electron microscopy analysis further showed that MBKO muscle contained more abnormal mitochondria that spanned a full sarcomere (Figs 5D and 5E). Moreover, mitochondria were larger in MBKO muscle than in Fl/Fl mice, and the size increase was exacerbated after repetitive exercise training (Fig 5E).
Mitochondrial structure is highly plastic and undergoes regulated fusion, fission, and mitophagy in response to different stresses (48). Chronic exercise improves mitochondrial quality by regulating mitochondrial dynamics (49). To determine whether the abnormal mitochondrial architecture in exercise-trained MBKO mice may be the result of defective mitochondrial dynamics, we examined the key markers of autophagy, mitochondrial fusion, mitochondrial fission, and mitophagy. Although chronic exercise training in Fl/Fl mice enhanced the phosphorylation of Thr172 in AMPK and Ser555 in ULK1, key events in acute exercise-initiated autophagy (37), these phosphorylation events were reduced in MBKO mice (Fig 5A). LC3 lipidation in the muscles after exercise training was also lower in MBKO mice than in Fl/Fl mice (Fig 5A). Similarly, exercise training also increased the amount of Parkin in Fl/Fl muscle but not in MBKO muscle (Fig 5A). The diminished Parkin abundance found in MBKO muscle may be due to impaired PPARδ activation because inhibiting PPARδ with GSK3787 blocked the BDNF-induced increase in Prkn expression (Fig 3F). In contrast, exercise training in MBKO mice induced a robust accumulation in muscle of full-length PTEN-induced kinase 1 (PINK1), a mitochondrial serine/threonine-protein kinase that is crucial for Parkin tethering to damaged mitochondria (50), an effect not seen in Fl/Fl muscle (Fig 5A). The increase in PINK1 abundance in MBKO muscle could be a compensatory response to the low Parkin level. The phosphorylation of Ser616 in dynamin-related protein 1 (DRP1), an activation marker of mitochondrial fission (51), was significantly increased in Fl/Fl muscle after exercise training. This finding, when combined with the increased Parkin abundance in muscle, support the view that mitochondrial fission and mitophagy are functionally coupled (52). In contrast, exercise failed to induce the phosphorylation of Ser616 in DPR1 in MBKO mice (Fig 5A), suggesting compromised exercise-induced mitochondrial fission. AMPK-mediated phosphorylation of Ser146 in mitochondrial fission factor (MFF), a mitochondrial membrane protein that recruits DRP1 to the mitochondria (53), was also enhanced by exercise training in Fl/Fl muscle but not in MBKO muscle (Fig 5A). We also examined the amount of key mitochondrial fusion factors, mitofusin 1 (MFN1), mitofusion 2 (MFN2), and optic atrophy protein 1 (OPA1) (54). Although exercise training increased MFN1 accumulation in the muscle of both genotypes (Fig 5A), exercised MBKO muscle accumulated more MFN2 and OPA1 (Fig 5A), which could underlie mitochondrial enlargement (Fig 5C). Together, our data demonstrate that muscle-generated BDNF promotes changes in gene expression and protein abundance that may underlie endurance exercise-induced mitochondrial biogenesis, mitochondrial fission, and mitophagy.
Consumption of a BDNF mimetic enhances exercise performance
To provide further evidence for the importance of BDNF signaling in muscle for exercise-mediated metabolic adaptation and muscle performance, we mimicked the exercise-induced BDNF secretion in muscle by feeding C57BL/6 mice with the bioavailable BDNF mimetic 7,8-DHF, which activates TrkB and AMPK signaling in cultured myotubes and mouse muscle (23, 24, 55). WT C57BL/6 mice administrated 7,8-DHF ad libitum for 3 months showed significant increases in total running distance, total running time, maximal running speed (Fig 6A), and hanging ability (Fig 6B), suggesting 7,8-DHF can boost muscle endurance. In contrast, 7,8-DHF treatment did not alter individual muscle mass, myofiber composition (Fig S4, A and B), or muscle strength (Fig 6C). Consistent with our previous report that 7,8-DHF increases mitochondrial biogenesis in C2C12 myotubes (24) (26), 7,8-DHF-treated mice had increased abundance of mitochondrial proteins such as SDHA in their muscles (Fig 6D), which was associated with enhanced expression of Nrf1, Tfam, and Ppargc1a (Fig 6E). We also detected increases in AMPK phosphorylation and LC3 lipidation but a decrease in p62 content in the muscle of 7,8-DHF-treated mice (Fig 6D), suggesting that autophagy was augmented. The muscle of 7,8-DHF-treated mice showed increased phosphorylation of Ser616 in DRP1, suggesting that 7,8-DHF consumption could enhance mitochondrial fission in skeletal muscle (Fig 6D). Moreover, increased Parkin abundance suggested enhanced mitophagy in the muscle of 7,8-DHF-treated mice (Fig 6D). In addition, 7,8-DHF treatment significantly increased the amount of PPARδ in muscle (Fig 6D), which was associated with augmented expression of the metabolic genes in FA mobilization (Cd36 and Fabp3), β-oxidation (Acadl and Cpt1b), and metabolic flexibility (Pdk4) (Fig 6E). Lastly, we tested if chronic consumption of 7,8-DHF rescued the functional impairment of MBKO muscle. As anticipated, the impairments in running time, total running distance, and maximal running speed of MBKO mice were attenuated after 7,8-DHF treatment for 3 months, and their exercise performance became comparable to that of naive Fl/Fl mice (Fig 6G). Together, our results indicate that 7,8-DHF treatment may be sufficient to enhance metabolic remodeling and muscle performance in normal mice.
Fig. 6. Consumption of BDNF mimetic 7,8-DHF enhances exercise performance.
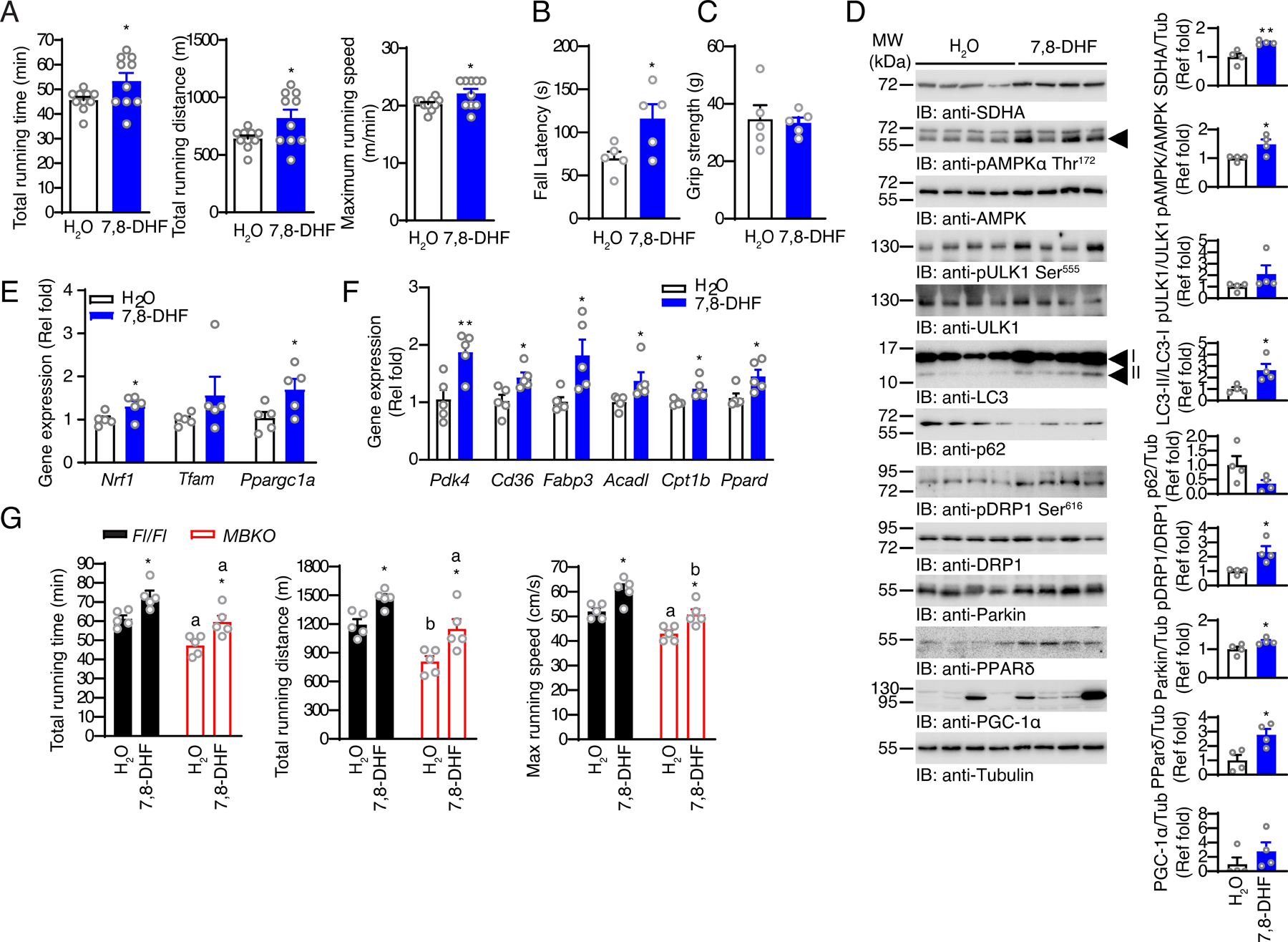
(A) The running capacity of mice that consumed 7,8-DHF for 12 weeks (*: P<0.05, Student’s t-test, n=10 mice per group). (B) Up-side down hanging time of mice that consumed 7,8-DHF for 12 weeks (*: P<0.01, n=5 mice per group). (C) Total muscle strength of mice that consumed 7,8-DHF for 12 weeks as determined by the grip-strength test (n=5 mice/group, Student’s t-test). (D) Immunoblotting analysis of signaling proteins in the gastrocnemius muscle of mice that consumed 7,8-DHF for 12 weeks. Bar graphs show quantification of immunoblot signals (*: P<0.05, **: P<0.01, Student’s t-test, n=4 mice per group). (E) Expression of genes involved in mitochondrial biogenesis in the gastrocnemius muscle of mice that consumed 7,8-DHF for 12 weeks (*: P<0.05, Student’s t-test, n=5 mice per group). (F) Expression of PPARδ-regulated genes in the gastrocnemius muscle of mice that consumed 7,8-DHF for 12 weeks (*: P<0.05, **: P<0.01, Student’s t-test, n=5 mice per group). (G) The running capacity of Fl/Fl and MBKO mice that consumed 7,8-DHF for 12 weeks (*: P<0.05 compared to H2O group of the same genotype; a: P<0.01, b: P<0.001 compared to Fl/Fl mice receiving the same treatment, two-way ANOVA, n=5 mice per group).
DISCUSSION
Although regular exercise has widespread health benefits for nearly all systems in the body, it also induces metabolic stresses, which include high ATP demand, depletion of glycogen and lipid reserves, and elevated ROS production (56). These stresses are resolved by increasing metabolic flux and regulating metabolic gene expression after exercise. PPARs partially contribute to these adaptations (14, 15). Because PPARδ activity is also essential to maintaining FA uptake (57), it seems logical to hypothesize that PPARδ plays a role in post-exercise IMTG recovery. Intramyocellular FAs have been suggested to induce Ppard expression, but this hypothesis is contradicted by FA incubation failing to alter Ppard expression in cultured myotubes (12, 41). Hence, the upstream factor that promotes Ppard expression after exercise remains unknown (12, 58). In this study, we found that post-exercise synthesis of BDNF was crucial for inducing Ppard expression in female mice to maintain metabolic homeostasis. Pharmacological inhibition of PPARδ activity in myotubes abolished the stimulatory effect of BDNF on the expression of genes involved in FA uptake, lipogenesis, FAO, and metabolic flexibility (Fig 3I), suggesting PPARδ is the major downstream effector of BDNF for regulating lipid metabolism in skeletal muscle. Consistent with this notion, exercise failed to induce the expression of Ppard or that of its downstream targets in MBKO muscle (Fig 4E), which might explain the delayed functional and metabolic recovery in muscle after repetitive exercise challenge (Figs 2E, 2G and 2F). The expression level of Ppard was comparable in the muscle of sedentary Fl/Fl and MBKO mice (Figs 3D and E), which conflicts with the lower Ppard expression after Bdnf knockdown in C2C12 myotubes (Fig 3B). The enhanced production of other PPARδ stimulators, such as fibroblast growth factor 21 (FGF21) (25, 59), might compensate for the loss of basal BDNF-induced Ppard expression in MBKO mice. The secretion of BDNF from non-muscle cells, such as motor neurons and immune cells, might also contribute to maintaining basal Ppard expression (60, 61). Nevertheless, the production of BDNF in muscle is essential to acute and chronic exercise-induced Ppard expression (Figs 3D and 4E). Together with the findings that Bdnf depletion in C2C12 myotubes suppresses mitochondrial respiration only upon FA overload (26) and that mitochondrial lipid oxidation in muscle is impaired by fasting in MBKO mice (25), BDNF production in muscle might be a protective mechanism against various metabolic stresses but may not be involved in maintaining basal metabolic activities.
The cumulative effect of transient changes in gene transcription during post-exercise recovery from each exercise session is critical to long-term cellular adaptation elicited by exercise training (9). Bdnf deficiency in muscle compromises exercise training-induced changes in the expression of genes involved in remodeling mitochondrial structure, β-oxidation, and metabolism, which is associated with failure to improve endurance (Figs 4E and 5C–E). However, chronic activation of BDNF-TrkB signaling by 7,8-DHF is sufficient to enhance exercise endurance and the expression of genes involved in mitochondrial dynamics and lipid metabolism (Figs 6A–F). The failure of exercise training to induce metabolic gene expression in MBKO muscle could be attributed to inactivation of PPARδ signaling. Because PPARδ is also important in promoting the expression of PGC-1α (the master regulator of mitochondrial biogenesis) in skeletal muscle and heart (62, 63), the reduced amount of PPARα in MBKO muscle might explain the lower amounts of proteins involved in mitochondrial biogenesis in the tissue (Fig 5A–B). In agreement with this notion, we showed that BDNF failed to induce Ppargc1a expression when PPARδ was inhibited in vitro (Fig 3I). However, whether the impaired PPARδ signaling in the MBKO muscle is involved in the dysregulated mitochondrial fission remains uncertain because the role of PPARδ in skeletal muscle mitochondrial dynamics has not been convincingly confirmed. We demonstrated that BDNF promoted Prkn expression through PPARδ (Fig 3I), suggesting the PPARδ could transcriptionally regulate mitochondrial dynamics. In support of this notion, PPARδ promotes Mfn2 expression in cardiomyocytes (64), and pharmacological activation of PPARδ triggers mitophagy to ameliorate mitochondrial damage in astrocytes (65). Given the role of dysregulated mitochondrial dynamics in myopathies (66), further study is warranted to uncover the role of PPARδ in mitochondrial quality maintenance.
Although BDNF in muscle controls lipid homeostasis during fasting (25) and after exercise (Figs 2E–G and 4E–G), it is also responsible for promoting mitochondrial clearance because suppressing Bdnf expression causes the accumulation of defective mitochondria in obese muscle (26). Hence, it is reasonable that MBKO mice have more faulty mitochondria in muscle after exercise training, which is associated with reduced abundance of proteins involved in mitochondrial fission and mitophagy (Fig 5A). In contrast to the enhanced PINK1 cleavage in the muscle of MBKO mice fed a high-fat diet (26), more full-length PINK1 was retained in MBKO muscle after exercise training (Fig 5A). Moreover, less Parkin was found in MBKO muscle than in Fl/Fl muscle after repeated bouts of exercise, but the amount of Parkin is comparable between the two genotypes after high-fat diet feeding (26). BDNF might activate distinct intracellular pathways to modulate mitochondrial behavior in response to different metabolic challenges. Given the role of BDNF in the energy homeostasis in exercise and fasting, it is tempting to suggest that enhanced BDNF production is key to synergy between fasting and aerobic exercise training in body weight reduction and muscle endurance improvement as reported in obese subjects (67, 68).
BDNF in muscle is an exercise-responsive gene. BDNF production has been suggested to promote FAO by activating AMPK during exercise (19). However, our findings suggest that Bdnf expression and AMPK activation were uncoupled because AMPK phosphorylation and BDNF production in muscle were not temporally linked (Fig 1E) and AMPK activity was comparable in Fl/Fl and MBKO mice during exercise (Fig 2A). Similar to mice, AMPK activation occurs during exercise in rat and human muscle, but the expression of BDNF in muscle is a post-exercise response (19, 35, 69), further supporting that BDNF is not upstream of AMPK in exercising muscles. Other signaling factors, such as Ca2+-calmodulin kinases (70–72) and increases in intracellular AMP levels (73), may be more prominent inducers of AMPK activation in contracting muscle. Although AMPK phosphorylation was slightly reduced in the muscle of exercise-trained MBKO mice (Fig 5A), this effect might be a consequence of lower PPARδ activity because activation of PAPRδ promotes AMPK phosphorylation (74–76). Nevertheless, although BDNF is unquestionably an upstream regulator of AMPK in many tissues (19, 77, 78), a functional link between BDNF and AMPK in muscle might be more critical in counteracting nutritional stresses such as fasting and obesity (25, 26).
MBKO mice have lower exercise capacity than Fl/Fl mice even after exercise training (Figs 2B–D and 4A–C). This observation contradicts the report that Bdnf ablation in muscle results in type IIB-to-type IIx myofiber transition and augments running performance (79). It is not clear why opposite phenotypes were detected in MBKO mice from different sources, but our conclusion that BDNF is beneficial for exercise performance aligns with several other reports. Firstly, chronic exercise training rewires the metabolic machinery towards a higher reliance on FAO in skeletal muscle, which is associated with improved endurance performance (80), which agrees with our finding that BDNF stimulation promotes FAO (25) and reprograms metabolic gene expression by inducing Ppard expression to favor lipid oxidation (Figs 3I–J and 4E). Secondly, we and others have shown that BDNF signaling stimulates mitochondrial biogenesis (81–83), which positively correlates with exercise capacity (84). Thirdly, administering recombinant BDNF in mice for 5 weeks increases exercise capacity and the expression of genes involved in lipid catabolism (77), which agrees with our results using a BDNF mimetic (Fig 6A–F). Lastly, the BDNF/TrkB signaling pathway is a crucial mechanism for exercise-induced rescue of muscle dysfunction in animals with heart failure (85).
The efficacy of exercise-induced Bdnf expression between sexes has not been compared. Indeed, sex-dimorphic effects are common with many BDNF-promoted neurological activities, such as pain sensation and stress response (86, 87). We have shown previously that fasting promotes Bdnf expression in muscle exclusively in female mice (25). Consequently, Bdnf muscle ablation changes body weight and lipid metabolism only in female mice (25, 26). A comparison of the exercise-induced skeletal muscle proteome showed no differences in BDNF protein abundance in the vastus lateralis muscle between men and women (88). These discrepancies could be caused by differences in the exercise protocols used. Ppar expression shows sexual dimorphism in humans, such that women have higher Ppard and Ppara expression in muscle, suggesting women might have higher pre-translational abundance of the genes responsible for lipid metabolism downstream of PPARs (89). This finding might explain the higher FA mobilization in women during endurance exercise (90). Moreover, women have a higher post-exercise recovery rate of lipid content, which is supported by decreased loss of fat mass in women compared to men during physical training (91). It is tempting to hypothesize that the BDNF-PPARδ signaling in the skeletal muscle might play a more critical role in females to maintain constant energy substrate stores during recovery from exercise.
In summary, our results indicate that BDNF is a crucial myokine for exercise-induced changes in gene expression that support metabolic reprogramming, mitochondrial remodeling, and recovery in skeletal muscle by activating the CaMKK2-PPARδ cascade (Fig 7). Hence, chronic activation of the BDNF signaling in the skeletal muscle will not only ameliorate the health condition of obese subjects (24, 26) but also improve exercise endurance in normal individuals.
Fig 7. Proposed role of BDNF in reprogramming muscle metabolism during recovery from endurance exercise.
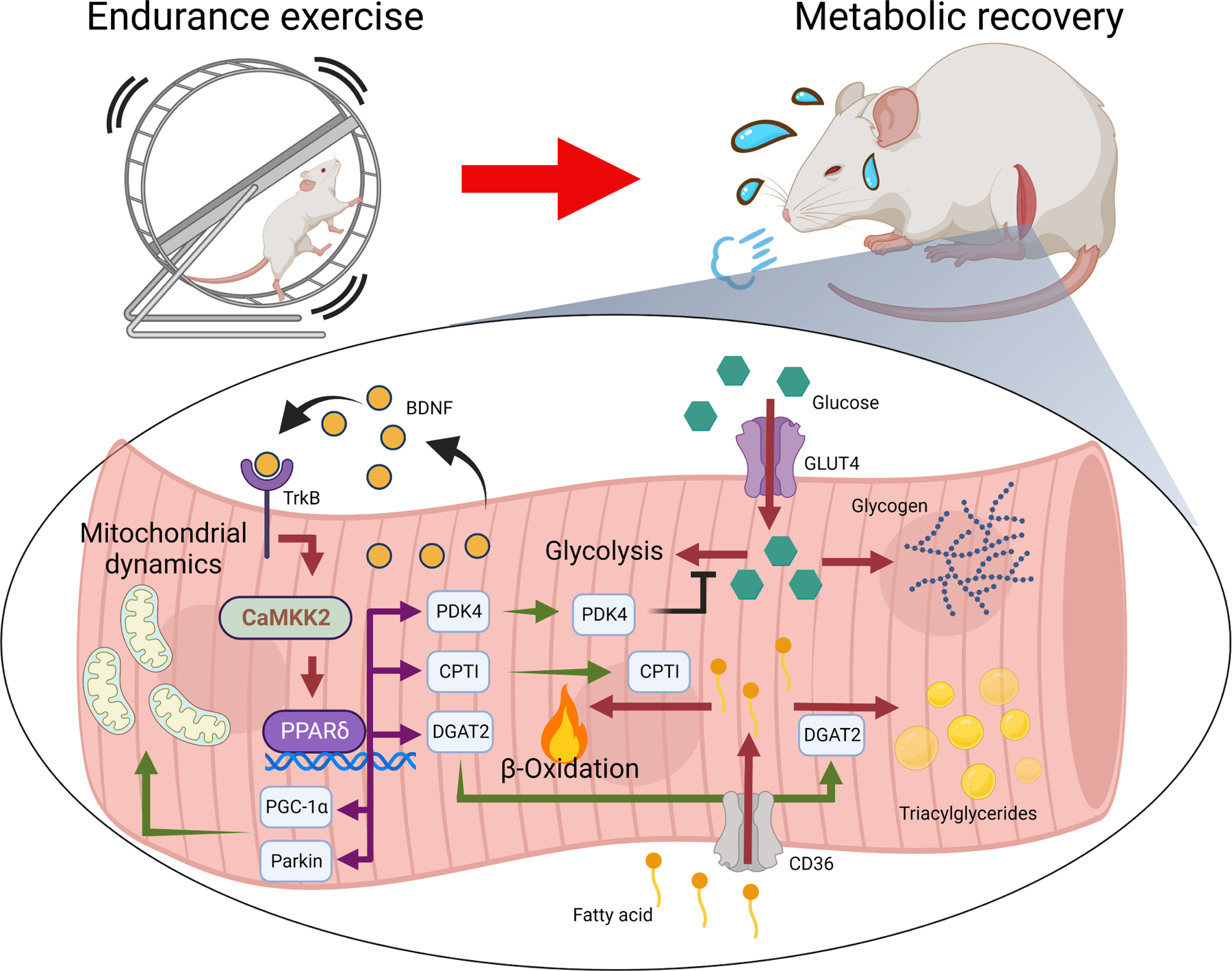
Expression of Bdnf in skeletal muscle is induced during the late recovery phase to change fuel utilization preference and restock intramyocellular lipid reserves through PPARδ-induced gene expression. It also promotes mitochondrial biogenesis and recycling after repeated exercise training, leading to improvements in oxidative metabolism and muscle performance.
MATERIALS AND METHODS
Chemicals
Recombinant human BDNF protein (GF029) was purchased from Merck. 7,8-DHF (TCI D1916) was purchased from Tokyo Chemical Industry Co., Ltd. The PPARδ inhibitor GSK3787 (ab144575) was purchased from Abcam. Antibodies against BDNF (ab108319), PGC-1α (ab54481), MFN1 (ab104274), myosin heavy chain IIx/b (ab91506) were obtained from Abcam. Antibodies against AMPKα phosphorylated at Thr172 (2535S), ULK1 phosphorylated at Ser555 (5869), ULK (8054), LC3A/B (12741P), p62 (5114S), SDHA (11998), VDAC (4661), Parkin (4211), PINK1 (6946), DRP1 phosphorylated at Ser616 (4494), DRP1 (8570), MFF phosphorylated at Ser146 (49281), MFF (84580), MFN2 (11925), OPA1 (80471), Tom 20 (42406) were ordered from Cell Signaling Technology. Anit-AMPKα (sc-25792) was purchased from Santa Cruz Biotechnology. Anti-Tubulin (T6074) and anti-myosin heavy chain type I (MAB1628) were obtained from Sigma-Aldrich. The siGENOME siRNA pool against mouse Camkk2 was purchased from Horizon Discovery (USA). Control adenovirus (Ad-Ctr) (VB161026-1127kyf) and Ad-shBDNF were customized by GeneScript as previously reported (29).
Animals
BDNF Fl/Fl mice containing loxP sequences that flank the Bdnf exon IX were purchased from the Jackson Laboratory (004339). MBKO mice were generated by crossing BDNF Fl/Fl mice with transgenic mice carrying a Cre gene regulated by the human α-skeletal actin promoter (006149, the Jackson Laboratory) as reported (25). Genotypes were confirmed by PCR using genomic DNA extracted from the tail. All experimental procedures on mice were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR) of the University of Hong Kong. Mice were kept in a standard housing area with a 12-hour light/dark cycle and ad libitum access to a chow diet and water. Due to the sex dimorphic action of BDNF in muscle metabolism (25, 26), only female mice were used for experiments when they reached 3 months of age. Their whole-body weight, fat mass, and lean mass were measured by NMR (Bruker). At the end of the experiments, animals were euthanized by an anesthetic overdose, and the tissues were snap frozen in liquid nitrogen. Parts of the tissues were fixed in 4% paraformaldehyde for histological analysis.
Exercise training protocol
Mice were randomly designated to either a non-trained group or an exercise-trained group. Mice in the exercise-trained group were subjected to treadmill training, which were trained at a speed of 12 m/min at a slope of 0° for 5 consecutive days/week for 4 weeks as previously reported (30). The number of electrical stimulations during the training was recorded every day to assess the animal’s fatigue level.
Endurance capacity test, grip strength test, and four-limb hanging test
One day before the exercise test, the mice were placed on the non-operating treadmill for 30 mins to become familiar with the sight and smell of the apparatus and the exercise training room. Then, the treadmill was turned on at a low speed (9.6 m/min) to allow the mouse to walk/run slowly for two 15-minute sessions separated by a 5-minute break. On the day of the test, the mice were placed on the treadmill to warm up at a speed of 10 m/min for 10 min. Mice were exercised to fatigue by increasing the treadmill speed for 1 m/min every 3 min until the animal was exhausted. The exhaustion of an animal was judged by its refusal to run on the treadmill belt and stay in the shock grid for more than 10s. The highest speed, total running time, total running distance, and the number of electrical stimulations were calculated.
The mouse grip strength was measured by a grip strength meter (BIO-GS3, BIOSEB). All four limbs of a mouse were placed on the grid before the measurement, and the tail was pulled horizontally until all four limbs came off the grid. The maximum grip force was recorded by the meter. Five replicate measurements were taken from each mouse continuously without a break, and the average grip strength was calculated for comparison.
In the four-limb hanging test, mice were hung up-side-down from a wire grid placed at 20 cm above the ground, and their hanging time was recorded. Three replicate measurements were made for each mouse, and the mouse was allowed to rest for 1 minute between each trial. The maximum time in all trials was used for comparison.
7,8-DHF feeding
The 7,8-DHF solution was prepared by dissolving 0.13 g/L 7,8-DHF in distilled water and was stirring overnight at room temperature. pH was adjusted to 7.6–7.8 by adding 1M NaOH. The solution was sterile-filtered and was given to female C57BL/6 mice ad libitum for 3 months (24).
Tissue and blood metabolite analysis
Muscle tissues were snap frozen in liquid nitrogen upon collection, and serum samples were prepared by centrifuging the coagulated blood at 2000 x g for 20 minutes. The content of TG, free FA, and glycogen concentrations were determined using the Triglyceride Colorimetric Assay Kit (10010303), Glycogen Assay Kit (700480), and Free Fatty Acid Quantification Colorimetric/Fluorometric Kit (700310), respectively, according to the manufacturer’s (Cayman) instructions. Serum BDNF was determined by PicoKine ELISA (EK0309, Boster Biological Technology).
Real-time PCR
Total RNA from tissue and cells was extracted using RNAiso Plus reagent (9109, Takara). Complementary DNA was synthesized using iScript™ Advanced cDNA Synthesis Kit (1725038, Bio-Rad) with oligo (dT) primer according to the manufacturer’s instructions. Real-time PCR was performed using iTaq™ Universal SYBR® Green Supermix (1725124, Bio-Rad) and detected by the LightCycler® 96 Instrument (Roche Life Science). Gene expression was normalized to Rpl7 gene, which encodes ribosomal protein L7 and is an optimal housekeeping gene for myocytes (31). Primer sequences used in real-time PCR are listed in Supplementary Table 1.
Fatty Acid oxidation
The 14C-palmitate oxidation assay was performed as previously described (32). Freshly isolated gastrocnemius muscle was minced in STE Buffer (0.25M sucrose, 10mM Tris-HCl, 1mM EDTA, pH 7.4) and homogenized on ice by a Dounce homogenizer. After centrifuging the tissue lysate at 450 x g for 10 minutes, intact mitochondria were collected in the supernatant. The Bradford assay was used to normalize mitochondrial amounts between samples. Lysates were incubated with 7% BSA-coupled 14C-palmitate (0.4 μCi per reaction, NEC075H050UC, Perkin Elmer) at 37°C for 30 minutes with frequent mixing. The reaction was quenched by adding 1 M perchloric acid, and the 14CO2 released was captured by 1M NaOH-saturated Whatman paper disc. The amount of 14CO2 in the paper disc was determined by scintillation counting.
AMPK activity
AMPK activity in the skeletal muscle was determined using the CycLex AMPK kinase Assay Kit (CY-1182, MBL International) as previously reported (33).
Cell culture and subcellular fractionation
C2C12 cells were purchased from the American Type Culture Collection (ATCC). Before differentiation, C2C12 myoblast was cultured in complete growth medium [15% calf serum (16010159, Thermo Fischer Scientific), 5% fetal bovine serum (SV30160.03, Cytiva) penicillin (100 IU/ml) and streptomycin (100 μg/ml) (SV30010, Cytiva) in DMEM (12800017, Thermo Fischer Scientific)]. For differentiation into myotubes, the cells were grown to 100% confluency before being switched to differentiation medium [2% horse serum (16050130, Thermo Fischer Scientific), penicillin (100 IU/ml), and streptomycin (100 μg/ml) in DMEM]. The differentiation medium was changed daily for 4 days until C2C12 cells fused into long myotubes. Mitochondria in C2C12 myotubes were isolated as previously reported (34). Briefly, the cells were homogenized using a glass douncer, then serially centrifuged at 800 x g for 15 min and 11,000 x g for 10 min at 4 °C. The pellet was suspended in lysis buffer and used for immunoblotting.
Oil Red O staining
An identical number of C2C12 myoblasts (2×106/well) were seeded in a 6-well plate and fully differentiated into myotubes. After adenovirus infection, cells were fixed in 4% paraformaldehyde and stained with Oil Red O (O0625, Sigma) solution for 4 h. After washing with PBS, Oil Red O was dissolved in isopropanol and added to cells, which were measured at O.D. 490 nm.
Immunoblotting
Tissues and cells were lysed in low-salt lysis buffer (50mM Tris-base (pH 7.4), 40mM NaCl, 1mM EDTA, 50mM Na3VO4, 10mM Na4P2O7, 10mM, C3H7Na2O6P, 0.5% Triton X-100) and centrifuged. Protein concentrations were determined by the Bradford assay. Proteins were separated by SDS-PAGE before being transferred to a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (1704150, BioRad). After blocking with 5% skim milk or 3% BSA for 1 h at room temperature, membranes were blotted with primary antibodies at 4°C overnight. The next day, membranes were blotted with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Chemiluminescence signals were developed using UltraScence Pico Ultra Western Substrate (CCH345-B600ML, Bio-Helix) and visualized in a G:BOX Chemi XRQ Imager (Syngene). Band intensity was measured with ImageJ (NIH). When the number of protein samples analyzed exceeded the capacity of a single SDS-PAGE gel, they were resolved in 2 gels. These gels underwent transfer and immunoblotting procedures simultaneously and chemiluminescence signals were determined by the same imager together.
Immunofluorescence staining and electron microscopy
Immunofluorescence staining was done in 6 μm paraffin-embedded muscle sections. After deparaffinization and rehydration, slides were boiled in 10 mM citric acid (pH 6.0) for antigen retrieval. Slides were permeabilized and blocked with blocking solution (1% BSA, 0.2% Tween-20, 0.3M glycine, 10% goat serum in PBS) for 1 h at room temperature. The slides were then stained overnight with anti-Tom20 antibodies. The next day, the slides were incubated with Alexa Fluor 488 Goat anti-Rabbit IgG (H+L) (A11008, Thermo Fischer Scientific). DAPI (14564, Affymetrix USB) was used to stain nuclei. Tissue sections were mounted with Fluoromount-G® Mounting Medium (0100-01, SouthernBiotech). Images were captured by the confocal microscope with a 60X magnification objective lens (Carl Zeiss LSM 880). Two to three images were taken from each animal sample, and images representative of 3 different mice for each genotype were shown. For electron microscopy, gastrocnemius muscles were fixed overnight in 4% paraformaldehyde, cut, and stored in 2.5% glutaraldehyde (GTA) buffer. The tissues were scanned by transmission electron microscope (Philips CM100 TEM, FEI Company). The morphology and area of the mitochondria were analyzed using ImageJ.
Metabolic flux analysis
The oxidative and glycolytic phenotypes of differentiated C2C12 myotubes were determined using the Agilent Seahorse XF Cell Energy Phenotype Test (103325-100, Agilent). The cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) before and after the addition of oligomycin (1 µM) and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 2µM) in the stressed state was measured by the Agilent Seahorse XFe24 Analyzer.
Statistical analysis
Results are expressed as the mean ± SEM and were considered significant when P < 0.05. Student’s t-test, one-way ANOVA, or two-way ANOVA followed by Tukey’s multiple comparison test for statistical analysis using GraphPad Prism (GraphPad Software). Only statistical outliers identified by the Grubbs’ test with α=0.05 threshold were omitted from the analysis.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Electron Microscope Unit and the Department of Pathology from the University of Hong Kong for the histology service and analysis. We also thank Dr. Jane Jie Zhao (School of Public Health, LKS Faculty of Medicine) at the University of Hong Kong for the statistical analysis consultation.
Funding
This work is supported by the Hong Kong Health and Medical Research Fund (HMRF06171836) and HKU Seed Fund for Basic Research to CBC, and NIH grant 5U2C-DK093000 to J.K.K. We would like to thank the National Mouse Metabolic Phenotyping Center (MMPC) at the University of Massachusetts Medical School for their assistance in performing the exercise performance test.
Footnotes
COMPETING INTEREST
The authors declare that they have no competing interests.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplemental Materials.
REFERENCES AND NOTES
- 1.Ehrenborg E, Krook A, Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol Rev 61, 373–393 (2009); published online EpubSep ( 10.1124/pr.109.001560). [DOI] [PubMed] [Google Scholar]
- 2.Lundsgaard AM, Fritzen AM, Kiens B, The Importance of Fatty Acids as Nutrients during Post-Exercise Recovery. Nutrients 12, (2020); published online EpubJan 21 ( 10.3390/nu12020280). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burke L, Fasting and recovery from exercise. Br J Sports Med 44, 502–508 (2010); published online EpubJun ( 10.1136/bjsm.2007.071472). [DOI] [PubMed] [Google Scholar]
- 4.Betts JA, Williams C, Short-term recovery from prolonged exercise: exploring the potential for protein ingestion to accentuate the benefits of carbohydrate supplements. Sports Med 40, 941–959 (2010); published online EpubNov 1 ( 10.2165/11536900-000000000-00000). [DOI] [PubMed] [Google Scholar]
- 5.van Loon LJ, Koopman R, Stegen JH, Wagenmakers AJ, Keizer HA, Saris WH, Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance-trained males in a fasted state. J Physiol 553, 611–625 (2003); published online EpubDec 1 ( 10.1113/jphysiol.2003.052431). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodpaster BH, He J, Watkins S, Kelley DE, Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86, 5755–5761 (2001); published online EpubDec ( 10.1210/jcem.86.12.8075). [DOI] [PubMed] [Google Scholar]
- 7.Décombaz J, Nutrition and recovery of muscle energy stores after exercise. Schweizerische Zeitschrift fur Sportmedizin und Sporttraumatologie 51, (2003); published online Epub01/01 ( [Google Scholar]
- 8.Kiens B, Richter EA, Utilization of skeletal muscle triacylglycerol during postexercise recovery in humans. Am J Physiol 275, E332–337 (1998); published online EpubAug ( 10.1152/ajpendo.1998.275.2.E332). [DOI] [PubMed] [Google Scholar]
- 9.Pilegaard H, Ordway GA, Saltin B, Neufer PD, Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab 279, E806–814 (2000); published online EpubOct ( 10.1152/ajpendo.2000.279.4.E806). [DOI] [PubMed] [Google Scholar]
- 10.van Hall G, Sacchetti M, Radegran G, Saltin B, Human skeletal muscle fatty acid and glycerol metabolism during rest, exercise and recovery. J Physiol 543, 1047–1058 (2002); published online EpubSep 15 ( 10.1113/jphysiol.2002.023796). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muoio DM, MacLean PS, Lang DB, Li S, Houmard JA, Way JM, Winegar DA, Corton JC, Dohm GL, Kraus WE, Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. J Biol Chem 277, 26089–26097 (2002); published online EpubJul 19 ( 10.1074/jbc.M203997200). [DOI] [PubMed] [Google Scholar]
- 12.Spangenburg EE, Brown DA, Johnson MS, Moore RL, Alterations in peroxisome proliferator-activated receptor mRNA expression in skeletal muscle after acute and repeated bouts of exercise. Mol Cell Biochem 332, 225–231 (2009); published online EpubDec ( 10.1007/s11010-009-0195-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA, Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J 17, 2299–2301 (2003); published online EpubDec ( 10.1096/fj.03-0269fje). [DOI] [PubMed] [Google Scholar]
- 14.Fan W, Waizenegger W, Lin CS, Sorrentino V, He MX, Wall CE, Li H, Liddle C, Yu RT, Atkins AR, Auwerx J, Downes M, Evans RM, PPARdelta Promotes Running Endurance by Preserving Glucose. Cell Metab 25, 1186–1193 e1184 (2017); published online EpubMay 2 ( 10.1016/j.cmet.2017.04.006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunmair B, Staniek K, Dorig J, Szocs Z, Stadlbauer K, Marian V, Gras F, Anderwald C, Nohl H, Waldhausl W, Furnsinn C, Activation of PPAR-delta in isolated rat skeletal muscle switches fuel preference from glucose to fatty acids. Diabetologia 49, 2713–2722 (2006); published online EpubNov ( 10.1007/s00125-006-0357-6). [DOI] [PubMed] [Google Scholar]
- 16.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM, Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2, e294 (2004); published online EpubOct ( 10.1371/journal.pbio.0020294). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedersen BK, Febbraio MA, Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8, 457–465 (2012); published online EpubApr 3 ( 10.1038/nrendo.2012.49). [DOI] [PubMed] [Google Scholar]
- 18.Huh JY, The role of exercise-induced myokines in regulating metabolism. Arch Pharm Res 41, 14–29 (2018); published online EpubJan ( 10.1007/s12272-017-0994-y). [DOI] [PubMed] [Google Scholar]
- 19.Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH, Penkowa M, Hojman P, Zankari A, Watt MJ, Bruunsgaard H, Pedersen BK, Febbraio MA, Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52, 1409–1418 (2009); published online EpubJul ( 10.1007/s00125-009-1364-1). [DOI] [PubMed] [Google Scholar]
- 20.Ogborn DI, Gardiner PF, Effects of exercise and muscle type on BDNF, NT-4/5, and TrKB expression in skeletal muscle. Muscle Nerve 41, 385–391 (2010); published online EpubMar ( 10.1002/mus.21503). [DOI] [PubMed] [Google Scholar]
- 21.Wang CS, Kavalali ET, Monteggia LM, BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 185, 62–76 (2022); published online EpubJan 6 ( 10.1016/j.cell.2021.12.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR, Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol 88, 2187–2195 (2002); published online EpubNov ( 10.1152/jn.00152.2002). [DOI] [PubMed] [Google Scholar]
- 23.Chan CB, Tse MC, Liu X, Zhang S, Schmidt R, Otten R, Liu L, Ye K, Activation of muscular TrkB by its small molecular agonist 7,8-dihydroxyflavone sex-dependently regulates energy metabolism in diet-induced obese mice. Chem Biol 22, 355–368 (2015); published online EpubMar 19 ( 10.1016/j.chembiol.2015.02.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood J, Tse MCL, Yang X, Brobst D, Liu Z, Pang BPS, Chan WS, Zaw AM, Chow BKC, Ye K, Lee CW, Chan CB, BDNF mimetic alleviates body weight gain in obese mice by enhancing mitochondrial biogenesis in skeletal muscle. Metabolism 87, 113–122 (2018); published online EpubOct ( 10.1016/j.metabol.2018.06.007). [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Brobst D, Chan WS, Tse MCL, Herlea-Pana O, Ahuja P, Bi X, Zaw AM, Kwong ZSW, Jia WH, Zhang ZG, Zhang N, Chow SKH, Cheung WH, Louie JCY, Griffin TM, Nong W, Hui JHL, Du GH, Noh HL, Saengnipanthkul S, Chow BKC, Kim JK, Lee CW, Chan CB, Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci Signal 12, (2019); published online EpubAug 13 ( 10.1126/scisignal.aau1468). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahuja P, Ng CF, Pang BPS, Chan WS, Tse MCL, Bi X, Kwan HR, Brobst D, Herlea-Pana O, Yang X, Du G, Saengnipanthkul S, Noh HL, Jiao B, Kim JK, Lee CW, Ye K, Chan CB, Muscle-generated BDNF (brain derived neurotrophic factor) maintains mitochondrial quality control in female mice. Autophagy, 1–18 (2021); published online EpubOct 25 ( 10.1080/15548627.2021.1985257). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fulgenzi G, Hong Z, Tomassoni-Ardori F, Barella LF, Becker J, Barrick C, Swing D, Yanpallewar S, Croix BS, Wess J, Gavrilova O, Tessarollo L, Novel metabolic role for BDNF in pancreatic beta-cell insulin secretion. Nat Commun 11, 1950 (2020); published online EpubApr 23 ( 10.1038/s41467-020-15833-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamanaka M, Tsuchida A, Nakagawa T, Nonomura T, Ono-Kishino M, Sugaru E, Noguchi H, Taiji M, Brain-derived neurotrophic factor enhances glucose utilization in peripheral tissues of diabetic mice. Diabetes Obes Metab 9, 59–64 (2007); published online EpubJan ( 10.1111/j.1463-1326.2006.00572.x). [DOI] [PubMed] [Google Scholar]
- 29.Jeanblanc J, Logrip ML, Janak PH, Ron D, BDNF-mediated regulation of ethanol consumption requires the activation of the MAP kinase pathway and protein synthesis. Eur J Neurosci 37, 607–612 (2013); published online EpubFeb ( 10.1111/ejn.12067). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SH, Kim BJ, Park DR, Kim UH, Exercise induces muscle fiber type switching via transient receptor potential melastatin 2-dependent Ca(2+) signaling. J Appl Physiol (1985) 124, 364–373 (2018); published online EpubFeb 1 ( 10.1152/japplphysiol.00687.2017). [DOI] [PubMed] [Google Scholar]
- 31.Perez LJ, Rios L, Trivedi P, D’Souza K, Cowie A, Nzirorera C, Webster D, Brunt K, Legare J-F, Hassan A, Kienesberger PC, Pulinilkunnil T, Validation of optimal reference genes for quantitative real time PCR in muscle and adipose tissue for obesity and diabetes research. Sci Rep 7, 3612–3612 (2017) 10.1038/s41598-017-03730-9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynh FK, Green MF, Koves TR, Hirschey MD, Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol 542, 391–405 (2014) 10.1016/B978-0-12-416618-9.00020-0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tse MCL, Herlea-Pana O, Brobst D, Yang X, Wood J, Hu X, Liu Z, Lee CW, Zaw AM, Chow BKC, Ye K, Chan CB, Tumor Necrosis Factor-alpha Promotes Phosphoinositide 3-Kinase Enhancer A and AMP-Activated Protein Kinase Interaction to Suppress Lipid Oxidation in Skeletal Muscle. Diabetes 66, 1858–1870 (2017); published online EpubJul ( 10.2337/db16-0270). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahuja P, Ng CF, Pang BPS, Chan WS, Tse MCL, Bi X, Kwan HR, Brobst D, Herlea-Pana O, Yang X, Du G, Saengnipanthkul S, Noh HL, Jiao B, Kim JK, Lee CW, Ye K, Chan CB, Muscle-generated BDNF (brain derived neurotrophic factor) maintains mitochondrial quality control in female mice. Autophagy 18, 1367–1384 (2022); published online EpubJun ( 10.1080/15548627.2021.1985257). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuppini R, Sartini S, Agostini D, Guescini M, Ambrogini P, Betti M, Bertini L, Vallasciani M, Stocchi V, Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch Ital Biol 145, 99–110 (2007); published online EpubMay ( [PubMed] [Google Scholar]
- 36.Haapasalo A, Sipola I, Larsson K, Akerman KE, Stoilov P, Stamm S, Wong G, Castren E, Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 277, 43160–43167 (2002); published online EpubNov 8 ( 10.1074/jbc.M205202200). [DOI] [PubMed] [Google Scholar]
- 37.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z, Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8, 548 (2017); published online EpubSep 15 ( 10.1038/s41467-017-00520-9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watt MJ, Heigenhauser GJ, LeBlanc PJ, Inglis JG, Spriet LL, Peters SJ, Rapid upregulation of pyruvate dehydrogenase kinase activity in human skeletal muscle during prolonged exercise. J Appl Physiol (1985) 97, 1261–1267 (2004); published online EpubOct ( 10.1152/japplphysiol.00132.2004). [DOI] [PubMed] [Google Scholar]
- 39.Hargreaves M, Spriet LL, Skeletal muscle energy metabolism during exercise. Nat Metab 2, 817–828 (2020); published online EpubSep ( 10.1038/s42255-020-0251-4). [DOI] [PubMed] [Google Scholar]
- 40.Fritzen AM, Lundsgaard AM, Kiens B, Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat Rev Endocrinol 16, 683–696 (2020); published online EpubDec ( 10.1038/s41574-020-0405-1). [DOI] [PubMed] [Google Scholar]
- 41.Chen M, Zhou L, Chen S, Shangguan R, Qu Y, Sun J, Acute and chronic effects of high-intensity interval training (HIIT) on postexercise intramuscular lipid metabolism in rats. Physiol Res 70, 735–743 (2021); published online EpubNov 29 ( 10.33549/physiolres.934722). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narendra D, Tanaka A, Suen DF, Youle RJ, Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803 (2008); published online EpubDec 1 ( 10.1083/jcb.200809125). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE, The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol 17, 2477–2493 (2003); published online EpubDec ( 10.1210/me.2003-0151). [DOI] [PubMed] [Google Scholar]
- 44.Shearer BG, Wiethe RW, Ashe A, Billin AN, Way JM, Stanley TB, Wagner CD, Xu RX, Leesnitzer LM, Merrihew RV, Shearer TW, Jeune MR, Ulrich JC, Willson TM, Identification and characterization of 4-chloro-N-(2-{[5-trifluoromethyl)-2-pyridyl]sulfonyl}ethyl)benzamide (GSK3787), a selective and irreversible peroxisome proliferator-activated receptor delta (PPARdelta) antagonist. J Med Chem 53, 1857–1861 (2010); published online EpubFeb 25 ( 10.1021/jm900464j). [DOI] [PubMed] [Google Scholar]
- 45.Koh JH, Hancock CR, Terada S, Higashida K, Holloszy JO, Han DH, PPARbeta Is Essential for Maintaining Normal Levels of PGC-1alpha and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab 25, 1176–1185 e1175 (2017); published online EpubMay 2 ( 10.1016/j.cmet.2017.04.029). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen IKN, Tusubira D, Ashrafi H, Dyrstad SE, Hansen L, Liu XZ, Nilsson LIH, Lovsletten NG, Berge K, Wergedahl H, Bjorndal B, Fluge O, Bruland O, Rustan AC, Halberg N, Rosland GV, Berge RK, Tronstad KJ, Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion 49, 97–110 (2019); published online EpubNov ( 10.1016/j.mito.2019.07.009). [DOI] [PubMed] [Google Scholar]
- 47.Moore TM, Zhou Z, Cohn W, Norheim F, Lin AJ, Kalajian N, Strumwasser AR, Cory K, Whitney K, Ho T, Ho T, Lee JL, Rucker DH, Shirihai O, van der Bliek AM, Whitelegge JP, Seldin MM, Lusis AJ, Lee S, Drevon CA, Mahata SK, Turcotte LP, Hevener AL, The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol Metab 21, 51–67 (2019); published online EpubMar ( 10.1016/j.molmet.2018.11.012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisner V, Picard M, Hajnoczky G, Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20, 755–765 (2018); published online EpubJul ( 10.1038/s41556-018-0133-0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Philp AM, Saner NJ, Lazarou M, Ganley IG, Philp A, The influence of aerobic exercise on mitochondrial quality control in skeletal muscle. J Physiol 599, 3463–3476 (2021); published online EpubJul ( 10.1113/JP279411). [DOI] [PubMed] [Google Scholar]
- 50.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S, PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107, 378–383 (2010); published online EpubJan 5 ( 10.1073/pnas.0911187107). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K, Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282, 11521–11529 (2007); published online EpubApr 13 ( 10.1074/jbc.M607279200). [DOI] [PubMed] [Google Scholar]
- 52.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS, Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446 (2008); published online EpubJan 23 ( 10.1038/sj.emboj.7601963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K, Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191, 1141–1158 (2010); published online EpubDec 13 ( 10.1083/jcb.201007152). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC, Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20, 3525–3532 (2009); published online EpubAug ( 10.1091/mbc.E09-03-0252). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K, A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 107, 2687–2692 (2010); published online EpubFeb 9 ( 10.1073/pnas.0913572107). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baar K, Nutrition and the Adaptation to Endurance Training. Sports Medicine 44, 5–12 (2014); published online Epub2014/05/01 ( 10.1007/s40279-014-0146-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holst D, Luquet S, Nogueira V, Kristiansen K, Leverve X, Grimaldi PA, Nutritional regulation and role of peroxisome proliferator-activated receptor delta in fatty acid catabolism in skeletal muscle. Biochim Biophys Acta 1633, 43–50 (2003); published online EpubJul 4 ( 10.1016/s1388-1981(03)00071-4). [DOI] [PubMed] [Google Scholar]
- 58.Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL, Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol 588, 4795–4810 (2010); published online EpubDec 1 ( 10.1113/jphysiol.2010.199448). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S, Wang N, Guo X, Li J, Zhang T, Ren G, Li D, Fibroblast growth factor 21 regulates glucose metabolism in part by reducing renal glucose reabsorption. Biomed Pharmacother 108, 355–366 (2018); published online EpubDec ( 10.1016/j.biopha.2018.09.078). [DOI] [PubMed] [Google Scholar]
- 60.Je HS, Yang F, Ji Y, Nagappan G, Hempstead BL, Lu B, Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc Natl Acad Sci U S A 109, 15924–15929 (2012); published online EpubSep 25 ( 10.1073/pnas.1207767109). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kruse N, Cetin S, Chan A, Gold R, Luhder F, Differential expression of BDNF mRNA splice variants in mouse brain and immune cells. J Neuroimmunol 182, 13–21 (2007); published online EpubJan ( 10.1016/j.jneuroim.2006.09.001). [DOI] [PubMed] [Google Scholar]
- 62.Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, Desvergne B, Wahli W, Chambon P, Metzger D, PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab 4, 407–414 (2006); published online EpubNov ( 10.1016/j.cmet.2006.10.003). [DOI] [PubMed] [Google Scholar]
- 63.Wang P, Liu J, Li Y, Wu S, Luo J, Yang H, Subbiah R, Chatham J, Zhelyabovska O, Yang Q, Peroxisome proliferator-activated receptor delta is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res 106, 911–919 (2010); published online EpubMar 19 ( 10.1161/CIRCRESAHA.109.206185). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Yin R, Liu J, Wang P, Wu S, Luo J, Zhelyabovska O, Yang Q, Peroxisome proliferator-activated receptor delta regulates mitofusin 2 expression in the heart. J Mol Cell Cardiol 46, 876–882 (2009); published online EpubJun ( 10.1016/j.yjmcc.2009.02.020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ji J, Li S, Jiang Z, Yu J, Sun Y, Cai Z, Dong Y, Sun X, Activating PPARbeta/delta Protects against Endoplasmic Reticulum Stress-Induced Astrocytic Apoptosis via UCP2-Dependent Mitophagy in Depressive Model. Int J Mol Sci 23, (2022); published online EpubSep 16 ( 10.3390/ijms231810822). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Mario A, Gherardi G, Rizzuto R, Mammucari C, Skeletal muscle mitochondria in health and disease. Cell Calcium 94, 102357 (2021); published online EpubMar ( 10.1016/j.ceca.2021.102357). [DOI] [PubMed] [Google Scholar]
- 67.Bhutani S, Klempel MC, Kroeger CM, Trepanowski JF, Varady KA, Alternate day fasting and endurance exercise combine to reduce body weight and favorably alter plasma lipids in obese humans. Obesity (Silver Spring) 21, 1370–1379 (2013); published online EpubJul ( 10.1002/oby.20353). [DOI] [PubMed] [Google Scholar]
- 68.Real-Hohn A, Navegantes C, Ramos K, Ramos-Filho D, Cahue F, Galina A, Salerno VP, The synergism of high-intensity intermittent exercise and every-other-day intermittent fasting regimen on energy metabolism adaptations includes hexokinase activity and mitochondrial efficiency. PLoS One 13, e0202784 (2018) 10.1371/journal.pone.0202784). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mason RR, Meex RC, Lee-Young R, Canny BJ, Watt MJ, Phosphorylation of adipose triglyceride lipase Ser(404) is not related to 5’-AMPK activation during moderate-intensity exercise in humans. Am J Physiol Endocrinol Metab 303, E534–541 (2012); published online EpubAug 15 ( 10.1152/ajpendo.00082.2012). [DOI] [PubMed] [Google Scholar]
- 70.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG, Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metabolism 2, 9–19 (2005) 10.1016/j.cmet.2005.05.009). [DOI] [PubMed] [Google Scholar]
- 71.Woods A, Dickerson K, Heath R, Hong S-P, Momcilovic M, Johnstone SR, Carlson M, Carling D, Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metabolism 2, 21–33 (2005) 10.1016/j.cmet.2005.06.005). [DOI] [PubMed] [Google Scholar]
- 72.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA, The Ca2+/Calmodulin-dependent Protein Kinase Kinases Are AMP-activated Protein Kinase Kinases * . Journal of Biological Chemistry 280, 29060–29066 (2005) 10.1074/jbc.M503824200). [DOI] [PubMed] [Google Scholar]
- 73.Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG, 5’-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem 270, 27186–27191 (1995); published online EpubNov 10 ( 10.1074/jbc.270.45.27186). [DOI] [PubMed] [Google Scholar]
- 74.Kramer DK, Al-Khalili L, Perrini S, Skogsberg J, Wretenberg P, Kannisto K, Wallberg-Henriksson H, Ehrenborg E, Zierath JR, Krook A, Direct activation of glucose transport in primary human myotubes after activation of peroxisome proliferator-activated receptor delta. Diabetes 54, 1157–1163 (2005); published online EpubApr ( 10.2337/diabetes.54.4.1157). [DOI] [PubMed] [Google Scholar]
- 75.Barroso E, Eyre E, Palomer X, Vazquez-Carrera M, The peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) agonist GW501516 prevents TNF-alpha-induced NF-kappaB activation in human HaCaT cells by reducing p65 acetylation through AMPK and SIRT1. Biochem Pharmacol 81, 534–543 (2011); published online EpubFeb 15 ( 10.1016/j.bcp.2010.12.004). [DOI] [PubMed] [Google Scholar]
- 76.Salvado L, Barroso E, Gomez-Foix AM, Palomer X, Michalik L, Wahli W, Vazquez-Carrera M, PPARbeta/delta prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 57, 2126–2135 (2014); published online EpubOct ( 10.1007/s00125-014-3331-8). [DOI] [PubMed] [Google Scholar]
- 77.Matsumoto J, Takada S, Furihata T, Nambu H, Kakutani N, Maekawa S, Mizushima W, Nakano I, Fukushima A, Yokota T, Tanaka S, Handa H, Sabe H, Kinugawa S, Brain-Derived Neurotrophic Factor Improves Impaired Fatty Acid Oxidation Via the Activation of Adenosine Monophosphate-Activated Protein Kinase-a - Proliferator-Activated Receptor-r Coactivator-1a Signaling in Skeletal Muscle of Mice With Heart Failure. Circ Heart Fail 14, e005890 (2021); published online EpubJan ( 10.1161/CIRCHEARTFAILURE.119.005890). [DOI] [PubMed] [Google Scholar]
- 78.Genzer Y, Chapnik N, Froy O, Effect of brain-derived neurotrophic factor (BDNF) on hepatocyte metabolism. Int J Biochem Cell Biol 88, 69–74 (2017); published online EpubJul ( 10.1016/j.biocel.2017.05.008). [DOI] [PubMed] [Google Scholar]
- 79.Delezie J, Weihrauch M, Maier G, Tejero R, Ham DJ, Gill JF, Karrer-Cardel B, Ruegg MA, Tabares L, Handschin C, BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc Natl Acad Sci U S A 116, 16111–16120 (2019); published online EpubAug 6 ( 10.1073/pnas.1900544116). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR, Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol 265, E380–391 (1993); published online EpubSep ( 10.1152/ajpendo.1993.265.3.E380). [DOI] [PubMed] [Google Scholar]
- 81.Cheng A, Wan R, Yang J-L, Kamimura N, Son T, Ouyang X, Luo Y, Okun E, Mattson M, Involvement of PGC-1α in the Formation and Maintenance of Neuronal Dendritic Spines. Nature communications 3, 1250 (2012); published online Epub12/04 ( 10.1038/ncomms2238). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Colitti M, Montanari T, Brain-derived neurotrophic factor modulates mitochondrial dynamics and thermogenic phenotype on 3T3-L1 adipocytes. Tissue Cell 66, 101388 (2020); published online EpubOct ( 10.1016/j.tice.2020.101388). [DOI] [PubMed] [Google Scholar]
- 83.Wang Z, Wang S.-p., Shao Q, Li P.-f., Sun Y, Luo L.-z., Yan X.-q., Fan Z.-y., Hu J, Zhao J, Hang P.-z., Du Z.-m., Brain-derived neurotrophic factor mimetic, 7,8-dihydroxyflavone, protects against myocardial ischemia by rebalancing optic atrophy 1 processing. Free Radical Biology and Medicine 145, 187–197 (2019); published online Epub2019/12/01/ ( 10.1016/j.freeradbiomed.2019.09.033). [DOI] [PubMed] [Google Scholar]
- 84.Broskey N, Boss A, Fares E-J, Greggio C, Gremion G, Schlüter L, Hans D, Kreis R, Boesch C, Amati F, Exercise efficiency relates with mitochondrial content and function in older adults. Physiological reports 3, (2015); published online Epub06/01 ( 10.14814/phy2.12418). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Z, Wang B, Fei A, BDNF contributes to the skeletal muscle anti-atrophic effect of exercise training through AMPK-PGC1alpha signaling in heart failure mice. Arch Med Sci 15, 214–222 (2019); published online EpubJan ( 10.5114/aoms.2018.81037). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moy JK, Szabo-Pardi T, Tillu DV, Megat S, Pradhan G, Kume M, Asiedu MN, Burton MD, Dussor G, Price TJ, Temporal and sex differences in the role of BDNF/TrkB signaling in hyperalgesic priming in mice and rats. Neurobiol Pain 5, 100024 (2019); published online EpubJan-Jul ( 10.1016/j.ynpai.2018.10.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Advani T, Koek W, Hensler JG, Gender differences in the enhanced vulnerability of BDNF+/− mice to mild stress. Int J Neuropsychopharmacol 12, 583–588 (2009); published online EpubJun ( 10.1017/S1461145709000248). [DOI] [PubMed] [Google Scholar]
- 88.Landen S, Jacques M, Hiam D, Alvarez-Romero J, Schittenhelm RB, Shah AD, Huang C, Steele JR, Harvey NR, Haupt LM, Griffiths LR, Ashton KJ, Lamon S, Voisin S, Eynon N, Sex differences in muscle protein expression and DNA methylation in response to exercise training. Biol Sex Differ 14, 56 (2023); published online EpubSep 5 ( 10.1186/s13293-023-00539-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maher AC, Fu MH, Isfort RJ, Varbanov AR, Qu XA, Tarnopolsky MA, Sex differences in global mRNA content of human skeletal muscle. PLoS One 4, e6335 (2009); published online EpubJul 22 ( 10.1371/journal.pone.0006335). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Montero D, Madsen K, Meinild-Lundby AK, Edin F, Lundby C, Sexual dimorphism of substrate utilization: Differences in skeletal muscle mitochondrial volume density and function. Exp Physiol 103, 851–859 (2018); published online EpubJun ( 10.1113/EP087007). [DOI] [PubMed] [Google Scholar]
- 91.Hausswirth C, Le Meur Y, Physiological and nutritional aspects of post-exercise recovery: specific recommendations for female athletes. Sports Med 41, 861–882 (2011); published online EpubOct 1 ( 10.2165/11593180-000000000-00000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplemental Materials.