

Abstract
Abstract
Inherited bone marrow failure syndromes (IBMFSs) are a group of disorders typified by impaired production of 1 or several blood cell types. The telomere biology disorders dyskeratosis congenita (DC) and its severe variant, Høyeraal-Hreidarsson (HH) syndrome, are rare IBMFSs characterized by bone marrow failure, developmental defects, and various premature aging complications associated with critically short telomeres. We identified biallelic variants in the gene encoding the 5′-to-3′ DNA exonuclease Apollo/SNM1B in 3 unrelated patients presenting with a DC/HH phenotype consisting of early-onset hypocellular bone marrow failure, B and NK lymphopenia, developmental anomalies, microcephaly, and/or intrauterine growth retardation. All 3 patients carry a homozygous or compound heterozygous (in combination with a null allele) missense variant affecting the same residue L142 (L142F or L142S) located in the catalytic domain of Apollo. Apollo-deficient cells from patients exhibited spontaneous chromosome instability and impaired DNA repair that was complemented by CRISPR/Cas9-mediated gene correction. Furthermore, patients' cells showed signs of telomere fragility that were not associated with global reduction of telomere length. Unlike patients' cells, human Apollo KO HT1080 cell lines showed strong telomere dysfunction accompanied by excessive telomere shortening, suggesting that the L142S and L142F Apollo variants are hypomorphic. Collectively, these findings define human Apollo as a genome caretaker and identify biallelic Apollo variants as a genetic cause of a hitherto unrecognized severe IBMFS that combines clinical hallmarks of DC/HH with normal telomere length.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2575.
Disclosures
Associate Editor Irene Roberts declares no competing financial interests. CME questions author Laurie Barclay owns stock, stock options, or bonds from the following ineligible company: AbbVie Inc (former).
Learning objectives
Upon completion of this activity, participants will:
-
1
Describe clinical features of 3 unrelated patients with biallelic variants in the gene encoding the 5′-to-3′ DNA exonuclease Apollo/SNM1B (Apollo), according to a case series
-
2
Determine genetic features of 3 unrelated patients with biallelic Apollo variants, according to a case series
-
3
Identify clinical and research implications of clinical and genetic features of 3 unrelated patients with biallelic Apollo variants, according to a case series
Release date: April 21, 2022; Expiration date: April 21, 2023
Key Points
-
•
Biallelic Apollo variants cause a bone marrow failure syndrome with clinical hallmarks of dyskeratosis congenita but normal telomere length.
-
•
Apollo is a genome caretaker critical for the proper development of the immunohematological system in humans.

Introduction
Inherited bone marrow failure syndromes (IBMFSs) represent heterogeneous Mendelian diseases having in common an impaired production of 1 or several blood cell lineages.1 Growth delay, mucocutaneous abnormalities, developmental defects, and cancer predisposition are other clinical outcomes that can manifest in IBMFS.1 Dyskeratosis congenita (DC) and its severe variant Høyeraal-Hreidarsson (HH) syndrome are rare IBMFSs. DC is mainly characterized by progressive bone marrow failure, premature aging manifestations, and increased cancer predisposition, whereas HH can associate with early-onset bone marrow failure, intrauterine growth retardation (IUGR), microcephaly, and/or cerebellar hypoplasia, and immunodeficiency.2, 3 Because DC and HH are caused by genetic defects that affect the integrity and/or the length of telomeres, they belong to a heterogeneous group of conditions termed either telomere biology disorders (TBDs), telomeropathies, or short telomere syndromes.2, 3, 4, 5 Telomeres are constituted by double-stranded TTAGGG repeats terminated by a 3′ single-stranded sequence called G-overhang. Telomeres are decorated by a complex named shelterin6 composed of 6 proteins (TRF1, TRF2, TIN2, RAP1, TPP1, and POT1) among which TRF1 and TRF2 bind directly to the duplex telomeric DNA and POT1 binds to the single-strand G-overhang.6 Shelterin is essential for the protection of chromosomes from degradation and/or fusion and for maintaining telomere length.7 To date, variants in 11 factors (TERT, TERC, dyskerin, NOP10, NHP2, TCAB1, TIN2, TPP1, CTC1, RTEL1, and PARN) that participate in telomere biology have been found to cause DC and HH.2, 3, 8 In DC and HH, the severity and onset of symptoms are generally correlated to the degree of telomere length reduction.9, 10 Thus, telomere length determination is an effective approach to diagnose DC/HH in patients with IBMFS.2, 10, 11
Apollo (SNM1B), encoded by the DNA cross-link repair 1B (DCLRE1B/Apollo; NC_000001.11) gene, is a 5′-to-3′ DNA exonuclease that functions within the Fanconi anemia (FA) pathway and is involved in the repair of both mitomycin C (MMC)-induced DNA interstrand crosslinks (ICL) and DNA double-strand breaks (DSB)12 as well as the stabilization of stalled replication forks and S-phase checkpoint activation.13, 14, 15, 16, 17 Moreover, the identification of single nucleotide polymorphisms in the DCLRE1B/Apollo locus associated with breast cancers and cutaneous melanoma supported a protective role of Apollo in genome integrity.18, 19
Apollo also participates in telomere protection via an interaction between its telomeric repeat factors homology (TRFH)–binding motif (TBM) and the TRFH domain of TRF2.20, 21, 22, 23 Apollo KO mouse embryonic fibroblasts (MEFs) exhibited impaired production of G-overhangs and frequent telomere fusions at the newly-replicated leading-end telomeres. This observation suggested that the nuclease activity of Apollo is involved in the generation of G-overhang that avoids fusion of leading telomeres.24, 25, 26 In human cells, the role of Apollo at telomeres is less clear since its depletion induces telomere fragility causing multiple telomeric signals (MTS), that is however not associated with impaired G-overhang, increased telomere fusion or telomere shortening.20, 21 Nonetheless, it has been demonstrated that human Apollo together with TRF2 and the topoisomerase Topo-IIafunction in DNA replication of telomeric sequences by alleviating topological stress.23
We previously described a HH patient expressing an aberrantly spliced Apollo transcript leading to the production of a truncated Apollo that exerted a dominant negative effect on the stability of telomeres without affecting their global length.27 Nonetheless, because we failed to identify the origin of the splice anomaly, we were unable to demonstrate a causal link between the truncated form of Apollo and the patient's clinical features.27 Thus, although Apollo appears to be important for telomere stability and DNA repair, its relative contribution to telomere maintenance and genome integrity remains elusive, especially in humans.
Here, we identified biallelic Apollo/DCLRE1B variants in children exhibiting clinical features akin to DC/HH that are, however, not associated with decreased telomere length. Our study defines human Apollo as a genome caretaker.
Materials and methods
Patients
Informed consent was obtained from the families in accordance with the Declaration of Helsinki. The institutional INSERM and Assistance Publique–Hôpitaux de Paris review boards approved this study.
Cells
Cells used in this study are described in supplemental Materials and Methods.
Telomere length measurements
The procedures for telomere length measurements are detailed in supplemental Materials and Methods.
Whole-exome sequencing and gene targeted sequencing
Sequencing approaches are detailed in supplemental Materials and Methods.
TeSLA
The telomere shortest length assay (TeSLA) method, performed as described by Lai et al,28 is described in supplemental Materials and Methods.
Telomeric FISH
The telomeric FISH procedure is described in supplemental Materials and Methods.
Detection of telomere dysfunction–induced foci (TIFs) and β-galactosidase activity
Procedures are described in supplemental Materials and Methods.
In-gel G-overhang assay
The in-gel G-overhang assay was performed as described elsewhere,29 with minor modifications detailed in supplemental Materials and Methods.
CRISPR/Cas9-mediated Apollo gene correction
The CRISPR/Cas9-mediated Apollo gene (DCLRE1B; Entrez Gene: 64858) correction procedure is detailed in supplemental Materials and Methods.
Expression vectors
Vectors are described in supplemental Materials and Methods.
Sister chromatid exchange detection
The sister chromatid exchange detection procedure is detailed in supplemental Materials and Methods.
Western blotting and coimmunoprecipitation
Western blotting, coimmunoprecipitation and antibodies are detailed in supplemental Materials and Methods.
Sensitivity to genotoxics assay
Procedures to analyze the cellular sensitivity are detailed in supplemental Materials and Methods.
Statistical analyses
Statistics procedures are detailed in supplemental Materials and Methods.
Results
Clinical features of patients
We studied 3 unrelated families, each having a single child with clinical features evocative of IBMFS. Patient 1 (P1) is a 7-year-old Caucasian boy presenting growth retardation (−3 SD weight and height) and microcephaly (−2.5 SD) (Table 1). At the age of 4 months, laboratory tests, performed because of multiple petechiae, revealed a trilinear cytopenia. Bone marrow aspirate showed hypocellularity with rare megakaryocytes and moderate dyserythropoiesis. Immunological evaluation revealed profound B and NK cell lymphopenia. The proportions of recent thymus emigrant T lymphocytes and central memory T cells were reduced suggesting dysfunctional T lymphocyte homeostasis (supplemental Table 1). The patient needed transfusion support with platelet and erythrocyte concentrates at the age of 11 months. He had undergone allogeneic hematopoietic stem cell transplantation (HSCT) at 15 months of age. Other clinical features included facial dysmorphia (hypotelorism), delayed speech, and esophageal strictures that appeared at the age of 5 years.
Table 1.
Clinical features of patients
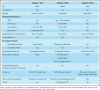 |
Patient 2 is an 11-year-old Portuguese girl born from consanguineous parents. Intrauterine growth retardation (IUGR) was diagnosed in the 3rd trimester. At birth, the patient was hypotrophic with weight at 1,685 g, height at 40.5 cm and head circumference of 31 cm (−2 SD) (Table 1). She presented with skin bruising and petechiae. Her hemogram revealed pancytopenia. Bone marrow aspirate revealed marked hypocellularity, reduced percentage of myeloid precursors, rare neutrophils with hyposegmented nuclei, and signs of dyserythropoiesis. Immunological evaluation showed a virtual absence of B and NK cells (supplemental Table 1). She received transfusion support with platelet and erythrocyte concentrates. As severe progressive pancytopenia developed, allogeneic HSCT was successfully performed at 8 months of age. At 8 years of age she developed mucocutaneous features including hyperpigmented areas of the trunk and limbs, sparse scalp hair, leukoplakia, and dystrophic nails (supplemental Figure 1).
Patient 3 is a 6-year-old Argentinian boy (Table 1) presenting with progressive bone marrow failure. Immunological evaluation evidenced profound B and NK cell lymphopenia (supplemental Table 1). T lymphocyte count was normal but a slight reduction of recent thymus emigrant T lymphocytes was noted that was however not accompanied by a bias in T-cell repertoire, as inferred by T cell receptor γ gene rearrangement analysis (supplemental Figure 2).30 He receives transfusion support (platelet and erythrocyte concentrates) and immunoglobulin replacement since the age of 3 months. He developed an inflammatory bowel disease at 10 months of age. Endoscopy showed a severe ulcerative pancolitis with signs of chronicity. Until now, the patient does not present any mucocutaneous or neurodevelopmental features.
Overall, the association of early-onset bone marrow failure, immunodeficiency, developmental defects and premature aging features in the 3 patients were consistent with DC/HH.2, 3, 4, 5
Patients exhibit normal global telomere length
Since DC/HH are classically associated with abnormally short telomeres,2, 10 we assessed telomere length in patients. Surprisingly, telomere restriction fragment (TRF) analysis did not unveil a significant telomere length defect in patients' blood samples (obtained before HSCT) as compared with their parents (Figure 1A), and in contrast to HH patients with RTEL1 or PARN deficiency (Figure 1B).31, 32, 33, 34 Flow-FISH10 further confirmed the normal telomere length in blood cells from P1 and P3 (P2's blood sample before HSCT was not available) (Figure 1C). The telomere length in P1 and P3′s blood cells sharply contrasted with the critically short telomeres measured in 13 DC/HH patients carrying pathogenic variants in TBD-causing genes (Figure 1C). Collectively, these results indicate that the 3 patients, although presenting clinical features akin to DC/HH, did not exhibit critically short telomeres as classically observed in these diseases.
Figure 1.

Telomere length in blood cells from patients. (A) Telomere length determined by telomere restriction fragment (TRF) assay in DNA from blood cells from patients P1, P2, P3 and their parents (P3′s father's sample was not available). (B) Graphic representation of TRF data obtained in panel A as well as in 8 age-matched controls, 8 RTEL1-deficient patients, and 2 PARN-deficient patients described elsewhere.31, 32, 33, 34 (C) Relative telomere length in comparison with the tetraploid control cell line (1301) measured by Flow-FISH in blood cells from P1, P3, P3′s mother and 13 DC/HH patients with pathogenic variants in DKC1, TERT, TERC, RTEL1, or TINF2. Lines represent the 1st, 10th, 50th and 90th percentiles of telomere length of healthy controls. (D) (Left) Detection of the shortest telomeres by TeSLA performed in blood cells from P1 and an age-matched healthy donor. (Right) Graphic representation of TeSLA data with statistical analyses reveals a significant increase in telomere loss events in P1's blood sample. A 2-tailed Student t-test was used for statistical analyses of band size and a χ2 test was used for analysis of fraction < 1.5kb.
Subtle telomere instability resulting from rare sudden telomere loss that does not translate to global telomere shortening has been previously reported.35 Telomere Shortest Length Assay (TeSLA), a highly sensitive method to detect the shortest telomeres,28 highlighted a significant increase in the frequency of very short telomeres in P1's blood cells (Figure 1D). Thus, the combination of TeSLA analysis and telomere length measurement by TRF and Flow-FISH suggested telomere instability in patients' cells that does not translate into global telomere length reduction.
Biallelic Apollo variants in patients
To determine the molecular etiology of this atypical TBD we performed whole-exome sequencing in P1 and P3 and Haloplex targeted sequencing of 10 telomere-related genes in P2. This analysis singled out Apollo/DCLRE1B as the only common gene carrying biallelic variants in the 3 patients. Sanger sequencing confirmed the Apollo variants which corresponded in P1 to a c.364C>T substitution leading to a premature stop codon (p.Arg122*; NP_073747.1) inherited from the healthy mother, and a c.426A>T substitution causing a p.Leu142Phe missense variant (thereafter noted L142F) inherited from the healthy father (Figure 2A; supplemental Figure 3). P2 carried a homozygous Apollo missense variant that, as in P1, affected the amino-acid p.Leu142 but changing it toward a serine (c.425T>C; p.Leu142Ser, thereafter noted L142S). As expected in a context of consanguinity, both P2's healthy parents carried the c.425T>C; L142S variant at a heterozygous status (supplemental Figure 3). P3 carried compound heterozygous Apollo variants consisting in a c.472C>T substitution producing a premature stop codon p.Arg158* inherited from his healthy mother and, as in P2, a substitution c.425T>C; L142S inherited from her father (Figure 2A; supplemental Figure 3).
Figure 2.

Genetic analysis identified Apollo variants in the patients. (A) Pedigrees and Apollo variants identified in individuals P1, P2 and P3 by Sanger sequencing. (B) Domain architecture of the human Apollo with the localization of the identified variants of P1, P2 and P3. NLS: nuclear localization signal; TBM: TRFH binding motif. (C) Ribbon representation of the 3D structure of human Apollo catalytic domain (pdb 5AHO,39 with 2 coordinated zinc ions and 2 tartrate molecules at the active site. Details of the region surrounding L142 are given in the box at left, highlighting the hydrophobic core in which L142 participates, in the vicinity of motif A D145 (bond with motif B H247, itself bound to a tartrate molecule, which may represent the phosphodiester cleaved by the nuclease39). (D) Co-immunoprecipitation of endogenous TRF2 with WT or mutated forms of FLAG-Apollo. Picture representative of 3 independent experiments.
The CADD36 scores and the American College of Medical Genetics and Genomics standards37 predicted the 4 Apollo variants to be deleterious/pathogenic. Apollo L142F and L142S variants were absent in gnomAD database, while both Apollo p.Arg122* and p.Arg158* variants were present at a very low frequency (supplemental Table 2). Of note, according to gnomAD, loss-of-function (LOF) variant observed/expected ratio for Apollo is of 0.2, suggesting intolerance of LOF variants. Direct sequencing of Apollo cDNA from P1's fibroblasts detected the c.346C>T;p.R122* variant indicating that it does not cause nonsense-mediated decay (NMD) (supplemental Figure 4). However, because the low endogenous expression of Apollo precludes its detection by specific antibodies,20, 21 we could not test whether the c.346C>T;p.R122* variant produces a severely truncated form of Apollo. P3′s cells were not available to assess whether the c.472C>T; p.R158* variant causes NMD.
Apollo belongs to the β-CASP family of proteins characterized by the presence of a specific clamp (the β-CASP domain) inserted into the β-lactamase domain and covering the nuclease active site.38 The Apollo residue L142, located in the β-lactamase domain (Figure 2B), is highly conserved across species and in other β-CASP proteins (supplemental Figure 5). Apollo's 3D structure highlights that L142 participates in the hydrophobic core of the metallo-β-lactamase domain (Figure 2C). It is located nearby the conserved D145 residue (supplemental Figure 5) that binds the amino acid H276 that is believed to bind the phosphodiester cleaved by the nuclease.39 L142F and L142S substitutions are thus predicted to indirectly impact the Apollo nuclease activity by structural disturbance nearby the active site.
Telomere localization of Apollo depends on the interaction of its TBM domain with the TRFH domain of TRF2.20, 21, 22 To know whether the L142S and L142F variants could impact Apollo's stability/expression and/or its interaction with TRF2, we performed immunoblots and co-IP with extracts from HEK293T cells transfected with wild type (WT) or mutant FLAG-tagged Apollo-expressing vectors. The similar detection of WT and Apollo mutants indicated that the L142S and L142F variants did not modify the Apollo expression/stability, at least not in the context of overexpression (Figure 2D). However, a consistent reduced amount of endogenous TRF2 was co-immunoprecipitated with the Apollo mutants (Figure 2D) suggesting that, although at a distance from the TBM (Figure 2B), both variants partially impaired Apollo interaction with TRF2.
Altogether, these results indicate that the 3 patients carry biallelic variants in Apollo. At least 1 allele in each patient corresponds to a missense variant affecting the amino acid L142 that does not impact Apollo expression/stability but partially reduces its capacity to interact with TRF2 and is predicted to impact the catalytic activity of Apollo.
Patients' cells exhibit some telomere aberrations but not global telomere shortening
To examine the functional consequences of Apollo variants on telomere stability we analyzed the phenotype of primary fibroblasts obtained from P1 and P2. TRF analysis did not reveal abnormal telomere length in patients' primary fibroblasts (Figure 3A), an observation congruent with the results obtained in patients' blood cells (Figure 1). Furthermore, unlike primary fibroblasts from TBDs patients with short telomeres (eg, RTEL1-, TIN2- and PARN-deficient fibroblasts31, 33, 34, 40), or even RTEL1-deficient patient's primary fibroblasts with normal telomere length,41 P1 and P2's primary fibroblasts did not exhibit an increase in TIFs42, 43 and senescence-associated β-galactosidase activity (supplemental Figure 6A-B) suggesting that telomeres in patients' fibroblasts are not overwhelmingly deprotected. Next, TeSLA performed in patients' primary fibroblasts uncovered a slight increase in the frequency of very short telomeres that was however not statistically significant (supplemental Figure 6C). Even though we precisely quantified the input DNA used for TeSLA analysis, we consistently noticed a reduction of the number of bands (corresponding to PCR-amplified products) in samples from both patients' fibroblasts compared with controls (supplemental Figure 6C). Since TeSLA depends on annealing of a primer to the telomeric G-overhang,28 this suggested a reduced availability of G-overhang in patients' cells, which would be in accordance with the role of Apollo in the generation of G-overhang from the leading telomere described in murine models.24, 25, 26 In-gel hybridization assays confirmed an overall reduction of 30% to 50% of the G-overhang in patients' cells (supplemental Figure 6D). Since the calculated G-overhang value corresponds to the portion of the telomere that is single stranded, we also corrected for the effect of the telomere length by multiplying the native/denatured signal by the mean TRF length (MTL). Also these values, reflecting the absolute G-overhang length, were reduced as compared with the WT controls (supplemental Figure 6D, experiment 3). We next performed telomeric FISH on metaphase spreads of simian virus 40 (SV40)–transformed fibroblasts to detect putative telomere aberrations (Figure 3B). P1 and P2 patients' cells exhibited a slight but significant increase of telomere-telomere fusions. P2's fibroblasts also had a statistically significant increase of MTS reflecting telomere fragility,44 while P1's cells exhibited a significant, although moderate, increase of dicentric chromosomes and sister chromatid fusions (Figure 3B-C). Other telomere aberrations were not overrepresented in patients' cells.
Figure 3.

Telomere phenotype in patients' fibroblasts. (A) Telomere length determined by TRF in primary fibroblasts from 2 healthy controls and patients P1 and P2. Population doubling is indicated in brackets. (B) Representative pictures of normal chromosomes and chromosomes with the indicated telomeric aberrations detected by FISH. (C) Quantitative analysis of telomeric aberrations detected by FISH in SV40-transformed fibroblasts from 2 age-matched healthy controls and P1 and P2 at similar passages (from passage 3 to 8). Ctl1 fibroblasts are from a healthy male individual, and Ctl2 are from a healthy female individual). Results from 5 independent experiments for P1 and 2 for P2 (counted chromosomes: Ctl1: n = 1,945; Ctl2: n = 1,462; P1: n = 3,325; P2: n = 2,631). Averages are shown and χ2 tests were applied to compare Ctls with either P1 or P2. *P < .05, **P < .01, ***P < .001, ****P < .0001.
We concluded from these analyses that the Apollo-mutated patients' cells exhibit some signs of telomere instability and reduced G-overhang that are not associated with global telomere shortening and hallmarks of dysfunctional telomeres (ie, TIFs and senescence), a situation different from the one observed in classic TBDs.31, 33, 34, 40, 45
Chromosome instability and DNA repair defect in patients' cells
Next, we assessed whether patients' Apollo-deficient cells could exhibit genome instability and impaired DNA repair. Metaphase spreads in patients' SV40-transformed fibroblasts highlighted a significant increase in spontaneous radial chromosomes (Figure 4A-B), which represent intermediates of recombination associated with genomic instability. The frequency of radial chromosomes was further increased in MMC-treated patients' cells (Figure 4B). As compared with WT cells, MMC-patients' cells also showed a significant increase in chromosome breaks (Figure 4A-C) and sister chromatid exchanges (supplemental Figure 7), suggesting a defect in ICL-repair. The reduced cell survival of MMC-treated patients' Apollo-mutated fibroblasts confirmed an impaired ICL repair, which was however not as pronounced as cells from a FA patient (Figure 4D), and not associated with defective FANCD2 ubiquitination (supplemental Figure 8). The DNA repair defect was not restricted to ICL since P1 and P2's fibroblasts also exhibited an increased sensitivity to aphidicolin that causes replicative stress by inhibiting DNA polymerase (Figure 4E), as well as to phleomycin, a DNA double-strand break-inducer (Figure 4F). These results indicate that DNA repair of several distinct DNA injuries is impaired in patients' Apollo-mutated cells.
Figure 4.

Genome instability and DNA repair defect in patients' cells. (A) Representative picture of normal chromosomes, radial chromosomes, and chromosome breaks found in metaphase. (B-C) Quantitative analysis of radial chromosomes (B) and chromosome breaks (C) in untreated and MMC-treated cells. Cells from a Fanconi (FANC-G deficient) patient are used as a sensitive control. Percentage of events per chromosomes is indicated (2 independent experiments); Counted chromosomes: Ctrl -MMC: n = 583; Ctl +MMC: n = 929; P1 -MMC: n = 789; P1 +MMC: n = 620; P2 -MMC: n = 710; P2 +MMC: n = 674; Fanconi -MMC: n = 811; Fanconi +MMC: n = 965). (D) MMC sensitivity of SV40-transformed fibroblasts from P1 and P2, a Fanconi patient, and control. Mean and standard deviation of triplicates are represented. Result representative of 4 independent experiments. A 2-tailed standard t-test was used. (E) Aphidicolin sensitivity of SV40-transformed fibroblasts from P1 and P2, control, and control with ATR inhibitor (ATRi, 10µM). Mean and standard deviation of triplicates are represented. Result representative of 3 independent experiments. A 2-tailed standard t-test was used. (F) Phleomycin sensitivity of SV40-transformed fibroblasts from P1 and P2, a Cernunnos-deficient patient,59 and a healthy control. Mean and standard deviation of triplicates are represented. Result representative of 5 independent experiments. A 2-tailed standard t-test was used. (G) Schematic representation of the correction of 1 Apollo variant in P2's cells via CRISPR/Cas9 generating a heterozygous Apollo-mutated P2 cell line (noted P2L142S/WT). (H) Phleomycin sensitivity of hTERT SV40-fibroblasts from P2, P2L142S/WT and a healthy control. Mean and standard deviation of triplicates are represented. Result representative of 3 independent experiments. A 2-tailed standard t-test was used. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Next, we used CRISPR/Cas9-mediated homology-directed repair to correct the homozygous L142S Apollo variant by a WT sequence in P2's hTERT-immortalized SV40-fibroblasts. We obtained a P2's SV40-hTERT cellular clone carrying a WT Apollo sequence in 1 allele, therefore corresponding to a heterozygous carrier of the L142S Apollo variant (noted P2L142S/WT; Figure 4G; supplemental Figure 9). While the native P2's clone with the homozygous L142S Apollo variant (noted P2L142S/L142S) exhibited reduced cell survival upon phleomycin treatment, P2L142S/WT cells behaved similarly to WT cells (Figure 4H). This result provides evidence of a causal link between impaired DNA repair and the presence of biallelic Apollo variants in patients' cells.
Collectively, our results indicate that the biallelic Apollo variants identified in patients cause telomere fragility, spontaneous genomic instability, and impaired DNA repair.
Telomere phenotype in human Apollo KO cell lines
The complete loss of function of Apollo is embryonic lethal in mouse24, 26, 46 and Apollo KO MEFs exhibit sharp telomere abnormalities, mostly characterized by leading telomere fusions.24, 26, 46 This severe phenotype contrasts with the rather limited telomere defect observed in patients' cells. We thus asked whether a complete lack of Apollo in human cell lines could have a stronger impact on telomere stability than the one found in patients' cells. We generated 2 HT1080 Apollo KO cell lines (#1 and #2) by using CRISPR/Cas9 with 2 distinct RNA guides (supplemental Figure 10). Unlike patients' cells, HT1080 Apollo KO cell lines exhibited telomere dysfunction as inferred by the significant increase in TIFs (Figure 5A). In addition, both HT1080 Apollo KO cell lines exhibited a significant augmentation of telomere-telomere fusion and dicentric chromosomes (Figure 5B), while other aberrations was not augmented (data not shown). Furthermore, TeSLA pointed out a significant increase in frequency of very short telomeres in Apollo KO cell lines (Figure 5C) and, as observed in primary fibroblasts from patients, a reduced number of bands amplified by TeSLA, suggesting defects in G-overhang formation (Figure 5C; supplemental Figure 6C). Accordingly, in-gel hybridization highlighted a ∼50% reduction of the relative G-overhang signal in the HT1080 Apollo KO cell line 1 (Figure 5D). Strikingly, in-gel G-overhang assay also highlighted a sharp reduction of global telomere length in the Apollo KO cell line (Figure 5D). Correction for the effect of the global telomere length resulted in even a greater reduction in the absolute G-overhang length to 17% of the WT control (Figure 5D). The assessment of global telomere length by Southern analysis of TRF confirmed that both HT1080 Apollo KO cellular clones exhibit critically short telomeres as compared with a WT HT1080 cellular clone at a similar passage (Figure 5E).
Figure 5.

Telomere defects in HT1080 Apollo KO cell lines. (A) Quantitative analysis of TIFs in the 2 HT1080 Apollo KO clones and WT cells. (WT: n = 88, KO #1: n = 55; KO #2: n = 66). (B) Quantitative analysis of telomeric aberrations detected by FISH. Percentage of events per chromosomes (counted chromosomes: WT: n = 715; KO #1: n = 887; KO #2: n = 528). Averages and χ2 tests were applied to compare Ctl with either P1 or P2. (C) (Top) Detection of the shortest telomeres by TeSLA performed in WT HT1080 cell line and in the 2 Apollo KO HT1080 cell lines #1 and #2. (Bottom) Graphic representation of TeSLA data with statistical analyses. A 2-tailed student t-test was used for statistical analyses of band size and a χ2 test was used for analysis of fraction < 1kb. (D) Measurement of G-overhang signal by native (Nat) versus denatured (den) in-gel hybridization method with C-rich telomeric probe in WT and Apollo KO #1 HT1080 cell lines. The value of the 3′ overhang was normalized to the control. Values after correction for the effect of the telomere length are also indicated (noted MTL*Nat/den). (E) Telomere length determined by telomere restriction fragment (TRF) assay with DNA from the WT and the Apollo KO HT1080 cell lines #1 and #2. **P < .01, ***P < .001, ****P < .0001.
Collectively, these analyses reveal that the complete loss of function of Apollo in HT1080 human cell line leads to a marked telomere phenotype more severe than the one observed in Apollo-deficient patients' cells. This result supports the idea that the Apollo L142F and L142S missense variants found in patients are hypomorphic.
Discussion
Herein we provide evidence that biallelic variants in Apollo cause a severe form of IBMFS characterized by early onset bone marrow failure, immunodeficiency (mostly B and NK lymphopenia), mucocutaneous anomalies, and several developmental defects (including microcephaly and intrauterine growth retardation). These clinical features are indistinguishable from the telomere biology disorders DC and HH.2, 3 Remarkably, however, unlike patients with classical DC/HH, cells from Apollo-deficient patients did not exhibit excessive overall telomere shortening. This finding defines biallelic variants in Apollo as a cause of a hitherto unrecognized IBMFS, which shares many hallmarks with DC/HH but is distinguished by normal overall telomere length. Since telomere length measurement represents an accurate and reliable mean to diagnose short telomere syndromes,10 we propose that Apollo should be considered as a candidate gene in patients presenting clinical features of TBDs but normal telomere length. Since DC/HH are cancer-prone diseases,2, 47, 48, 49 Apollo patients might be at risk to develop cancer. Furthermore, heterozygous carriers of pathogenic variants in classical TBD-causing genes are predisposed to prematurely develop fibrosis and other age-related ailments.2, 50, 51 The Apollo-deficient patients' parents, who are heterozygous carriers of Apollo variants, are still young and healthy so far. A careful attention should be paid to ensure that they would not prematurely develop age-related diseases.
Consistent with the reported role of Apollo in DNA repair,13 we showed that patients' Apollo-deficient fibroblasts exhibited spontaneous chromosome anomalies, impaired repair of ICL and DSBs, and increased sensitivity to replicative stress. Importantly, CRISPR/Cas9-mediated correction of 1 Apollo-mutated allele complemented the DNA repair defect in P2's cells, demonstrating the causal link between Apollo deficiency and impaired DNA repair in these cells. Notably, we noticed that patients' Apollo-deficient cells shared several features with cells from FA patients, including increased frequency of radial chromosomes and defective ICL repair. These observations, congruent with previous studies conducted in Apollo-depleted human cells, support the idea that Apollo functions in the FA pathway.14, 15, 52 The normal FANCD2 ubiquitination in MMC-treated Apollo-deficient cells suggests that Apollo functions downstream of this step in the FA/BRCA pathway as previously suggested in Apollo-depleted cells.12, 14 However, the extremely severe phenotype of Apollo-deficient patients (requiring HSCT before the age of 18 months in 2 of them) contrasted with the progressive bone marrow failure usually detected between 5-10 years of age in FA patients.1 Furthermore, unlike blood cells from FA patients,53 blood lymphocytes from the 3 Apollo-deficient patients did not exhibit increased chromosomal breakages in the presence of the ICL-inducer diepoxybutane (Table 1). Interestingly, sensitivity to ICL-inducer restricted to fibroblasts but not to blood cells has also been observed in RTEL1-deficient HH patients,54, 55 raising the possibility that RTEL1 and Apollo may participate in a common DNA repair pathway. Together, these observations rule out a diagnosis of Fanconi anemia in Apollo-deficient patients and suggest that Apollo-deficiency causes defects in biological processes that differ from and expand beyond the classical FA/BRCA pathway.
Recently, Mendez-Bermudez and colleagues have proposed that some factors originally devoted to warrant proper DNA replication and repair have evolved to be recruited and act at telomeres.56 Interestingly, phylogenetic analyses suggested that Apollo might be one of these factors since its TBM domain coemerged with the 2 telomeric paralogues TRF1 and TRF2, presumably to direct the specific binding of Apollo to TRF2.27, 57 The importance of the interaction between Apollo and TRF2 has been further substantiated by a comparative genomic analysis that pinpointed the existence of variants in the TBM of Apollo and in the TRFH domain of TRF2 in long-lived Galapagos giant tortoises compared with short-lived turtles.58 This finding argues that variation in the strength of TRF2-Apollo interaction could influence aging and organismal lifespan. The fact that both missense Apollo variants L142S and L142F reduce the interaction with TRF2 supports the notion that they might contribute to the premature aging phenotype observed in patients. Furthermore, the decreased interaction between TRF2 and the Apollo L142S and L142F mutants suggests that the β-lactamase domain containing the L142 residue might possess a second interface that stabilizes the TBM-dependent Apollo-TRF2 interaction. This hypothesis will require further investigations.
Studies conducted in Apollo KO MEFs established that the nuclease activity of Apollo participated in the generation of the 3′ G-overhang of the leading telomere.24, 25, 26 The 30% to 50% reduction of G-overhang signal observed in Apollo-deficient patients' cells was similar to the one found in Apollo KO MEFs, supporting the idea that the patients' variants impair Apollo nuclease activity. However, several clues suggest that the L142S and L142F Apollo are hypomorphic rather than complete loss-of-function mutants. First, Apollo KO mice are unviable24, 26, 46 and none of the 3 patients carry biallelic null Apollo alleles, but all carry at least 1 missense Apollo variant affecting the residue L142. Furthermore, we noticed that the frequency of telomere fusions in Apollo-deficient patient cells was less pronounced than in Apollo KO MEFs. Along the same line, telomere anomalies detected in patients' cells were not accompanied by an increase in TIFs and premature senescence, in contrast to Apollo KO MEFs.20, 21, 24, 26 Lastly, human Apollo KO HT1080 cell lines exhibit a more pronounced telomere phenotype than patients' cells. In particular, the telomerase positive Apollo KO human HT1080 cell lines exhibited a severe telomere length defect contrasting with the normal telomere length measured in patients' Apollo-deficient cells. This raises the possibility that the critically short telomeres caused by the absence of Apollo could result from the high frequency of sudden telomere loss detected by TeSLA and/or uncontrolled telomere degradation by undetermined nuclease that could not be compensated by telomerase activity. Alternatively, one cannot exclude that Apollo contributes to the recruitment and/or activity of telomerase to telomeres. In these 2 last hypotheses we assume that the presence of Apollo at telomeres, even at a low level (eg, in depleted cells by siRNA/shRNA) or with defective nuclease activity, would suffice to warrant telomere maintenance and partial telomere protection. This surmise is further supported by the observation that a nuclease-dead Apollo mutant expressed in Apollo KO MEFs exhibit a telomeric protective effect despite an impaired G-overhang generation at leading telomeres.26 Collectively, these findings led us to propose that the L142F and L142S Apollo mutants, although deleterious, retain some protective function compatible with life. Future studies will be necessary to further determine the structural and functional impact of the L142F and L142S Apollo variants and figure out what governs the phenotypic differences between patients' Apollo-deficient cells and murine and human Apollo KO cell lines.
We conclude from this study that biallelic Apollo variants leading to both impaired DNA repair and telomere instability are responsible for a hitherto unrecognized IBMFS akin to Høyeraal-Hreidarsson syndrome that is however not associated with excessive telomere shortening. These findings define Apollo as a genome caretaker critical for the proper development of the immunohematological system in humans.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank the individuals P1, P2, and P3 and their families for their contribution to this study. P.R. thanks Yanick Crow for the kind gift of Bloom-deficient SV40-transformed fibroblasts.
This work was supported by institutional grants from INSERM, Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue ‘LIGUE 2020' to P.R. and ‘LIGUE 2021' to V.G.), CEREDIH (Centre de Référence Déficits Immunitaires Héréditaires) and state funding from the Agence Nationale de la Recherche under “Investissements d'avenir” program (ANR-10-IAHU-01) and (ANR-21-CE12-APOthesis). This work was supported by the Israel Science Foundation grant [2071/18] to Y.T. and the Israel Ministry of Science and Technology (Navon Fellowship) to A.A. This work was also supported by the STEP-GTP Fellowship to A.A. and by the Israel-UK-Palestine GROWTH Fellowship Scheme, the British Council, to R.S. This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001PR. P.R. is a scientist from the Centre National de la Recherche Scientifique (CNRS).
Authorship
Contribution: L.K. carried out most of the experimental work; D.C. performed TeSLA analysis; A.A. and R.S. performed in-gel hybridization experiments; L.R. participated to the initial characterization of the shortest telomeres; P.R., C.K., S.D., and G.S. performed genetic analysis and identified Apollo variants; A.B., S.A.-B., S.H., E.C., M.O., F.T., S.D., and G.S. identified the affected patients and assisted with related clinical and laboratory studies; E.L. performed Flow-FISH; M.M. provided intellectual input; C.A. performed TCR repertoire analysis; I.C. performed structural analysis; and P.R. conceived the project and wrote the manuscript with editing contributions from J-P.d.V., F.T., I.C., V.G., and Y.T.
Footnotes
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
DNA cross-link repair 1B (DCLRE1B), also known as Apollo, is a multifaceted protein that acts as a DNA exonuclease in DNA repair and a protector of telomeres during S phase. In this Plenary Paper, Kermasson et al report on the discovery of biallelic mutations of DCLRE1B in 3 unrelated children who presented with hematologic, immunologic, and mucocutaneous features suggestive of dyskeratosis congenita, but with unexpectedly normal telomere lengths. Their work establishes Apollo deficiency as a new bone marrow failure syndrome and extends our understanding of telomeropathies.
Supplementary Material
REFERENCES
- 1.Savage SA, Dufour C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol. 2017;54(2):105–114. doi: 10.1053/j.seminhematol.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Savage SA. Beginning at the ends: telomeres and human disease. F1000 Res. 2018;7:7. doi: 10.12688/f1000research.14068.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170(4):457–471. doi: 10.1111/bjh.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armanios M, Blackburn EH. The telomere syndromes [published correction appears in Nat Rev Genet. 2013 Mar;14(3):235] Nat Rev Genet. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lange T. Shelterin-mediated telomere protection. Annu Rev Genet. 2018;52(1):223–247. doi: 10.1146/annurev-genet-032918-021921. [DOI] [PubMed] [Google Scholar]
- 7.MacNeil DE, Bensoussan HJ, Autexier C. Telomerase regulation from beginning to the end. Genes (Basel). 2016;7(9):64. doi: 10.3390/genes7090064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benyelles M, O'Donohue MF, Kermasson L, et al. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Høyeraal-Hreidarsson syndrome. Hum Mol Genet. 2020;29(6):907–922. doi: 10.1093/hmg/ddaa011. [DOI] [PubMed] [Google Scholar]
- 9.Hao LY, Armanios M, Strong MA, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123(6):1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 10.Alder JK, Hanumanthu VS, Strong MA, et al. Diagnostic utility of telomere length testing in a hospital-based setting [published correction appears in Proc Natl Acad Sci U S A. 2018;115(18):E4312] Proc Natl Acad Sci USA. 2018;115(10):E2358–E2365. doi: 10.1073/pnas.1720427115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–359. doi: 10.3324/haematol.2011.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demuth I, Digweed M, Concannon P. Human SNM1B is required for normal cellular response to both DNA interstrand crosslink-inducing agents and ionizing radiation. Oncogene. 2004;23(53):8611–8618. doi: 10.1038/sj.onc.1207895. [DOI] [PubMed] [Google Scholar]
- 13.Schmiester M, Demuth I. SNM1B/Apollo in the DNA damage response and telomere maintenance. Oncotarget. 2017;8(29):48398–48409. doi: 10.18632/oncotarget.16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae JB, Mukhopadhyay SS, Liu L, et al. Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene. 2008;27(37):5045–5056. doi: 10.1038/onc.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demuth I, Bradshaw PS, Lindner A, et al. Endogenous hSNM1B/Apollo interacts with TRF2 and stimulates ATM in response to ionizing radiation. DNA Repair (Amst). 2008;7(8):1192–1201. doi: 10.1016/j.dnarep.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason JM, Das I, Arlt M, et al. The SNM1B/APOLLO DNA nuclease functions in resolution of replication stress and maintenance of common fragile site stability. Hum Mol Genet. 2013;22(24):4901–4913. doi: 10.1093/hmg/ddt340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason JM, Sekiguchi JM. Snm1B/Apollo functions in the Fanconi anemia pathway in response to DNA interstrand crosslinks. Hum Mol Genet. 2011;20(13):2549–2559. doi: 10.1093/hmg/ddr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michailidou K, Hall P, Gonzalez-Neira A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353–361. doi: 10.1038/ng.2563. 361e351-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang XS, Pfeiffer RM, Wheeler W, et al. Genetic variants in DNA repair genes and the risk of cutaneous malignant melanoma in melanoma-prone families with/without CDKN2A mutations. Int J Cancer. 2012;130(9):2062–2066. doi: 10.1002/ijc.26231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenain C, Bauwens S, Amiard S, Brunori M, Giraud-Panis MJ, Gilson E. The Apollo 5′ exonuclease functions together with TRF2 to protect telomeres from DNA repair. Curr Biol. 2006;16(13):1303–1310. doi: 10.1016/j.cub.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 21.van Overbeek M, de Lange T. Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr Biol. 2006;16(13):1295–1302. doi: 10.1016/j.cub.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 22.Freibaum BD, Counter CM. hSnm1B is a novel telomere-associated protein. J Biol Chem. 2006;281(22):15033–15036. doi: 10.1074/jbc.C600038200. [DOI] [PubMed] [Google Scholar]
- 23.Ye J, Lenain C, Bauwens S, et al. TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell. 2010;142(2):230–242. doi: 10.1016/j.cell.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 24.Lam YC, Akhter S, Gu P, et al. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 2010;29(13):2230–2241. doi: 10.1038/emboj.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu P, Takai H, de Lange T. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell. 2012;150(1):39–52. doi: 10.1016/j.cell.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu P, van Overbeek M, Rooney S, de Lange T. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol Cell. 2010;39(4):606–617. doi: 10.1016/j.molcel.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Touzot F, Callebaut I, Soulier J, et al. Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2010;107(22):10097–10102. doi: 10.1073/pnas.0914918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai TP, Zhang N, Noh J, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun. 2017;8(1):1356. doi: 10.1038/s41467-017-01291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Awad A, Glousker G, Lamm N, et al. Full length RTEL1 is required for the elongation of the single-stranded telomeric overhang by telomerase. Nucleic Acids Res. 2020;48(13):7239–7251. doi: 10.1093/nar/gkaa503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armand M, Derrieux C, Beldjord K, et al. A new and simple TRG multiplex PCR assay for assessment of T-cell clonality: a comparative study from the EuroClonality Consortium. HemaSphere. 2019;3(3):e255. doi: 10.1097/HS9.0000000000000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benyelles M, Episkopou H, O'Donohue MF, et al. Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models. EMBO Mol Med. 2019;11(7):e10201. doi: 10.15252/emmm.201810201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Touzot F, Kermasson L, Jullien L, et al. Extended clinical and genetic spectrum associated with biallelic RTEL1 mutations. Blood Adv. 2016;1(1):36–46. doi: 10.1182/bloodadvances.2016001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Guen T, Jullien L, Touzot F, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22(16):3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- 34.Jullien L, Kannengiesser C, Kermasson L, et al. Mutations of the RTEL1 helicase in a Hoyeraal-Hreidarsson syndrome patient highlight the importance of the ARCH domain. Hum Mutat. 2016;37(5):469–472. doi: 10.1002/humu.22966. [DOI] [PubMed] [Google Scholar]
- 35.Takai H, Jenkinson E, Kabir S, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 2016;30(7):812–826. doi: 10.1101/gad.276873.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Callebaut I, Moshous D, Mornon JP, de Villartay JP. Metallo-beta-lactamase fold within nucleic acids processing enzymes: the beta-CASP family. Nucleic Acids Res. 2002;30(16):3592–3601. doi: 10.1093/nar/gkf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allerston CK, Lee SY, Newman JA, Schofield CJ, McHugh PJ, Gileadi O. The structures of the SNM1A and SNM1B/Apollo nuclease domains reveal a potential basis for their distinct DNA processing activities. Nucleic Acids Res. 2015;43(22):11047–11060. doi: 10.1093/nar/gkv1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Touzot F, Gaillard L, Vasquez N, et al. Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J Allergy Clin Immunol. 2012;129(2):473–482. doi: 10.1016/j.jaci.2011.09.043. 482 e471-473. [DOI] [PubMed] [Google Scholar]
- 41.Lamm N, Ordan E, Shponkin R, Richler C, Aker M, Tzfati Y. Diminished telomeric 3′ overhangs are associated with telomere dysfunction in Hoyeraal-Hreidarsson syndrome. PLoS One. 2009;4(5):e5666. doi: 10.1371/journal.pone.0005666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13(17):1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 43.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 44.Sfeir A, Kosiyatrakul ST, Hockemeyer D, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138(1):90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA. 2013;110(36):E3408–E3416. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akhter S, Lam YC, Chang S, Legerski RJ. The telomeric protein SNM1B/Apollo is required for normal cell proliferation and embryonic development. Aging Cell. 2010;9(6):1047–1056. doi: 10.1111/j.1474-9726.2010.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schratz KE, Haley L, Danoff SK, et al. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood. 2020;135(22):1946–1956. doi: 10.1182/blood.2019003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanley SE, Armanios M. The short and long telomere syndromes: paired paradigms for molecular medicine. Curr Opin Genet Dev. 2015;33:1–9. doi: 10.1016/j.gde.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia CK. Insights from human genetic studies of lung and organ fibrosis. J Clin Invest. 2018;128(1):36–44. doi: 10.1172/JCI93556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Juge PA, Borie R, Kannengiesser C, et al. FREX consortium Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017;49(5):1602314. doi: 10.1183/13993003.02314-2016. [DOI] [PubMed] [Google Scholar]
- 52.Salewsky B, Schmiester M, Schindler D, Digweed M, Demuth I. The nuclease hSNM1B/Apollo is linked to the Fanconi anemia pathway via its interaction with FANCP/SLX4. Hum Mol Genet. 2012;21(22):4948–4956. doi: 10.1093/hmg/dds338. [DOI] [PubMed] [Google Scholar]
- 53.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668(1-2):4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Speckmann C, Sahoo SS, Rizzi M, et al. Clinical and molecular heterogeneity of RTEL1 deficiency [published correction appears in Front Immunol. 2017;8:1250] Front Immunol. 2017;8:449. doi: 10.3389/fimmu.2017.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mendez-Bermudez A, Giraud-Panis MJ, Ye J, Gilson E. Heterochromatin replication goes hand in hand with telomere protection. Nat Struct Mol Biol. 2020;27(4):313–318. doi: 10.1038/s41594-020-0400-1. [DOI] [PubMed] [Google Scholar]
- 57.Myler LR, Kinzig CG, Sasi NK, Zakusilo G, Cai SW, de Lange T. The evolution of metazoan shelterin. Genes Dev. 2021;35(23-24):1625–1641. doi: 10.1101/gad.348835.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quesada V, Freitas-Rodríguez S, Miller J, et al. Giant tortoise genomes provide insights into longevity and age-related disease. Nat Ecol Evol. 2019;3(1):87–95. doi: 10.1038/s41559-018-0733-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buck D, Malivert L, de Chasseval R, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.