

Abstract
High-mobility group box 1 (HMGB1) is involved in the tumor-associated activation of regulatory T cells (Treg), but the mechanisms remain unknown. In a mouse tumor model, silencing HMGB1 in tumor cells or inhibiting tumor-derived HMGB1 not only dampened the capacity of tumor cells to produce thymic stromal lymphopoietin (TSLP), but also aborted the tumor-associated modulation of Treg-activating DC. Tumor-derived HMGB1 triggered the production of TSLP by tumor cells. Importantly, both tumor-derived HMGB1 and TSLP were necessary for modulating DC to activate Treg in a TSLP receptor (TSLPR)-dependent manner. In the therapeutic model, intratumorally inhibiting tumor-derived HMGB1 (causing downstream loss of TSLP production) attenuated Treg activation, unleashed tumor-specific CD8 T cell responses, and elicited CD8α+/CD103+DC- and T cell-dependent antitumor activity. These results suggest a new pathway for the activation of Treg involving in tumor-derived HMGB1 and TSLP, and have important implications for incorporating HMGB1 inhibitors into cancer immunotherapy.
Electronic supplementary material
The online version of this article (10.1007/s00262-017-2087-7) contains supplementary material, which is available to authorized users.
Keywords: HMGB1, TSLP, DC, Treg, Cancer immunotherapy, Mouse tumor model
Introduction
Soluble factors produced by growing tumor cells directly activate Treg or indirectly modulate other cells (e.g., DC) to activate Treg [1–4]. As Treg play an important role in tumor immunity by suppressing CTL and other cells associated with antitumor immunity, identifying those tumor-derived factors and better understanding how they activate Treg would be a key issue for successful cancer immunotherapy [1–4].
HMGB1, a conserved protein with various post-translation modifications in tumor cells and the tumor microenvironment (TME) [5], is thought to stimulate immune responses as a danger signal. HMGB1 released from dying tumor cells following some chemo-, viro- or irradiation-therapies triggers antitumor immunity through innate pathways, such as the TLR pathway in DC [6–8]. Recombinant or isolated HMGB1 kills cancer cells [9]. Antibody blockade or pharmacological inhibition of HMGB1 dampens tumor progression [10, 11].
HMGB1 is a powerful tumor-promoting factor directly tied to hallmarks of tumor progression, including angiogenesis, inflammation, adaptive immunity, invasion, and metastasis [5, 12–14]. This suggests the possibility of immunosuppression pathway(s) via tumor-derived HMGB1 during tumor progression. HMGB1 derived from tumor cells exhibits an inhibitory effect on both mouse and human DC [15]. HMGB1 interacts with T-cell immunoglobulin and mucin-domain-containing molecule-3 (TIM-3) on tumor-infiltrating DC to suppress nucleic acid-mediated antitumor immunity [16], and contributes to immunosuppression by inducing and activating MDSC [17, 18]. HMGB1 is also involved in the tumor-associated activation of Treg even though the mechanisms remain unknown [19–21].
Regarding the mechanism(s) of action, tumor-derived HMGB1 may regulate yet-to-be-identified factor(s) within tumors and subsequently cooperate with them in the TME to initiate immunosuppression, for example, by activating Treg. Among tumor-derived factors, TSLP facilitates a CD4 T helper 2 (Th2)-mediated protumor immune response directly, or indirectly (via DC) [22–28]. TSLP is produced upon injury, following TLR ligation by microbial byproducts, and in response to other cytokines [29, 30]. Although the signals that regulate TSLP in tumor cells are largely unknown, myeloid cell-derived IL-1β may be a candidate [31].
In this study, tumor-derived HMGB1 triggered TSLP production by tumor cells. Both tumor-derived HMGB1 and TSLP were required for modulating DC to activate Treg. In vivo inhibition of HMGB1 in the TME reduced TSLP production and tumor-associated Treg activation, unleashed tumor-specific CD8 T cell responses, and elicited CD103+/CD8α+DC- and T cell-dependent therapeutic antitumor immunity.
Materials and methods
Mice, cell lines and reagents
BALB/c-wild type (WT), -Batf3−/−, -TLR4−/− or -Foxp3-GFP mice and C57BL/6 (B6)-WT or -Batf3−/− mice [female, 6–8 weeks (wks)] were purchased from JAX or Taconic. BALB/c-CD4−/− or -TSLPR−/− mice (kindly provided by Dr. Steven F. Ziegler at Benaroya Research Institute) were reported previously [32, 33]. Mice were housed under specific pathogen-free conditions in the University of Pittsburgh animal facility, and handled under aseptic conditions per an Institutional Animal Care and Use Committee-approved protocol, and in accordance with recommendations for the proper care and use of laboratory animals.
Mouse breast cancer cell lines 4T1.2-Neu and short hairpin RNA (shRNA)-mediated genetic stable knockdown (KD) of HMGB1 or control (Ctl) vector in 4T1.2-Neu (i.e., 4T1.2-Neu-HMGB1 KD, 4T1.2-Neu-Ctl KD) were described previously [19], and cultured in DMEM (IRVINE Scientific) supplemented with 10% FBS (Hyclone), 2 mmol/l glutamine (Invitrogen), 1 × antibiotic/antimycotic solution (Sigma) and G-418 (500 µg/ml) (Invitrogen). Mouse melanoma B16 cell line (ATCC) was cultured in the aforementioned media excluding G-418.
BamHI/XhoI-digested mouse (m) TSLP was inserted into BamHI/XhoI-digested pLenti6-EGFP-Trip vector [33] following standard molecular cloning techniques, resulting in pLenti6-mTSLP. Insertion of mTSLP was confirmed by DNA sequencing. A Ctl vector, for pLenti6-mTSLP, pLenti6 was made by digesting pLenti6-EGFP-Trip with BamHI/XhoI, blunting the stick ends and self-ligating the vector. All DNA plasmids were purified using the EndoFree plasmid kit (Qiagen). 4T1.2-Neu-HMGB1 KD cells were transfected with pLenti6-mTSLP, or pLenti6 Ctl using Lipofectamine 2000 (Invitrogen) and selected with Blasticidin (Invivogen) in the 4T1.2-Neu culture media. Selected TSLP- or Ctl-knockin (KI)-4T1.2-Neu-HMGB1 KD cell lines (i.e., TSLP KI-4T1.2-Neu-HMGB1 KD, Ctl KI-4T1.2-Neu-HMGB1 KD) were maintained in 4T1.2-Neu culture media containing Blasticidin.
Tumor-derived HMGB1/TSLP (i.e., HMGB1+TSLP+: HMGB1- and TSLP-containing tumor cell supernatants [19, 23]) were used to prepare tumor-derived HMGB1 (i.e., HMGB1+TSLP−: HMGB1-containing TSLP-depleted tumor cell supernatants), TSLP (i.e., HMGB1−TSLP+: HMGB1-depleted TSLP-containing tumor cell supernatants), and Ctl (i.e., HMGB1−TSLP−: HMGB1- and TSLP-depleted tumor cell supernatants) using anti-HMGB1 and/or -TSLP Abs and Dynabeads® protein G following the protocol [35] with modifications: 4T1.2-Neu (1 × 106) were cultured in 2 ml RPMI 1640 (IRVINE Scientific) 3% FBS for 48 h (h). In a single preparation, 2 ml tumor cell supernatants were concentrated to 200 µl using Centricon®15 (Millipore). Then, HMGB1 and/or TSLP in tumor cell supernatants were depleted by anti-HMGB1, -TSLP or -HMGB1 and TSLP Abs (25–50 µg/200 µl/each) [19, R&D Systems] and Dynabeads® protein G following a standard protocol (Life technologies). Multiple batches of tumor-derived HMGB1/TSLP, HMGB1, TSLP, and Ctl were pooled, examined by western blot (WB) to ensure the presence of HMGB1 and/or TSLP in those tumor cell supernatants [19, see below], aliquoted, and stored at − 80 °C.
TSLP production
4T1.2-Neu, 4T1.2-Neu-HMGB1 KD, 4T1.2-Neu-Ctl KD, TSLP KI-4T1.2-Neu-HMGB1 KD, or Ctl KI-4T1.2-Neu-HMGB1 KD cells (5 × 104) were cultured in 200 µl RPMI 1640 with 10% FBS for 36–48 h. Comparable cell numbers among groups were seen after 48 h of culture [19, data not shown]. Tumor cell supernatants were harvested and the concentrations of total proteins were measured using a standard Bradford assay. Due to the absence of an internal (housekeeping gene encoded proteins, e.g., β-actin) control in tumor cell culture supernatants, supernatants containing equal amounts of total proteins were loaded to run WB with two different anti-mouse TSLP Abs (AF555 from R&D Systems; Cat#3872-100 from BioVision), following the protocol described previously [19]. The relative TSLP levels in the supernatants were measured by ImageJ software (https://imagej.nih.gov/ij/).
4T1.2-Neu-HMGB1 KD (4T1.2-Neu as Ctl) (2.5 × 104) were cultured in 200 µl RPMI 1640 3% FBS (untreated) or 190 µl RPMI 1640 3% FBS supplemented with 10 µl of tumor-derived HMGB1 (i.e., HMGB1+TSLP−) or Ctl (i.e., HMGB1−TSLP−) for 48 h.
BALB/c mice (3–5/group) were subcutaneously (s.c.) inoculated with 4T1.2-Neu, 4T1.2-Neu-HMGB1 KD, or 4T1.2-Neu-Ctl KD cells (1 × 105) at the 4th mammary fat pad at day (d) 0 [19]. 10 d later, sera were harvested. Simultaneously, primary tumors were removed and digested in HBSS buffer supplemented with 2% FBS, 2.7% collagenase, 0.25% hyaluronidase and 20 Unit/ml DNase (Sigma) for 2 h in a 37 °C incubator. Cell suspensions were passed through 70-μm Nylon mesh. Equal amounts of live cells, determined by Trypan blue staining, from primary tumors were cultured in RPMI 1640 10% FBS (5 mg/ml) for 24 h.
Glycyrrhizin (GL) (Calbiochem) was dissolved in 50 mM NaOH (80 μg/μl) (pH 7.4 adjusted with 1 M Tris–HCl: vehicle solution) [36]. 4T1.2-Neu cells (5 × 104) were cultured with or without GL (0.5 µg/µl) or an equal volume of vehicle solution in 200 µl RPMI 1640 10% FBS for 48 h. This dose of GL or vehicle solution was shown to be nontoxic, as comparable cell numbers among groups were seen after 48 h of culture (data not shown).
BALB/c mice (3–5/group) were inoculated s.c. with 4T1.2-Neu (1 × 105) as described above. 6–8 d later, mice bearing tumors (mean sizes of tumors around 3 mm in diameter) were intratumorally (i.t.) injected daily with 10 μl of GL (800 μg) or vehicle solution for 2 wks. In the independent experiments, tumor-bearing mice were i.t. injected with 20 μl of Box A from HMGB1 (HM-014, HMGBiotech) (50 μg), ethyl pyruvate (EP) (E47808, Sigma-Aldrich) (80 mg/kg), or vehicle solution (1XPBS) every 3 d for four times. On d 21 (GL) or 25 (Box A or EP), sera from mice were collected.
The concentrations of TSLP in tumor cell culture supernatants, sera, or primary tumor cell culture supernatants were measured by ELISA (eBioscience).
Flow cytometry analysis
BALB/c mice were s.c. inoculated with 4T1.2-Neu as described above. 10 d later, single-cell suspensions of tumor-draining lymph nodes (TDLN) or LN from age-matched naïve mice were preincubated with anti-CD16/32 and then stained with anti-CD11c-APC (HL3) and either anti-TLR2-PE (TL2.1), -TLR4-PE (UT41), or -RAGE (MAB11795) (followed by anti-Rat IgG 2nd Ab-PE) [isotype (ISO) Abs for Ctl staining] (eBioscience, BD Biosciences, R&D Systems). Propidium iodide (BD Biosciences) was used to check cell viability. Forward scatter and side scatter were used to exclude cell debris. After three final washes using FACS staining buffer, the cells were resuspended in 500 μl 1% PFA. Sample data were acquired on a BD LSRII with CellQuest software (BD Biosciences) and analyzed with FlowJo software (Tree Star).
DC modulation
CD11c+DC were purified from spleens/LN of naïve BALB/c mice (5/group) using anti-mouse CD11c (N418) microbeads (Miltenyi Biotec). DC (4 × 105) were cultured with 4T1.2-Neu (1 × 104) in 200 µl of RPMI 1640 plus 10% FBS, with or without anti-HMGB1 or rabbit IgG (20 µg/ml) [19]. 3 d later, DC were pooled and stained with anti-CD11c-APC (HL3) and sorted using a BD FACSAria High Speed Cell Sorter (BD Biosciences). Sorted DC (1 × 104) were cocultured with Treg (2 × 105) that were sorted stringently from spleens/LN of naïve BALB/c-Foxp3-GFP mice in 200 µl RPMI 1640 plus 10% FBS for 3 d. To avoid possible contamination, sorted GFP+ cells were stained using the Treg staining kit (eBioscience) to ensure actual Foxp3 expression by naïve GFP+CD25+CD4+ T cells in each experiment. Foxp3+CD25+CD4+ T cells > 95% of GFP+ cells were used in experiments.
CD11c+DC (1 × 106) purified from spleens/LN of naïve BALB/c-WT, -TSLPR−/−, or -TLR4−/− mice were cultured in 450 µl of RPMI 1640 plus 3% FBS plus 50 µl of tumor-derived HMGB1, TSLP, or HMGB1 + TSLP (25 µl each). 3 d later, harvested DC (1 × 105) were cocultured with naïve Treg (2 × 106), sorted from naïve BALB/c-Foxp3-GFP mice, in 200 µl RPMI 1640 plus 10% FBS for 3 d.
BALB/c mice (5/group) were inoculated with 4T1.2-Neu, 4T1.2-Neu-HMGB1 KD, or 4T1.2-Neu-Ctl KD [19]. Some of 4T1.2-Neu-bearing mice were left untreated, or treated with GL or vehicle solution as described above. After 10 d (4T1.2-Neu, 4T1.2-Neu-HMGB1 KD, or 4T1.2-Neu-Ctl KD inoculated mice) or 21 d (4T1.2-Neu-bearing mice untreated or treated with GL or vehicle solution), CD11c+DC were sorted from pooled TDLN (i.e., TDLN DC). Sorted TDLN DC (5 × 104) were cocultured with naïve Treg (2 × 105), sorted from spleens/LN of BALB/c-Foxp3-GFP mice, in 200 μl RPMI 1640 plus10%FBS for 3 d.
Treg were purified from pooled DC–Treg cocultures using anti-mouse CD4 microbeads (Miltenyi Biotec) for the assays of their suppressive activity.
Treg activity
4T1.2-Neu-bearing BALB/c-Foxp3-GFP mice (3/group) were left untreated, or treated with GL or vehicle solution as described above. At d21, Treg (GFP+) in the TDLN and spleens were sorted as described above. The ability of Treg, activated by tumors in vivo or by Treg-activating DC ex vivo (described above), to suppress T-cell activation was measured as reported previously [37]. Briefly, 4T1.2-Neu-primed CD4 T cells (2 × 105), 4T1.2-Neu lysate-loaded naïve BALB/c splenic DC (2 × 105), and naïve BALB/c splenic CD8 T cells (2 × 105) were cultured with or without Treg (2 × 105) for 3 d. Then, concentrations of IFN-γ in the cell culture supernatants were measured by ELISA (BD Biosciences).
Therapeutic antitumor immunity
BALB/c-WT, -Batf3−/−, -TLR4−/− or -CD4−/− mice were s.c. inoculated with 4T1.2-Neu as described above. 6–8 d later, mice bearing tumors (mean sizes of tumors around 3 mm in diameter) were injected i.t. daily with 10 μl of GL (800 μg) or vehicle solution for 2 wks. In independent experiments, tumor-bearing mice were injected i.t. with 20 μl of Box A from HMGB1 (50 μg), EP (80 mg/kg), or vehicle solution (1XPBS) every 3 d for four times. In some experiments, anti-mouse CD8 mAbs (53-6.7, Bio X Cell) were intraperitoneally (i.p.) injected (200 µg/injection) into 4T1.2-Neu-bearing mice on d 7, 9, and 14 post-tumor inoculation [19]. B6-WT or -Batf3−/− mice were inoculated s.c. with melanoma B16 (5 × 104). 8 d later, B16-bearing mice were treated with 10 μl of GL or vehicle solution as described above. Tumor sizes were measured using an electronic caliper every 3 d to determine the two perpendicular diameters, and the mean sizes of tumors were calculated [(length + width)/2] [19, 37]. Mice were euthanized if tumors exceeded 10 mm in diameter, when tumors became ulcerated or bled, or when mice displayed signs of disease-associated distress. Lung metastases were examined by weighing the lungs, or by fixing the lungs with Bouin’s solution (Sigma) and counting tumor foci [32]. In some experiments, 28 d after tumor cell inoculation, CD8 T cells were purified from TDLN and spleens using anti-CD8 microbeads (Miltenyi Biotec), and restimulated with 4T1.2-Neu lysate- (or irrelevant CT26 tumor cell lysate-) pulsed CD8− splenocytes (SPC) (serving as antigen-presenting cells: APC) from naïve BALB/c for 3 d [19, 37]. Then, concentrations of IFN-γ in the cell culture supernatants were measured by ELISA.
Statistical analysis
Differences between groups were analyzed using a Student’s t test (Graph Pad Prism version 6). p < 0.05 was considered statistically significant; *p < 0.05; **p < 0.01; ***p < 0.001; NS: not significant.
Results
Silencing HMGB1 in tumor cells, or inhibiting tumor-derived HMGB1, dampens the capacity of tumor cells to produce TSLP
To explore the impact of HMGB1 on TSLP, which has been shown to be a tumor-promoting cytokine [22–28], 4T1.2-Neu-WT, -HMGB1 KD, or -Ctl KD tumor cells were cultured in vitro or inoculated into mice. As shown in Fig. 1a–e, silencing HMGB1 in tumor cells in vitro and in vivo (d 10 post-tumor cell inoculation) reduced their production of TSLP. To verify this finding, 4T1.2-Neu tumor cells were cultured in vitro in the presence of GL (a specific inhibitor of extracellular HMGB1) [36], and mice bearing 4T1.2-Neu tumors were injected i.t. with GL. The HMGB1 inhibitor GL decreased the production of TSLP by tumor cells in vitro and in tumor-bearing mice (d 21 post-tumor cell inoculation) (Fig. 1f–g). To further confirm the influence of HMGB1 on TSLP in vivo, mice bearing 4T1.2-Neu tumors were injected i.t. with either a potent HMGB1 inhibitor EP, which reduces HMGB1 levels and was explored in cancer treatments [11, 38], or the antagonist for HMGB1 (i.e., Box A) [17, 38, 39]. Both Box A and EP reduced the production of TSLP in vivo (Fig. 1h). To directly demonstrate the effect of HMGB1 on TSLP production by tumor cells in vitro, 4T1.2-Neu-derived HMGB1 was added to cultures of 4T1.2-Neu-HMGB1 KD tumor cells. As shown in Fig. 1i, tumor-derived HMGB1 significantly restored the production of TSLP by HMGB1 silenced tumor cells (which had substantially reduced TSLP production, Fig. 1a–c), indicating that tumor-derived HMGB1 triggers the production of TSLP by tumor cells. The result may also explain why HMGB1-silenced tumor cells (which lack extracellular HMGB1) [19] were ineffective in producing TSLP (Fig. 1a–c). Taken together, the results suggest that tumor-derived HMGB1 may act as an extracellular signal to trigger TSLP production by tumor cells.
Fig. 1.
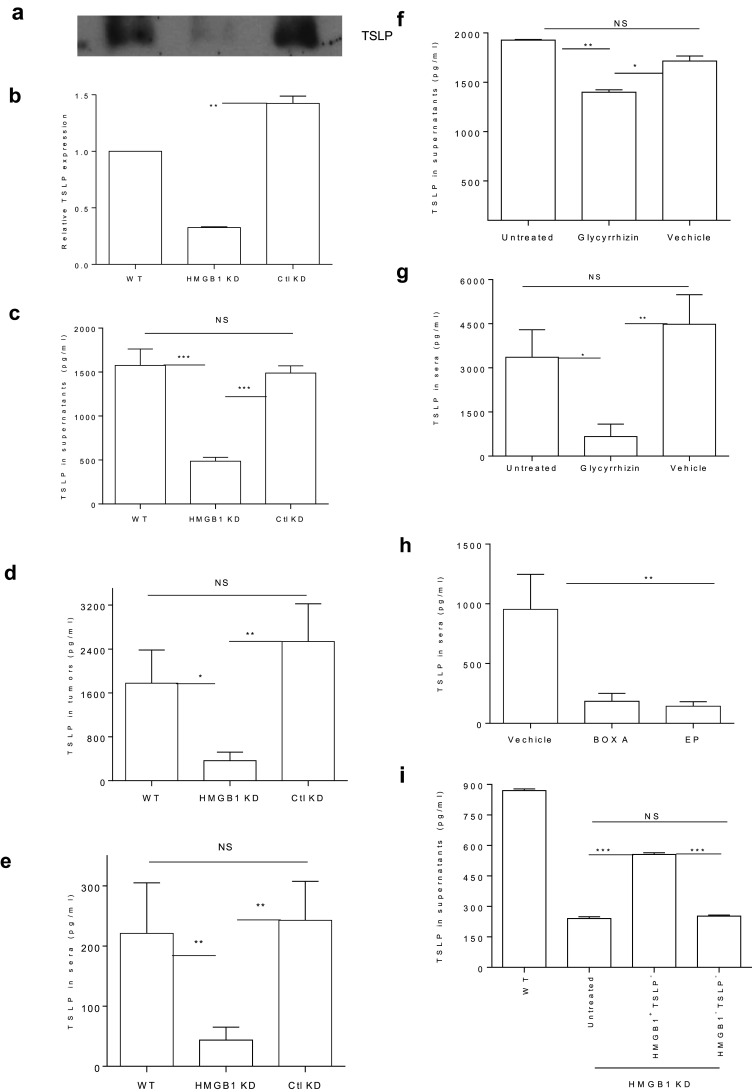
a–c Silencing HMGB1 in tumor cells reduces their production of TSLP in vitro. 4T1.2-Neu-WT, -HMGB1 KD or -Ctl KD were cultured for 48 h. Tumor cell supernatants were harvested and the concentrations of whole proteins in the supernatants were measured. Supernatants containing equal amounts of whole proteins were loaded to run WB using anti-mouse TSLP Ab. One representative of three independent experiments is shown (a). The relative TSLP levels in the supernatants of WT-, HMGB1 KD- and Ctl KD-tumor cells from three independent experiments are shown (b). The TSLP level in WT-tumor cell supernatants was normalized to be 1. The concentrations of TSLP in tumor cell supernatants were measured by ELISA (c). d, e Silencing HMGB1 in tumor cells impairs TSLP production in vivo. BALB/c mice were s.c. inoculated with 4T1.2-Neu-WT, -HMGB1 KD or -Ctl KD at d 0. 10 d later, sera were harvested (e). Simultaneously, equal amounts of live cells of primary tumors were cultured for 24 h (d). f–h I.t. inhibiting HMGB1 reduces TSLP production in vitro and in vivo. 4T1.2-Neu tumor cells were cultured with or without GL or equal volumes of vehicle solution for 48 h (f). BALB/c mice were s.c. inoculated with 4T1.2-Neu. 6–8 d later, 4T1.2-Neu-bearing mice were untreated or treated with either GL (g) or EP and Box A (h) or vehicle solution. 21 (g) or 25 (h) d later, sera were collected. i Tumor-derived HMBG1 triggers TSLP production by tumor cells. 4T1.2-Neu-HMGB1 KD were cultured in the absence or presence of tumor-derived HMGB1 (i.e., HMGB1+TSLP−: HMGB1-included TSLP-depleted tumor cell supernatants) or Ctl (i.e., HMGB1−TSLP−: HMGB1- and TSLP-depleted tumor cell supernatants) for 48 h. 4T1.2-Neu-WT served as Ctl. The concentrations of TSLP in tumor cell supernatants (c, f, i), sera (e, g, h) or primary tumor cell supernatants (d) were measured by ELISA. Data from two (g, h) or three (b–f, i) independent experiments are shown and were statistically analyzed using a Student’s t test. HMGB1 KD vs. Ctl KD: p < 0.01 (b); WT or Ctl KD vs. HMGB1 KD: p < 0.001(c); WT vs. HMGB1 KD: p < 0.05, Ctl KD vs. HMGB1 KD: p < 0.01(d); WT or Ctl KD vs. HMGB1 KD: p < 0.01(e); WT vs. Ctl KD: NS (c–e); Untreated vs. GL: p < 0.01, Vehicle vs. GL: p < 0.05 (f); Untreated vs. GL: p < 0.05, Vehicle vs. GL: p < 0.01 (g); Untreated vs. Vehicle: NS (f, g); Vehicle vs. Box A or EP: p < 0.01 (h); Untreated or Ctl vs. HMGB1: p < 0.001, Untreated vs. Ctl: NS (i)
Silencing or inhibiting tumor-derived HMGB1 (causing downstream loss of TSLP production) abrogates the tumor-associated modulation of DC that activates Treg
Tumor-derived HMGB1 has been linked to tumor-induced activation of Treg in in vivo mouse tumor models and in vitro human cancer models [19–21]. Tumor-derived HMGB1 enhanced the production of IL-10 by tumor-associated Treg but could not directly activate Treg in vitro [19], suggesting that tumor-derived HMGB1 may indirectly activate Treg through other immune cells, such as DC. To test this hypothesis, tumor cells were cocultured with DC isolated from naïve mice, those tumor cell-DC were cocultured with Treg sorted from naïve mice, and the suppressive activity of those tumor cell-DC-activated Treg was measured (Fig. 2a). The suppressive activity of Treg activated by tumor cell-modulated DC was determined by their capacity to suppress T-cell activation as measured by IFN-γ production in vitro [37]. As shown in Fig. 2b, syngeneic naïve Treg were activated by tumor cell-modulated DC (tumor cell-DC), and neutralizing tumor-derived HMGB1 in tumor cell-DC cultures impaired the capacity of tumor cells to modulate DC to activate Treg. To confirm this finding in vivo, DC isolated from mice bearing 4T1.2-Neu-HMGB1 KD tumors, or 4T1.2-WT tumors treated by HMGB1 inhibitor GL or vehicle, were cocultured with Treg isolated from naïve mice, and the suppressive activity of those DC-activated Treg was measured (Fig. 2c). As shown in Fig. 2d, e, silencing HMGB1 in tumor cells or i.t. inhibiting tumor-derived HMGB1 in vivo [causing downstream loss of TSLP production (Fig. 1a–g)] reduced the capacity of tumor cells in vivo to modulate DC to activate Treg. Taken together, the results suggest that tumor-derived HMGB1 and TSLP are involved in modulating DC to activate naïve Treg.
Fig. 2.
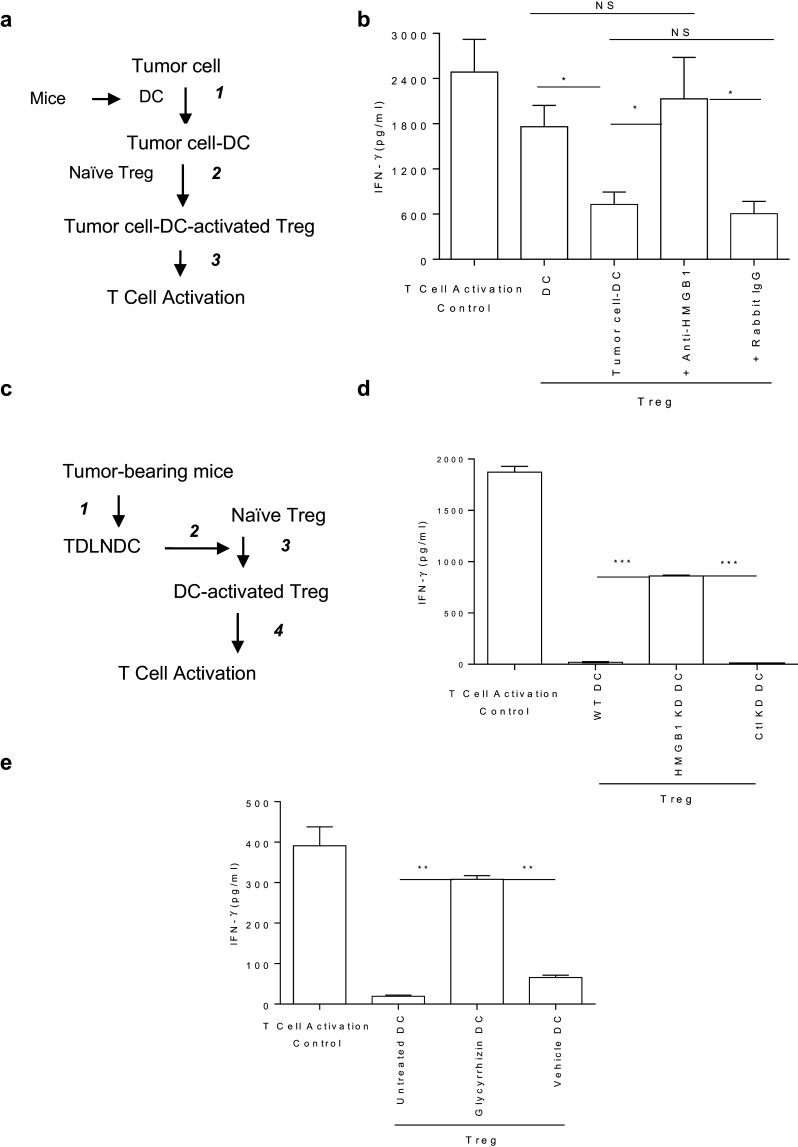
a, b Neutralizing HMGB1 impairs the ability of tumor cells to in vitro modulate DC to activate Treg. Experimental outline (a): (1) DC purified from naïve BALB/c mice were cultured with 4T1.2-Neu with or without anti-HMGB1 Ab or rabbit IgG; (2) 3 d later, sorted DC were cocultured with Treg sorted from naïve BALB/c-Foxp3-GFP mice for 3 d, and then Treg were purified from pooled DC–Treg coculture; (3) The ability of those Treg to suppress T-cell activation was measured. Data from three independent experiments are presented and were statistically analyzed using a Student’s t test (b). DC vs. tumor cell-DC: p < 0.05; Tumor cell-DC + anti-HMGB1 vs. tumor cell-DC or tumor cell-DC + rabbit IgG: p < 0.05; DC vs. tumor cell-DC + anti-HMGB1: NS; tumor cell-DC vs. tumor cell-DC + rabbit IgG: NS. c–e Silencing HMGB1 in tumor cells or i.t. inhibiting HMGB1 in tumors impairs the ability of tumor cells to in vivo modulate DC to activate Treg. Experimental outline (c): (1) BALB/c mice were inoculated with 4T1.2-Neu-WT, -HMGB1 KD or -Ctl KD (d) and some of 4T1.2-Neu-WT-bearing mice were untreated or treated with GL or vehicle solution (e); 2) 10 (d) or 21 (e) d later, DC were sorted from TDLN and Treg were sorted from naive BALB/c-Foxp3-GFP mice; (3) Sorted TDLN DC were cocultured with sorted naïve Treg for 3 d, and then Treg were purified from pooled DC–Treg coculture; (4) The ability of those Treg to suppress T cell activation was measured. Experiments were performed independently three times and results were statistically analyzed using a Student’s t test (d, e). HMGB1 KD vs. WT or Ctl KD: p < 0.001 (d); GL vs. untreated or vehicle: p < 0.01 (e)
Tumor-derived HMGB1 and TSLP are required for modulating DC to activate Treg in a TSLPR-dependent manner
To determine the impact of tumor progression on the expression of putative HMGB1 receptors on DC (e.g., TLR4, TLR2 and RAGE) [13, 40, 41], single-cell suspensions of TDLN from tumor-bearing mice were stained and analyzed by flow cytometry. Tumor progression increased the expression of TLR4 and TLR2 (p < 0.05), and RAGE (p = 0.077) on DC in the TDLN (Fig. 3a, b). It also slightly increased the expression of receptors of TSLP [IL-7Rα, TSLP receptor (TSLPR)] on those DC (data not shown). To examine the requirements of tumor-derived HMGB1, TSLP, or both, and the corresponding receptors for modulating Treg-activating DC, DC isolated from naïve WT, TSLPR−/−, and TLR4−/− mice were cultured in the presence of tumor-derived HMGB1, TSLP, or both. While neither tumor-derived HMGB1 nor TSLP alone could modulate DC to activate Treg, they worked together to modulate DC to activate Treg (Fig. 3c). In experiments with tumor-derived HMGB1/TSLP-based modulation of DC, TSLPR- but not TLR4-deficient DC lost the capacity to activate Treg (Fig. 3d). These results indicate that tumor-derived HMGB1 cooperates with TSLP to modulate DC to activate Treg in a TSLPR-dependent manner.
Fig. 3.
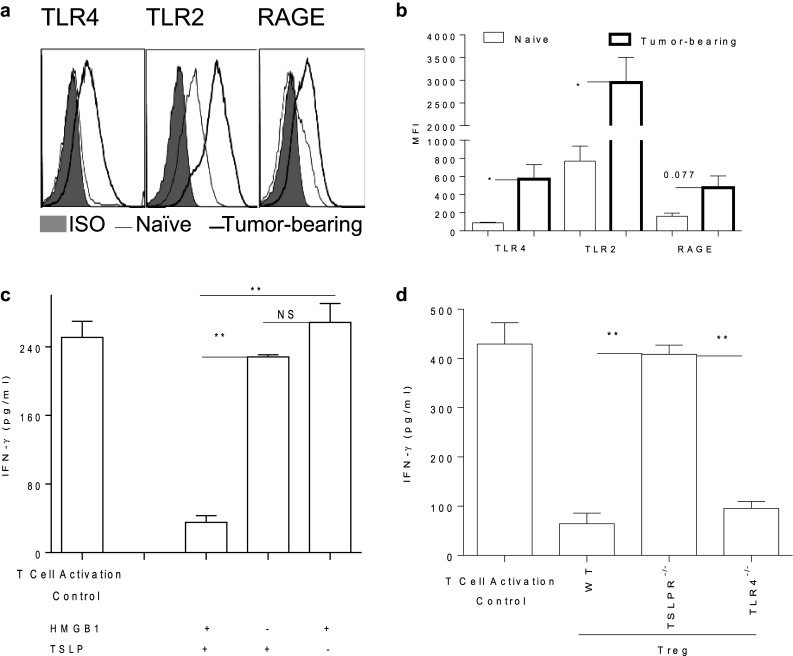
Tumor progression up-regulates the expression of HMGB1 receptors on DC. BALB/c mice were inoculated with 4T1.2-Neu. 10 d later, single-cell suspensions of TDLN or LN from naïve mice were stained with anti-TLR2, -TLR4 or -RAGE or ISO Ab and analyzed by flow cytometry. Data shown the expression of RAGE, TLR2 or TLR4 on gated CD11c+DC are representative of three independent experiments (a). The data of median fluorescence intensity (MFI) from three independent experiments are presented and were statistically analyzed using a Student’s t test (b). Tumor-bearing vs. naïve: p < 0.05 (TLR4, TLR2), p = 0.077 (RAGE). c Both tumor-derived HMGB1 and TSLP are needed to modulate DC to activate Treg. CD11c+DC purified from spleen/LN of naïve BALB/c mice were cultured in the presence or absence of tumor-derived HMGB1, TSLP or HMGB1 + TSLP for 3 d and then cocultured with Treg sorted from naïve BALB/c-Foxp3-GFP mice. 3 d later, the ability of those Treg purified from pooled coculture of DC–Treg to suppress T cell activation was measured. d TSLPR but not TLR4 on DC is required for the HMGB1/TSLP-mediated modulation of Treg-activating DC. Experiments were performed as described (c) except that DC were purified from LN/spleen of naïve BALB/c-WT, -TSLPR−/− or TLR4−/− mice. Data from two independent experiments are shown and were statistically analyzed using a Student’s t test (c, d). HMGB1 + TSLP vs. HMGB1 or TSLP: p < 0.01, HMGB1 vs. TSLP: NS (c); TSLPR−/− vs. WT or TLR4−/−: p < 0.01 (d)
To restore TSLP production in HMGB1 KD tumor cells, TSLP gene was reintroduced into HMGB1-silenced tumor cells (TSLP KI-HMGB1 KD). TSLP KI-HMGB1 KD tumor cells significantly restored their TSLP production (Fig. 4a). To examine the impact of TSLP restoration in the HMGB1 KD tumor cells on tumor immunity, TSLP KI-HMGB1 KD tumor cells were inoculated s.c. into mice. TSLP in HMGB1-silenced tumor cells enhanced the HMGB1 KD-provoked antitumor activity (Fig. 4b), indicating that TSLP in the HMGB1-lacking tumors promotes antitumor responses. The data further suggest an important role of HMGB1 in tumor progression, probably through tumor-derived HMGB1 cooperating with TSLP to modulate Treg-activating DC (Fig. 3c).
Fig. 4.
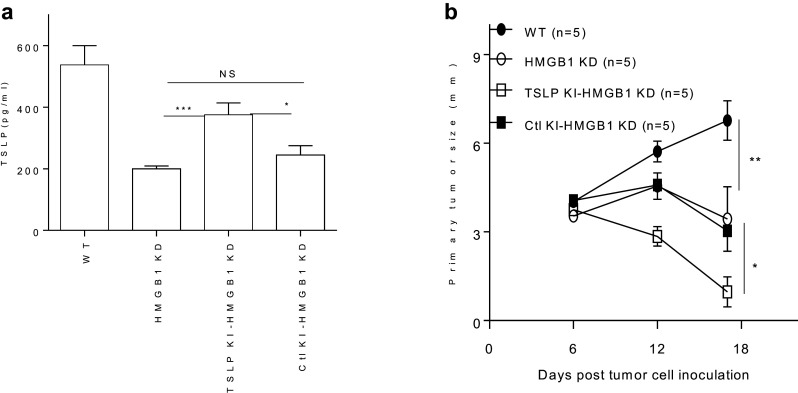
a TSLP KI restores the production of TSLP in HMGB1 KD tumor cells. 4T1.2-Neu-WT, 4T1.2-Neu-HMGB1 KD, TSLP KI-4T1.2-Neu-HMGB1 KD, or Ctl KI-4T1.2-Neu-HMGB1 KD were cultured for 36 h. The concentrations of TSLP in tumor cell supernatants were measured by ELISA. Data from three independent experiments are shown and were statistically analyzed using a Student’s t test. TSLP KI-HMGB1 KD vs. HMGB1 KD: p < 0.001; TSLP KI-HMGB1 KD vs. Ctl KI-HMGB1 KD: p < 0.05; HMGB1 KD vs. Ctl KI-HMGB1 KD: NS. b TSLP in HMGB1 KD tumor cells enhances HMGB1 KD-provoked antitumor activity. BALB/c mice were s.c. inoculated with 4T1.2-Neu-WT, 4T1.2-Neu-HMGB1 KD, TSLP KI-4T1.2-Neu-HMGB1 KD, or Ctl KI-4T1.2-Neu-HMGB1 KD. Tumor growth was monitored. Experiments were performed independently twice and results were statistically analyzed using a Student’s t test. TSLP KI-HMGB1 KD vs. Ctl KI-HMGB1 KD or HMGB1 KD: p < 0.05; WT vs. HMGB1 KD or Ctl KI-HMGB1 KD: p < 0.01
Intratumoral inhibition of HMGB1 (causing downstream loss of TSLP production) attenuates tumor-associated Treg activation, unleashes tumor-specific CD8 T-cell responses, and elicits CD8α+/CD103+DC- and T cell-dependent therapeutic antitumor immunity
Silencing HMGB1 in tumor cells leads to a substantial reduction in extracellular HMGB1 and CD8 T cell-dependent antitumor responses [19]. To determine the effect of intratumorally inhibiting HMGB1 on antitumor responses, mice with established 4T1.2-Neu tumors were injected i.t. with the HMGB1 inhibitor GL [36]. Intratumoral injection of GL did not directly inhibit tumor progression at an early stage shown by comparable tumor growth within 5 d of the 1st treatment (vehicle vs. GL) (Fig. 5a). However, this treatment attenuated tumor-associated Treg activation (Fig. 5b), enhanced tumor-specific CD8 T-cell responses (Fig. 5c), and generated therapeutic antitumor immunity to control tumor growth at later stages and lung metastases (Fig. 5a, Supplementary Fig. 1a). This effective therapeutic antitumor activity of GL was lost in Batf3-deficient, CD4 T cell-deficient, or CD8 T cell-depleted mice (Fig. 5a, d, e). This finding was further supported in a distinct B16 melanoma model (Supplementary Fig. 1b, c).
Fig. 5.
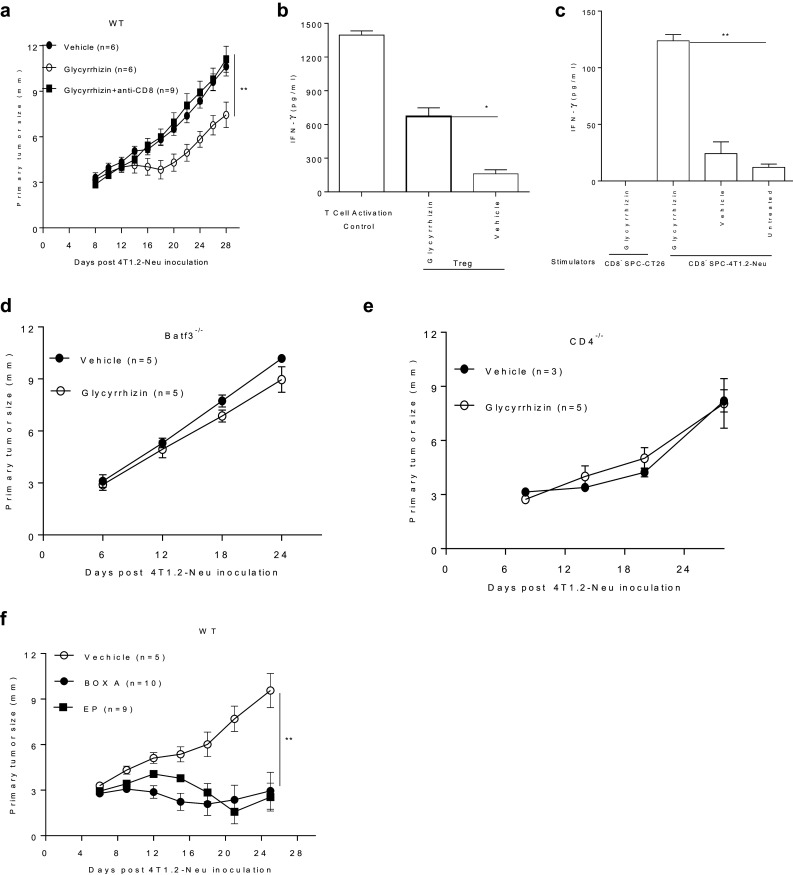
a I.t. inhibiting HMGB1 by GL generates CD8 T cell-dependent antitumor immunity. BALB/c mice were s.c. inoculated with 4T1.2-Neu. On d 8, tumor-bearing mice were i.t. injected daily with GL or vehicle solution for 2 wks. Anti-mouse CD8 Abs were i.p. injected into some tumor-bearing mice on d 7, 9 and 14. Tumor growth was monitored. Data present two independent experiments and were statistically analyzed using a Student’s t test. GL vs. GL + anti-CD8 or vehicle: p < 0.01. b I.t. inhibiting HMGB1 by GL reduces the tumor-associated Treg activation. Tumor-bearing BALB/c-Foxp3-GFP mice were treated as listed (a). D 21, Treg (GFP+) in TDLN were sorted. The ability of those Treg to suppress T cell activation was measured. IFN-γ in culture supernatants were measured by ELISA. Data present three independent experiments and were statistically analyzed using a Student’s t test. GL vs. vehicle: p < 0.05. c I.t. inhibiting HMGB1 by GL induces tumor-specific CD8 T cell responses. Tumor-bearing BALB/c mice were untreated or treated (a). D 28, CD8 T cells purified from spleen were restimulated with 4T1.2-Neu lysates (irrelevant CT26 tumor cell lysates as tumor-specific control)-pulsed CD8− SPC (served as APC) of naïve BALB/c for 3 d. the concentrations of IFN-γ in culture supernatants were measured by ELISA. Data present three independent experiments and were statistically analyzed using a Student’s t test. GL vs. vehicle or untreated: p < 0.01. d, e I.t. inhibiting HMGB1 by GL elicits CD8α+/CD103+DC- and CD4 T cell-dependent antitumor immunity. 4T1.2-Neu-bearing BALB/c-Batf3−/− or -CD4−/− mice were treated with GL or vehicle solution and tumor growth was monitored (a). f I.t. inhibiting HMGB1 by Box A or EP controls tumor growth. BALB/c mice were s.c. inoculated with 4T1.2-Neu. On d 6, tumor-bearing mice were i.t. injected with Box A, EP or vehicle every 3 d for 4 times. Tumor growth was monitored. Data present two independent experiments and were statistically analyzed using a Student’s t test. Box A or EP vs. vehicle: p < 0.01
The potent inhibitor of HMGB1 (i.e., EP), or the antagonist for HMGB1 (i.e., Box A), were demonstrated to control tumor growth [11, 38]. Especially in a similar 4T1 breast tumor model, intraperitoneal injection of Box A inhibits tumor growth [17]. To confirm the antitumor activity provoked by i.t. inhibiting HMGB1, tumor-bearing mice were injected i.t. with EP or Box A. As shown in Fig. 5f and Supplementary Fig. 1d, intratumoral injection of either EP or Box A effectively inhibited the growth of primary tumors and lung metastases. Indeed, inhibiting HMGB1 with i.t. EP or Box A also significantly decreased the levels of TSLP in sera (Fig. 1h).
Taken together, the data suggest that in vivo inhibition of HMGB1 in the TME reduces tumor-associated Treg activation, unleashes tumor-specific CD8 T-cell responses and elicits CD8α+/CD103+DC- and T cell-dependent therapeutic antitumor immune responses.
Discussion
HMGB1 has been documented to play paradoxical roles in tumor models [5–21]. The protumor effect of HMGB1 during tumor progression has been emerging [5, 10–21, 30–43]. HMGB1 could be involved in tumor-associated immune cells, such as DC, MDSC, macrophages, T cells, and Treg in a direct or indirect manner, even though the mechanisms remain elusive [15–21, 42, 43]. HMGB1 acts via multiple pathways and is therefore complex and somewhat controversial [5–21, 42–54]: it is specifically structurally modified, binds to multiple receptors and associates with its ‘partner’ molecules under various physiological or disease conditions.
Silencing HMGB1 expression in tumor cells, or inhibiting tumor-derived HMGB1, either in vitro or in vivo, impeded the capacity of tumor cells to produce TSLP, which could be triggered by tumor-derived HMGB1. Silencing TSLP in tumor cells delays growth and lung metastasis but does not result in tumor-rejection [23]. However, silencing HMGB1 (causing downstream loss of TSLP production) in tumor cells does lead to impaired Treg activation and tumor-rejection [19]. In the current study, TSLP KI in HMGB1 KD tumor cells has no effect on initial growth of the tumor in vivo (just as HMGB1 KD tumor cells), but then suddenly has an effect from d6–10, inducing antitumor immunity. This observation might be explained by the possibility that spontaneous antitumor immune responses have not yet been generated during the initial growth of the tumor (d1–6) after inoculation. TSLP may strengthen the spontaneous antitumor immunity induced by HMGB1 KD tumor cells, leading to the robust inhibition of TSLP KI-HMGB1 KD tumors when compared to HMGB1 KD tumors from d6–10. Indeed, TSLP has been indicated to be associated with antitumor responses [55–57]. However, tumor cell (or other cells, such as myeloid cell)-derived TSLP promotes tumor progression by facilitating tumor-promoting Th2-inducing DC [22–24, 58]. TLR2 is linked to TSLP regulation through transcription factor NF-κB signaling in epithelial cells [59]. Tumor cells express TLR2 (Supplementary Fig. 2). Whether tumor-derived HMGB1 directly acts as an extracellular signal on tumor cells via TLR2 needs further studies. Alternatively, HMGB1 may indirectly induce other signals (e.g., IL-1β) on tumor or other cells including myeloid cells to heighten TSLP production by tumor cells [31, 58].
HMGB1 produced by cells may be oxidized through reactive oxygen species, and oxidized HMGB1 induces immune tolerance [35]. HMGB1 released from dying tumor cells after some therapies triggers antitumor immune responses via TLRs, such as TLR2 or TLR4 on DC [6, 7]. Oxidized HMGB1, which might have altered receptor-binding activity, does not interact with TLR4 [60]. HMGB1 binds with low amounts of TLR ligands including LPS to exert its immune stimulation through TLR including TLR4 [53]. TSLP was shown to induce Treg activation by DC in human thymus [61]. Tumor-derived TSLP alone failed to modulate Treg-activating DC. Both tumor-derived HMGB1 and TSLP were necessary for modulating Treg-activating DC in a TSLPR-dependent manner. HMGB1 easily attaches to other molecules, especially glycosylated proteins. Accumulated data indicate that HMGB1 may bind with its ‘partner’ molecule via the partner molecule receptor to exert its biological function [42–54]. Whether tumor-derived HMGB1 complexes with TSLP to modulate DC via TSLPR, especially under physiological conditions, remains to be determined. Alternatively, tumor-derived HMGB1 may interact with its receptor(s) (e.g., RAGE, TLR2) in cooperation with the TSLP-TSLPR axis on DC, thereby modulating DC to activate Treg. Silencing or inhibiting HMGB1 (causing downstream loss of TSLP production) reduced the capacity of tumor cells to modulate Treg-activating DC, suggesting they are the main players in activating Treg in the TME. The fact that tumors did not regress in CD8α+/CD103+DC-deficient Batf3−/− mice, indicates that CD8α+/CD103+DC may not be responsive to HMGB1/TSLP signaling. The exact DC subset that is modulated by tumor-derived HMGB1/TSLP for the activation of Treg needs to be identified in the future.
Anti-CD4 treatment depletes all CD4 T cells including Treg and CD4 T helper 1 (Th1). The question remains why Treg depletion (via anti-CD4 treatment) had no impact on the antitumor response. One possible explanation is that Th1 depletion impairs the induction of effective antitumor CD8 T cells. Although tumor progression induced the expression of putative receptors of HMGB1 (i.e., TLR4, RAGE, TLR2) on tumor-associated Treg (Supplementary Fig. 3), deficiency of TLR4 did not significantly affect the production of IL-10 by tumor-associated Treg, or the growth of tumors (Supplementary Fig. 4). Tumor-derived HMGB1/TSLP-based modulation of Treg-activating DC was TLR4-independent. The data indicate that TLR4 on Treg and DC may be not the major receptor for tumor-derived HMGB1 in this tumor model.
As a potent inhibitor of HMGB, EP was used to reduce HMGB1 levels as experimental cancer treatments [11, 38]. Box A from HMGB1, an antagonist for HMGB1, was also used to block tumor-derived HMGB1 for antitumor responses [38]. In a similar 4T1 model, intraperitoneal injection of Box A inhibits tumor growth, and tumor-derived HMGB1 is suggested to enhance immune suppression through promoting MDSC [17]. While GL, the specific inhibitor for the activity of extracellular HMGB1 [36], exhibits the ability to suppress multiple cell signaling pathways (e.g., down-regulating transcription factors, growth factors, enzymes, cell-surface adhesion molecules, and chemokines), inhibiting cancer cells (e.g., breast cancer and melanoma) and promoting immune cell (e.g., natural killer cells and T cells) activity against cancer [62–70], results from this study suggest a novel functionality of GL in impeding tumor-derived HMGB1-mediated activation of Treg. Mechanisms of action (e.g., cell death, autophagy and cross-presentation) involved in T cell-dependent antitumor immunity caused by inhibiting HMGB1 i.t. need further investigation. Also, it is worthwhile to examine whether i.t. inhibiting tumor-derived HMGB1 may prevent lung metastases by controlling primary tumor growth. In consideration of the paradigm in which HMGB1 promotes TSLP induction that then develops a feed-forward autocrine loop associated with DC licensing of Treg, HMGB1 inhibition may be an alternative approach, possibly superior to Treg (or MDSC) depletion/antagonism, to prevent Treg and promote Type-1 responses in cancer immunotherapy. HMGB1 inhibitors, such as GL, EP, and Box A, may enhance the efficacy of immunotherapeutic approaches including vaccination or checkpoint blockade.
In summary, this study suggests a new pathway for the activation of Treg involving in tumor-derived HMGB1 and TSLP, and has important implications for incorporating HMGB1 inhibitors into cancer immunotherapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are indebted to Steven F. Ziegler (Benaroya Research Institute) for providing critical experimental materials which were prepared by Whitney Xu at the laboratory of Steven F. Ziegler, Dewayne Falkner at Flow Cytometry Core Facility (University of Pittsburgh) for assisting in flow cytometry and cell sorting, and Stephen C. Balmert at the laboratory of Louis D. Falo, Jr. for editing the manuscript. This work was supported by Department of Dermatology at The University of Pittsburgh School of Medicine and NIH Grants R21CA191522 (Zhaoyang You) and P50CA121973 (Louis D. Falo, Jr.).
Abbreviations
- APC
Antigen-presenting cells
- Box A
An antagonist for HMGB1
- EP
Ethyl pyruvate
- GL
Glycyrrhizin
- HMGB1
High-mobility group box 1
- I.t.
Intratumorally
- KD
Knockdown
- KI
Knockin
- shRNA
Short hairpin RNA
- SPC
Splenocytes
- TDLN
Tumor-draining lymph nodes
- Th1 CD4+
T helper 1
- Th2 CD4+
T helper 2
- TME
Tumor microenvironment
- Treg Foxp3+
T regulatory cells
- TSLP
Thymic stromal lymphopoietin
- TSLPR
TSLP receptor
- WB
Western blot
- WT
Wild type
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Yi Zhang, Zuqiang Liu and Xingxing Hao are contributed equally to this work.
References
- 1.Zou W. Regulatory T cells, tumor immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 2.Beyer M, Schultze JL. Regulatory T cells: major players in the tumor microenvironment. Curr Pharm Des. 2009;15:1879–1892. doi: 10.2174/138161209788453211. [DOI] [PubMed] [Google Scholar]
- 3.Savage PA. Basic principles of tumor-associated regulatory T cell biology. Trends Immunol. 2013;34:33–40. doi: 10.1016/j.it.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28:401–409. doi: 10.1093/intimm/dxw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–2848. doi: 10.1158/1078-0432.CCR-06-1953. [DOI] [PubMed] [Google Scholar]
- 6.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 7.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, Muhammad AK, Clark MC, Arditi M, Comin-Anduix B, Ribas A, Lowenstein PR, Castro MG. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerriero JL, Ditsworth D, Catanzaro JM, Sabino G, Furie MB, Kew RR, Crawford HC, Zong WX. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. 2011;186:3517–3526. doi: 10.4049/jimmunol.1003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gdynia G, Sauer SW, Kopitz J, Fuchs D, Duglova K, Ruppert T, Miller M, Pahl J, Cerwenka A, Enders M, Mairbäurl H, Kamiński MM, Penzel R, Zhang C, Fuller JC, Wade RC, Benner A, Chang-Claude J, Brenner H, Hoffmeister M, Zentgraf H, Schirmacher P, Roth W. The HMGB1 protein induces a metabolic type of tumour cell death by blocking aerobic respiration. Nat Commun. 2016;7:10764. doi: 10.1038/ncomms10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Blockade of RAGE-amphoterin signaling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 11.Liang X, Chavez AR, Schapiro NE, Loughran P, Thorne SH, Amoscato AA, Zeh HJ, Beer-Stolz D, Lotze MT, de Vera ME. Ethyl pyruvate administration inhibits hepatic tumor growth. J Leukoc Biol. 2009;86:599–607. doi: 10.1189/jlb.0908578. [DOI] [PubMed] [Google Scholar]
- 12.Campana L, Bosurgi L, Rovere-Querini P. HMGB1: a two-headed signal regulating tumor progression and immunity. Curr Opin Immunol. 2008;20:518–523. doi: 10.1016/j.coi.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 14.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer Cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusume A, Sasahira T, Luo Y, Isobe M, Nakagawa N, Tatsumoto N, Fujii K, Ohmori H, Kuniyasu H. Suppression of dendritic cells by HMGB1 is associated with lymph node metastasis of human colon cancer. Pathobiology. 2009;76:155–162. doi: 10.1159/000218331. [DOI] [PubMed] [Google Scholar]
- 16.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, Tracey KJ, Ostrand-Rosenberg S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74:5723–5733. doi: 10.1158/0008-5472.CAN-13-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su Z, Ni P, She P, Liu Y, Richard SA, Xu W, Zhu H, Wang J. Bio-HMGB1 from breast cancer contributes to M-MDSC differentiation from bone marrow progenitor cells and facilitates conversion of monocytes into MDSC-like cells. Cancer Immunol Immunother. 2016;66:391–401. doi: 10.1007/s00262-016-1942-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Liu Z, Falo LD, Jr, You Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J Immunol. 2011;187:118–125. doi: 10.4049/jimmunol.1003378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wild CA, Brandau S, Lotfi R, Mattheis S, Gu X, Lang S, Bergmann C. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012;48:409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Wild CA, Bergmann C, Fritz G, Schuler P, Hoffmann TK, Lotfi R, Westendorf A, Brandau S, Lang S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24:485–494. doi: 10.1093/intimm/dxs051. [DOI] [PubMed] [Google Scholar]
- 22.Pedroza-Gonzalez A, Xu K, Wu TC, Aspord C, Tindle S, Marches F, Gallegos M, Burton EC, Savino D, Hori T, Tanaka Y, Zurawski S, Zurawski G, Bover L, Liu YJ, Banchereau J, Palucka AK. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J Exp Med. 2011;208:479–490. doi: 10.1084/jem.20102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olkhanud PB, Rochman Y, Bodogai M, Malchinkhuu E, Wejksza K, Xu M, Gress RE, Hesdorffer C, Leonard WJ, Biragyn A. Thymic stromal lymphopoietin is a key mediator of breast cancer progression. J Immunol. 2011;186:5656–5662. doi: 10.4049/jimmunol.1100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdmann RB, Gartner JG, Leonard WJ, Ellison CA. Lack of functional TSLP receptors mitigates Th2 polarization and the establishment and growth of 4T1 primary breast tumours but has different effects on tumour quantities in the lung and brain. Scand J Immunol. 2013;78:408–418. doi: 10.1111/sji.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo Kuan E, Ziegler SF. Thymic stromal lymphopoietin and cancer. J Immunol. 2014;193:4283–4288. doi: 10.4049/jimmunol.1400864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barooei R, Mahmoudian RA, Abbaszadegan MR, Mansouri A, Gholamin M. Evaluation of thymic stromal lymphopoietin (TSLP) and its correlation with lymphatic metastasis in human gastric cancer. Med Oncol. 2015;32:217. doi: 10.1007/s12032-015-0653-4. [DOI] [PubMed] [Google Scholar]
- 28.De Jesus-Carrion S, Ziegler SF. Control of tumor growth by TSLP during colorectal cancer. J Immunol. 2016;196:73.3. [Google Scholar]
- 29.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11:289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Redhu NS, Gounni AS. Function and mechanisms of TSLP/TSLPR complex in asthma and COPD. Clin Exp Allergy. 2012;42:994–1005. doi: 10.1111/j.1365-2222.2011.03919.x. [DOI] [PubMed] [Google Scholar]
- 31.Wu TC, Xu K, Banchereau J, Palucka AK. Cancer cell-derived transforming growth factor (TGF)-β and myeloid cell-derived interleukin (IL)-1β synergize to promote type 2 inflammation and breast cancer progression. J Immunol. 2016;196:73.8. doi: 10.4049/jimmunol.1501729. [DOI] [Google Scholar]
- 32.Zhang Y, Tian S, Liu Z, Zhang J, Zhang M, Bosenberg MW, Kedl RM, Waldmann TA, Storkus WJ, Falo LD, Jr, You Z. Dendritic cell-derived interleukin-15 is crucial for therapeutic cancer vaccine potency. OncoImmunology. 2014;3:e959321. doi: 10.4161/21624011.2014.959321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, Gyarmati D, Aye T, Campbell DJ, Ziegler SF. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 34.He Y, Zhang J, Mi Z, Robbins P, Falo LD., Jr Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol. 2005;174:3808–3817. doi: 10.4049/jimmunol.174.6.3808. [DOI] [PubMed] [Google Scholar]
- 35.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, Agresti A, Trisciuoglio L, Musco G, Bianchi ME. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14:431–441. doi: 10.1016/j.chembiol.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z, Kim JH, Falo LD, Jr, You Z. Tumor regulatory T cells potently abrogate antitumor immunity. J Immunol. 2009;182:6160–6167. doi: 10.4049/jimmunol.0802664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141:347–357. doi: 10.1016/j.pharmthera.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Yang H, Oceani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaima R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton M, Anderson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: the role of multiligand/rage axis. Carcinogenesis. 2010;31:334–341. doi: 10.1093/carcin/bgp322. [DOI] [PubMed] [Google Scholar]
- 42.He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013;73:629–639. doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 43.Cottone L, Capobianco A, Gualteroni C, Monno A, Raccagni I, Valtorta S, Canu T, Di Tomaso T, Lombardo A, Esposito A, Moresco RM, Maschio AD, Naldini L, Rovere-Querini P, Bianchi ME, Manfredi AA. Leukocytes recruited by tumor-derived HMGB1 sustain peritoneal carcinomatosis. Oncoimmunology. 2016;5:e1122860. doi: 10.1080/2162402X.2015.1122860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Müller S, Iannacone M, Traversari C, Bianchi ME, Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 46.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Bianchi ME, Manfredi AA. High mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 48.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 49.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 50.Kang R, Zhang Q, Zeh HJ, 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both. Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsan MF. Heat shock proteins and high mobility group box 1 protein lack cytokine function. J Leukoc Biol. 2011;89:847–853. doi: 10.1189/jlb.0810471. [DOI] [PubMed] [Google Scholar]
- 52.Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209:551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hreggvidsdóttir HS, Lundberg AM, Aveberger AC, Klevenvall L, Andersson U, Harris HE. High mobility group box protein 1 (HMGB1)-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol Med. 2012;18:224–230. doi: 10.2119/molmed.2011.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Piazza M, Nowell CS, Koch U, Durham AD, Radtke F. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell. 2012;22:479–493. doi: 10.1016/j.ccr.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 56.Demehri S, Turkoz A, Manivasagam S, Yockey LJ, Turkoz M, Kopan R. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell. 2012;22:494–505. doi: 10.1016/j.ccr.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Demehri S, Cunningham TJ, Manivasagam S, Ngo KH, Moradi Tuchayi S, Reddy R, Meyers MA, DeNardo DG, Yokoyama WM. Thymic stromal lymphopoietin blocks early stages of breast carcinogenesis. J Clin Invest. 2016;126:1458–1470. doi: 10.1172/JCI83724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lo Kuan E, Ziegler SF. Thymic stromal lymphopoietinpromotes interplay between breast tumor cells, neutrophils and Ly6Chi monocytes to regulate breast tumor progression. J Immunol. 2016;196:72. doi: 10.4049/jimmunol.1500702. [DOI] [PubMed] [Google Scholar]
- 59.Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NF-κB. Proc Natl Acad Sci USA. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ. Hassall’s corpuscles instruct dendritic cells to induce CD4 + CD25 + regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki F, Schmitt DA, Utsunomiya T, Pollard RB. Stimulation of host resistance against tumors by glycyrrhizin, an active component of licorice roots. In Vivo. 1992;6:589–596. [PubMed] [Google Scholar]
- 63.Hsiang CY, Lai IL, Chao DC, Ho TY. Differential regulation of activator protein 1 activity by glycyrrhizin. Life Sci. 2002;70:1643–1656. doi: 10.1016/S0024-3205(01)01556-9. [DOI] [PubMed] [Google Scholar]
- 64.Kobayashi M, Fujita K, Katakura T, Utsunomiya T, Pollard RB, Suzuki F. Inhibitory effect of glycyrrhizin on experimental pulmonary metastasis in mice inoculated with B16 melanoma. Anticancer Res. 2002;22:4053–4058. [PubMed] [Google Scholar]
- 65.Hibasami H, Iwase H, Yoshioka K, Takahashi H. Glycyrrhizin induces apoptosis in human stomach cancer KATO III and human promyelotic leukemia HL-60 cells. Int J Mol Med. 2005;16:233–236. [PubMed] [Google Scholar]
- 66.Rossi T, Benassi L, Magnoni C, Ruberto AI, Coppi A, Baggio G. Effects of glycyrrhizin on UVB-irradiated melanoma cells. In Vivo. 2005;19:319–322. [PubMed] [Google Scholar]
- 67.Niwa K, Lian Z, Onogi K, Yun W, Tang L, Mori H, Tamaya T. Preventive effects of glycyrrhizin on estrogen-related endometrial carcinogenesis in mice. Oncol Rep. 2007;17:617–622. [PubMed] [Google Scholar]
- 68.Thirugnanam S, Xu L, Ramaswamy K, Gnanasekar M. Glycyrrhizin induces apoptosis in prostate cancer cell lines DU-145 and LNCaP. Oncol Rep. 2008;20:1387–1392. [PubMed] [Google Scholar]
- 69.Raphael TJ, Kuttan G. Effect of naturally occurring triterpenoids ursolic acid and glycyrrhizic acid on the cell-mediated immune responses of metastatic tumor-bearing animals. Immunopharmacol Immunotoxicol. 2008;30:243–255. doi: 10.1080/08923970701675044. [DOI] [PubMed] [Google Scholar]
- 70.Sharma G, Kar S, Palit S, Das PK. 18β-Glycyrrhetinic acid induces apoptosis through modulation of Akt/FOXO3a/Bim pathway in human breast cancer MCF-7 cells. J Cell Physiol. 2012;227:1923–1931. doi: 10.1002/jcp.22920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.