

Abstract
The development of a number of different solid tumours is associated with over-expression of ErbB1, or the epidermal growth factor receptor (EGFR), and this over-expression is often correlated with poor prognosis of patients. Therefore, this receptor tyrosine kinase is considered to be an attractive target for antibody-based therapy. Indeed, antibodies to the EGFR have already proven their value for the treatment of several solid tumours, especially in combination with chemotherapeutic treatment regimens. Variable domains of camelid heavy chain-only antibodies (called Nanobodies™) have superior properties compared with classical antibodies in that they are small, very stable, easy to produce in large quantities and easy to re-format into multi-valent or multi-specific proteins. Furthermore, they can specifically be selected for a desired function by phage antibody display. In this report, we describe the successful selection and the characterisation of antagonistic anti-EGFR Nanobodies. By using a functional selection strategy, Nanobodies that specifically competed for EGF binding to the EGFR were isolated from ‘immune’ phage Nanobody repertoires. The selected antibody fragments were found to efficiently inhibit EGF binding to the EGFR without acting as receptor agonists themselves. In addition, they blocked EGF-mediated signalling and EGF-induced cell proliferation. In an in vivo murine xenograft model, the Nanobodies were effective in delaying the outgrowth of A431-derived solid tumours. This is the first report describing the successful use of untagged Nanobodies for the in vivo treatment of solid tumours. The results show that functional phage antibody selection, coupled to the rational design of Nanobodies, permits the rapid development of novel anti-cancer antibody-based therapeutics.
Keywords: EGFR, Nanobody, Tumour, Therapy, Signalling
Introduction
The epidermal growth factor (EGF) receptor (EGFR, or ErbB1) is a receptor tyrosine kinase (RTK) belonging to a family of four receptors (ErbB1 to 4). The EGFR has an important role in the regulation of growth and differentiation of a large number of different cell types (for review, see [55]). Many models also predict the EGFR to be at the convergence point of several signal transduction pathways: e.g. the proliferative signalling of different G-protein coupled receptors has been shown to be dependent on EGFR [11]. Because of the role of the EGFR in proliferation, cell-survival and angiogenesis, over-expression of the receptor confers advantages to tumour cells at different stages of tumour development (initiation, progression and neo-vascularisation). Indeed, the EGFR is frequently found to be over-expressed in a large number of epithelial tumours, including carcinomas of the head and neck, breast, colon, lung, prostate, kidney, ovary, brain, pancreas and bladder (reviewed in [26]). In addition, this over-expression is often correlated with poor prognosis of patients [52]. For these reasons, the EGFR constitutes an attractive target for cancer therapy. Two approaches are currently being undertaken to inhibit EGFR signalling, i.e. the development of small molecule inhibitors of the intra-cellular tyrosine kinase and the isolation of monoclonal antibodies directed to the extra-cellular domain of the EGFR [38, 45]. In recent years, several antibodies directed to the EGFR, such as Cetuximab (Erbitux) [24, 47], EMD72000 and ABX-EGF [17, 46], have already been successfully introduced into pre-clinical and clinical development.
Heavy-chain antibodies (HcAb’s) have been described in species belonging to the family of camilidae (i.e. Dromedary, Camel and Llama) as a second class of antibodies next to the conventional (four-chain) antibody repertoire [21]. Since these antibodies are composed of two identical heavy chains and are devoid of light chains, their antigen-binding part is composed of only one single immunoglobulin (Ig) variable region (termed VHH, or Nanobody ™). These antigen-specific antibody fragments have many inherent, favourable characteristics, such as high solubility and the capacity to refold after denaturation while retaining their binding capacity [14, 16]. In addition, the genes encoding these fragments can easily be engineered to obtain multi-valent and multi-specific formats, or can be re-cloned as fusion to other (effector) proteins (for review, see [39]). And since these fragments are composed of only a single Ig fold, they do not have the disadvantage of partial unfolding, thereby exposing hydrophobic patches, as have single-chain Fv (scFv) fragments. Furthermore, the absence of protease-sensitive linker sequences makes them more stable than scFv’s.
Ever since the first description of phage display of peptides [50] and of antibody fragments [35], this technique has been successfully used to isolate antibodies against a broad range of antigens (for review, see [28, 29]). Because of the ease of cloning Nanobody-encoding genes, they can easily (and efficiently) be displayed on filamentous phage. Indeed, phage display of Nanobodies has already proven to yield specific antibody fragments directed to both hapten- and protein antigens [1, 32]. Finally, Nanobodies have already been shown to be potent vehicles for the targeting of tumours [10] and for antibody-based therapy of cancer [9]. Therefore, the Nanobody format provides a superior scaffold for the development of antibody-based anti-cancer molecules (for review, see [42]).
In this report, we describe the use of functional phage antibody selection using competitive elution with the ligand EGF to develop antagonistic anti-EGFR Nanobodies for cancer therapy. This selection for function resulted in the isolation of a panel of Nanobodies that inhibited binding of EGF to its receptor without acting as receptor agonists themselves. These Nanobodies performed excellently in vitro in inhibiting EGF-induced signalling and EGF-induced cell proliferation, and they were efficient in inhibiting tumour outgrowth in an in vivo model for solid tumours. These results show the great potential of combining functional phage antibody selection strategies with the favourable characteristics of Nanobodies for the development of antibody-based cancer therapeutics.
Materials and methods
Cell lines
Human epidermoid squamous carcinoma cell line A431 [19], carrying an amplification of the EGFR gene [36], was obtained from the ATCC (cat. nr. CRL-1555). NIH 3T3, clone 2.2 murine fibroblasts were selected for low endogenous EGFR expression [27]. Her14 cells are derived from NIH 3T3 fibroblasts and stably express roughly 105 copies of the human EGFR on their cell surface [27]. The cervix adenocarcinoma cell line Hela was obtained from the ATCC (cat. nr. CCL-2). All cells were cultured in Dulbecco’s Modification of Eagle medium (DMEM: Gibco, Invitrogen, Paisley, UK) containing 7.5% (v/v) foetal calf serum and 2 mM l-glutamine in a humidified atmosphere at 37°C under 5% CO2. Cells were regularly tested for the presence of mycoplasma and consistently found to be mycoplasma-free.
Immunisation of Llama glama with EGFR-containing cell preparations and construction of phage Nanobody repertoires
All animal experiments were conducted with the approval of the Ethical Committee of the Faculty of Veterinary Medicine (University of Ghent, Belgium). To induce a humoral immune response directed towards the EGFR in Llama glama, animals were injected with EGFR-containing cell preparations. Two animals were injected with intact human A431 cells (approximately 108 cells per injection), while A431-derived membrane vesicles (prepared from the same number of cells, according to the method described by Cohen et al. [8]) were administered to two other llamas. Each animal received seven doses of subcutaneously administered antigen at weekly intervals. Pre-immune and immune sera were collected at days 0 (before immunisation), and after 4 and 6 weeks of immunisation. Four days after the last antigen injection, a 150 ml blood sample was collected, and periferal blood lymphocytes (PBLs) were purified by density gradient centrifugation on Ficoll-Paque™ PLUS gradients (Amersham Biosciences, Little Chalfont, UK), resulting in the isolation of approximately 108 PBLs. As an alternative source of B-cells, a small biopsy was taken from the lymph node draining the site of immunisation. Total RNA was extracted from these tissues as described [6] and transcribed into cDNA using an oligo-dT primer and the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA) according to the manufacturers’ recommendations. Next, cDNA was treated with RNAse H to deplete for residual RNA prior to purification with the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). The purified cDNA was then used as template to amplify the repertoire of Ig heavy chain-encoding gene segments with the use of a framework 1 (FR1) specific primer and an oligo-dT primer. This amplification procedure introduces a SfiI restriction site at the 5′ end of FR1 and results in PCR fragments of approximately 1.6 kb (representing conventional IgGs) and fragments of 1.3 kb (comprising heavy-chain IgGs that lack a CH1 domain). The two classes of heavy chain-encoding genes were then size-separated on agarose gels and genes encoding heavy-chain only IgG were purified. Since a BstEII restriction site naturally occurs in approximately 90% of the FR4 of Nanobody genes, the repertoire of PCR-amplified genes was cut with SfiI and BstEII and the resulting 300–400 bp cDNA fragments were purified by gel electrophoresis. cDNA fragments were finally ligated in a phagemid vector for display on filamentous bacteriophage [12] and electro-transformed to Escherichia coli TG1 (K12, Δ(lac-pro), supE, thi, hsdD5/F’traD36, proA + B +, lacIq, lacZΔM15). This resulted in ‘immune’ Nanobody repertoires of approximately 107 transformants each.
Testing pre-immune and immune llama sera for the presence of anti-EGFR antibodies by (cell-)ELISA
Her14 cells and NIH 3T3 clone 2.2 cells were seeded in gelatine-coated (0.25% (w/v) in PBS) 96 wells tissue culture plates at roughly 10,000 cells per well and cultured overnight. The next day, cells were washed with PBS, fixed with 4% (w/v) formaldehyde in PBS for 30 min at room temperature (RT) and non-reacted fixative was quenched with 100 mM glycine in PBS for 10 min at RT. Non-specific binding was prevented by blocking with 2% skimmed milk powder (Marvell) in PBS (2% MBPS) for 30 min at RT and serial dilutions of pre-immune and immune sera were added in 2% MPBS. All further incubations were carried out for 1 h at RT and after every incubation, plates were washed four times with PBS. Detection of bound antibody was performed by incubation with a rabbit anti-llama Ig antiserum (1:1,000 in 2% MPBS) and peroxydase-conjugated donkey anti-rabbit Ig (1:5,000 in 2% MPBS; Jackson Immunoresearch, West Grove, PA, USA). Substrate used was o-phenylenediamine and the OD of the resulting stain was measured at 490 nm. Purified EGFR was purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands) and coated overnight at 4°C at 50 ng/ml in PBS in ELISA plates (Nunc, Rochester, MN, USA). Next day, plates were washed with PBS, blocked with 2% MPBS and the assay was performed as described above.
Selection of antagonistic Nanobody fragments by ligand-specific elution
To select Nanobody fragments that would effectively compete with ligand (EGF) binding to the EGFR, the method of competitive elution [37] was employed. Briefly, A431-derived membrane vesicles were coated to 96 wells immunosorp plates (Costar, Corning, NY, USA) at 5 μg/ml overnight at 4°C. Phage (approximately 1010 colony forming units (cfu)), prepared from the ‘immune’ libraries as described [34], were then panned for binding to immobilised EGFR. After extensive washing with PBS, phage that could compete for EGF binding were eluted by the addition of 1 mM of EGF (Molecular Probes, Invitrogen) for 15 min at RT. Displaced phage were used to infect exponentially growing E. coli TG1 and bacteria were plated on LB agar plates containing 2% (w/v) glucose and 100 μg/ml ampicillin.
Re-cloning, expression and purification of anti-EGFR Nanobody fragments
Expression of recombinant Nanobody fragments in the periplasm of E. coli and purification by means of immobilised metal ion affinity chromatography (IMAC) were performed as has been described for Fab antibody fragments [43]. In order to obtain biotinylated Nanobodies, Nanobody-encoding gene segments were re-cloned as SfiI-BstEII fragments in vector pUR5850 [54]. This vector allows expression of c-Myc- and His6-tagged protein in the periplasmic space of E. coli and it adds a biotinylation sequence (LRSIFEAQKMEW) between the c-Myc and His6 tag. The lysine in this sequence is biotinylated upon expression of the Nanobody in an E. coli strain that over-expresses the BirA gene (AVB101; Avidity, Denver, CO, USA). Bi- and trivalent Nanobody constructs were synthesised essentially as described [15]. However, Nanobody fragments were separated by the N-terminal part of the long hinge region found in IgG3 HcAb’s, followed by three alanine residues encoded by the Not1 site (EPKTPKPQPAAA). To make trivalent, bispecific Nanobody constructs, the construct containing a bivalent Nanobody fragment in vector pAX011 was PstI-digested. After agarose gel electrophoresis and subsequent gel extraction, the 400 bp PstI fragment (corresponding to one Nanobody-encoding gene followed by the linker) was ligated in the PstI site of pAX011 containing the anti-mouse serum albumin (MSA) Nanobody MSA21 (a kind gift of R. Klooster, Utrecht University, The Netherlands). The latter construct was obtained by ligation of MSA21 as a PstI-BstEII fragment in pAX011. This procedure allows cloning of (repeating units of) Nanobody-linker encoding segments amino-terminally to the MSA21 Nanobody.
Immuno-precipitation of EGFR using biotinylated Nanobody
Hela cells were seeded at 800,000 cells per dish in Ø 10 cm tissue culture dishes (Costar, Corning, NY, USA) and grown for 24 h. Total cell lysates were prepared in 750 μl lysis buffer (50 mM Tris–HCl, pH 7.5; 150 mM NaCl; 5 mM Na-EDTA, pH 8.0; 1% (v/v) Triton X100), containing a mix of protease-inhibitors (Complete™, EDTA-free: Roche, Mannheim, Germany) by scraping the cells off the plate. Nuclei were spun down (14,000 rpm, 5 min, 4°C) and an aliquot of the lysate was set apart. Streptavidin-coated agarose beads (50 μl per precipitation; Uptima (Interchim), Montluçon, France) were washed twice with lysis buffer and saturated with biotinylated Nanobody (10 μg per precipitation). Beads were washed again with lysis buffer twice, added to the cellular lysate and incubated for 1 h at 4°C with gentle mixing. Beads were washed four times with lysis buffer and boiled in 2× Laemmli sample buffer [30]. Proteins were then size-separated on 8% (w/v) poly-acrylamide gels (SDS-PAGE), blotted to PVDF membrane (Roche, Mannheim, Germany) and blots were stained for the EGFR as described below.
Inhibition of EGF binding to EGFR by selected Nanobodies and functionality of the trivalent, bispecific format
To determine whether the selected EGFR-specific Nanobodies were able to prevent binding of EGF to the EGFR, a competition ELISA was designed. A431-derived membrane vesicles (5 μg/ml, as determined by the bicinchoninic acid (BCA) protein assay; Perbio Science, Etten-Leur, The Netherlands) were immobilised overnight at 4°C in 96 wells immunosorp plates (Costar, Corning, NY, USA). Plates were washed with PBS containing 0.05% (v/v) Tween-20 and subsequently blocked with PBS containing 1% (w/v) casein for 2 h at RT. After washing, equal volumes of 8 ng/ml (1.6 nM) biotinylated EGF (Peprotech, New York, NY, USA) mixed with serial dilutions of purified soluble Nanobody (in final concentrations of 160 nM to 9 pM) were simultaneously added in PBS containing 0.1% (w/v) casein and incubated for 1 h at room temperature. Receptor-bound EGF-biotin was finally detected with an extravidine-alkaline phosphatase conjugate (Sigma-Aldrich, Zwijndrecht, The Netherlands), followed by staining using para-nitrophenyl phosphate (PNP).
To demonstrate the simultaneous reactivity of the two antigen specificities present in the bispecific, trivalent Nanobody constructs, a sandwich ELISA was performed. Coating of A431 vesicles and blocking were as described above. 10 nM of the trivalent, bispecific molecules was then incubated for an hour at RT in PBS containing 0.1% casein and bound Nanobody was detected with biotinylated MSA (100 nM in PBS containing 0.1% casein) and alkaline phosphatase-coupled extravidin as described earlier.
Staining of different cell lines by fluorescein-labelled Nanobodies using FACS
Purified, recombinant, monovalent Nanobodies (approximately 200 μg per reaction) were labelled with fluorescein using a commercially available fluorescein labelling kit (Roche, Mannheim, Germany). Fluorescence-labelled Nanobody (100 nM) was then used to stain EGFR-expressing (Her-14 and A431) cell lines and the EGFR-negative cell line 3T3 clone 2.2. Staining was performed on live, non-fixed cells for 30 min at 4°C in DMEM. Excess Nanobody was then removed by washing with Dulbecco’s balanced salt solution (four times) and cells were analyzed on a FACSVantage fluorescence-activated cell sorter (Becton & Dickinson, San Jose, CA, USA).
Activation of the EGFR and inhibition of EGF-induced EGFR activation by multi-valent anti-EGFR Nanobodies
Her14 cells (approximately 105 cells per well) were seeded in 12-wells tissue culture clusters (Costar, Corning, NY, USA) and allowed to adhere. After 8 h, cells were rinsed once with DMEM containing 0.1% (v/v) FCS and serum-starved overnight in the same medium. The day of the assay, medium was refreshed with the same medium containing 1% (w/v) BSA and EGF (50 ng/ml, corresponding to approximately 8 nM) or trivalent, bispecific Nanobodies (1 μM) were added at 37°C. After 15 min of incubation, cells were quickly cooled down on ice and washed twice with ice-cold PBS. Total cell lysates were prepared by scraping the cells off the plate in 50 μl 2× Laemmli protein sample buffer and by boiling for 5 min at 100°C. Proteins were size-separated on 8% (w/v) poly-acrylamide gels and blotted to PVDF membrane (Roche, Mannheim, Germany). Blots were then stained for the total amount of EGFR with a rabbit polyclonal antiserum to the receptor (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and for phosphorylated receptor using a mouse monoclonal anti-EGFR phospho-tyrosine 1068 antibody (Cell Signalling, Beverly, MA, USA), followed by the respective peroxidase-conjugated secondary antibodies (donkey anti-rabbit and donkey anti-mouse; Jackson Immunoresearch, West Grove, PA, USA). As a loading control, the lower parts of the blots were stained for actin with a monoclonal anti-actin antibody (ICN Biomedicals, Irvine, CA, USA). Bound antibody was visualised by enhanced chemoluminescence using Western Lightning™ substrate (Perkin Elmer Life Sciences, Wellesley, MA, USA). To measure the capacity of the different bispecific, trivalent anti-EGFR Nanobodies to inhibit EGF-induced signalling, dilutions of Nanobodies (1,000, 100, 10 or 1 nM) were mixed with 8 nM of human EGF and these were added to serum-starved Her-14 cells for 15 min. EGF receptor phosphorylation was then measured by means of Western blotting as described earlier.
Inhibition of EGF-induced cell proliferation by anti-EGFR Nanobodies
Her14 cells were seeded at 1,000 cells/well in 96 wells tissue culture clusters (Costar, Corning, NY, USA) and grown for 2 days to mid-log phase. Medium was then replaced by medium containing 0.1% FCS and cells were serum-starved overnight. Next day (day 0), dilutions of bispecific, trivalent Nanobodies (0, 1, 10, 100 and 1,000 nM) were mixed with 8 nM of EGF in medium containing 0.1% FCS and mixtures were added to the cells in hexa-duplicate. After 4 days of growth, total cellular protein was precipitated by the addition of 5% (w/v) of trichloro-acetic acid (TCA) and stained with sulpho-rhodamine B (SRB) as described [49]. OD was read at 540 nm and the number of cells was measured relative to the number at day 0. In addition, a bio-assay was used in which cell-proliferation was determined by measuring the uptake of radio-active thymidine: A431 cells were seeded at approximately 4,000 cells/well in 100 μl ITS medium (DMEM supplemented with insulin/transferring/selenium; Gibco, Invitrogen) in 96-wells tissue culture clusters and incubated overnight. After 24 h, the medium was refreshed and dilutions of Nanobodies (0, 1, 10, 100 and 1,000 nM) were added together with 1 ng/ml (corresponding to approximately 170 pM) EGF. After 2 days of incubation, cells were pulsed with 1–2 μCi [3H]-thymidine and incubated for an additional 16 h, prior to freezing at −20°C. Cells were subsequently thawed and embedded on glass fiber membranes using a cell harvester (Perkin Elmer Life Sciences, Wellesley, MA, USA). After several washing with MiliQ-grade water, filters were air-dried and counted using a γ-counter (Perkin Elmer Life Sciences).
Efficacy of trivalent, bispecific anti-EGFR Nanobodies in in vivo tumour therapy
Bispecific, trivalent Nanobody fragments were expressed and endotoxin purified. At day 0, 1 day before the start of therapy, the left flank of each mouse was subcutaneously injected with 107 A431 carcinoma cells. Eight female athymic (nude) mice (NMRI: nu/nu) were used per treatment. As a placebo control, mice were treated with PBS; antibody fragments and placebo were administered intra-peritoneally. Animals were treated twice a week at indicated days (1, 4, 8, 11, 15, 18, 22 and 25) with 1 mg of Nanobody per mouse and per injection, independent of body weight. Tumour growth was monitored twice a week, starting at day 4 by measuring the two perpendicular diameters of the tumour with a vernier caliper. When tumour mass exceeded 10% of body weight, mice were sacrificed. Tumour volume (in cm3) was calculated as (π/6)ab 2, where a and b are length and width (in cm) of the tumour, respectively, and a ≥ b. The mean relative tumour volume (RTV) for each group of mice was followed for 37 days.
Results
Induction of a humoral anti-EGFR response in Llama glama
To obtain antagonistic Nanobodies specific for the EGF receptor, we used phage antibody display with competitive elution [37] using the ligand EGF. As an optimally rich source of such antibodies, ‘immune’ repertoires [7] of phage-displayed Nanobodies were constructed. Llama glama were immunised with the human epidermoid carcinoma cell line A431 [19], containing an amplification of the EGFR gene [36] and thus a high expression of the protein (roughly 106 receptor molecules per cell), or with A431-derived membrane vesicles, made according to the method described by Cohen et al. [8]. The induction of a humoral immune response was followed by testing sera of the animals before and after immunisation by (whole cell-) ELISA. Both immunisation strategies resulted in the induction of a specific response towards the EGFR (Fig. 1). When whole, live A431 cells were used for immunisation, the induced antibody response was also directed towards other cell surface-exposed epitopes than the EGFR, witnessed by the reactivity of immune sera with the EGFR-negative cell line 3T3, clone 2.2 (Fig. 1a, b). Immunisation with A431-derived membrane vesicles resulted in a more specific response towards the EGFR (Fig. 1c, d), although the titer of anti-EGFR antibodies in these immune sera seemed to be slightly lower (compare Fig. 1a with c). In addition, immune sera were also reactive with purified EGF receptor, which is shown for immune sera taken from animals that were immunised with whole cells (Fig. 1e). These data clearly demonstrate the successful induction of a humoral immune response towards the EGFR.
Fig. 1.

Immunisation of Llama glama with EGFR-containing cell preparations induces a strong anti-EGFR humoral immune response. The reactivity of pre-immune (day 0: diamonds) and immune sera (day 28: squares and day 42: triangles) of animals immunised with whole, intact A431 cells (a, b, e), or with A431-derived membrane vesicles (c, d) towards A431 cells (a, c), towards EGFR-negative 3T3 2.2 cells (b, d) and towards purified EGF receptor (e) was determined by whole cell ELISA (a–d) or conventional ELISA (e) as described in Materials and methods
Selection and characterisation of antagonistic Nanobodies
Phage Nanobody repertoires were then synthesised by RT-PCR from two different lymphoid sources obtained from immunised animals: peripheral blood lymphocytes (PBLs) and a biopsy from a lymph node draining the site of immunisation. This resulted in libraries of approximately 107 transformants each. These were then panned to immobilised, A431-derived membrane vesicles (coated to microtiter plates) and Nanobodies competing with EGF binding were specifically eluted with a pulse of human EGF. After two rounds of selection, single clones were screened for EGFR reactivity by whole cell-ELISA on EGFR-negative 3T3 2.2 cells and EGFR-expressing A431 or Her14 cells. Approximately 30% of the clones tested were found to react with EGFR-positive cells and not to show reactivity towards EGFR-negative cells (data not shown). DNA fingerprinting using the restriction enzyme Hinf1 was then used to identify possibly different Nanobodies and these were grouped according to their restriction profile. Representative clones from each group were re-tested in ELISA using purified phage. A strong reactivity of Nanobody Ia1 was found towards EGFR-positive cell line Her14, whereas the signal on EGFR-negative cell line 3T3 clone 2.2 was negligible (Fig. 2a). No reaction was observed for control phage that did not express any Nanobody. These results provide proof of the successful isolation of specific anti-EGFR Nanobodies. Because in this ELISA assay the cells were fixed prior to antibody incubation, the selected Nanobodies were also tested for their reactivity with surface-exposed EGFR on non-fixed, living cells by means of FACS staining. FITC was directly conjugated to purified Nanobodies and labelled proteins were tested in FACS staining of EGFR-positive and -negative cells. As was found in whole cell-ELISA, selected antibody fragments were reactive with the EGFR-expressing cell lines, A431 and Her14, and not with the EGFR-negative cell line 3T3 2.2 (shown for Nanobody Ia1 in Fig. 2b). Finally, to prove the EGFR-specificity of the selected Nanobodies by a different method, immuno-precipitations were performed using site-specifically biotinylated Nanobodies and streptavidin-coated agarose beads. The cell lysates prepared from Hela cells that were used for the different immuno-precipitations all contained the same quantity of EGFR (Fig. 2c, left panel). Nanobody Ia1 efficiently precipitated the EGFR, resulting in a strong enrichment of the receptor (Fig. 2c, right panel). Empty streptavidin beads, or beads loaded with a control (anti-GST) Nanobody failed to precipitate the EGFR (Fig. 2c). Results for the different anti-EGFR Nanobodies isolated were similar (data not shown).
Fig. 2.
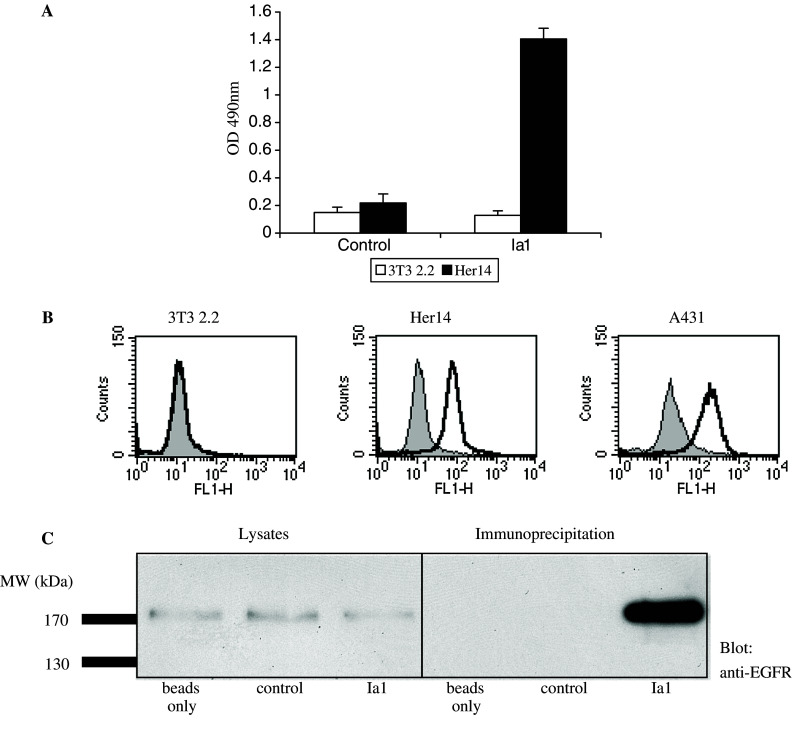
Selected Nanobodies specifically recognise the EGFR. a The binding of anti-EGFR Nanobody Ia1 displayed on phage and of control phage (not expressing any antibody fragment) to EGFR-negative cell line 3T3 2.2 and EGFR-expressing cell line Her14 was determined by cell-ELISA. Error bars indicate the standard deviation of three independent results. b FACS staining of cell lines expressing EGFR (A431 and Her14) and of the EGFR-negative cell line 3T3 2.2 with FITC-coupled anti-EGFR Nanobody Ia1 (black line) or control Nanobody (grey fill). c Immuno-precipitation (IP) of EGFR from Hela cell lysates using biotinylated anti-EGFR Nanobody Ia1. Lanes 1–3 total cell lysates used for IP. Lanes 4–6 IP with streptavidin beads only, with an anti-glutathion S transferase (control) Nanobody and with the anti-EGFR Nanobody Ia1, respectively. Blots were stained for EGFR as described in Materials and methods
Since the obtained Nanobodies were specifically selected for their ability to compete for EGF binding to the EGFR, their potency in inhibiting binding of EGF to the receptor was tested. A431-derived membrane vesicles were immobilised and the binding of biotinylated EGF to immobilised EGFR was detected in the presence of an increasing amount of purified, monovalent Nanobody. The three Nanobodies that most efficiently blocked EGF binding to the EGFR (named Ia1, IIIa3 and L2–3.40; shown in Fig. 3a) were selected for further characterisation. These selected Nanobodies inhibited binding of EGF to the receptor with IC50 values in the low nanomolar range (approximately 5 nM for the Ia1 and L2–3.40 Nanobody and 20 nM for the IIIa3 Nanobody: Fig. 3a). When tested for their epitope-specificity, none of these three Nanobodies competed with the whole monoclonal anti-EGFR antibody Erbitux for binding to the EGFR, but they recognised overlapping epitopes on the receptor, as judged by competition ELISA (data not shown).
Fig. 3.
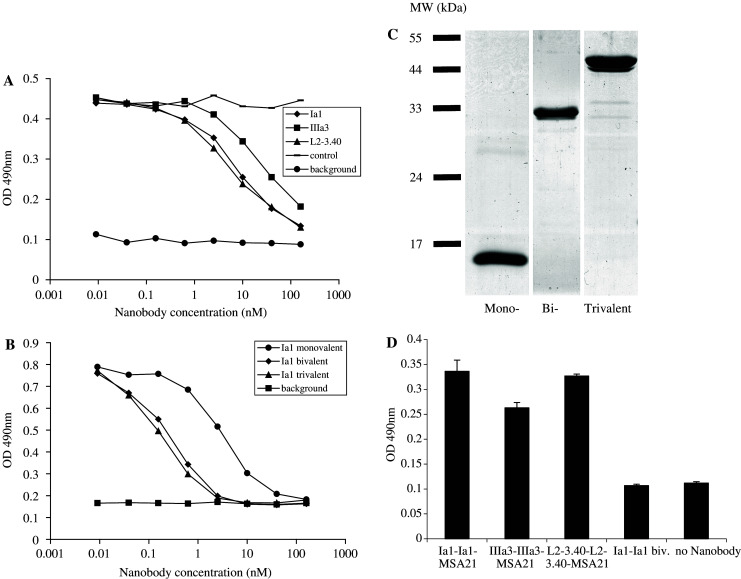
Anti-EGFR Nanobodies compete for EGF binding to the EGFR and trivalent bispecific Nanobodies react simultaneously with EGFR and MSA. a By means of ELISA, the binding of biotinylated EGF to immobilised EGFR was measured in the presence of increasing amounts of a control Nanobody (stripes) or of the anti-EGFR Nanobodies Ia1 (diamonds), IIIa3 (squares) or L2-3.40 (triangles). Background staining was defined as no biotinylated EGF being added (rounds). b Monovalent (rounds), bivalent (diamonds) and trivalent (triangles) variants of anti-EGFR Nanobody Ia1 were tested for their ability to block EGF binding to the EGFR in ELISA. c 3 μg of purified mono-, bi- and trivalent Ia1 Nanobody was size-separated on a 15% poly-acrylamide gel and the gel was stained with coomassie brilliant blue to visualise the proteins. d Trivalent, bispecific Nanobodies (of Ia1, IIIa3 and L2–3.40), or bivalent Ia1 were tested for simultaneous binding to EGFR (coated to the ELISA plate) and biotinylated MSA (used to detect bound Nanobody with alkaline-phosphatase coupled extravidin)
To investigate the effect of increased apparent affinity (through avidity) on the capacity of the selected Nanobodies to block EGF binding to the receptor, bi- and trivalent molecules were synthesised. Two, or three Nanobody-encoding genes were re-cloned in-frame and expressed as a single molecule (Table 1). The different Nanobodies were separated by a flexible sequence: the N-terminal part of the long hinge region found in IgG3 HcAb’s (EPKTPKPQP). Purified, multi-valent proteins (Fig. 3c) were then tested for their potency to block EGF binding to the receptor. The addition of a second antigen-binding site to the same molecule decreased the IC50 value with an order of magnitude, as is shown for the Ia1 Nanobody in Fig. 3b (an IC50 value of approximately 5 nM for monovalent Ia1 and 0.5 nM for the bivalent molecule). However, addition of a third Nanobody to the molecule did not significantly further decrease the IC50 value for inhibition of EGF binding. Similar results were obtained for the IIIa3 and L2–3.40 Nanobodies (data not shown).
Table 1.
Schematic illustration of the different Nanobody-derived constructs

Bio biotinylation tag; Hinge N-terminal part of the long hinge region found in IgG3 HcAb’s; His6 hexahistidine tag; MSA21 cDNA encoding the anti-mouse serum albumin (MSA) Nanobody clone 21; Myc cMyc-derived epitope tag; pLacZ LacZ promoter; B BstEII site; N Not1 site; P Pst1 site; S Sfi1 site
Re-formatting of Nanobodies for in vivo use and in vitro characterisation of these formats
Monovalent Nanobodies have an in vivo half-life (T½) of approximately 1.5 h in blood [10]. Therefore, this Nanobody format (being a 15 kDa protein) is not optimal for the in vivo use in cancer treatment. Even bi- or trivalent molecules (of approximately 30 and 45 kDa, respectively) are below the renal threshold for first-pass clearance and will therefore have a very short in vivo half-life. To increase the in vivo half-life of bivalent anti-EGFR Nanobodies, trivalent, bispecific Nanobody constructs were designed. In these molecules, two antigen-binding sites are directed towards the EGFR and one Nanobody is specific for mouse serum albumin (MSA21; Table 1). These 45–50 kDa proteins were first shown to be able to simultaneously bind to immobilised EGFR and biotinylated MSA in a sandwich-ELISA (Fig. 3d). In addition, their serum half-life was determined and found to be significantly longer than that of a control trimeric Nanobody of the same size (T½ of approximately 44 h, compared to 1 h: G. van Dongen, personal communication).
The ability of these trivalent, bispecific anti-EGFR Nanobodies to inhibit EGF-mediated signalling was subsequently tested: EGF-induced EGFR phosphorylation was measured in the presence or absence of the different Nanobodies. As a measure of mitogenic signalling, the phosphorylation status of tyrosine (Y-)1068 of the EGFR was determined, as this tyrosine is the docking site for Grb2 and its phosphorylation is the initiation of signalling towards Ras [5]. Addition of an increasing amount of the trivalent, bispecific Nanobodies to a constant quantity (8 nM) of EGF could completely block the EGF-induced phosphorylation of Y-1068 of the EGFR (Fig. 4a). This effect was already significant by addition of an equimolar quantity of antibody fragment. Furthermore, when the antibody fragments themselves were used as ligand at high concentration (1 μM), only a marginal phosphorylation of the EGFR Y-1068 could be observed (Fig. 4b), showing that the fragments themselves did not act as receptor agonists.
Fig. 4.

Anti-EGFR Nanobodies block EGF-induced EGFR phosphorylation but do not act as receptor agonists. a EGF (50 ng/ml, corresponding to approximately 8 nM) was mixed with increasing amounts of purified, trivalent Nanobodies and added to serum-starved Her14 cells for 15 min. EGFR phosphorylation was then measured in cell lysates by Western blotting. Upper panel staining for phosphorylated tyrosine 1068 of EGFR; middle panel staining for total quantity of EGFR; lower panel staining for total amount of actin. b EGF (8 nM) or trivalent, bispecific Nanobodies (1 μM) were added to serum-starved Her14 cells for 15 min and EGFR phosphorylation was measured in total cell lysates by Western blotting. Blots were stained as described in a
The next test performed in the development of these antibody fragments for cancer therapy was to measure their effect on EGF-induced cell proliferation. Increasing quantities of the three selected anti-EGFR trivalent Nanobodies were mixed with a constant quantity of EGF and the mitogenic effect of these mixtures was then tested on serum-starved Her14 cells. Both the Ia1 and L2–3.40 Nanobodies inhibited proliferation for more than 80% at high doses (Fig. 5). The IIIa3 Nanobody was less potent, giving a reduction in proliferation of approximately 40% at the highest dose tested (1 μM; Fig. 5b). The observed growth effect was dependent on EGF, since control cells not receiving any EGF stimulation did not significantly proliferate in the time span of the assay (4 days; Fig. 5). Crucial for the application of Nanobodies in cancer therapy is their effect on the growth of tumour cells. This effect was measured using the A431 cell line, both in vitro and in vivo in a murine xenograft model. The effect of the selected Nanobodies on A431 cell proliferation was assessed by measuring [3H]-thymidine incorporation. In the presence of a low concentration of EGF (170 pM), all three trivalent antibody fragments inhibited the growth of A431 cells (Fig. 6a–c). When added in excess, all three Nanobodies could completely block the EGF-induced proliferation of A431 cells, whereas a large excess of a control Nanobody had no significant effect (data not shown). Having the in vitro characteristics of true EGFR antagonistic molecules with suitable in vivo half-lives, these trivalent, bispecific Nanobodies were then tested for their in vivo efficacy in tumour treatment.
Fig. 5.

Trivalent, bispecific Nanobodies inhibit EGF-dependent growth of Her14 cells in vitro. Increasing quantities (0. 1, 10, 100 and 1,000 nM) of purified trivalent Nanobodies were mixed with EGF (8 nM) and added to serum-starved Her14 cells for 4 days. Total cellular protein was then precipitated with tri-chloro acetic acid (TCA) and stained with sulpho-rhodamine B (SRB) as a measure of total cell number. Background proliferation was determined in the absence of EGF. a trivalent Ia1; b trivalent IIIa3 and c trivalent L2–3.40
Fig. 6.
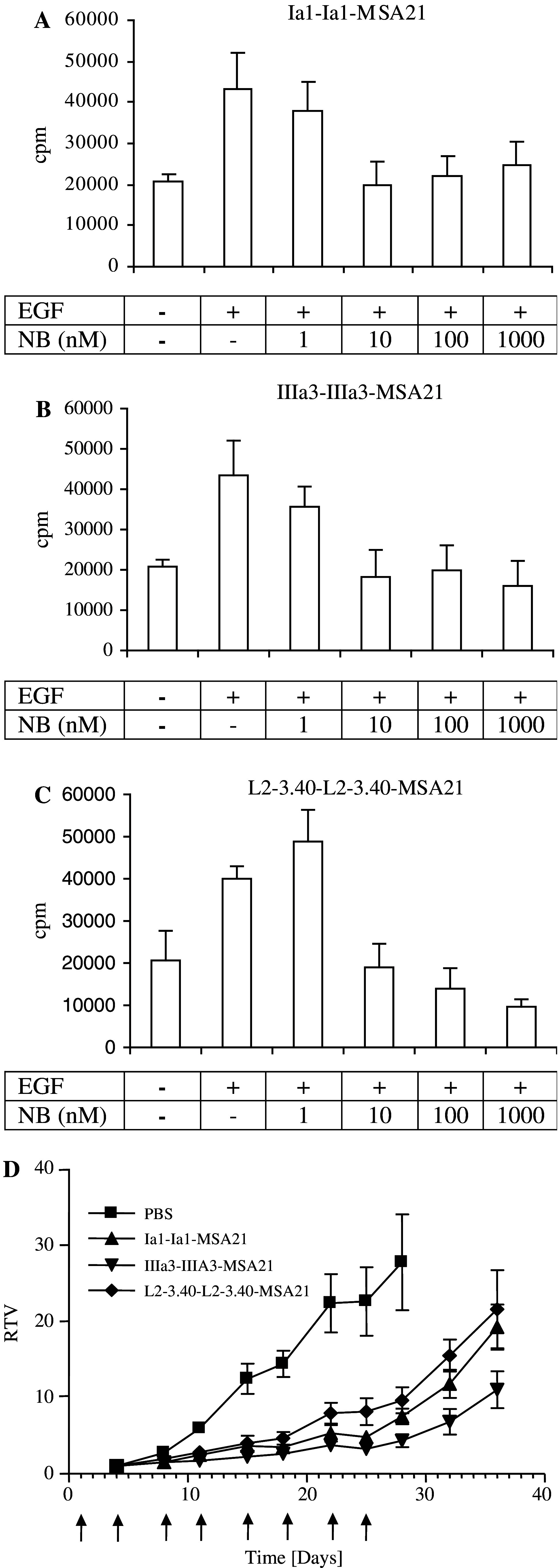
Trivalent, bispecific Nanobodies inhibit A431 tumour cell growth in vitro and growth of A431 xenografts in athymic mice. a–c A431 epidermoid carcinoma cells were grown in serum-free medium supplemented with insulin, transferrin and selenium. After 24 h, medium was refreshed and dilutions (100, 10, 1 and 0 nM) of trivalent Nanobodies (a Ia1; b IIIa3; c L2–3.40) were added, together with 1 ng/ml (approximately 170 pM) of human EGF. After 2 days of culture, cells were pulsed with [3H]- thymidine and assayed 24 h later for the incorporation of radioactivity in genomic DNA. d Groups of 8 mice were subcutaneously injected with A431 cells (107 cells per mouse). One day later, treatment was started by intra-peritoneal injection of 1 mg of purified trimeric Nanobody. Mice were treated twice weekly with the same quantity of Nanobody for up to 4 weeks (indicated by arrows). Relative tumour volume (RTV) was measured as function of time. Squares solvent control (PBS); triangles IaI; inverse triangles IIIa3 and diamonds L2–3.40. Error bars indicate the standard error of the mean of 8 mice
In vivo efficacy of bispecific, trivalent anti-EGFR Nanobodies
To test the effect of the selected anti-EGFR Nanobodies on in vivo tumour development, an efficacy study was performed to determine the inhibition of tumour outgrowth in athymic (nude) mice. Mice were subcutaneously injected with A431 tumour cells and subsequently treated with high doses of antibody (1 mg per mouse per injection, twice a week for 4 weeks). This treatment schedule, in combination with the long in vivo half-life of the antibody fragments, was designed to ensure a continuous high blood level of Nanobody. Treatment with all three Nanobodies resulted in a significant delay of tumour outgrowth (Fig. 6d). Since Nanobody-treatment was abrogated after 4 weeks, most tumours started growing at control rate from that time onwards. However, this was surprisingly not the case for tumours treated with the IIIa3 Nanobody (Fig. 6d). During treatment, there was no statistically significant difference between the different Nanobodies tested. In conclusion, the selected Nanobodies were effective both in vitro and in vivo in inhibiting tumour (cell) growth.
Discussion
In this paper, we report on the successful isolation and the characterisation of a panel of antagonistic anti-EGFR Nanobodies. These antibody fragments were shown to potently inhibit EGF binding to the receptor, to block EGF-induced EGFR signalling and to inhibit EGF-dependent cell proliferation. Importantly, to our knowledge, this is the first report describing the successful in vivo use of unconjugated Nanobodies to inhibit solid tumour outgrowth. These antibody fragments may prove to be useful building blocks for further rational design of anti-cancer therapeutics.
Several reports have already described the isolation of recombinant antibody fragments specific for the EGFR [23, 51]. However, this is the first report describing EGFR-specific Nanobodies. Furthermore, by employing a functional selection approach, EGFR-specific Nanobodies could readily be identified that indeed blocked EGFR signalling by competing for EGF binding. This obviated the need to screen EGFR-specific Nanobodies for their capacity to block EGF binding to the receptor. However, true EGFR antagonism still had to be confirmed (Fig. 4), since the selected Nanobodies could have been able to stimulate the tyrosine kinase activity of the receptor.
To synthesise ‘immune’ Nanobody repertoires for the EGFR, Llama glama were first immunised with EGFR-containing cell extracts. The successful induction of a humoral immune response in the animals was then demonstrated by whole cell-ELISA (Fig. 1). However, this assay detects conventional, as well as HcAb’s. Earlier reports describe significant contributions of HcAb’s to the total antibody response induced by immunisation of camelids, especially for protein antigens [32]. For hapten antigens, however, conflicting data have been reported, ranging from the successful induction of HcAb titers [18] to a complete failure to detect hapten-specific HcAb’s in the serum of immunised animals [31]. Therefore, the serum titers shown in Fig. 1 are merely a strong indication of the successful induction of an HcAb-response. However, when polyclonal phage, prepared from one of the immune phage Nanobody libraries, was tested in an ELISA on purified EGFR, specific binding was observed (data not shown). This means that there probably was a relatively high percentage of anti-EGFR Nanobodies present in the ‘immune’ repertoires, indicative of a strong humoral immune response directed towards the EGFR. Such a strong response is remarkable in view of the low quantity of antigen used per immunisation (108 A431 cells contain roughly 60 μg of EGFR).
To improve the affinity of the selected anti-EGFR Nanobodies, multi-valent formats were synthesised. When these molecules were tested for their potency to block EGF binding to the receptor, bivalent and trivalent molecules seemed almost equally effective, whereas bivalent molecules were significantly better than their monovalent counterparts (Fig. 3). Conflicting data have been obtained when measuring the effect of increased apparent affinity of antibody fragments through the addition of a second antigen-binding site to the same molecule [44, 53]. One explanation for this is that this effect heavily depends on the format of the assay used to determine the apparent affinity. In the assay used to determine the capacity of multi-valent Nanobodies to block EGF binding to the receptor, the density of coated EGFR molecules used might be so low that trivalent binding was spatially impossible. Therefore, trivalency for the EGFR may still prove to be advantageous for in vivo therapy. In addition, the introduction of more flexibility in the trivalent molecules by changing the length and composition of the linker sequence joining two Nanobody molecules may positively affect their therapeutic capacity.
Nanobodies are small proteins and will therefore have a very short half-life in vivo that is not compatible with their use in tumour therapy. To increase the in vivo half-life of small proteins that are below the renal threshold for first-pass clearance, addition of polyethylene glycol (PEG; [48]) has often been used. However, since Nanobodies are very small, there is a considerable danger that such chemical modification may diminish the immuno-reactivity of the molecule [13]. Therefore, a mouse serum albumin-specific Nanobody was linked to the bivalent anti-EGFR Nanobodies to prolong their in vivo half-life. The three ‘heads’ in such a trivalent molecule function independently of each other. This is exemplified by the fact that the IC50 values for EGF competition did not differ between bivalent Nanobodies and their trivalent counterparts containing the anti-MSA Nanobody (data not shown). Indeed, the half-life of these trivalent Nanobodies was significantly longer and much more adequate for treatment purposes. However, this half-life is still less than that of a whole IgG (being 10–14 days).
The immunogenicity of Nanobodies is something that still needs to be thoroughly investigated. This is also important for the pharmacokinetic behaviour of these molecules, as immunogenicity will cause the formation of immune complexes and a diminished half-life in vivo. However, the large sequence homology between Nanobodies and human VH genes of the VH III family [40] indicates that this may not be a major problem. In addition, no B- or T-cell responses have been detected in mice treated with Nanobodies, giving further support to the notion that these molecules may ultimately be safely administered to patients.
An important advantage of this phage display approach is the large number of different anti-EGFR Nanobodies selected. Subsequent selection criteria, based on the in vitro, but also the in vivo characteristics of the molecules are then very important to determine the candidate having the greatest therapeutic potential. For example, it is not always possible to extrapolate in vitro data, e.g. blocking of ligand binding and inhibition of (EGF-dependent) cell growth, to in vivo performance (i.e. inhibition of tumour growth). This is exemplified by the fact that the Nanobody displaying the weakest capacity to block EGF binding to the EGFR (IIIa3; Fig. 3) seemed most potent in inhibition of in vivo tumour growth (Fig. 6d). Whether the observed reduced tumour growth after treatment with Nanobody IIIa3 had been stopped (Fig. 6d) is significant remains to be determined. It is possible that treatment with Nanobody IIIa3 caused an immune response generating immunological memory (e.g. through the induction of an anti-idiotype (Id) cascade). A recent report [2] describes anti-tumour effects of an anti-EGFR whole human monoclonal antibody that are probably mediated by Fc receptor-bearing immune effector cells (ADCC). However, whether these immune effector functions will have important beneficial effects in patients remains to be determined. Indeed, an antibody devoid of effector functions (ABX-EGF, IgG2) has shown promising pre-clinical results [17] giving further support to the notion that blocking EGFR signalling on itself can have potent anti-tumour effects.
Recombinant anti-EGFR antibody fragments have also been used to generate fusion proteins designed to re-direct immune effector cells to tumours [22] or to deliver cytotoxic agents [3, 4, 33] or genes [20] to tumour cells. The observed anti-tumour effects of the Nanobodies described in this report can be solely attributed to their antagonistic mode of action. In addition, they are internalised in target cells over-expressing the receptor (data not shown). Therefore, the re-formatting of these fragments, e.g. as immuno-toxin, may significantly enhance their anti-tumour effect. Because the antibody fragments described here are already effective in delaying tumour outgrowth, we are confident that they will prove to be useful building blocks for the rational design of anti-cancer therapeutics. An especially promising treatment modality may be the combination of chemo- or radio-therapeutic regimens in combination with antibody treatment [25, 41].
In conclusion, the data presented here demonstrate that the selection of Nanobodies for a particular function using antibody phage display is a very powerful tool to quickly generate biologically active antibody-based cancer therapeutics. In addition, the favourable characteristics of Nanobodies for cancer therapy [42] make these molecules ideal candidates for the development of antibody-based immuno-therapeutics.
Acknowledgments
We thank prof. Dr. Guus van Dongen (VUMC, Amsterdam, The Netherlands) for pharmacokinetic data of trivalent, bispecific Nanobody fragments and for critically reading the manuscript. The term Nanobody™ was used with the kind permission of Ablynx N.V.
References
- 1.Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997;414:521. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- 2.Bleeker WK, van Lammerts Bueren JJ, van Ojik HH, Gerritsen AF, Pluyter M, Houtkamp M, Halk E, Goldstein J, Schuurman J, van Dijk MA, van de Winkel JG, Parren PW. Dual mode of action of a human anti-epidermal growth factor receptor monoclonal antibody for cancer therapy. J Immunol. 2004;173:4699. doi: 10.4049/jimmunol.173.7.4699. [DOI] [PubMed] [Google Scholar]
- 3.Bremer E, Samplonius DF, van Genne L, Dijkstra MH, Kroesen BJ, de Leij LF, Helfrich W. Simultaneous inhibition of EGFR signaling and enhanced activation of TRAIL-R-mediated apoptosis induction by an scFv:sTRAIL fusion protein with specificity for human EGFR. J Biol Chem. 2005;280(11):10025–10033. doi: 10.1074/jbc.M413673200. [DOI] [PubMed] [Google Scholar]
- 4.Bruell D, Stocker M, Huhn M, Redding N, Kupper M, Schumacher P, Paetz A, Bruns CJ, Haisma HJ, Fischer R, Finnern R, Barth S. The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA’ suppresses growth of a highly metastatic pancreatic carcinoma cell line. Int J Oncol. 2003;23:1179. doi: 10.3892/ijo.23.4.1179. [DOI] [PubMed] [Google Scholar]
- 5.Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 1993;73:611. doi: 10.1016/0092-8674(93)90146-H. [DOI] [PubMed] [Google Scholar]
- 6.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 7.Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624. doi: 10.1038/352624a0. [DOI] [PubMed] [Google Scholar]
- 8.Cohen S, Ushiro H, Stoscheck C, Chinkers M. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J Biol Chem. 1982;257:1523. [PubMed] [Google Scholar]
- 9.Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, Revets H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64:2853. doi: 10.1158/0008-5472.CAN-03-3935. [DOI] [PubMed] [Google Scholar]
- 10.Cortez-Retamozo V, Lauwereys M, Hassanzadeh Gh G, Gobert M, Conrath K, Muyldermans S, De Baetselier P, Revets H. Efficient tumor targeting by single-domain antibody fragments of camels. Int J Cancer. 2002;98:456. doi: 10.1002/ijc.10212. [DOI] [PubMed] [Google Scholar]
- 11.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 12.De Haard HJ, Bezemer S, Ledeboer AM, Muller WH, Boender PJ, Moineau S, Coppelmans MC, Verkleij AJ, Frenken LG, Verrips CT. Llama antibodies against a lactococcal protein located at the tip of the phage tail prevent phage infection. J Bacteriol. 2005;187:4531. doi: 10.1128/JB.187.13.4531-4541.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgado C, Pedley RB, Herraez A, Boden R, Boden JA, Keep PA, Chester KA, Fisher D, Begent RH, Francis GE. Enhanced tumour specificity of an anti-carcinoembrionic antigen Fab’ fragment by poly(ethylene glycol) (PEG) modification. Br J Cancer. 1996;73:175. doi: 10.1038/bjc.1996.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolk E, van Vliet C, Perez JM, Vriend G, Darbon H, Ferrat G, Cambillau C, Frenken LG, Verrips T. Induced refolding of a temperature denatured llama heavy-chain antibody fragment by its antigen. Proteins. 2005;59:555. doi: 10.1002/prot.20378. [DOI] [PubMed] [Google Scholar]
- 15.Els Conrath K, Lauwereys M, Wyns L, Muyldermans S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J Biol Chem. 2001;276:7346. doi: 10.1074/jbc.M007734200. [DOI] [PubMed] [Google Scholar]
- 16.Ewert S, Cambillau C, Conrath K, Pluckthun A. Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry. 2002;41:3628. doi: 10.1021/bi011239a. [DOI] [PubMed] [Google Scholar]
- 17.Foon KA, Yang XD, Weiner LM, Belldegrun AS, Figlin RA, Crawford J, Rowinsky EK, Dutcher JP, Vogelzang NJ, Gollub J, Thompson JA, Schwartz G, Bukowski RM, Roskos LK, Schwab GM. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys. 2004;58:984. doi: 10.1016/j.ijrobp.2003.09.098. [DOI] [PubMed] [Google Scholar]
- 18.Frenken LG, van der Linden RH, Hermans PW, Bos JW, Ruuls RC, de Geus B, Verrips CT. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae . J Biotechnol. 2000;78:11. doi: 10.1016/S0168-1656(99)00228-X. [DOI] [PubMed] [Google Scholar]
- 19.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 20.Haisma HJ, Grill J, Curiel DT, Hoogeland S, van Beusechem VW, Pinedo HM, Gerritsen WR. Targeting of adenoviral vectors through a bispecific single-chain antibody. Cancer Gene Ther. 2000;7:901. doi: 10.1038/sj.cgt.7700198. [DOI] [PubMed] [Google Scholar]
- 21.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi H, Asano R, Tsumoto K, Katayose Y, Suzuki M, Unno M, Kodama H, Takemura S, Yoshida H, Makabe K, Imai K, Matsuno S, Kumagai I, Kudo T. A highly effective and stable bispecific diabody for cancer immunotherapy: cure of xenografted tumors by bispecific diabody and T-LAK cells. Cancer Immunol Immunother. 2004;53:497. doi: 10.1007/s00262-003-0465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heitner T, Moor A, Garrison JL, Marks C, Hasan T, Marks JD. Selection of cell binding and internalizing epidermal growth factor receptor antibodies from a phage display library. J Immunol Methods. 2001;248:17. doi: 10.1016/S0022-1759(00)00340-9. [DOI] [PubMed] [Google Scholar]
- 24.Herbst RS, Arquette M, Shin DM, Dicke K, Vokes EE, Azarnia N, Hong WK, Kies MS. Phase II multicenter study of the epidermal growth factor receptor antibody Cetuximab and Cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23:5578. doi: 10.1200/JCO.2005.07.120. [DOI] [PubMed] [Google Scholar]
- 25.Herbst RS, Langer CJ. Epidermal growth factor receptors as a target for cancer treatment: the emerging role of IMC-C225 in the treatment of lung and head and neck cancers. Semin Oncol. 2002;29:27. doi: 10.1053/sonc.2002.31525. [DOI] [PubMed] [Google Scholar]
- 26.Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99. doi: 10.1016/S0014-4827(02)00099-X. [DOI] [PubMed] [Google Scholar]
- 27.Honegger AM, Dull TJ, Felder S, Van Obberghen E, Bellot F, Szapary D, Schmidt A, Ullrich A, Schlessinger J. Point mutation at the ATP binding site of EGF receptor abolishes protein-tyrosine kinase activity and alters cellular routing. Cell. 1987;51:199. doi: 10.1016/0092-8674(87)90147-4. [DOI] [PubMed] [Google Scholar]
- 28.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 29.Hoogenboom HR, de Bruine AP, Hufton SE, Hoet RM, Arends JW, Roovers RC. Antibody phage display technology and its applications. Immunotechnology. 1998;4:1. doi: 10.1016/S1380-2933(98)00007-4. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Lange IG, Daxenberger A, Meyer HH. Studies on the antibody response of Lama glama—evaluation of the binding capacity of different IgG subtypes in ELISAs for clenbuterol and BSA. Vet Immunol Immunopathol. 2001;83:1. doi: 10.1016/S0165-2427(01)00376-2. [DOI] [PubMed] [Google Scholar]
- 32.Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Holzer W, De Genst E, Wyns L, Muyldermans S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. Embo J. 1998;17:3512. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003;63:3154. [PubMed] [Google Scholar]
- 34.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581. doi: 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 35.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 36.Merlino GT, Xu YH, Ishii S, Clark AJ, Semba K, Toyoshima K, Yamamoto T, Pastan I. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science. 1984;224:417. doi: 10.1126/science.6200934. [DOI] [PubMed] [Google Scholar]
- 37.Meulemans EV, Slobbe R, Wasterval P, Ramaekers FC, van Eys GJ. Selection of phage-displayed antibodies specific for a cytoskeletal antigen by competitive elution with a monoclonal antibody. J Mol Biol. 1994;244:353. doi: 10.1006/jmbi.1994.1735. [DOI] [PubMed] [Google Scholar]
- 38.Mosesson Y, Yarden Y. Oncogenic growth factor receptors: implications for signal transduction therapy. Semin Cancer Biol. 2004;14:262. doi: 10.1016/j.semcancer.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 2001;74:277. doi: 10.1016/s1389-0352(01)00021-6. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen VK, Hamers R, Wyns L, Muyldermans S. Camel heavy-chain antibodies: diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. Embo J. 2000;19:921. doi: 10.1093/emboj/19.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raben D, Helfrich B, Chan DC, Ciardiello F, Zhao L, Franklin W, Baron AE, Zeng C, Johnson TK, Bunn PA., Jr The effects of cetuximab alone and in combination with radiation and/or chemotherapy in lung cancer. Clin Cancer Res. 2005;11:795. [PubMed] [Google Scholar]
- 42.Revets H, De Baetselier P, Muyldermans S. Nanobodies as novel agents for cancer therapy. Expert Opin Biol Ther. 2005;5:111. doi: 10.1517/14712598.5.1.111. [DOI] [PubMed] [Google Scholar]
- 43.Roovers RC, Henderikx P, Helfrich W, van der Linden E, Reurs A, de Bruine AP, Arends JW, de Leij L, Hoogenboom HR. High-affinity recombinant phage antibodies to the pan-carcinoma marker epithelial glycoprotein-2 for tumour targeting. Br J Cancer. 1998;78:1407. doi: 10.1038/bjc.1998.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roovers RC, van der Linden E, de Bruine AP, Arends JW, Hoogenboom HR. In vitro characterisation of a monovalent and bivalent form of a fully human anti Ep-CAM phage antibody. Cancer Immunol Immunother. 2001;50:51. doi: 10.1007/s002620000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowinsky EK. The erbB family: targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Annu Rev Med. 2004;55:433. doi: 10.1146/annurev.med.55.091902.104433. [DOI] [PubMed] [Google Scholar]
- 46.Rowinsky EK, Schwartz GH, Gollob JA, Thompson JA, Vogelzang NJ, Figlin R, Bukowski R, Haas N, Lockbaum P, Li YP, Arends R, Foon KA, Schwab G, Dutcher J. Safety, pharmacokinetics, and activity of ABX-EGF, a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol. 2004;22:3003. doi: 10.1200/JCO.2004.11.061. [DOI] [PubMed] [Google Scholar]
- 47.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 48.Savoca KV, Abuchowski A, van Es T, Davis FF, Palczuk NC. Preparation of a non-immunogenic arginase by the covalent attachment of polyethylene glycol. Biochim Biophys Acta. 1979;578:47. doi: 10.1016/0005-2795(79)90111-9. [DOI] [PubMed] [Google Scholar]
- 49.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 50.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 51.Souriau C, Rothacker J, Hoogenboom HR, Nice E. Human antibody fragments specific for the epidermal growth factor receptor selected from large non-immunised phage display libraries. Growth Factors. 2004;22:185. doi: 10.1080/08977190412331279872. [DOI] [PubMed] [Google Scholar]
- 52.Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Benamouzig R, Attar A, Benichou J, Martin A, Morere JF, Raphael M, Penault-Llorca F, Breau JL, Fagard R, Khayat D, Wind P. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16:102. doi: 10.1093/annonc/mdi006. [DOI] [PubMed] [Google Scholar]
- 53.Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods. 2001;248:47. doi: 10.1016/S0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 54.Verheesen P, ten Haaft MR, Lindner N, Verrips CT, de Haard JJ. Beneficial properties of single-domain antibody fragments for application in immunoaffinity purification and immuno-perfusion chromatography. Biochim Biophys Acta. 2003;1624:21. doi: 10.1016/j.bbagen.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 55.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]