

Abstract
This study focused on the question of how monocyte-derived dendritic cells (Mo-DCs) that capture dead tumor cells (Mo-DCs-Tum) secrete interleukin 12 (IL-12) and tumor necrosis factor α (TNF-α). Mo-DCs-Tum showed higher secretions of IL-12 and TNF-α than were shown by Mo-DCs. Enhanced nuclear factor-kappa B (NF-κB) activation was also induced in Mo-DCs-Tum within 6 h. The NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), suppressed both IL-12 and TNF-α secretions from Mo-DCs-Tum. Administration of recombinant TNF-α or IL-12 enhanced IL-12 or TNF-α secretion respectively in Mo-DCs-Tum. Addition of anti-TNF-α or anti-IL-12 neutralizing antibody decreased NF-κB activation and IL-12 or TNF-α secretion in Mo-DCs-Tum. These results suggest that TNF-α or IL-12 secretion induces NF-κB activation, and it stimulates further TNF-α and IL-12 secretions, i.e., an IL-12/TNF-α/NF-κB autocrine loop, in Mo-DCs-Tum. Thus, Mo-DCs-Tum secrete a large amount of IL-12 and TNF-α through accelerated NF-κB activation induced by the IL-12/TNF-α/NF-κB autocrine loop.
Keywords: Dendritic cells, Human, Transcription factors, Tumor immunity
Introduction
Dendritic cells (DCs) play a pivotal role in T and B cell–mediated immune responses [3, 5]. Recent advances in biotechnology have made it possible to generate monocyte-derived DC-like cells (Mo-DCs) in vitro from peripheral blood monocytes with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin 4 (IL-4) [23]. Clinical trials of Mo-DCs pulsed with autologous tumor cells, i.e., Mo-DC–based immunotherapies, are underway for patients with a variety of malignancies including malignant melanoma, B-cell lymphoma, renal cell carcinoma, gastrointestinal carcinoma, and thyroid carcinoma [9, 13, 16, 22, 24]. However, Mo-DC–based tumor immunotherapies have had limited success. One reason is that the level of specific antigen-MHC complexes on tumors may be low and thus only a few T-cell clones that recognize tumor antigens may be activated. It has been shown that Mo-DCs which capture necrotic tumor cells (N) or apoptotic tumor cells (A) become mature, i.e., mature Mo-DCs, and they produce tumor necrosis factor α (TNF-α) and interleukin 12 (IL-12) [3]. Efficient secretion of IL-12 from Mo-DCs is helpful to overcome this problem because Th1-type helper T cells are preferentially induced by IL-12 and play an important role in the antitumor immune response [6, 8]. Unfortunately, IL-12 secretion of Mo-DCs generated from patients with advanced cancer is weaker than that of Mo-DCs from healthy volunteers [18]. These findings indicate that we should prepare Mo-DCs which secrete larger amounts of IL-12 as DC-vaccine source. However, details regarding the molecular mechanism of how Mo-DCs that capture dead tumor cells (Mo-DCs-Tum) secrete IL-12 remain unclear.
Nuclear factor-kappa B (NF-κB) is responsible for maturation of DCs and is a major regulator of the antigen-presenting function of DCs [21]. In resting cells, NF-κB is located in the cytoplasm as heterodimers of the structurally related proteins p50, p52, RelA, c-Rel, and RelB, both of which are noncovalently associated with the cytoplasmic inhibitor, inhibitory NF-κB (IκB) [17, 25]. Activation of NF-κB is preceded by phosphorylation of IκB by IκB kinase, which is followed by proteolytic removal of IκB and movement of NF-κB to the nucleus. Nuclear translocation of NF-κB is thought to reflect activation of NF-κB [1]. After translocation to the nucleus, NF-κB stimulates transcription of cytokines such as TNF-α and IL-12 [7, 20].
In the present study, we focused on the IL-12 and TNF-α secretions from Mo-DCs-Tum and their molecular mechanism.
Materials and methods
Generation of Mo-DCs
Cultures of human peripheral blood mononuclear cells (PBMCs) were maintained in RPMI 1640 (Sanko Pure Chemicals, Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS; Filtron Pty, Brooklyn, Victoria, Australia), 100 U/ml penicillin (Meijiseika, Tokyo, Japan), and 100 μg/ml of streptomycin (Meijiseika) (hereafter referred to as RPMI medium). Mo-DCs were generated from the adherent fraction of PBMCs as described previously [18] but with minor modifications. In brief, PBMCs were isolated from heparinized peripheral blood from healthy volunteers by Ficol-Paque (Life Technologies, Gaithersburg, MD, USA) density gradient centrifugation. PBMCs were resuspended in RPMI medium, plated at a density of 2×106 cells/ml, and allowed to adhere in 24-well culture plates (Nalge Nunk International, Chiba, Japan). After an overnight incubation at 37°C, the nonadherent cells were removed, and the adherent cells were harvested and cultured in RPMI medium supplemented with GM-CSF (200 ng/ml; Genetech, China) and IL-4 (500 U/ml; Osteogenetics, Wuerzburg, Germany). On day 6, nonadherent cell fractions were collected as immature Mo-DCs and examined. In the present study, Mo-DCs from ten healthy donors were prepared and used.
Induction of apoptosis and necrosis in tumor cells
A human gastric carcinoma cell line, GCTM-1, was established in our laboratory and maintained in RPMI medium [15]. Complete disruption of the necrotic cells into fragments was confirmed by light microscopy after five cycles of freezing with liquid nitrogen and thawing at 37°C. UV-triggered apoptosis of tumor cells was induced by 2 mJ/cm2 of irradiation with a 120-mJ UVB lamp (Amersham Biosciences, Piscataway, NJ, USA). Eight hours after irradiation, induction of apoptosis was confirmed by double staining with Hoechst 33342 (Wako Pure Chemical, Osaka, Japan) and propidium iodide (PI; Sigma, St Louis, MO, USA). Under fluorescence microscopy, PI-negative and Hoechst 33432-positive cells with characteristically condensed or fragmented nuclei were defined as being apoptotic. More than 99% of UV-irradiated GCTM-1 cells had the PI-negative and Hoechst 33432-positive phenotype and typical apoptotic morphology. PI-positive cells were defined as secondary necrotic cells. Necrosis- or apoptosis-induced tumor cell cultures were centrifuged at 6,000 g for 10 min, the supernatant was removed, and the pellets were saved. No contamination of LPS into dead tumor cell fraction was confirmed with Limulus amebocyte lysate test (Wako, Tokyo, Japan).
Capture of dead tumor cells by Mo-DCs
Apoptotic or necrotic tumor cells were incubated with Mo-DCs at a cell ratio of 1:1 at 4°C or 37°C. Four hours after incubation, cells were collected, washed with posphate-buffered saline (PBS), and stained with Giemsa (Kokusaishinyaku, Kobe, Japan) to determine the number of Mo-DCs-Tum that captured N (Mo-DCs-Tum-N) or A (Mo-DCs-Tum-A). One hundred Mo-DCs were examined under a light microscope at ×400 magnification, and the percentage of Mo-DCs-Tum (%Mo-DCs-Tum) was calculated. Alternatively, the membrane components of N or A tumor cells were labeled with the PKH 26 red fluorescent cell linker kit (Sigma), and Mo-DCs were labeled with the PKH 67 green fluorescent cell linker kit (Sigma) according to the manufacturer’s protocol. Fluorescence-labeled Mo-DCs and dead tumor cells were cocultured at an original cell ratio of 1:1 for 4 h at 4°C or 37°C, washed, and then applied to a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The fluorescence intensity was analyzed with CELLQuest software (Becton Dickinson). PKH 26-positive cells in gated Mo-DCs populations were defined as Mo-DCs-Tum-A or Mo-DCs-Tum-N.
Cytokine secretions by Mo-DCs
Ten thousand Mo-DCs and dead tumor cells obtained from 1×104 GCTM-1 cells were suspended in 200 μl of RPMI 1640 containing 1% human albumin. Alternatively, these cell mixtures were incubated with or without 1 μg/ml of anti-TNF-α neutralizing IgG (R&D Systems), 200 U/ml of recombinant human TNF-α (Dainippon Pharmaceutical, Osaka, Japan), 20 μg/ml of anti-IL-12 neutralizing IgG (R&D Systems), 100 μg/ml of recombinant IL-12 (Dainippon Pharmaceutical) or 100 μM of pyrrolidine dithiocarbamate (PDTC, Sigma). Twenty-four hours after the initial culture, cell-free supernatants were collected by centrifugation and stored at −80°C until use. The concentrations of IL-12 p40, IL-12 p70, and TNF-α in the culture supernatants were measured by ELISA kit specific for IL-12 p40, IL-12 p70, or TNF-α (Biosource, Camarillo, CA, USA). The detection limit of each kit was 10 pg/ml.
Preparation of nuclear extract of Mo-DCs
Monocyte-derived dendritic cells cocultured with dead GCTM-1 cells for various durations with or without various agents, including anti-TNF-α neutralizing mAb, anti-IL-12 neutralizing mAb, or PDTC, were collected and washed once with PBS. Cells were then homogenized in 400 μl of hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.1% Nonidet P-40, and 5% protease inhibitor cocktail [0.2 mM DTT, 10 mM benzamidine, 7 μg/ml leupeptin, 50 μg/ml soybean trypsin inhibitor, 2 μg/ml aprotinin, 2 μg/ml antipain, 0.7 μg/ml pepstatin, 0.5 mM phenylmethylsulfonyl fluoride, and 0.5 mM 4-(2-aminoethyl) benzenesulfonylfluoride] [Sigma]), and then incubated for 10 min on ice. Nuclei collected by centrifugation at 800 g for 5 min were washed once with 200 μl of hypotonic buffer and resuspended in 20 μl of low-salt buffer (20 mM HEPES [pH 7.9], 0.02 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, and protease inhibitor cocktail). An equal volume of high-salt buffer (same composition as low-salt buffer but containing 800 mM KCl) was added with vortex mixing. Nuclei were incubated for 30 min at 4°C and centrifuged at 18,000 g for 30 min, and the supernatants were collected.
Electrophoretic mobility shift assay (EMSA)
Nuclear protein extracts of Mo-DCs were analyzed by electrophoretic mobility shift assay (EMSA) for NF-κB nuclear translocation as described previously [11]. Briefly, nuclear protein extracts of 1×106 cells were incubated for 30 min at 37°C with binding buffer (60 mM HEPES [pH 7.5], 180 mM KCl, 15 mM MgCl2, 0.6 mM EDTA, and 24% glycerol), poly (dI-dC) (Amersham Pharmacia Biotech AB, Uppsala, Sweden), and 32 P-labeled double-stranded oligonucleotide containing the binding motif of NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) (Promega, Madison, WI, USA). These mixtures were loaded onto a 4% polyacrylamide gel and separated by electrophoresis in 0.25×TBE running buffer. The oligomerprotein complexes were visualized by autoradiography. The intensity of the NF-κB band was estimated with the use of NIH Image version 1.60 software (NIH Division of Computer Research and Technology, Bethesda, MD, USA).
Statistical analysis
The Fisher exact probability test was used for statistical analyses. Calculations were carried out with StatView software (Abacus Concepts, Berkeley, CA, USA). All results with a p value less than 0.05 were considered statistically significant.
Results
Mo-DCs capture dead tumor cells
Capture of dead tumor cells by Mo-DCs was essentially determined with Giemsa staining. When the ratio of Mo-DCs-Tum to total Mo-DCs (%Mo-DCs-Tum) was calculated with a light microscope as described in “Materials and methods,” %Mo-DCs-Tum was not affected by cellular death pattern, i.e., necrosis or apoptosis, of tumor cells (Fig. 1a). The data are representative of three independent experiments using Mo-DCs generated from ten different donors. Since evaluation with microscopy may be dependent on the investigator’s subjective response, %Mo-DCs-Tum was also examined with a flow cytometer. As described in “Materials and methods,” Mo-DCs and dead tumor cells were prestained with green fluorescence PKH-67 and red fluorescence PKH-26, respectively. As a result, Mo-DCs-Tum become double-positive cells (upper right rectangle in Fig. 1b). The percentage of double-positive cells in Mo-DCs-Tum-N and Mo-DCs-Tum-A was 87.3% and 72.6%, respectively. Uptake was profoundly reduced when dead cells were cocultured with Mo-DCs at 4°C, indicating that nonspecific binding of dead cells to Mo-DCs was minimal (Fig. 1b). The data are representative of three independent experiments using Mo-DCs generated from three different donors. Both Fig. 1a, b, indicate that phagocytotic ability of Mo-DCs was not affected by death pattern of tumor cells.
Fig. 1a,b.

Analysis of dead tumor cells by Mo-DCs. a Dead tumor cells were cocultured with an equivalent number of Mo-DCs for 4 h and then stained with Giemsa. One hundred Mo-DCs were examined under a light microscope, and the percentage of Mo-DCs-Tum was calculated (%Mo-DCs-Tum). Results are presented as mean ± SE (bars). The data are representative of three independent experiments using Mo-DCs generated from ten different donors. b Membrane components of dead tumor cells were labeled with PKH 26, and Mo-DCs were labeled with PKH 67. Fluorescence-labeled Mo-DCs were cocultured with dead tumor cells at a ratio of 1:1 for 4 h at 4°C or 37°C. Data are the log fluorescence intensity of Mo-DCs gated by cellular size and granulation phenotype. Fluorescence profiles of Mo-DCs-Tum at 4°C or 37°C are shown. The percentage of double-positive (PKH 26+, PKH 67+) Mo-DCs is shown in the upper right corner of each figure. The data are representative of three independent experiments using Mo-DCs generated from three different donors
Mo-DCs-Tum secrete TNF-α and IL-12
We first investigated whether Mo-DCs-Tum secrete IL-12 or TNF-α. Mo-DCs-Tum secreted both TNF-α and IL-12 p40 (Fig. 2). IL-12 p40 secretion (321 ± 26 pg/ml) by Mo-DCs-Tum-N was significantly higher than that (228 ± 10 pg/ml) of Mo-DCs-Tum-A (p=0.008). TNF-α secretion (1,390 ± 60 pg/ml) of Mo-DCs-Tum-N was also significantly higher than that (1,004 ± 42 pg/ml) of Mo-DCs-Tum-A (p=0.047). Concentrations of IL-12 p70 in the culture medium were below the limits of detection in both Mo-DCs-Tum-A and Mo-DCs-Tum-N. These data suggest that the death pattern of tumor cells may affect the production of these cytokines from Mo-DCs-Tum. The data are representative of two independent experiments using Mo-DCs generated from ten different donors.
Fig. 2.

Secretion of IL-12 p40, TNF-α, and IL-12 p70 by Mo-DCs. Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentrations of of IL-12 p40, TNF-α, and IL-12 p70 were measured by ELISA (n=10). Data are the mean ± SE (bars); p values for Mo-DCs-Tum-A vs Mo-DCs-Tum-N are listed. The data are representative of two independent experiments using Mo-DCs generated from ten different donors
TNF-α and IL-12 secreted from Mo-DCs-Tum induce further secretion of these cytokines
We next investigated the relationship between TNF-α secretion and IL-12 secretion in Mo-DCs-Tum (Fig. 3). When anti-TNF-α neutralizing antibody was added into the mixture of Mo-DCs and dead tumor cells, IL-12 p40 secretion was significantly decreased (Fig. 3a). Addition of recombinant TNF-α enhanced IL-12 p40 secretion by Mo-DCs-Tum-A but not Mo-DCs-Tum-N (Fig. 3b). Similarly, when anti-IL-12 neutralizing antibody was added into the mixture of Mo-DCs and A, TNF-α secretion was significantly decreased. However, TNF-α secretion by Mo-DCs-Tum-N has not altered significantly (Fig. 3c). Addition of recombinant IL-12 significantly enhanced TNF-α secretion by Mo-DCs-Tum (Fig. 3d). Neither cell viability nor total cell number was changed significantly by the assay (data not shown). These data suggest that TNF-α and IL-12 stimulate each other’s cytokine production, i.e., TNF-α/IL-12 autocrine loop. The data are representative of three independent experiments using Mo-DCs generated from ten different donors.
Fig. 3.

a IL-12 p40 secretion by captured Mo-DCs in the presence of anti-TNF-α neutralizing antibody (1 μg/ml). Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of of IL-12 p40 was measured by ELISA. Data are mean ± SE (bars). b IL-12 p40 secretion by captured Mo-DCs in the presence of recombinant TNF-α (200 U/ml). Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of of IL-12 p40 was measured by ELISA. Data are mean ± SE (bars). c TNF-α secretion by Mo-DCs-Tum in the presence of anti-IL-12 (20 μg/ml). Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of TNF-α was measured by ELISA. Data are mean ± SE (bars). d TNF-α secretion by captured Mo-DCs in the presence of recombinant IL-12 (100 μg/ml). Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of TNF-α was measured by ELISA. Data are mean ± SE (bars). The data are representative of three independent experiments using Mo-DCs generated from ten different donors
Both IL-12 and TNF-α secreted from Mo-DCs-Tum induce NF-κB activation in Mo-DCs
Since both IL-12 and TNF-α are target genes of NF-κB, induction of NF-κB activation in Mo-DCs-Tum was examined (Fig. 4). NF-κB activation was estimated as translocation of NF-κB p65 to the nuclei of Mo-DCs by EMSA. Enhanced nuclear translocation of NF-κB was observed in Mo-DCs-Tum. Nuclear translocation of NF-κB was stronger in Mo-DCs-Tum-N than Mo-DCs-Tum-A (Fig. 4a). In the presence of anti-TNF-α neutralizing antibody, nuclear translocation of NF-κB in Mo-DCs-Tum was suppressed (Fig. 4b). In the presence of anti-IL-12 neutralizing antibody, nuclear translocation of NF-κB in Mo-DCs-Tum was also suppressed (Fig. 4c). These data suggest the close relationship between TNF-α, IL-12, and NF-κB. The data are representative of three independent experiments using Mo-DCs generated from three different donors.
Fig. 4.
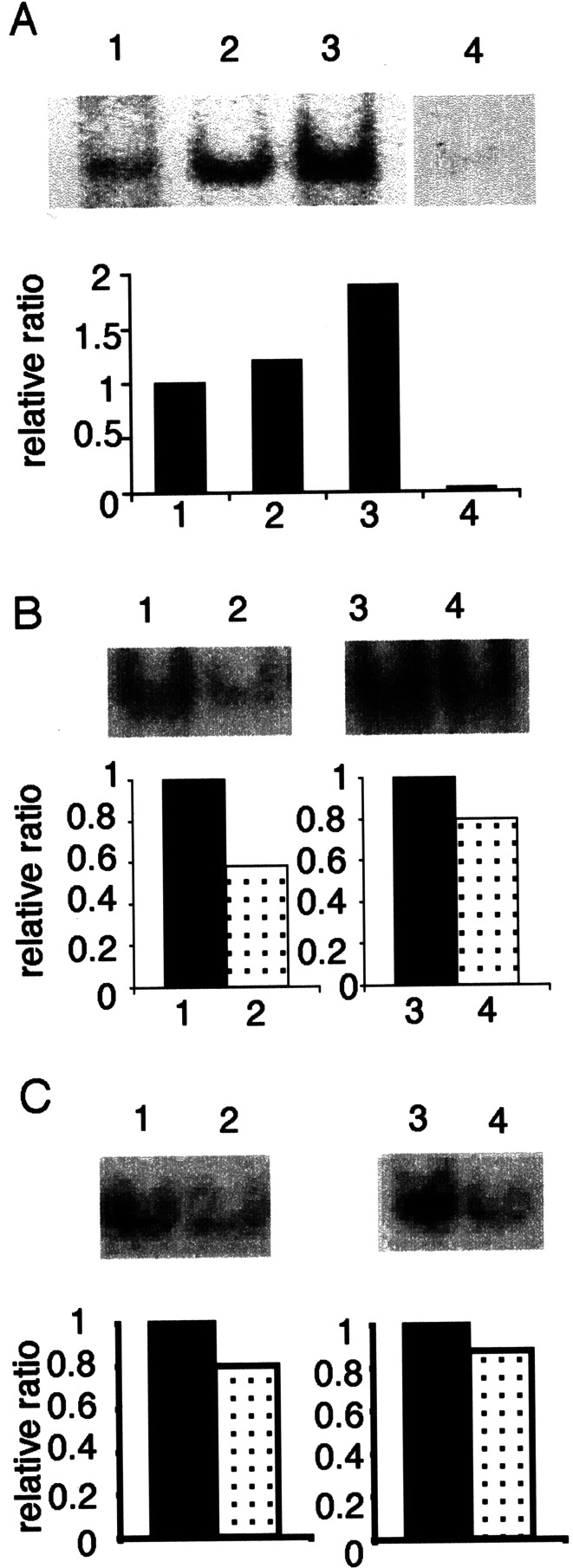
a NF-κB activation in Mo-DCs-Tum. Six hours after GCTM-1 cell pulsation, nuclear translocation of NF-κB in Mo-DCs was detected by EMSA. Lane 1 Mo-DCs (control), Lane 2 Mo-DCs-Tum-A, Lane 3 Mo-DCs-Tum-N, Lane 4 competition assay by the addition of 100 times NF-κB oligonucleotide. Data are the ratio of the intensity of Mo-DCs-Tum to control Mo-DCs as determined by NIH image. b NF-κB activation in Mo-DCs-Tum in the presence of anti-TNF-α neutralizing antibody (1 μg/ml). Twelve hours after GCTM-1 cell pulsation, nuclear translocation of NF-κB p65 in Mo-DCs-Tum was analyzed by EMSA. Lane 1 Mo-DCs-Tum-A, Lane 2 Mo-DCs-Tum-A in the presence of anti-TNF-α neutralizing antibody, Lane 3 Mo-DCs-Tum-N, Lane 4 Mo-DCs-Tum-N in the presence of anti-TNF-α neutralizing antibody. Data are the ratio of Mo-DCs-Tum with anti-TNF-α neutralizing antibody to Mo-DCs-Tum without anti-TNF-α neutralizing antibody as determined by NIH image. c NF-κB activation in Mo-DCs-Tum in the presence of anti-IL-12 neutralizing antibody (20 μg/ml). Twelve hours after GCTM-1 cell pulsation, nuclear translocation of NF-κB p65 in Mo-DCs-Tum was analyzed by EMSA. Lane 1 Mo-DCs-Tum-A, Lane 2 Mo-DCs-Tum-A in the presence of anti-IL-12 neutralizing antibody, Lane 3 Mo-DCs-Tum-N, Lane 4 Mo-DCs-Tum-N in the presence of anti-IL-12 neutralizing antibody. Data are the ratio of Mo-DCs-Tum with anti-IL-12 neutralizing antibody to Mo-DCs-Tum without anti-IL-12 neutralizing antibody as determined by NIH image. The data are representative of three independent experiments using Mo-DCs generated from three different donors
NF-κB inhibitor PDTC suppresses both TNF-α and IL-12 secretion by Mo-DCs-Tum
To confirm the relationship between TNF-α, IL-12, and NF-κB activation, a specific NF-κB inhibitor PDTC was used. In the presence of PDTC, nuclear translocation of NF-κB in Mo-DCs-Tum was certainly suppressed (Fig. 5a). The data are representative of three independent experiments using Mo-DCs generated from three different donors. IL-12 p40 secretion by Mo-DCs-Tum in the presence of PDTC was decreased significantly in comparison to that of Mo-DCs cultured in the absence of PDTC (Fig. 5b; p<0.001). The data are representative of three independent experiments using Mo-DCs generated from ten different donors. Addition of recombinant TNF-α could not ameliorate the decreased IL-12 secretion in PDTC-treated Mo-DCs-Tum (Fig. 5b). Similarly, TNF-α secretion by Mo-DCs-Tum-N or Mo-DCs-Tum-A in the presence of PDTC was decreased significantly in comparison with that in the absence of PDTC (p<0.001 and p=0.002, respectively; Fig. 5c). The data are representative of three independent experiments using Mo-DCs generated from ten different donors. These data indicate the TNF-α/IL-12 /NF-κB autocrine loop in Mo-DCs-Tum.
Fig. 5.

a NF-κB activation in Mo-DCs-Tum in the presence of 100 μM of PDTC. Six hours after the GCTM-1 cell pulsation, nuclear translocation of NF-κB in Mo-DCs was examined by EMSA. Lane 1 Mo-DCs-Tum-A, Lane 2 Mo-DCs-Tum-A treated with 100 μM PDTC, Lane 3 Mo-DCs-Tum-N, Lane 4 Mo-DCs-Tum-N treated with 100 μM PDTC. Data are the ratio of Mo-DCs-Tum in the presence of PDTC to Mo-DCs-Tum in the absence of PDTC as calculated by NIH image. The data are representative of three independent experiments using Mo-DCs generated from three different donors. b IL-12 p40 secretion by Mo-DCs-Tum treated with 100 μM of PDTC with or without 200 U/ml of recombinant TNF-α. Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of TNF-α was measured by ELISA. Data are mean ± SE (bars). The data are representative of three independent experiments using Mo-DCs generated from ten different donors. c TNF-α secretion by Mo-DCs-Tum in the presence of 100 μM of PDTC. Mo-DCs (104) were cocultured with 1×105 dead GCTM-1 cells for 24 h. Cell-free culture supernatants were collected, and the concentration of TNF-α was measured by ELISA. Data are mean ± SE (bars). The data are representative of three independent experiments using Mo-DCs generated from ten different donors
Discussion
We showed that Mo-DCs-Tum secrete considerable amounts of IL-12 and TNF-α through the IL-12/TNF-α/NF-κB autocrine loop.
We first showed a possibility that TNF-α secreted from Mo-DCs-Tum induces further IL-12 secretion from them, and similarly IL-12 secreted from captured Mo-DCs induces further TNF-α secretion from them (Fig. 3). These findings lead to speculation regarding the existence of a TNF-α/IL-12 autocrine loop in Mo-DCs-Tum. Antibodies against TNF-α or IL-12, however, could not completely suppress the secretion of these cytokines. In addition, recombinant TNF-α did not induce significant IL-12 secretion from Mo-DCs-Tum-N. Therefore, our data also indicate that some other mechanisms different from the TNF-α/IL-12 autocrine loop play roles in cytokine secretion from Mo-DCs-Tum. Why do Mo-DCs-Tum secrete both TNF-α and IL-12? It has been shown that transcription of TNF-α is regulated by NF-κB activation [20] and that TNF-α acts in an autocrine manner to induce NF-κB activation in Mo-DCs [23]. In addition, it has been reported that IL-12 secretion of DCs from mice with a double knockout of the NF-κB subunits p50 and cRel was impaired [19]. IL-12 is also a target gene of NF-κB in humans [7]. These data indicate that NF-κB activation plays a key role in secretion of both TNF-α and IL-12 in Mo-DCs-Tum. This possibility may be strongly supported by data showing that an NF-κB inhibitor, PDTC, significantly reduced secretion of both TNF-α and IL-12 in Mo-DCs-Tum (Fig. 5). The next question is what the initial NF-κB activation factor is. Many factors such as lipopolysaccharides, microbial stimuli, viral infection, proinflammatory cytokines, double-stranded DNA, and heat shock proteins (HSPs) have been reported to be activators of NF-κB [2, 4, 10, 14, 26–28]. We have shown that both secretion of IL-12 and activation of NF-κB induced by a streptococcal preparation OK-432 were suppressed when Mo-DCs were pretreated with an inhibitor of endocytosis, cytochalasin B [12], suggesting a possibility that NF-κB activation may be induced in Mo-DCs by capture of dead tumor cells. We are now speculating on at least two possibilities. One possibility is that capture of dead tumor cells induces first TNF-α secretion and then TNF-α secretion stimulates NF-κB activation. Another possibility is that the capture of dead tumor cells directly induces NF-κB activation.
In conclusion, it seems likely that captured Mo-DCs secrete a large amount of IL-12 and TNF-α through the IL-12/TNF-α/NF-κB autocrine loop (Fig. 6). Briefly, NF-κB activation is induced in Mo-DCs-Tum (step 1). NF-κB activation induces secretions of both TNF-α and IL-12 (step 2). Both IL-12 and TNF-α secretions induce further NF-κB activation (step 3). Enhanced NF-κB activation induces further secretion of IL-12 and TNF-α. Thus, IL-12, TNF-α, and NF-κB form an autocrine loop in Mo-DCs-Tum. Such an enhanced NF-κB activation in Mo-DCs-Tum induces them to secrete a large amount of IL-12 and TNF-α. However, it is still unknown to what degree the IL-12/TNF-α/NF-κB autocrine loop plays a role in IL-12 and TNF-α secretions from Mo-DCs-Tum. Identification of an initial signal or key molecule (step 1 in Fig. 6) operating the IL-12/TNF-α/NF-κB autocrine loop might provide new insights that could foster development of an effective antitumor immunotherapy.
Fig. 6.

Possible model of IL-12 and TNF-α secretions through IL-12/TNF-α/NF-κB autocrine loop. NF-κB activation is induced in Mo-DCs-Tum (step 1). NF-κB activation induces secretion of both TNF-α and IL-12 (step 2). Both IL-12 and TNF-α secretions induce further NF-κB activation (step 3). Enhanced NF-κB activation induces further secretion of IL-12 and TNF-α. Thus, IL-12, TNF-α, and NF-κB form an autocrine loop in Mo-DCs-Tum
Acknowledgements
This study was supported by a Grant-in-Aid for General Scientific Research (12557106 and 13470240) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Kaori Nomiyama for skillful technical assistance.
References
- 1.Baeuerle Annu Rev Immunol. 1994;12:141. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin Annu Rev Immunol. 1996;14:649. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau Nature. 1998;392:245. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Basu Int Immunol. 2000;12:1539. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 5.Cella Curr Opin Immunol. 1997;9:10. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 6.Gerosa J Exp Med. 1996;183:2559. doi: 10.1084/jem.183.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grohmann Immunity. 1998;9:315. doi: 10.1016/s1074-7613(00)80614-7. [DOI] [PubMed] [Google Scholar]
- 8.Heufler Eur J Immunol. 1996;26:659. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 9.Hsu Nat Med. 1996;2:52. [Google Scholar]
- 10.Ishi J Immunol. 2001;167:2602. doi: 10.4049/jimmunol.167.5.2602. [DOI] [PubMed] [Google Scholar]
- 11.Kojima Oncogene. 2000;19:1225. doi: 10.1038/sj.onc.1203427. [DOI] [PubMed] [Google Scholar]
- 12.Kuppner Eur J Immunol. 2001;31:1602. doi: 10.1002/1521-4141(200105)31:5<1602::AID-IMMU1602>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 13.Kuroki Cancer Immunol Immunother. 2003;52:561. doi: 10.1007/s00262-003-0394-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marten Cancer Immunol Immunother. 2002;51:637. doi: 10.1007/s00262-002-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morisaki Anti Cancer Res. 2000;20:3363. [PubMed] [Google Scholar]
- 16.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 17.Neumann Int J Oncol. 1997;11:1335. doi: 10.3892/ijo.11.6.1335. [DOI] [PubMed] [Google Scholar]
- 18.Onishi Clin Immunol. 2002;105:286. doi: 10.1006/clim.2002.5293. [DOI] [PubMed] [Google Scholar]
- 19.Ouaaz Immunity. 2002;16:257. doi: 10.1016/S1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 20.Prieschl J Immunol. 1996;157:2645. [PubMed] [Google Scholar]
- 21.Rescigno J Exp Med. 1998;188:2175. doi: 10.1084/jem.188.11.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadanaga Clin Cancer Res. 2001;7:2277. [PubMed] [Google Scholar]
- 23.Sallusto J Exp Med. 1994;179:1109. [Google Scholar]
- 24.Schott J Clin Endocrinol Metab. 2001;86:4965. doi: 10.1210/jcem.86.10.7949. [DOI] [PubMed] [Google Scholar]
- 25.Thanos Cell. 1995;80:529. [Google Scholar]
- 26.Todryk J Immunol. 1999;163:1398. [PubMed] [Google Scholar]
- 27.Vabulas J Biol Chem. 2002;277:20847. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 28.Zheng J Immunol. 2001;167:6731. doi: 10.4049/jimmunol.167.12.6731. [DOI] [PubMed] [Google Scholar]