

Key Points
-
•
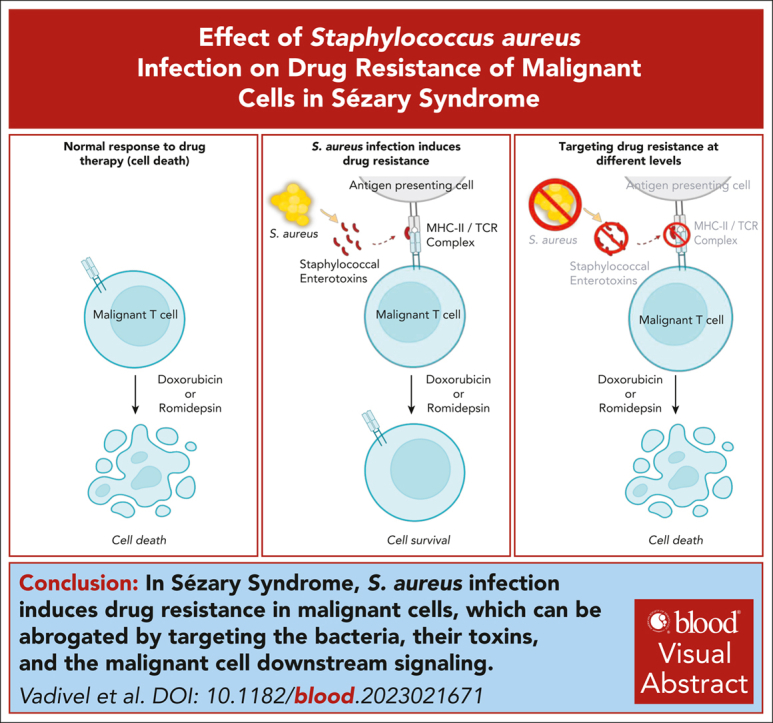
Enterotoxins from S aureus bacteria induce drug resistance in primary malignant T cells in SS.
-
•
Targeting bacteria, their toxins, and downstream signaling pathways in malignant T cells abrogate the induction of drug resistance.
Visual Abstract
Abstract
Patients with Sézary syndrome (SS), a leukemic variant of cutaneous T-cell lymphoma (CTCL), are prone to Staphylococcus aureus infections and have a poor prognosis due to treatment resistance. Here, we report that S aureus and staphylococcal enterotoxins (SE) induce drug resistance in malignant T cells against therapeutics commonly used in CTCL. Supernatant from patient-derived, SE-producing S aureus and recombinant SE significantly inhibit cell death induced by histone deacetylase (HDAC) inhibitor romidepsin in primary malignant T cells from patients with SS. Bacterial killing by engineered, bacteriophage-derived, S aureus–specific endolysin (XZ.700) abrogates the effect of S aureus supernatant. Similarly, mutations in major histocompatibility complex (MHC) class II binding sites of SE type A (SEA) and anti-SEA antibody block induction of resistance. Importantly, SE also triggers resistance to other HDAC inhibitors (vorinostat and resminostat) and chemotherapeutic drugs (doxorubicin and etoposide). Multimodal single-cell sequencing indicates T-cell receptor (TCR), NF-κB, and JAK/STAT signaling pathways (previously associated with drug resistance) as putative mediators of SE-induced drug resistance. In support, inhibition of TCR-signaling and Protein kinase C (upstream of NF-κB) counteracts SE-induced rescue from drug-induced cell death. Inversely, SE cannot rescue from cell death induced by the proteasome/NF-κB inhibitor bortezomib. Inhibition of JAK/STAT only blocks rescue in patients whose malignant T-cell survival is dependent on SE-induced cytokines, suggesting 2 distinct ways SE can induce drug resistance. In conclusion, we show that S aureus enterotoxins induce drug resistance in primary malignant T cells. These findings suggest that S aureus enterotoxins cause clinical treatment resistance in patients with SS, and antibacterial measures may improve the outcome of cancer-directed therapy in patients harboring S aureus.
Patients with Sézary syndrome (SS) are prone to Staphylococcus aureus infections and have treatment resistance which leads to poor prognosis. Vadivel and colleagues link these observations, suggesting that staphylococcal enterotoxins (SE) induce drug resistance in SS T cells through induction of several T-cell signaling pathways. Treatment with antibacterials as well as downstream mediators triggered by SE abrogate drug resistance, suggesting that antistaphylococcal antibiotics might improve outcomes of SS-directed therapy.
Introduction
Sézary syndrome (SS) is a malignant disorder of T cells that belongs to the spectrum of cutaneous T-cell lymphoma (CTCL), a non-Hodgkin lymphoma.1, 2, 3, 4, 5 The etiology of CTCL remains unresolved, and studies of the mutational landscape have been unrevealing.6, 7, 8, 9, 10, 11, 12 Dysregulation of JAK/STAT, PLCγ, and NF-κΒ signaling has repeatedly been linked to malignant cell proliferation, apoptosis resistance, and inflammation in CTCL.13, 14, 15, 16, 17, 18
CTCL is characterized by heightened vulnerability to bacterial infections, contributing significantly to disease-related morbidity and mortality.19, 20, 21, 22, 23, 24, 25, 26, 27 Skin barrier defects induced by malignant T cells are likely ports of entrance for bacterial infection.28, 29, 30
For decades, staphylococcal enterotoxin (SE)–producing Staphylococcus aureus has been suspected to play an important role in the pathogenesis of CTCL.26,31, 32, 33 In support, many patients with severe disease (particularly patients with SS) display skin colonization by SE-producing S aureus, and treatment with antibiotics has a beneficial clinical effect in SS and erythrodermic mycosis fungoides.33, 34, 35, 36 These findings suggest that S aureus can affect the malignant cells (directly or via the tumor microenvironment) and promote disease aggravation, which can be inhibited by antibiotics.
The mechanisms underlying the effects of S aureus on CTCL pathogenesis remains to be fully elucidated, and the impact of S aureus eradication on disease activity and cancer treatment outcomes remains an open question. Here, we show, to the best of our knowledge, for the first time that SE-producing S aureus can induce drug resistance in malignant T cells, providing a novel rationale for antibacterial therapy targeting S aureus in patients with CTCL.
Materials and methods
Patient material
This study includes 20 patients with SS. The patient characteristics are mentioned in Table 1. Blood samples were collected from 17 patients with SS after obtaining written consent from all patients as well as institutional approvals from Denmark (Committee on Health Research Ethics, H-16025331) and Germany (University Hospital Würzburg, 115/15). Experiments were performed in accordance with the Declaration of Helsinki.
Table 1.
Patient characteristics
| Patient | Sex | Age, y | Diagnosis | Year of diagnosis | Stage/classification | Treatment | Previous treatment | S aureus infection∗ |
|---|---|---|---|---|---|---|---|---|
| SS1 | M | 75 | SS | 2015 | IVA1 (T4, N0, M0, B2) | Doxorubicin | PUVA, ECP, IFN-α, bexarotene, mogamulizumab | Yes† |
| SS2 | M | 71 | SS | 2017 | IVA1 (T4, N1, M0, B2) | Vorinostat | ECP, UVB, IFN-α, mogamulizumab, brentuximab vedotin | Yes† |
| SS3 | M | 64 | SS | 2021 | IVA1 (T4, N1, B2, M0) | ECP, alitretinoin | UVB, acitretin, brodalumab, topical corticosteroids | Yes† |
| SS4 | M | 85 | SS | 2012 | NA | ECP, IFN-α | PUVA, IFN-α, bexarotene | Yes‡ |
| SS5 | M | NA | SS | 2013 | B2 | Blood collected before ECP treatment | NA | NA |
| SS6 | M | 67 | SS | 2016 | B2 | ECP, IFN-α, bexarotene, TSEI | Acitretin, chloroquine, doxycycline, prednisolone | Yes‡ |
| SS7 | M | 65 | SS | 2016 | B2 | ECP, IFN-α, prednisolone | Ifliximab, ustekinumab, secukinumab, adalimumab, acitretin, alitretinoin, prednisolone | Yes‡ |
| SS8 | F | 74 | SS | 2016 | B2 | ECP | Prednisolone, methotrexate, PUVA | No‡ |
| SS9 | M | 79 | SS | 2016 | B2 | ECP | Ifliximab, etanercept, adalimumab, ustekinumab, cyclosporin, PUVA | Yes‡ |
| SS10 | M | 58 | SS | 2018 | B2 | ECP, IFN-α, prednisolone | Prednisolone, methotrexate | Yes‡ |
| SS11 | M | 82 | SS | 2018 | B2 | ECP | NA | Yes‡ |
| SS12 | M | 77 | SS | 2017 | B2 | ECP, brentuximab vedotin | PUVA, UVB, methotrexate, IFN-α, bexarotene | Yes‡ |
| SS13 | M | 71 | SS/leukemic MF | 2014 | IVA2 (pT4, N3, B2, M0) | Gemcitabine | UVB, ECP, bexarotene, methotrexate | NA |
| SS14 | M | 80 | SS | 2011 | IVB (pT4p, N3, M1) | ECP, brentuximab vedotin | UVB, methotrexate, bexarotene, PUVA, gemcitabine, prednisolone, domatinostat (4SC-202), doxorubicin, brentuximab vedotin, ECP, bendamustine, dimethyl fumarate | Yes† |
| SS15 | F | 85 | SS | 2014 | IVA1 (pT4, N0, B2, M0) | Bexarotene | Methotrexate | NA |
| SS16 | F | 60 | SS | 2020 | IVA1 (T4, N1, B2, M0) | ECP, methotrexate | Methotrexate | NA |
| SS17 | M | 77 | SS | 2022 | III | Prednisolone, topical corticosteroid, grade IV | No previous treatment | Yes‡ |
| SS18 | M | 84 | SS | 2022 | IVA (T4, N0, M0, B2) | Alemtuzumab | ECP | Yes‡ |
| SS19 | F | 71 | SS | 2023 | IVA (T4, N0, M0, B2) | Acitretin, ECP | No previous treatment | Yes‡ |
| SS20 | M | 67 | SS | 2023 | IVA1 (T4, N2, B2, M0) | ECP, mogamulizumab | CHOP, brentuximab vedotin, cisplatin, cytarabine | Yes† |
Treatment indicates the patient clinical treatments at the time point of blood collection where PBMCs were isolated and used for experiments.
ECP, extracorporal photopheresis; IFN, interferon; F, female; M, male; MF, mycosis fungoides; NA, not available; PUVA, psoralen and ultraviolet A; SS, Sézary syndrome; TSEI, total skin electron irradiation; UVB, ultraviolet B.
S aureus infection during or before the sample collection.
Data obtained from clinical swabs.
Data based on the measure of immunoglobulin G levels against S aureus proteins in patient serum by enzyme-linked immunosorbent assay (supplemental Methods).
PBMC isolation, cell culture, inhibitors, and pulse experiments.
Peripheral blood mononuclear cells (PBMCs) from blood of patients with SS (SS PBMCs) were isolated by density gradient centrifugation following manufacturer’s protocol (StemCell Technologies). In most cases, PBMCs were cryopreserved (supplemental Methods, available on the Blood website) and used later for experiments. PBMCs were cultured in RPMI 1640 media (Sigma-Aldrich) supplemented with 10% pooled human serum (Copenhagen Hospital) and 1% antibiotics (penicillin and streptomycin) (Sigma-Aldrich). For SE stimulation, a pool of SE (SEA, SEB, SEC2, SED, and SEI) (Toxin Technologies) at 50 to 100 ng/mL were added to the cells before treatment with CTCL drugs. For cytokine experiments, a pool of interleukin-2 (IL-2) family cytokines, IL-2 (2 × 103 U/mL; Novartis), IL-4, IL-7, and IL-15 (10 ng/mL each; Peprotech), were added. For romidepsin pulse experiments, cells were treated with high concentration of romidepsin for 6 hours.37 Cells were washed and kept in complete media for 16 hours followed by incubation with and without SE pool for 3 days.
Patient bacterial isolation and culture
S aureus from patients with CTCL was isolated and validated for SE expression in our previous study.35 Supernatants from S aureus cultures were prepared as previously described.38 Briefly, S aureus was prepared from an overnight culture diluted to an optical density 600 nm of 0.01 in tryptic soy broth and regrown for 4 hours with or without 1 μg/mL XZ.700 (an engineered S aureus–specific endolysin39,40 from Micreos Pharmaceuticals AG, Baar, Switzerland). Bacterial supernatants were prepared by centrifugation (10 000g for 10 minutes) followed by sterile filtration (0.22 μm filter).
Flow cytometry and cell sorting
Primary conjugated monoclonal antibodies against CD3, CD4, CD7, CD8, CD14, CD26, CD45, TCRCβ1, TCRVβ1, TCRVβ2, TCRVβ8, and TCRVβ18 were purchased from BD Biosciences, BioLegend, Miltenyi Biotec, Beckman Coulter, and R&D Systems. Cultured cells were pelleted by centrifugation, washed using FACS-PBS buffer (phosphate-buffered saline [PBS] + 1% fetal bovine serum + 0.02% NaN3). Cell surface staining (30 minutes on ice) was performed using Brilliant Stain buffer (BD Biosciences). Malignant cells were gated as CD3+CD4+TCRVβ#+. In patients for whom the dominant TCRVβ clone could not be targeted, malignant cells were gated as CD3+CD4+CD7low and/or CD26– or CD3+CD4+CD26–TCRCβ1+/TCRCβ1– (depending on whether the T-cell receptor [TCR] of the malignant clone uses the TRBC1 or TRBC2 gene segment in its TCRβ chain). Flow cytometric analysis was conducted using a 5-laser BD LSR-Fortessa. Data were analyzed using FlowJo (TreeStar). Cell sorting was conducted on a 3-laser BD FACS Aria-II, using a 100-μm nozzle. Propidium Iodide (PI) was added to exclude dead cells while sorting. Purity of sorted cells was consistently >95%.
Apoptosis assay and western blotting
For apoptotic experiments, at the end of cell culture, cells were stained with Mitotracker red CMXRos (Thermo Fisher) and were incubated at 37°C for 30 minutes. Cells were washed, and flow cytometry procedure was followed. After cell surface staining, cells were stained with PI and annexin V suspended in annexin V binding buffer (BD Biosciences). Apoptotic cells were identified from nonpermeable (PI–) malignant or nonmalignant cells as MitotrackerlowAnnexin V+; that is, if a patient had dominant TCRVβ1 clone, then the apoptotic population of malignant cells would be gated as PI–CD3+CD4+TCRVβ1+MitotrackerlowAnnexin V+. Western blotting was performed as previously described41 and analyzed using Image Lab (BioRad).
CITE-seq
For cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) experiment, PBMCs from a patient with SS (SS17) were treated ex vivo with romidepsin in the presence or absence of SE for 36 hours. Each condition was multiplexed by tagging with a unique hashtag antibody together with a cocktail of oligo-conjugated antibodies targeting 101 different surface proteins (TotalSeq-C, BioLegend). CITE-seq antibodies were individually optimized and titrated using a previously described protocol.42 Live cells from each condition were sorted and pooled before loading on the 10X chromium using the Chromium Next GEM Single Cell 5′ reagent kits v2 (10X Genomics). For further description of procedures, sequencing, data processing, and analysis, please see supplemental Methods.
Statistics
Graphs and statistical analyses, except for CITE-seq experiments, were made using GraphPad Prism 9. Error bars represent the standard error of the mean, and the level of statistical significance was set at ∗P < .05.
Results
S aureus culture supernatants and SE induce drug resistance in malignant T cells
Figure 1A-C shows examples of patients with SS with S aureus skin colonization before and after treatment with antibiotics. Eradication of S aureus with antibiotics reduced disease activity in patients undergoing anticancer treatment with doxorubicin, vorinostat, and alitretinoin (Figure 1A-C). This confirms and extends previous data that eliminating bacterial colonization increased the efficacy of different types of ongoing anticancer treatment.35 This prompted us to hypothesize that S aureus and its toxins may directly induce drug resistance in malignant T cells. To address this hypothesis, we initially focused on the effect of S aureus on malignant T-cell responses to romidepsin, which is a potent inducer of apoptosis in malignant cells but not in nonmalignant CD4+ T cells (supplemental Figure 1A). Accordingly, we examined whether supernatant from SE-producing S aureus (isolated from affected skin of a patient with SS) could modulate romidepsin-induced apoptosis in primary malignant cells. Thus, we treated SS PBMCs with romidepsin for 72 hours in the presence or absence of supernatant followed by flow cytometric analysis of apoptosis in malignant cells. Supernatant from SE-producing S aureus drastically reduced romidepsin-induced apoptosis in malignant cells, whereas supernatant from endolysin (XZ.700)–treated S aureus did not have this effect (Figure 1D; supplemental Figure 1B). Because endolysin abrogates SE production and kills S aureus,38 we hypothesized that the antiapoptotic effect of S aureus supernatants was mediated by SE. To address this, we treated SS PBMCs with romidepsin in the presence or absence of supernatants from S aureus isolates derived from 2 patients that either produced or did not produce SE (supplemental Figure 1C). Intriguingly, only the S aureus supernatant that contained SE showed potent reduction in romidepsin-induced apoptosis. To test whether SE was sufficient to counteract the romidepsin-induced apoptosis, we treated SS PBMCs with romidepsin in the presence or absence of a pool of SE and analyzed apoptosis in malignant cells by assessing poly (ADP-ribose) polymerase (PARP) cleavage (Figure 1E) and by flow cytometry (supplemental Figure 1D). As hypothesized, romidepsin-induced apoptosis was markedly reduced in the presence of SE in a dose- and time-dependent manner (Figure 1F). The antiapoptotic effect of SE was not restricted to this treatment regimen. Thus, even when added 16 hours after pulse treatment with romidepsin, SE blocked the apoptotic effect of very high concentrations of romidepsin (25-100 nM; Figure 1G), whereas pretreatment with SE had little effect (supplemental Figure 1E). Analysis of S aureus supernatants identified SEA as the dominating type of SE produced by S aureus derived from patients.38 Consistent with these results, recombinant SEA (SEAwt) drastically diminished romidepsin-induced apoptosis (Figure 1H-I). In contrast, a mutated SEA (SEAF47A/D227A) that cannot bind to major histocompatibility complex (MHC) II43 had no effect (Figure 1H-I). As expected, the effect of SEA was abrogated by addition of a blocking anti-SEA antibody (Figure 1J-K). Together, this indicates that (1) SEA is sufficient to counteract treatment-induced apoptosis, (2) this effect is dependent on the superantigenic properties of SEA, and (3) the observation is not due to off-target effects or contaminants.
Figure 1.

S aureus culture supernatants and SE induce drug resistance in malignant T cells. (A-C) Photographs of affected skin from 3 patients with SS before and after antibiotic treatment with ongoing cancer-directed treatments: (A) doxorubicin (SS1), (B) vorinostat (SS2), and (C) alitretinoin (SS3). (D) Flow cytometric plots showing apoptotic fraction of malignant T cells from SS17 PBMCs after 72 hours of treatment with 2 nM romidepsin and bacterial culture supernatant (sup) from S aureus cultured in the presence or absence of endolysin. The used S aureus strain was originally isolated from lesional skin of a different patient with SS. For control, tryptic soy broth medium (Ctrl medium) was added to the PBMC culture. (E) Western blot showing cleaved and uncleaved PARP expression after 24- and 48-hour treatment of SS4 PBMCs with either SE and/or romidepsin. GAPDH is used as a loading control. (F) Apoptotic fraction of malignant cells from PBMCs of 5 patients with SS (SS4, SS5, SS8, SS9, and SS12) after 72 and 144 hours treated with increasing concentrations of romidepsin in the presence (SE) or absence (PBS) of SE. Statistical significance was assessed by 2-way analysis of variance (ANOVA) followed by the Šídák multiple comparisons test. ∗∗P < .01; ∗∗∗P < .0005. (G) Apoptotic fraction of malignant cells from PBMCs of 2 patients with SS (SS8 and SS12) after a different 6 hours pulse, 16 hours chase in vitro treatment regimen with high concentrations of romidepsin in the presence (SE) or absence (PBS) of SE. (H-I) Representative flow cytometric plots (H; SS13) and quantification (I) of apoptotic fraction of malignant cells from PBMCs of 5 patients with SS (SS4, SS8, SS12, SS13, and SS15) after 72 hours of treatment with 2 nM romidepsin in presence of wild-type SEA or mutant SEA (SEAF47A/D227A). (J-K) Representative flow cytometric plots (J; SS15) and quantification (K) of apoptotic fraction of malignant cells from 4 patients with SS (SS4, SS12, SS13, and SS15) after 72 hours of treatment with 2 nM romidepsin in presence of SEA or SEA and a blocking anti-SEA antibody. For panels I-K, statistical significance was assessed by repeated measures 1-way ANOVA followed by Tukey multiple comparison test. ∗P < .05. Ctrl, control; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
SE largely overrides the transcriptional effect of romidepsin in malignant T cells
In our SS cohort, malignant cells from 9 of the 10 romidepsin-naïve patients were sensitive to romidepsin as measured by a significant drop in cell survival after treatment (Figure 2A-B). Importantly, presence of SE drastically improved malignant cell survival after romidepsin treatment (Figure 2A-B). To explore this effect, we performed single-cell CITE-seq analysis of SS PBMCs treated with romidepsin in the presence or absence of SE (Figure 2C-H). Cell cultures were collected after 36 hours to capture the transcriptional profiles before the onset of cell death induced by romidepsin (sensitivity of the malignant cells to romidepsin was confirmed by flow cytometry after 72 hours culture; supplemental Figure 2A-B). Cell types were identified by their surface protein expression, and malignant cells were defined by their monoclonal TCR44 and expression of malignant-associated markers TOX45 and KIR3DL2,46 in concert with low surface levels of CD747 and CD2648 (Figure 2C-D; supplemental Figure 2C-D). Although romidepsin and SE individually induced strong transcriptional responses in the malignant cells (Figure 2E-F), treatment with romidepsin in the presence of SE largely resembled the response to SE alone, as evident by their differentially expressed genes and coclustering in transcriptional space. This trend was also evident from gene-set enrichment analysis and transcription factor activity analysis (Figure 2G-H; supplemental Figure 2E). These findings suggest that upon treatment with romidepsin in the presence of SE, the SE-induced gene-regulation largely overshadows the romidepsin-mediated changes in gene expression. Both gene-set enrichment analysis and transcription factor activity analysis identified induction of key signaling pathways including NF-κB signaling, cytokine-mediated JAK/STAT signaling, and TCR/B-cell receptor signaling upon SE exposure, both in isolation and in combination with romidepsin treatment (Figure 2G-H), and this activation profile largely resembles that observed in malignant T cells from skin (supplemental Figure 3).
Figure 2.

SE largely overrides the transcriptional effect of romidepsin in malignant T cells. (A-B) Representative flow cytometric plots (A; SS8) and quantification (B) of percentage of viable malignant cells from PBMCs of 10 patients with SS (SS4, SS7, SS8, SS10, SS11, SS12, SS13, SS15, SS16, and SS17) treated with 2 nM romidepsin for 72 hours in the presence (SE) or absence (PBS) of SE. Red triangle shows the PBMCs of a patient with SS, which did not respond to romidepsin treatment. Statistical significance was assessed by ordinary 1-way ANOVA followed by Tukey multiple comparison test. ∗∗P < .01; ∗∗∗P < .0005. (C-E) Integrated uniform manifold approximation and projection (UMAP) from the 4 sample conditions (DMSO, romidepsin, SE, and SE + romidepsin) based on both mRNA and surface protein expression using totalVI colored by cell types (C), TCRβ CDR3 clonotype (D), and sample culture conditions (E; SS17). (F) Heatmap showing relative mean expression of malignant T cells after 36 hours treatment with romidepsin in the presence or absence of SE in SS17. Genes were selected based on differential expression between treatment and PBS control with a false discovery rate (FDR) <0.05 and a log2 fold change >0.5 or <−0.5 among any of the 3 treatments, yielding 574 genes in total. (G) Gene-set enrichment analysis (GSEA) of immune system pathways included in the Reactome database comparing 3 treatment conditions after 36 hours of culture (SS17): romidepsin (Ro), SE, and SE + romidepsin with PBS-treated control. Plot shows top 15 pathways from each comparison based on highest absolute normalized enrichment scores (NES) having a q-value <0.05. Other pathways are included in supplemental Figure 2E. (H) Transcription factor activity analysis using DoRothEA transcription factor signatures comparing 3 treatment conditions after 36 hours of culture (SS17): romidepsin, SE, and SE + romidepsin with PBS-treated control. Plot shows top 10 differentially activated transcription factors from each comparison based on highest absolute log2 fold change having an FDR <0.05. DMSO, dimethyl sulfoxide; mRNA, messenger RNA; NK cells, natural killer cells.
SE-mediated drug resistance of malignant T cells is not limited to romidepsin
Because SE significantly reduced romidepsin-induced apoptosis of malignant cells, we tested whether SE also counteracted the effects of other clinically used histone deacetylase inhibitors (HDACi), vorinostat49 and resminostat.50 Similar to romidepsin, treatment of SS PBMCs with either vorinostat or resminostat induced potent cell death, and both were counteracted by the presence of SE, resulting in survival of malignant cells (Figure 3A-D). Our CITE-seq analysis indicated that SE modulated signaling pathways involved in the survival of malignant cells such as NF-κB and JAK/STAT. Thus, we hypothesized that the prosurvival effect of SE on malignant cells may not be limited to HDACi. Doxorubicin and etoposide are 2 chemotherapeutic agents that induce DNA damage and have been used as a treatment for CTCL.51,52 We selected PBMCs from patients with SS that responded to doxorubicin or etoposide ex vivo and tested whether presence of SE also counteracted the cytotoxic effect on malignant cells of these chemotherapeutics. Indeed, we found that SE exposure induced marked resistance in malignant cells toward both doxorubicin and etoposide (Figure 3E-H). Due to fluorescent properties of doxorubicin being incompatible with PI staining, we used Mitotracker red CMXRos to mark viable cells. Furthermore, the effect of SE-induced drug resistance was not limited solely to HDACi and drugs inducing DNA damage, because SE exposure also maintained malignant cell survival after treatment with the adenosine triphosphate synthase inhibitor oligomycin (supplemental Figure 4A-B). Together, this indicates that the presence of SE does not directly interfere with the mode of action of these agents but rather promote overall drug resistance and survival of the malignant cells. Moreover, SE did not induce efflux of drugs as judged by flow cytometry analysis of fluorescent drugs, doxorubicin and fluorophore-conjugated vorinostat (coumarin–suberoylanilide hydroxamic acid [c-SAHA]) (supplemental Figure 4C-E) and was not associated with an increased expression of drug transporters as judged from CITE-seq analysis (supplemental Figure 4F).
Figure 3.

SE-mediated drug resistance of malignant T cells is not limited to romidepsin. Representative flow cytometric plots (SS12 [A,C]; SS4 [E]; SS17 [G]; SS7 [I]) and quantifications of percentage of viable malignant cells from PBMCs of patients with SS cultured for 72 hours in the presence or absence of SE and treated with 1 μM vorinostat (A-B) (n = 5 [SS5, SS7, SS10, SS12, and SS17]), 2 μM resminostat (C-D) (n = 3 [SS4, SS10, and SS12]), 100 to 400 nM doxorubicin (E-F) (n = 4 [SS4, SS7, SS10, and SS17]; due to fluorescent properties of doxorubicin being incompatible with PI staining, we used Mitotracker red CMXRos to mark viable cells), 10 to 50 μM etoposide (G-H) (n = 5 [SS7, SS8, SS10, SS11, and SS17]), or 50 nM bortezomib (I-J) (n = 4 [SS7, SS10, SS11, and SS12]). Statistical significance was assessed by repeated measures 1-way ANOVA followed by Tukey multiple comparisons test. ∗P < .05; ∗∗P < .005; ∗∗∗P < .0005. DMSO, dimethyl sulfoxide; ns, not significant.
Because NF-κB signaling is enhanced by SE treatment (Figure 2G-H), we investigated whether using the clinically approved proteasome and NF-κB inhibitor bortezomib,14,53 could abrogate SE-induced drug resistance. We found that bortezomib induced potent malignant cell death that could not be rescued by SE (Figure 3I-J). Similarly, SE did not confer resistance to crystal violet (gentian violet,54 which has been shown to inhibit NF-κB in CTCL55) (supplemental Figure 4G). These findings suggests that SE-induced drug resistance may be driven by enhanced NF-κB-mediated cell survival of malignant cells.
SE-induced drug resistance is mediated by TCR signaling via LCK-PKC-NF-κB
NF-κB is known to be vital for the survival of malignant cells in CTCL and is directly induced by TCR activation56,57 Our CITE-seq analysis confirmed that SE triggered profound TCR-mediated activation of malignant cells (as shown by pathway enrichment and induction of surface CD25, CD69, and CD71 expression; Figure 2G; supplemental Figure 2C), suggesting that SE-induced drug resistance could be mediated through TCR engagement. Thus, we examined whether the blockage of TCR signaling could abrogate SE-induced drug resistance. TCR signaling is contingent upon early activation of protein tyrosine kinases such as lymphocyte cell-specific protein-tyrosine kinase (LCK) (SRC family of tyrosine kinases), a critical kinase in the initiation of TCR signaling after antigen presentation.58 Accordingly, we tested whether SRC inhibitor A-419259 would block SE-induced drug resistance. A-419259 treatment effectively blocked SE abrogation of romidepsin-induced cell death (Figure 4A-B). Similar results were obtained with dasatinib, a clinically approved inhibitor used against different cancers. Dasatinib, known to inhibit LCK,59 had little effect on malignant cell survival by itself but abrogated the SE-mediated resistance to romidepsin (Figure 4C-D). Of note, A-419259 inhibited activation and proliferation in both malignant and nonmalignant T cells (supplemental Figure 5A-B). Because TCR-induced NF-κB signaling is downstream of protein kinase C (PKC)-θ activation, we blocked PKC-θ with a clinical inhibitor, sotrastaurin.60 Sotrastaurin significantly inhibited SE-induced romidepsin resistance (Figure 4E-F). Next, we inhibited the MAP kinase cascade in TCR signaling because MAP kinases have previously been linked to HDACi resistance.61 Using 2 clinical MEK inhibitors, we found that neither inhibitor abrogated SE-induced romidepsin resistance (supplemental Figure 5C-D). TLRs can activate NF-κB,62 but inhibitors of TLR2 and TLR4 had no effect on SE-induced romidepsin resistance, suggesting that they are not involved (supplemental Figure 5E). Taken together, our findings support the hypothesis that SE-induced drug resistance is induced by TCR signaling through the PKC-θ pathway leading to NF-κB activation.
Figure 4.

SE-induced drug resistance is mediated by TCR signaling via LCK-PKC-NF-κB. Representative flow cytometric plots (SS4 [A]; SS13 [C,E]) and quantifications of percentage of viable malignant cells from PBMCs of patients with SS treated with 2 nM romidepsin for 72 hours in the presence or absence of SE and 2 μM A-419259 (A-B) (Src inhibitor) (n = 8 [SS4, SS5, SS8, SS10, SS12, SS13, SS14, and SS15]), 100 nM dasatinib (C-D) (n = 6 [SS4, SS8, SS10, SS12, SS13, and SS15]), and 1.5 μM sotrastaurin (E-F) (PKC inhibitor) (n = 5 [SS4, SS10, SS12, SS13, and SS15]). Statistical significance was assessed by repeated measures 1-way ANOVA followed by Tukey multiple comparison test. ∗P < .05; ∗∗P < .01; ∗∗∗∗P < .0001. DMSO, dimethyl sulfoxide.
SE-induced drug resistance can be both direct and indirect through release of IL-2 family cytokines
In addition to NF-κB signaling, our CITE-seq data indicated that JAK-STAT–mediated interleukin signaling and STAT3 activity was induced in malignant cells in the presence of SE (Figure 2G-H). IL-2 and its family members stimulate JAK1 and JAK3, leading to STAT3/5 activation.63 Both malignant and nonmalignant T cells express JAK/STAT activating cytokines upon SE stimulation (Figure 5A-B). To determine whether signaling through cytokines could facilitate SE-induced drug resistance, we treated SS PBMCs ex vivo with romidepsin in the presence or absence of a cocktail of IL-2 family cytokines (IL-2, IL-4, IL-7, and IL-15). We observed that the IL-2 family cytokines were sufficient to protect malignant cells from cell death induced by romidepsin (Figure 5C-D). To determine whether cytokine signaling is necessary for SE-induced drug resistance, we added either A-419259 (to block TCR signaling) or a clinically used JAK inhibitor, tofacitinib (known to block IL-2 family cytokine signaling64), while treating SS PBMCs with romidepsin in the presence or absence of either SE or IL-2 family cytokines. Consistent with the results above, both SE and cytokines induced romidepsin resistance in malignant T cells (Figure 5E). Importantly, A-419259 completely blocked SE-induced romidepsin resistance but did not affect cytokine-induced romidepsin resistance (Figure 5E). In contrast, although tofacitinib completely blocked cytokine-induced romidepsin resistance (Figure 5E), we found remarkable differences in the effect of tofacitinib on SE-induced romidepsin resistance between the patients (Figure 5E-H; supplemental Figure 6B-C). Tofacitinib had limited effect on cytokine secretion (supplemental Figure 6A). In PBMCs investigated from 3 of the 5 patients, we found only modest or no reduction in SE-induced romidepsin resistance in the presence of tofacitinib (JAK-independent; Figure 5E-F; supplemental Figure 6B). In PBMCs from the remaining 2 patients, SE-induced romidepsin resistance was almost completely abrogated by the presence of tofacitinib, suggesting that cytokine signaling was necessary for the effect in these patients (JAK-dependent; Figure 5G-H; supplemental Figure 6C). These individual differences in the impact of JAK/STAT on SE-induced HDACi resistance were not surprising because SS is a highly heterogeneous disease,65, 66, 67 and SE can induce STAT3 activation indirectly in malignant cells through IL-2 family cytokines released by nonmalignant, SE-responsive bystander CD4+ T cells.68
Figure 5.


SE-induced drug resistance can be either JAK-dependent or -independent. (A) Heatmaps showing cytokines secreted by PBMCs derived from SS4 after 24, 48, and 72 hours of treatment with 2 nM romidepsin in the presence or absence of SE. (B) Heatmap showing cytokine expression across the cell types identified in the CITE-seq data set from SS17 after 36 hours of treatment with 2 nM romidepsin in the presence or absence of SE. Expression is calculated as log1p transformed unique transcripts per million . (C-D) Representative flow cytometric plots (C; SS13) and quantification (D) of percentage of viable malignant cells from PBMCs of 7 patients with SS (SS4, SS5, SS6, SS8, SS12, SS13, and SS14) treated with 2 nM romidepsin for 72 hours in the presence or absence of cytokines (IL-2, IL-4, IL-7, and IL-15). Statistical significance was assessed by ordinary 1-way ANOVA followed by Tukey multiple comparison test. ∗∗∗∗P < .0001. (E-H) Representative flow cytometric plots (SS8 [E]; SS10 [G]) and quantifications of percentage of viable malignant cells from PBMCs of patients with SS treated with 2 nM romidepsin for 72 hours in the presence or absence of SE and JAK inhibitor (1 μM tofacitinib) of patients exhibiting JAK-independent (E-F) (n = 3 [SS8, SS12, and SS13]) or JAK-dependent (G-H) (n = 2 [SS4 and SS10]) resistance being induced by the presence of SE. (E) PBMCs from SS8 treated in the presence of cytokines (IL-2, IL-4, IL-7, and IL-15) as well as SRC inhibitor (2 μM A-419259). DMSO, dimethyl sulfoxide; NK cells, natural killer cells.
To determine whether TCR signaling is sufficient to induce drug resistance in patients who respond to SE in a JAK-dependent fashion and to investigate the dependence of nonmalignant cells, we sorted malignant T cells from the PBMCs of 3 patients with SS (based on CD3, CD4, CD8, CD7, CD26, and TCR-Cβ1 expression) (Figure 6A). Due to the lack of surface markers that define malignant T cells, sorted malignant cells will unavoidably contain a minute nonmalignant CD4+ T cells. Nonetheless, even in these highly enriched conditions, SE still induced potent drug resistance in sorted malignant T cells. For 2 patients, this resistance was abrogated by the presence of tofacitinib (JAK-dependent), whereas malignant cells from 1 patient showed JAK-independent resistance (Figure 6B-C; supplemental Figure 7).
Figure 6.

SE induce drug resistance in purified malignant T-cell cultures via JAK-dependent or -independent mechanisms. (A) Flow cytometric plot showing percentage of malignant T cells before and after sorting of PBMCs from SS4. (B-C) Flow cytometric plots showing percentage of viable malignant cells from sorted malignant T cells treated with 2 nM romidepsin for 72 hours in the presence or absence of SE and JAK inhibitor (1 μM tofacitinib) of patient exhibiting JAK-independent (SS13) (B) and JAK-dependent (SS4) (C) resistance being induced by the presence of SE. DMSO, dimethyl sulfoxide.
Discussion
In this study, we show that S aureus and its toxins induce resistance to drug-induced cell death in primary malignant T cells from patients with SS. Thus, supernatants from SE-expressing S aureus isolated from SS skin largely protected malignant cells from cell death induced by romidepsin, whereas endolysin-treated SE-producing S aureus and SE-negative S aureus had no effect. Because only live S aureus express exoproteins such as SE, these findings imply that the effect was mediated by SE. In support, the inhibitory effect on romidepsin-induced apoptosis was fully replicated by highly purified SEA and abrogated by antibody blocking and mutations in the MHC class II binding domain of SEA (SEAF47A/D227A), which prevent TCR–MHC class II crosslinking (a prerequisite for SEA-mediated, TCR activation43). For decades, it has been suspected but never proven that bacterial superantigens play a pathogenetic role in CTCL.26,69, 70, 71, 72 The present findings indicate that SE-mediated induction of drug resistance could be one of the missing links between S aureus and disease aggravation. Importantly, induction of resistance was not confined to romidepsin. On the contrary, SE also induced malignant T-cell resistance to other HDACi (vorinostat and resminostat), chemotherapeutic drugs (doxorubicin and etoposide), and metabolic inhibitors (oligomycin). However, the protective effect was not universal and unspecific, because SE could not rescue malignant cells from cell death induced by bortezomib and crystal violet, which have also been used for the treatment of CTCL.52,54 Importantly, the induction of drug resistance was dependent on continuous presence of SE, implying that drug resistance may also subside after eradication of S aureus in patients. This points to a possible beneficial clinical effect of a persistent control and prevention of skin colonization by S aureus in these patients.
Short-term pulse treatment of malignant cells with a high dose of romidepsin was equally potent at inducing malignant cell death. Importantly, presence of SE 16 hours after the removal of the romidepsin pulse was still sufficient to counteract the drug-induced cell death, showing that SE-induced drug resistance is not due to direct SE-drug interaction but mediated at a level downstream of the drug entering and initiating its effect. Consequently, SE-induced drug resistance was neither mediated through direct SE interaction with the drug nor blocking entrance or facilitating its removal. In line with a broad resistance mechanism, SE-induced resistance appears to be mediated through the cellular response to SE rather than direct interference with the treatment. In support, our CITE-seq analysis identified 3 potentially important pathways: NF-κB, TCR, and STAT3. NF-κB activity has been implicated in resistance to HDACi73 and chemotherapeutic drugs.74, 75, 76, 77 In vitro studies on different cancer cell lines have shown that inhibiting NF-κB enhanced sensitivity to treatment with doxorubicn74,75 and etoposide.76,77 Together with our present finding that SE activates NF-κB signaling in malignant cells and overrides most of the transcriptional changes induced by romidepsin, it is likely that SE induce drug resistance, at least partly, through the NF-κB pathway. This conclusion is consistent with our observations that SE could not rescue malignant cells from cell death induced by bortezomib and crystal violet, which has been shown to inhibit NF-κB activity in malignant cells from patients with CTCL.14,55
TCR signaling depends on activation of the SRC tyrosine kinase, LCK, resulting in NF-κB activation through a PKC-dependent pathway that promotes cell survival by inhibiting proapoptotic proteins.78,79 Accordingly, blockage of both early TCR signaling (dasatinib) and PKC activity (sotrastaurin) inhibited SE-induced drug resistance. The response was selective to the PKC pathway, because inhibition of MEK (which has been implicated in HDACi resistance61) was not sufficient to counteract SE-induced drug resistance. These differences between cancer and cell models suggest that HDACi resistance may be dependent on both cell-intrinsic features and factors in the tumor microenvironment.
Our CITE-seq data showed that SE enhanced STAT3 activation in HDACi-treated cultures. JAK/STAT signaling is also induced by cytokines including the IL-2 family, which stimulate JAK-dependent, STAT–mediated cell survival in malignant cells from patients with SS.38,68,69,71 It also showed that SE-induced expression of cytokines in malignant and nonmalignant T cells as confirmed by our analysis of secreted cytokines. Indeed, our results show that IL-2 family cytokines provided survival signals, which protected malignant cells from drug-induced apoptosis. These findings provide first evidence that cytokines, which are expressed in the tumor microenvironment,80 may play a role in drug resistance in patients with CTCL. Surprisingly, JAK-STAT activation was only necessary for SE-induced drug resistance in some patients but not all. As illustrated in Figure 7, these findings suggested that 2 pathways are involved: (1) direct TCR-dependent, JAK-independent pathway mediated through PKC-dependent NF-κB signaling (Figure 7, upper part); and (2) an indirect JAK-dependent pathway that relies on SE-induced cytokine production in bystander T cells or the malignant cells themselves in an auto/paracrine fashion (Figure 7, lower part). Experiments with sorted malignant cells confirmed that SE could induce drug resistance even in highly enriched cultures from both JAK-dependent and -independent malignant populations. At present, it is not known why different responses were observed in malignant T cells from different patients. Because SE can either directly (via TCRVβ interaction) or indirectly (via bystander T cells) activate malignant cells,68,81,82 this may explain why induction of drug resistance in malignant cells from some patients relies on cytokine signaling, which can be blocked by tofacitinib, whereas others do not. Interestingly, a study in chronic lymphocytic leukemia showed IL-6 induced resistance to vorinostat through STAT3 activation, and inhibition of STAT3 reversed vorinostat resistance.83 Several other lines of evidence support that STAT3 plays a role in HDACi resistance: (1) STAT3 phosphorylation was associated with vorinostat resistance,84 (2) JAK inhibition boosted the efficacy of romidepsin,85 and (3) Sézary cells carrying the constitutively active variant of STAT3 (Y640F) were less sensitive to romidepsin-induced apoptosis.86 Importantly, eradication of SE-producing S aureus by antibiotics inhibited STAT3 activation in vivo in patients with CTCL.35 Taken together, these findings support the hypothesis that SE-producing S aureus can induce STAT3-dependent resistance to drugs such as HDACi.
Figure 7.

S aureus induces drug resistance in malignant T cells through direct and indirect TCR activation.S aureus can induce drug resistance in malignant T cells through secretion of SE using 2 distinct pathways: (1) direct TCR-dependent, JAK-independent pathway mediated through PKC-dependent NF-κB signaling (upper part), and (2) an indirect JAK-dependent pathway, which relies on SE-induced cytokine production in bystander T cells or the malignant cells themselves in an auto/paracrine fashion (lower part).
Although we find a clear effect in patient with advanced SS, larger controlled clinical studies are warranted to establish the overall clinical implications of S aureus in CTCL and particularly in relation to disease stage, subtype, microbiome, and ethnic/geographical factors.87 Of note, Wang et al recently reported on disease-stage–dependent increased skin colonization by S aureus in a large cohort of Chinese patients with CTCL.88 Importantly, many patients harbored SE-producing S aureus confirming and extending the milestone study by Duvic et al.31 These data suggest that S aureus–mediated induction of drug resistance is a global problem in relation to the treatment of patients with CTCL.89
In conclusion, we demonstrate that S aureus and its toxins induce drug resistance in malignant T cells. These findings provide one of the missing links between S aureus and cancer and provide an explanation of how skin colonization by SE-producing bacteria can fuel disease activity in CTCL. Moreover, our results highlight the need to eradicate and prevent S aureus skin colonization in addition to targeting the malignant cells in patients with CTCL.
Conflict-of-interest disclosure: C.K.V., N.Ø., and T.B.B. are listed as inventors on a patent application related to findings in this study (patent application EP23207309.8). N.Ø. has received consulting honoraria from Mindera Corp, Micreos Human Health, PS Consulting, and Almirall. E.M.H.P is currently employed at Novo Nordisk A/S. T.L. is funded by LEO Pharma. S.B.K.’s laboratory has previously received funding from Micreos, Dracen Pharmaceuticals, Kymera Therapeutics, and Bristol-Myers Squibb. R.B. has received research grants from Kyowa Kirin, Takeda, and Recordati. L.I. is also an employee at MC2 Therapeutics A/S. S.E.B. reports research funding to institution from Pfizer Inc., Merck, Amgen, RenovoRx, Agio, and the Pancreatic Cancer Action Network; payment or honoraria as an advisory board member for Servier, Elmedix, Pegascy, and Ipsen; and participation on a data safety monitoring board for Acrivon. K.W. has participated in advisory boards or lectures for Astra Zeneca, Galderma, and Kyowa Kirin. A.O. has participated in advisory boards or lectures for AbbVie, Celgene/Amgen, Eli Lilly, Novartis, Pfizer, Meda, UCB Pharma, Janssen Cilag, Allmiral, Kyowa Kirin, Sanofi, Bristol-Myers Squibb, Theracos, Recordati Rare Diseases, and Leo Pharma. M.W. received honoraria and participated in advisory boards of Takeda, Kyowa Kirin, Stemline Therapeutics, and Recordati Rare Diseases. The remaining authors declare no competing financial interests.
Acknowledgments
The authors are grateful for the patients who participated and provided samples for this study. The authors are grateful for their fruitful discussions and collaboration with Bob de Rooij and Wouter Eijkelkamp at Micreos Pharmaceuticals AG (Baar, Switzerland) and the generous donation of endolysin XZ.700 from Micreos Pharmaceuticals. The authors thank Thomas Leanderson (Lund University, Lund, Sweden), Gunnar Hedlund (Immunopoint Consulting AB, Lund, Sweden), and Karin Leanderson (Lund University) for the generous donation of SEAwt and SEAF47A/D227A. The authors also thank Sana Ahmad for her excellent technical assistance. The authors thank the Core Facility for flow cytometry and single-cell analysis and Faculty of Health and Medical Sciences, University of Copenhagen, for their technical assistance with flow cytometry and CITE-seq experiments. The visual abstract and Figure 7 were created with BioRender.com.
This research was funded by LEO Foundation through the LEO Foundation Skin Immunology Research Center, LEO Foundation grant (LF-OC-20-000351; S.B.K.), the Danish Cancer Society (Kræftens Bekæmpelse; C.K.V. and N.Ø.) the Fight Cancer Program (Knæk Cancer), Novo Nordisk Research Foundation, Novo Nordisk Foundation Tandem Program, Lundbeck Foundation (A.W.-O.), the Danish Council for Independent Research (Danmarks Frie Forskningsfond, 2 project grants; N.Ø.), LINAK A/S, Nordborg, Aage Bangs Foundation, and National Cancer Institute–National Institutes of Health (R01CA271245; S.B.K.). The funding source had no influence on the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Authorship
Contribution: C.K.V., N.Ø., and T.B.B. conceived and designed the study; C.K.V., A.W.-O., and T.B.B. performed experiments; C.K.V. and T.B.B made figures; C.K.V. drafted the manuscript with inputs from N.Ø. and T.B.B.; N.Ø. obtained the funding; C.K.V. managed the study with supervision from N.Ø. and T.B.B.; M.R.J.N, Z.Z., L.Y., M.D., M.G., E.M.H.P., K.W., A.O., S.H., Y.-T.C., L.M.L., S.B.K., L.J.G., T.L., S.E.B., L.I., R.B., M.W, E.G., and M.R.K. provided administrative, technical, or material support; and all authors performed analysis or interpretation of data and critical revision of the manuscript for important intellectual content.
Footnotes
Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) data generated in this study have been deposited to FigShare under https://doi.org/10.6084/m9.figshare.23649780. CITE-seq data used in reanalysis of previously published cohort are deposited at Gene Expression Omnibus database (accession number GSE171811). Code used for analysis of CITE-seq data and figures is available at: https://github.com/Terkild/CTCL_SE_drug_resistance.
All other data are available upon reasonable request from the corresponding authors, Terkild B. Buus (terkild.buus@sund.ku.dk) and Niels Ødum (ndum@sund.ku.dk).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Niels Ødum, Email: ndum@sund.ku.dk.
Terkild B. Buus, Email: terkild.buus@sund.ku.dk.
Supplementary Material
References
- 1.Sézary A, Bouvrain Y. Erythrodermie avec présence de cellules monstrueuses dans le derme et le sang circulant. Bull Soc Fr Dermatol Syphiligr. 1938;45:254–260. [Google Scholar]
- 2.Brouet JC, Flandrin G, Seligmann M. Indications of the thymus-derived nature of the proliferating cells in six patients with Sézary's syndrome. N Engl J Med. 1973;289(7):341–344. doi: 10.1056/NEJM197308162890703. [DOI] [PubMed] [Google Scholar]
- 3.Dummer R, Vermeer MH, Scarisbrick JJ, et al. Cutaneous T cell lymphoma. Nat Rev Dis Primers. 2021;7(1):61. doi: 10.1038/s41572-021-00296-9. [DOI] [PubMed] [Google Scholar]
- 4.Lutzner M, Edelson R, Schein P, Green I, Kirkpatrick C, Ahmed A. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83(4):534–552. doi: 10.7326/0003-4819-83-4-534. [DOI] [PubMed] [Google Scholar]
- 5.Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004;350(19):1978–1988. doi: 10.1056/NEJMra032810. [DOI] [PubMed] [Google Scholar]
- 6.Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome - clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139(7):857–866. doi: 10.1001/archderm.139.7.857. [DOI] [PubMed] [Google Scholar]
- 7.Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011–1019. doi: 10.1038/ng.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bastidas Torres AN, Cats D, Mei H, et al. Genomic analysis reveals recurrent deletion of JAK-STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer. 2018;57(12):653–664. doi: 10.1002/gcc.22679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGirt LY, Jia P, Baerenwald DA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015;126(4):508–519. doi: 10.1182/blood-2014-11-611194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song X, Chang S, Seminario-Vidal L, et al. Genomic and single-cell landscape reveals novel drivers and therapeutic vulnerabilities of transformed cutaneous T-cell lymphoma. Cancer Discov. 2022;12(5):1294–1313. doi: 10.1158/2159-8290.CD-21-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tensen CP, Quint KD, Vermeer MH. Genetic and epigenetic insights into cutaneous T-cell lymphoma. Blood. 2022;139(1):15–33. doi: 10.1182/blood.2019004256. [DOI] [PubMed] [Google Scholar]
- 12.Weiner DM, Durgin JS, Wysocka M, Rook AH. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: current and future approaches. J Am Acad Dermatol. 2021;84(3):597–604. doi: 10.1016/j.jaad.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel VM, Flanagan CE, Martins M, et al. Frequent and persistent PLCG1 mutations in Sézary cells directly enhance PLCgamma1 activity and stimulate NFkappaB, AP-1, and NFAT signaling. J Invest Dermatol. 2020;140(2):380–389.e384. doi: 10.1016/j.jid.2019.07.693. [DOI] [PubMed] [Google Scholar]
- 14.Sors A, Jean-Louis F, Pellet C, et al. Down-regulating constitutive activation of the NF-kappaB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood. 2006;107(6):2354–2363. doi: 10.1182/blood-2005-06-2536. [DOI] [PubMed] [Google Scholar]
- 15.Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle. 2014;13(21):3331–3335. doi: 10.4161/15384101.2014.965061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Nowak I, Vonderheid EC, et al. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sézary syndrome. Proc Natl Acad Sci U S A. 1996;93(17):9148–9153. doi: 10.1073/pnas.93.17.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen M, Kaltoft K, Nordahl M, et al. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: Tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci U S A. 1997;94(13):6764–6769. doi: 10.1073/pnas.94.13.6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorger H, Dey S, Vieyra-Garcia PA, et al. Blocking STAT3/5 through direct or upstream kinase targeting in leukemic cutaneous T-cell lymphoma. EMBO Mol Med. 2022;14(12) doi: 10.15252/emmm.202115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Axelrod PI, Lorber B, Vonderheid EC. Infections complicating mycosis fungoides and Sézary syndrome. JAMA. 1992;267(10):1354–1358. [PubMed] [Google Scholar]
- 20.Mirvish ED, Pomerantz RG, Geskin LJ. Infectious agents in cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;64(2):423–431. doi: 10.1016/j.jaad.2009.11.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Seminario-Vidal L, Cohen L, et al. Alterations in the skin microbiota are associated with symptom severity in mycosis fungoides. Front Cell Infect Microbiol. 2022;12 doi: 10.3389/fcimb.2022.850509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hooper MJ, LeWitt TM, Veon FL, et al. Nasal dysbiosis in cutaneous T-cell lymphoma is characterized by shifts in relative abundances of non-Staphylococcus bacteria. JID Innov. 2022;2(5) doi: 10.1016/j.xjidi.2022.100132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen PB, Switchenko J, Ayers A, Kim E, Lechowicz MJ. Risk of bacteremia in patients with cutaneous T-cell lymphoma (CTCL) Leuk Lymphoma. 2020;61(11):2652–2658. doi: 10.1080/10428194.2020.1779259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posner LE, Fossieck BE, Jr., Eddy JL, Bunn PA., Jr. Septicemic complications of the cutaneous T-cell lymphomas. Am J Med. 1981;71(2):210–216. doi: 10.1016/0002-9343(81)90107-8. [DOI] [PubMed] [Google Scholar]
- 25.Bonin S, Tothova SM, Barbazza R, Brunetti D, Stanta G, Trevisan G. Evidence of multiple infectious agents in mycosis fungoides lesions. Exp Mol Pathol. 2010;89(1):46–50. doi: 10.1016/j.yexmp.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Willerslev-Olsen A, Krejsgaard T, Lindahl LM, et al. Bacterial toxins fuel disease progression in cutaneous T-cell lymphoma. Toxins (Basel) 2013;5(8):1402–1421. doi: 10.3390/toxins5081402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dehner CA, Ruff WE, Greiling T, et al. Malignant T cell activation by a Bacillus species isolated from cutaneous T-cell lymphoma lesions. JID Innov. 2022;2(2) doi: 10.1016/j.xjidi.2021.100084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gluud M, Pallesen EMH, Buus TB, et al. Malignant T cells induce skin barrier defects through cytokine-mediated JAK/STAT signaling in cutaneous T-cell lymphoma. Blood. 2023;141(2):180–193. doi: 10.1182/blood.2022016690. [DOI] [PubMed] [Google Scholar]
- 29.Thode C, Woetmann A, Wandall HH, et al. Malignant T cells secrete galectins and induce epidermal hyperproliferation and disorganized stratification in a skin model of cutaneous T-cell lymphoma. J Invest Dermatol. 2015;135(1):238–246. doi: 10.1038/jid.2014.284. [DOI] [PubMed] [Google Scholar]
- 30.Suga H, Sugaya M, Miyagaki T, et al. Skin barrier dysfunction and low antimicrobial peptide expression in cutaneous T-cell lymphoma. Clin Cancer Res. 2014;20(16):4339–4348. doi: 10.1158/1078-0432.CCR-14-0077. [DOI] [PubMed] [Google Scholar]
- 31.Jackow CM, Cather JC, Hearne V, Asano AT, Musser JM, Duvic M. Association of erythrodermic cutaneous T-cell lymphoma, superantigen-positive Staphylococcus aureus, and oligoclonal T-cell receptor V beta gene expansion. Blood. 1997;89(1):32–40. [PubMed] [Google Scholar]
- 32.Tokura Y, Yagi H, Ohshima A, et al. Cutaneous colonization with staphylococci influences the disease activity of Sézary syndrome: a potential role for bacterial superantigens. Br J Dermatol. 1995;133(1):6–12. doi: 10.1111/j.1365-2133.1995.tb02485.x. [DOI] [PubMed] [Google Scholar]
- 33.Emge DA, Bassett RL, Duvic M, Huen AO. Methicillin-resistant Staphylococcus aureus is an important pathogen in erythrodermic cutaneous T-cell lymphoma patients. J Am Acad Dermatol. 2019;81(4) doi: 10.1007/s00403-019-02015-7. Ab94-Ab94. [DOI] [PubMed] [Google Scholar]
- 34.Talpur R, Bassett R, Duvic M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br J Dermatol. 2008;159(1):105–112. doi: 10.1111/j.1365-2133.2008.08612.x. [DOI] [PubMed] [Google Scholar]
- 35.Lindahl LM, Willerslev-Olsen A, Gjerdrum LMR, et al. Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood. 2019;134(13):1072–1083. doi: 10.1182/blood.2018888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duvic M. Current treatment of cutaneous T-cell lymphoma. Dermatol Online J. 2001;7(1):3. [PubMed] [Google Scholar]
- 37.Luchenko VL, Litman T, Chakraborty AR, et al. Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin. Mol Oncol. 2014;8(8):1379–1392. doi: 10.1016/j.molonc.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pallesen EMH, Gluud M, Vadivel CK, et al. Endolysin inhibits skin colonization by patient-derived Staphylococcus aureus and malignant T cell activation in cutaneous T cell lymphoma. J Invest Dermatol. 2023;143(9):1757–1768.e3. doi: 10.1016/j.jid.2023.01.039. [DOI] [PubMed] [Google Scholar]
- 39.Eichenseher F, Herpers BL, Badoux P, et al. Linker-improved chimeric endolysin selectively kills Staphylococcus aureus in vitro, on reconstituted human epidermis, and in a murine model of skin infection. Antimicrob Agents Chemother. 2022;66(5) doi: 10.1128/aac.02273-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuiper JWP, Hogervorst JMA, Herpers BL, et al. The novel endolysin XZ.700 effectively treats MRSA biofilms in two biofilm models without showing toxicity on human bone cells in vitro. Biofouling. 2021;37(2):184–193. doi: 10.1080/08927014.2021.1887151. [DOI] [PubMed] [Google Scholar]
- 41.Vadivel CK, Gluud M, Torres-Rusillo S, et al. JAK3 is expressed in the nucleus of malignant T cells in cutaneous T cell lymphoma (CTCL) Cancers. 2021;13(2) doi: 10.3390/cancers13020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buus TB, Herrera A, Ivanova E, et al. Improving oligo-conjugated antibody signal in multimodal single-cell analysis. Elife. 2021;10 doi: 10.7554/eLife.61973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehindate K, Thibodeau J, Dohlsten M, Kalland T, Sékaly RP, Mourad W. Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression. J Exp Med. 1995;182(5):1573–1577. doi: 10.1084/jem.182.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrera A, Cheng A, Mimitou EP, et al. Multimodal single-cell analysis of cutaneous T-cell lymphoma reveals distinct subclonal tissue-dependent signatures. Blood. 2021;138(16):1456–1464. doi: 10.1182/blood.2020009346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Y, Su MW, Jiang X, Zhou Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T-cell lymphoma. Blood. 2015;125(9):1435–1443. doi: 10.1182/blood-2014-05-571778. [DOI] [PubMed] [Google Scholar]
- 46.Hurabielle C, Thonnart N, Ram-Wolff C, et al. Usefulness of KIR3DL2 to diagnose, follow-up, and manage the treatment of patients with Sézary syndrome. Clin Cancer Res. 2017;23(14):3619–3627. doi: 10.1158/1078-0432.CCR-16-3185. [DOI] [PubMed] [Google Scholar]
- 47.Vonderheid EC, Bigler RD, Kotecha A, et al. Variable CD7 expression on T cells in the leukemic phase of cutaneous T cell lymphoma (Sézary syndrome) J Invest Dermatol. 2001;117(3):654–662. doi: 10.1046/j.1523-1747.2001.01456.x. [DOI] [PubMed] [Google Scholar]
- 48.Jones D, Dang NH, Duvic M, Washington LT, Huh YO. Absence of CD26 expression is a useful marker for diagnosis of T-cell lymphoma in peripheral blood. Am J Clin Pathol. 2001;115(6):885–892. doi: 10.1309/U1Y6-J4AG-5M4M-7AYV. [DOI] [PubMed] [Google Scholar]
- 49.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 50.Resminostat for maintenance treatment of patients with advanced stage mycosis fungoides (MF) or Sézary syndrome (SS). ClinicalTrials.gov identifier: NCT02953301. https://ClinicalTrials.gov/show/NCT02953301 Updated 6 September 2023.
- 51.Wollina U, Dummer R, Brockmeyer NH, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer. 2003;98(5):993–1001. doi: 10.1002/cncr.11593. [DOI] [PubMed] [Google Scholar]
- 52.Purnak S, Azar J, Mark LA. Etoposide as a single agent in the treatment of mycosis fungoides: a retrospective analysis. Dermatol Ther. 2018;31(2):e12586. doi: 10.1111/dth.12586. [DOI] [PubMed] [Google Scholar]
- 53.Zinzani PL, Musuraca G, Tani M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(27):4293–4297. doi: 10.1200/JCO.2007.11.4207. [DOI] [PubMed] [Google Scholar]
- 54.Westergaard SA, Lechowicz MJ, Harrington M, Elsey J, Arbiser JL, Khan MK. Induction of remission in a patient with end-stage cutaneous T-cell lymphoma by concurrent use of radiation therapy, gentian violet, and mogamulizumab. JAAD Case Rep. 2020;6(8):761–765. doi: 10.1016/j.jdcr.2020.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J, Wood GS. Analysis of the effect of gentian violet on apoptosis and proliferation in cutaneous T-cell lymphoma in an in vitro study. JAMA Dermatol. 2018;154(10):1191–1198. doi: 10.1001/jamadermatol.2018.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang TP, Vancurova I. NFkappaB function and regulation in cutaneous T-cell lymphoma. Am J Cancer Res. 2013;3(5):433–445. [PMC free article] [PubMed] [Google Scholar]
- 57.Paul S, Schaefer BC. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013;34(6):269–281. doi: 10.1016/j.it.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci. 2018;43(2):108–123. doi: 10.1016/j.tibs.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schade AE, Schieven GL, Townsend R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111(3):1366–1377. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.A dose finding study of AEB071 assessing psoriasis area and severity index in patients with plaque psoriasis. ClinicalTrials.gov identifier: NCT00885196. https://ClinicalTrials.gov/show/NCT00885196 Updated 22 December 2020.
- 61.Chakraborty AR, Robey RW, Luchenko VL, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121(20):4115–4125. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 63.Hammarén HM, Virtanen AT, Raivola J, Silvennoinen O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 2019;118:48–63. doi: 10.1016/j.cyto.2018.03.041. [DOI] [PubMed] [Google Scholar]
- 64.Meyer DM, Jesson MI, Li X, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm. 2010;7:41. doi: 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borcherding N, Voigt AP, Liu V, Link BK, Zhang W, Jabbari A. Single-cell profiling of cutaneous T-cell lymphoma reveals underlying heterogeneity associated with disease progression. Clin Cancer Res. 2019;25(10):2996–3005. doi: 10.1158/1078-0432.CCR-18-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buus TB, Willerslev-Olsen A, Fredholm S, et al. Single-cell heterogeneity in Sézary syndrome. Blood Adv. 2018;2(16):2115–2126. doi: 10.1182/bloodadvances.2018022608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaydosik AM, Tabib T, Geskin LJ, et al. Single-cell lymphocyte heterogeneity in advanced cutaneous T-cell lymphoma skin tumors. Clin Cancer Res. 2019;25(14):4443–4454. doi: 10.1158/1078-0432.CCR-19-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woetmann A, Lovato P, Eriksen KW, et al. Nonmalignant T cells stimulate growth of T-cell lymphoma cells in the presence of bacterial toxins. Blood. 2007;109(8):3325–3332. doi: 10.1182/blood-2006-04-017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willerslev-Olsen A, Gjerdrum LMR, Lindahl LM, et al. Staphylococcus aureus induces signal transducer and activator of transcription 5‒dependent miR-155 expression in cutaneous T-cell lymphoma. J Invest Dermatol. 2021;141(10):2449–2458. doi: 10.1016/j.jid.2021.01.038. [DOI] [PubMed] [Google Scholar]
- 70.Willerslev-Olsen A, Buus TB, Nastasi C, et al. Staphylococcus aureus enterotoxins induce FOXP3 in neoplastic T cells in Sézary syndrome. Blood Cancer J. 2020;10(5):57. doi: 10.1038/s41408-020-0324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willerslev-Olsen A, Krejsgaard T, Lindahl LM, et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood. 2016;127(10):1287–1296. doi: 10.1182/blood-2015-08-662353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krejsgaard T, Willerslev-Olsen A, Lindahl LM, et al. Staphylococcal enterotoxins stimulate lymphoma-associated immune dysregulation. Blood. 2014;124(5):761–770. doi: 10.1182/blood-2014-01-551184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13(24):7237–7242. doi: 10.1158/1078-0432.CCR-07-2114. [DOI] [PubMed] [Google Scholar]
- 74.Chiao PJ, Na R, Niu J, Sclabas GM, Dong Q, Curley SA. Role of Rel/NF-kappaB transcription factors in apoptosis of human hepatocellular carcinoma cells. Cancer. 2002;95(8):1696–1705. doi: 10.1002/cncr.10829. [DOI] [PubMed] [Google Scholar]
- 75.Abdin SM, Tolba MF, Zaher DM, Omar HA. Nuclear factor-κB signaling inhibitors revert multidrug-resistance in breast cancer cells. Chem Biol Interact. 2021;340 doi: 10.1016/j.cbi.2021.109450. [DOI] [PubMed] [Google Scholar]
- 76.Morotti A, Cilloni D, Pautasso M, et al. NF-kB inhibition as a strategy to enhance etoposide-induced apoptosis in K562 cell line. Am J Hematol. 2006;81(12):938–945. doi: 10.1002/ajh.20732. [DOI] [PubMed] [Google Scholar]
- 77.Meley D, Spiller DG, White MR, McDowell H, Pizer B, See V. p53-mediated delayed NF-kappaB activity enhances etoposide-induced cell death in medulloblastoma. Cell Death Dis. 2010;1(5):e41. doi: 10.1038/cddis.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertolotto C, Maulon L, Filippa N, Baier G, Auberger P. Protein kinase C theta and epsilon promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J Biol Chem. 2000;275(47):37246–37250. doi: 10.1074/jbc.M007732200. [DOI] [PubMed] [Google Scholar]
- 79.Cheng J, Montecalvo A, Kane LP. Regulation of NF-kappaB induction by TCR/CD28. Immunol Res. 2011;50(2-3):113–117. doi: 10.1007/s12026-011-8216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abraham RM, Zhang Q, Odum N, Wasik MA. The role of cytokine signaling in the pathogenesis of cutaneous T-cell lymphoma. Cancer Biol Ther. 2011;12(12):1019–1022. doi: 10.4161/cbt.12.12.18144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tokura Y, Heald PW, Yan SL, Edelson RL. Stimulation of cutaneous T-cell lymphoma-cells with superantigenic staphylococcal toxins. J Invest Dermatol. 1992;98(1):33–37. doi: 10.1111/1523-1747.ep12494184. [DOI] [PubMed] [Google Scholar]
- 82.Vonderheid EC, Bigler RD, Hou JS. On the possible relationship between staphylococcal superantigens and increased Vbeta5.1 usage in cutaneous T-cell lymphoma. Br J Dermatol. 2005;152(4):825–826. doi: 10.1111/j.1365-2133.2005.06524.x. author reply 827. [DOI] [PubMed] [Google Scholar]
- 83.Lu K, Fang XS, Feng LL, et al. The STAT3 inhibitor WP1066 reverses the resistance of chronic lymphocytic leukemia cells to histone deacetylase inhibitors induced by interleukin-6. Cancer Lett. 2015;359(2):250–258. doi: 10.1016/j.canlet.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 84.Fantin VR, Loboda A, Paweletz CP, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–3794. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- 85.Cortes JR, Patrone CC, Quinn SA, et al. Jak-STAT inhibition mediates romidepsin and mechlorethamine synergism in cutaneous T-cell lymphoma. J Invest Dermatol. 2021;141(12):2908–2920.e7. doi: 10.1016/j.jid.2021.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Butler RM, McKenzie RC, Jones CL, et al. Contribution of STAT3 and RAD23B in primary Sézary cells to histone deacetylase inhibitor FK228 resistance. J Invest Dermatol. 2019;139(9):1975–1984.e2. doi: 10.1016/j.jid.2019.03.1130. [DOI] [PubMed] [Google Scholar]
- 87.Vermeer MH. Antibiotics can improve CTCL. Blood. 2019;134(13):1000–1001. doi: 10.1182/blood.2019002236. [DOI] [PubMed] [Google Scholar]
- 88.Liu X, Sun J, Gao Y, et al. Characteristics of Staphylococcus aureus colonization in cutaneous T-cell lymphoma. J Invest Dermatol. 2024;144(1):188–191. doi: 10.1016/j.jid.2023.06.205. [DOI] [PubMed] [Google Scholar]
- 89.Guenova E, Ødum N. Old sins cast long shadows: news on Staphylococcus aureus in cutaneous T cell lymphoma. J Invest Dermatol. 2024;144(1):8–10. doi: 10.1016/j.jid.2023.08.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.