
Fig. 1.
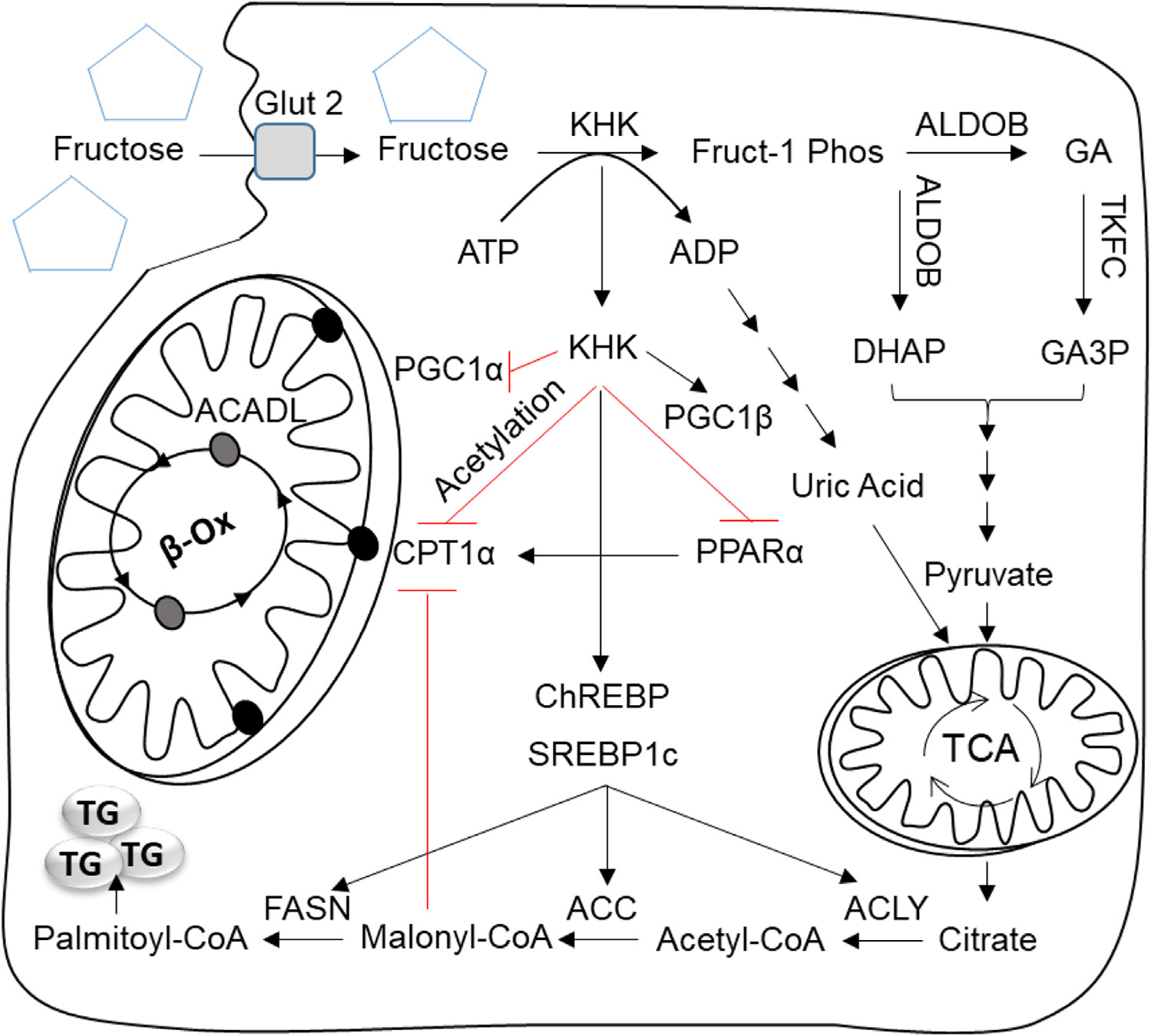
The effect of fructose on lipogenesis and mitochondrial oxidation. In the liver fructose is taken up by solute carrier family 2, facilitated glucose transporter, member 2 (SLC2A2 aka GLUT2) and phosphorylated by ketohexokinase (KHK) to fructose-1 phosphate (Fruct-1 Phos). KHK mediated fructose phosphorylation is rapid leading to depletion of adenosine triphosphate (ATP), accumulation of adenosine diphosphate (ADP) and eventually production of uric acid. Fruct-1 Phos is further metabolized by aldolase b (ALDOB) into dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA). GA is then phosphorylated by triokinase and FMN cyclase (TKFC) into glyceraldehyde-3 phosphate (GA3P). DHAP and GA3P are also intermediates of glycolysis pathway and they are metabolized into pyruvate. Pyruvate enters the tricarboxylic acid cycle (TCA) to produce electrons for energy production. When the cellular energy stores are plentiful citrate is transported out of mitochondria into the cytosol to serve as a substrate for de novo lipogenesis (DNL). Uric acid produced by fructose metabolism inhibits aconites, an enzyme in TCA cycle, to further increase citrate production. In the cytosol, citrate is converted to acetyl-CoA by the enzyme ATP citrate lyase (ACLY). Acetyl-CoA carboxylase (ACC1), then catalyzes the production of malonyl-CoA and fatty acid synthase (FASN) extends the growing fatty acid chain to synthesize palmitoyl-CoA, a major building block of triglyceride (TG) formation. Malonyl-CoA, an intermediate in DNL also inhibits carnitine palmitoyltransferase 1 alpha (CPT1α) the rate limiting enzyme of mitochondrial fatty acid oxidation (FAO). In addition to providing substrate for DNL fructose through KHK affects the function of transcription factors mediating DNL and FAO. KHK and carbohydrate responsive element binding protein (ChREBP) regulate each other via a bidirectional loop, and upregulated ChREBP increases the expression of genes involved in DNL. Fructose through KHK also upregulates another lipogenic transcription factor, sterol regulatory element-binding protein 1c (SREBP1c), either directly via PPARG coactivator 1 beta (PGC1β) or indirectly by inducing selective insulin resistance and hyperinsulinemia. We present additional evidence that fructose through KHK decreases FAO. These effects are mediated, in part, via lower peroxisome proliferator activated receptor alpha (PPARα), a transcription factor that regulates expression of FAO enzymes, leading to decreased CTP1α protein. Additionally, fructose decreases PGC1α, a cofactor required for optimal PPARα activity via increasing PGC1α acetylation. Lastly, our research indicates that fructose through KHK increases CPT1α acetylation and decreases its protein stability.