

Abstract
Resistance arteries and arterioles evolved as specialized blood vessels serving two important functions: (a) regulating peripheral vascular resistance and blood pressure and (b) matching oxygen and nutrient delivery to metabolic demands of organs. These functions require control of vessel lumen cross-sectional area (vascular tone) via coordinated vascular cell responses governed by precise spatial-temporal communication between intracellular signaling pathways. Herein, we provide a contemporary overview of the significant roles that redox switches play in calcium signaling for orchestrated endothelial, smooth muscle, and red blood cell control of arterial vascular tone. Three interrelated themes are the focus: (a) smooth muscle to endothelial communication for vasoconstriction, (b) endothelial to smooth muscle cell cross talk for vasodilation, and (c) oxygen and red blood cell in-terregulation of vascular tone and blood flow. We intend for this thematic framework to highlight gaps in our current knowledge and potentially spark interest for cross-disciplinary studies moving forward.
Keywords: endothelial, smooth muscle, red blood cell, redox, calcium, microcirculation
INTRODUCTION
The microcirculation encompasses a collection of small-diameter blood vessels: resistance arteries (50–200 μm), arterioles (20–50 μm), meta-arterioles branching to precapillary sphincters (10–20 μm), capillaries (<10 μm), and venules (10–250 μm). Resistance arteries and arterioles, the focus of this review, have two essential functions: (a) to govern peripheral vascular resistance and blood pressure and (b) modulate blood flow to capillaries for nutrient and oxygen (O2) delivery and waste product removal (e.g., CO2). Anatomically, resistance arteries and arterioles contain a single layer of endothelial cells (ECs) lining the vessel lumen and perpendicularly oriented contractile vascular smooth muscle cells (VSMCs) in single or multiple layers. ECs and VSMCs sandwich a thin internal elastic lamina matrix, which provides elasticity and adhesivity for cells. While resistance arteries and arterioles are surrounded by a thick VSMC wall, capillaries are encircled by thin multifunctional mural cells called pericytes, which have some contractile properties within certain organ systems (1). Maintenance of homeostatic blood pressure and blood flow in resistance arteries and arterioles requires constant modulation of vessel lumen cross-sectional area or vascular tone. Endocrine, paracrine, and autocrine signals continually trigger vascular tone adaptations to changing conditions within the organism. This constant on-demand control of vascular tone relies on direct cross-communication between ECs and VSMCs via input signals from red blood cells (RBCs), sympathetic nerves, and other circulating cells. Notably, resistance arteries and arterioles have evolved unique anatomical structures called myoendothelial junctions (MEJs) that protrude through the internal elastic lamina with diameters that are typically ~0.5 μm. MEJs directly connect ECs and VSMCs, serving as essential communication hubs for rapid electrical signaling and exchange of second messengers that regulate vascular tone in a highly organized and spatial-temporal fashion (2, 3). The density of MEJs inversely correlates with the luminal size of resistance arteries and arterioles (3). A central regulator of vascular tone is cytosolic Ca2+ ([Ca2+]cyto), which is essential for EC and VSMC function, albeit in a contrasting manner. In VSMCs, elevated [Ca2+]cyto causes vasoconstriction, and Ca2+ sparks generally cause vasodilation. By contrast, increases in EC [Ca2+]cyto elicit vasodilation. Vascular Ca2+ homeostasis is tightly controlled by redox signaling and redox switches that enable fine-tuning of [Ca2+]cyto signaling pathways for organ-specific vascular tone regulation. Redox switches arise from natural or imbalanced oxidative posttranslational modifications to proteins with metal centers or thiol bases, affecting pathways for folding, localization, quality control, and degradation of proteins. Importantly, redox switches represent rapid, low-energy mechanisms of sensing and regulation of cellular homeostasis or defense. Over the past decade, we and others have come to appreciate that vascular cells rely on precision redox signaling to provide a stable and efficient microenvironment for the right reactive oxygen species (ROS) and reactive nitrogen species (RNS), i.e., the right location, the right timing, the right level, and the right target (4). In this review, we aim to highlight important contributions made to the field by summarizing key contributions of redox switches to Ca2+ signaling and vascular tone control.
REACTIVE NITROGEN SPECIES/REACTIVE OXYGEN SPECIES: ORIGIN, REGULATION, AND TARGETS
RNS are continually produced in ECs and VSMCs, signaling at volumes determined by physiological and pathophysiological conditions. Endogenous RNS mostly originate from nitric oxide (NO) produced by neuronal, inducible, and endothelial nitric oxide synthase (eNOS) activity (5). NO is a lipophilic, membrane-permeable molecule that participates in near diffusion-limited reactions regulating physiological functions, including intestinal motility, platelet aggregation, proliferation, apoptosis, and vascular tone (6). NO is a free radical owing to its unpaired electron, making it highly reactive with other radicals and transition metals like ferrous iron–containing hemoproteins. NO exerts biological actions primarily through three cellular signaling cascades: (a) activation of its receptor soluble guanylyl cyclase (sGC), catalyzing the generation of cyclic guanosine 3′,5′-cyclic monophosphate (cGMP) for stimulating protein kinase G (PKG); (b) radical–radical reactions with superoxide, nitrogen dioxide, and lipid radicals, yielding RNS; and (c) posttranslational modification of cysteine (Cys) residues via indirect S-nitrosation, causing protein functional changes. In subsequent sections, we highlight cGMP-dependent and cGMP-independent signaling mechanisms in resistance arteries and arterioles.
ROS, such as hydrogen peroxide (H2O2), superoxide (O2·−), and hydroxyl radical (·OH), are major oxidants with specific signaling actions (7). Two major sources of ROS are mitochondria and oxidases. Mitochondria produce ROS via electron leak in the electron transport chain (complexes I, II, and III) and α-keto acid dehydrogenase complexes (8). The membrane-bound NADPH oxidases (NOXs) also produce ROS (i.e.,O2·− and H2O2) when electrons are transferred between NADPH and O2 (9). Endogenous and exogenous ROS are also produced as by-products of extracellular xanthine oxidoreductase activity (10). In disease states, eNOS can also generate ROS (O2·−) when its reductase and oxidase domains become uncoupled (11). Iron-sulfur cluster oxidation, DNA damage, and peroxynitrite formation are consequences of O2·− reactions (12), and protein thiols and transition metals, such as iron and copper, are targeted by H2O2 (13).
Cells have evolved enzymes for controlling levels of O2·− and H2O2. There are three superoxide dismutases (SOD1–3) for catalyzing dismutation of O2·− levels to H2O2: cytosolic expressed SOD1, mitochondria-localized SOD2, and extracellular-restricted SOD3 (14). Likewise, numerous degradation systems have evolved to control H2O2. Catalase is central to catabolizing cellular H2O2 to O2 and water in a two-step reaction (15). Glutathione (GSH) can act as a redox buffer against oxidative and electrophilic stress (16).When GSH reacts with H2O2,glutathione disulfide (GSSG) is formed, and GSH levels are restored by NADH-dependent GSH reductase (17). GSH can serve as a cosubstrate for glutathione peroxidase (GPx) degradation of H2O2 into H2O (18). Peroxiredoxins (Prx) are highly expressed antioxidant enzymes that can also degrade H2O2 and alkyl peroxide in collaboration with thioredoxins (Trx) (19).
Protein redox modifications can be categorized into two groups: irreversible and reversible. Irreversible modifications leading to protein carbonyls, nitrotyrosine, and sulfonic acid generally cause protein aggregation and degradation (20). Reversible modification is involved in signaling and regulation of protein function, occurring on cysteine residues of proteins: S-sulfenation, S-nitrosation, disulfide bond formation, S-glutathionylation, and methionine sulfoxide (21). Redox cycling of transition metals (e.g., iron and copper) also occurs (22). In subsequent sections, we summarize how resistance arteries and arterioles rely on redox balance and Ca2+ signaling to control vascular tone.
SMOOTH MUSCLE TO ENDOTHELIAL COMMUNICATION AND CONTROL OF VASOCONSTRICTION
Phase 1: Redox Regulation of Calcium Signaling in Vascular Smooth Muscle Cells and Contraction
Vascular tone is largely modulated through (a) G protein–coupled receptor (GPCR) activation, (b) shear stress on ECs, and (c) intraluminal pressure. These stimuli trigger extracellular Ca2+ entry or store [sarcoplasmic or endoplasmic reticulum (SR/ER) and lysosomes] operated Ca2+ release, increasing VSMC and EC [Ca2+]cyto for calmodulin-dependent activation of myosin regulatory light chain (RLC20) phosphorylation and actin-myosin crossbridge formation (23). Simultaneously, the activation of RhoA-dependent kinase inhibits myosin light chain phosphatase from dephosphorylating RLC20, thereby sustaining VSMC contraction.
In VSMCs, Ca2+ influx occurs via plasma membrane voltage-gated and nonvoltage-gated pathways in response to mechanical, electrical, or hormonal stimuli. Electrophysiology studies identified two voltage-gated Ca2+ currents (VGCCs) in isolated VSMCs: transient (T-type) and long-lasting (L-type) (24). T-type Ca2+ channel currents (TTCCs) become activated at a lower, hyperpolarized membrane potential and decreased intraluminal pressure than L-type Ca2+ channel currents (LTCCs); however, they become inactivated faster than LTCCs (25). LTCCs consist of five different subunits (α1, α2, δ, β, and γ) (26). The α1 subunit of LTCCs is encoded by CaV1 (CaV1.1–3) genes and contain voltage-sensing, pore, and gating domains. The α1C subunit, encoded by CaV1.2, is recognized as the primary voltage-gated Ca2+ influx pathway and is key to myogenic and receptor-mediated (α1-adrenergic and angiotensin 2) vasoconstriction in VSMCs (27). Inactivation of LTCC channel activity occurs by calmodulin binding to an isoleucine-glutamate domain, preventing cytotoxicity from Ca2+ overload (28). Protein kinases (PKA, PKC, and PKG) also affect LTCC activity in VSMCs, exemplified by NO-cGMP-PKG signaling for reduced LTCC current and VSMC vasodilatation (29). The Ca2+ activated K+ (BK) channel and voltage-gated K+ channels regulate LTCC activity by stimulating membrane potential change, as shown by LTCC deactivation with membrane depolarization (30). BK channel activity is increased by NO-cGMP-PKG signaling and through S-nitrosation. Redox sensing by LTCCs also regulates its activity, as shown by increased Ca2+ influx in ischemic human heart following H2O2 oxidization of CaV1.2 Cys543 and subsequent glutathionylation, effects that were reversible by thiol reducing agents (dithiothreitol or GSH) (31).
One of the major distinctions between the two VGCCs is that, unlike LTCCs, TTCCs are not associated with auxiliary subunits (32). TTCC currents are mediated by ion-conducting α1G,α1H, and α1I subunits encoded by CaV3 (CaV3.1–3) genes, the expression of which has been detected in all arteries (33). Regulation of TTCCs occurs via protein kinases, low concentrations of metal ions (Ni2+, Zn2+), and ROS and RNS (34, 35). NO-dependent cGMP/PKG signaling inhibits TTCC currents in cerebral arteries (35). H2O2 was shown to inhibit CaV3.2 currents, likely through the oxidization of histidine 191 in the channel’s extracellular IS3–IS4 domains (36). Amelioration of CaV3.2 currents and CaV3.2-ryanodine receptor (RyR)-BK signaling, which promote vasoconstriction, occurs following angiotensin 2 activation of NADPH oxidase (36). TTCC activation and myogenic constriction have also been shown to be inhibited by NO-triggered cGMP/PKG signaling and PKA activation in rat cerebral arteries (35, 37). There is likely cross talk between the ROS- and RNS-mediated regulation of both LTCCs and TTCCs to modify vascular tone (Figure 1a).
Figure 1.
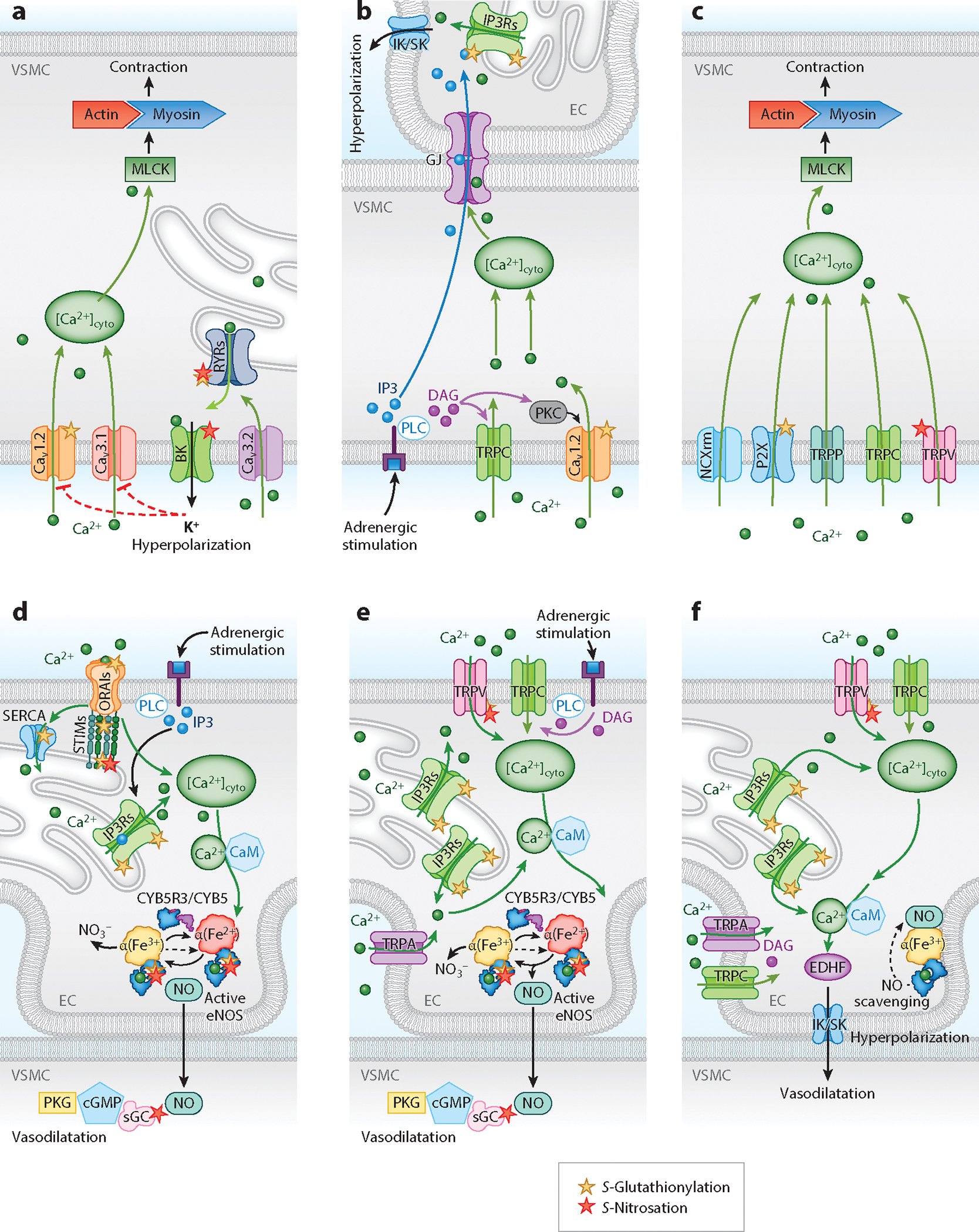
Ca2+ entry through voltage-gated Ca2+ channels (CaV1.2 and CaV3.1) triggers the VSMC contractile machine through the activation of MLCK to promote VSMC contraction. (a) Activation of CaV3.2 channels stimulates RyRs through CICR to trigger Ca2+ release from the SR/ER, potentiating BK channel–mediated hyperpolarization and vasodilatation. The red dotted line indicates inhibition of CaV1.2 and CaV3.1 channel function by hyperpolarization. (b) Adrenergic stimulation induces IP3 and DAG formation by PLC. DAG activates TRPC channels and PKC, which phosphorylates the CaV1.2 channel to increase [Ca2+]cyto and induce vasoconstriction in VSMCs. IP3 and Ca2+ can diffuse from VSMCs to the EC through the GJs of the myoendothelial junctions to increase EC [Ca2+]cyto and limit vasoconstriction by activating IK/SK channels. (c) Activation of NCXrm and P2X and TRPP, TRPC, and TRPV channels increases VSMC [Ca2+]cyto to induce vasoconstriction through the activation of MLCK/actin-myosin. (d) IP3R-mediated SR/ER Ca2+ release is induced by CICR or adrenergic stimulation through the generation of IP3 by PLC. The subsequent decrease in [Ca2+]SR/ER induces SOCE (mediated by STIM and ORAI) and ER store refilling (facilitated by the activity of SERCA) to increase [Ca2+]cyto for Ca2+–CaM-dependent activation of eNOS and generation of NO. NO diffuses to the VSMCs and causes vasodilatation through the activation of the NO-sGC-cGMP-PKG pathway. (e) Activation of TRPV, TRPC, and TRPA channels through mechanical stimuli and/or adrenergic activation of PLC and generation of DAG and subsequent activation of SR/ER Ca2+ release (IP3R) increase [Ca2+]cyto to activate eNOS and cause VSMC relaxation through the production of NO. (f) Ca2+ influx via TRPC, TRPV, and TRPA channels and/or Ca2+ release from the SR/ER through IP3Rs activates IK/SK channels via Ca2+–CaM. The activation of the IK/SK channels (EDHF) causes EC hyperpolarization and VSMC relaxation. eNOS-derived NO is scavenged by α-globin in the microcirculation. Abbreviations: Ca2+–CaM, calcium-calmodulin; [Ca2+]cyto, cytosolic Ca2+ concentration; [Ca2+]SR/ER, sarcoplasmic/endoplasmic reticulum Ca2+ concentration; CaV1.2, voltage-gated Ca2+ channel 1.2; CaV3.1, voltage-gated Ca2+ channel 3.1; cGMP, cyclic guanosine 3′,5′-cyclic monophosphate; CICR, Ca2+-induced Ca2+ release; CYB5, cytochrome B5; CYB5R3, cytochrome B5; reductase 3; DAG, diacylglycerol; EC, endothelial cell; EDHF, endothelial-derived hyperpolarizing factor; eNOS, endothelial nitric oxide synthase; GJ, gap junction; IK/SK, intermediate and small conductance Ca2+-activated K+ channels; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; MLCK, myosin light chain kinase; NCXrm, Na+/Ca2+ exchangers in reverse mode; NO, nitric oxide; ORAI, calcium release–activated calcium channel protein; P2X, purinergic P2X; PKG, protein kinase G; PLC, phospholipase C; RyR, ryanodine receptor; SERCA, SR/ER Ca2+ ATPase; sGC, soluble guanylyl cyclase; SOCE, store-operated Ca2+ entry; STIM, stromal interaction molecule; TRP, transient receptor potential channel; TRPA, ankyrin-rich protein TRP subfamily; TRPC, canonical TRP subfamily; TRPP, polycystic TRP subfamily; TRPV, vanilloid TRP subfamily; VMSC, vascular smooth muscle cell.
Signaling for VSMC contraction is also regulated by nonvoltage-gated Ca2+ channels through purinergic P2X receptor (P2XR) activation, transient receptor potential (TRP) channels, store-operated Ca2+ entry (SOCE) channels, Na+/Ca2+ exchangers in reverse mode (NCXrm), and Ca2+ release from intracellular stores (Figure 1c). Because an extensive review on the VSMC and EC Ca2+ signaling was recently published by Ottolini & Sonkusare (38), this review only covers the channels that are known or suspected to be redox regulated.
Purinergic signaling controls vascular remodeling and resistance. P2XRs are ubiquitously expressed nonselective cation channels that transfer monovalent and divalent cations released from perivascular nerve terminals at neuromuscular junctions or by pannexins in the VSMC/EC membranes (39, 40). Purinergic signaling is subject to modulation, as shown by studies wherein ATP-induced P2XR signaling increased [Ca2+]cyto in preglomerular VSMCs (41). Subsequent studies showed that H2O2 potentiated P2XR through the oxidation of its Cys430 thiol group in HEK293 cells (39).
In addition to purinergic channels, TRP channels are nonvoltage-gated cation channels regulating contractility and proliferation in VSMCs (42). Their major role is to promote global or local [Ca2+]cyto transients that induce membrane depolarization through ion channel activation. The TRP channel family consists of six subfamilies: canonical (TRPC), melastatin (TRPM), mucolipins (TRPML), vanilloid (TRPV), polycystic (TRPP), and ankyrin-rich protein (TRPA) (43). The most abundant TRP channels in VSMCs are TRPV (1 and 4), TRPC (1, 3 and 6), and TRPP (1 and 2).
Most vascular beds have TRPV channels in both VSMCs and ECs. TRPV channels generally conduct more Ca2+ than Na+ and are regulated by temperature, mechanical stimuli, or neurohormonal signals. The main endogenous regulators of TRPV1 channel activity are PKC, [Ca2+]cyto, and calcineurin (44). TRPV4 channels in VSMCs can promote both vasodilatation and vasoconstriction depending on the signaling pathways and the vascular bed (38). In VSMCs of mesenteric and cerebral arteries, TRPV4 mediated the Ca2+ influx-induced activity of RyR-BK signaling and hyperpolarization, promoting vasodilatation. TRPV4 channels have been shown to counteract angiotensin 2–induced vasoconstriction in cerebral arteries through enhancement of AKAP150 and PKCα interactions that phosphorylate TRPV4 channels in a proximity-dependent manner (45). By contrast, upregulation of TRPV4 channels in murine pulmonary arteries has been shown to enhance contractility and a hypertensive phenotype (46). These findings indicate that TRPV4 channels regulate systemic and pulmonary vasculature differently. While ROS-related activation of TRPV4 channels has been observed with H2O2-induced phosphorylation of Ser824 and inhibition by RNS through NO-induced S-nitrosation of Cys853, redox regulation of TRPV channels is more established in ECs and may be cell type dependent (47, 48).
TRPM channels are Ca2+ activated, Ca2+ impermeable, and nonselective. As cation channels, they are key regulators of pressure-induced depolarization of VSMC membranes (49). TRPMs are expressed in VSMCs and mostly transfer monovalent ions. Increased [Ca2+]cyto interacts with TRPM4 by calmodulin (CaM) binding, which facilitates channel membrane depolarization and LTCC activation in VSMCs (49). Redox modification of Cys1093 of TRPM4 channels caused reduced desensitization, possibly through increased CaM binding. H2O2 stimulated TRPM4 channels in human umbilical vein cells and through an as-yet-unknown mechanism increased its expression levels (50).
TRPC channels are nonselective cation channels with three types (TRPC1, 3, and 6) expressed in VSMCs. These channels are permeable to monovalent and divalent cations in an isoform-specific manner. TRPC3 and TRPC6 are both activated by diacylglycerol (DAG) in a PKC-independent manner (51). It has been found in cerebral arteries that the C-terminal tail of TRPC3 interacts with the N-terminal of inositol 1,4,5 trisphosphate receptor (IP3R) 1, which causes TRPC3 channel activation and membrane depolarization (52). Depolarization of VSMCs activates VGCCs, leading to vasoconstriction. For TRPC6, activity is increased by α-adrenergic stimulation and DAG-PLC signaling (Figure 1b). In the context of mechanical stimulation, TRPC6 channels can also participate in myogenic vasoconstriction. On a different note, there is controversy surrounding the role of TRPC1 and other SOCE proteins, such as stromal interaction molecule 1-calcium release–activated calcium channel protein 1 (STIM1–ORAI1), in VSMC Ca2+ signaling. TRPC1 has been reported to participate in SOCE in VSMCs, which is central to cellular Ca2+ homeostasis, and STIM–ORAI has been shown to mediate SOCE in rat aorta and pulmonary arteries. Knockdown of either STIM1 or ORAI1 has been shown to reduce SOCE and Ca2+ release–activated Ca2+ channel currents in response to Ca2+ store depletion or hypoxia, impairing VSMC migration and proliferation (53). Silencing of TRPC channels (TRPC1, 4, or 6) had no effect on SOCE in VSMCs (53). Although normal SOCE has been reported in aorta and cerebral arteries of TRPC1 null mice, anticipated induction of SOCE by thapsigargin was inhibited by STIM1 silencing (54). Additionally, native VSMCs expressed low levels of STIM–ORAI (55). In contrast, a molecular complex between TRPC1 and STIM1 was identified as the main regulator of SOCE in pulmonary arterial smooth muscle cells (56). These data suggest that the exact role of SOCE and its regulation by nonvoltage-gated channels need further study in the cardiovascular system. Redox regulation of STIM–ORAI IP3Rs by H2O2 are discussed in the next section.
Cytosolic Ca2+ transients are not only generated by transport of extracellular Ca2+ across the plasma membrane but also can be increased by second messenger-induced Ca2+ mobilization from intracellular stores by redox sensitive high-conductance cation channels (IP3Rs and RyRs). RyR is activated by Ca2+-induced Ca2+ release (CICR) or the binding of cyclic ADP ribose, and its activity is increased by thiol modification, such as S-glutathionylation and S-nitrosation (57), which can occur at the RyR Cys3635. Substitution of Cys3635 with alanine was found to cause loss of sensitivity to L-type Ca2+ channel voltage-gated activation, highlighting the redox sensor nature of Cys3635 in its role as mediator of RyR-dependent Ca2+ release.
Furthermore, a recent study proposed a close interplay between TRP channels and IP3Rs in cerebral arteries, wherein increased intraluminal pressure-induced TRPC6-mediated Ca2+ influx enhanced IP3R activity (58). SR/ER Ca2+ mobilization by IP3Rs was instrumental in activating TRPM4 channels for membrane depolarization and vasoconstriction. Ca2+ release from the SR/ER is inducible by activation of TRPML1, RyRs, or IP3Rs. Notably, α1-adrenergic signaling has been found to increase intracellular IP3, triggering [Ca2+]cyto signaling and VSMC constriction.
Phase 2: Redox Regulation of Calcium Signaling at the Myoendothelial Junction
Following receptor-dependent or -independent IP3 activation,Ca2+ release and vasoconstriction, resistance arteries and arterioles are known to subsequently release vasodilators to modulate the contractile response. A seminal study by Dora et al. (59) uncovered this phenomenon following phenylephrine or potassium chloride stimulation of arterioles, which triggered a rise in VSMC and EC calcium, leading to contraction and increased NO in arterioles. This feedback mechanism was proposed as a mechanism for arterioles to buffer the contractile response through a process thought to involve VSMC-to-EC signal transmission through the MEJ. Serving as a connective device between ECs and VSMCs, the MEJ was considered at the time to be a potential microsignaling domain due to its enrichment of ER, caveolae, and mitochondria (3). High-resolution electron microscopy revealed that EC and VSMC membranes are juxtaposed at the MEJ, opening up the possibility that gap junctions (GJs) may be positioned at these contact sites (60). Using a transwell coculture model system of ECs and VSMCs, Isakson & Duling (61) established that EC and VSMC contact at the MEJ stimulates organized GJ formation. Composed of connexins, which form a hexameric channel allowing small molecules less than 1 kDa to pass between cell types, these MEJs allowed trafficking of IP3 from VSMCs to ECs, causing activation of IP3R1 and release of local calcium at the MEJ (62). The MEJs are enriched with eNOS, an enzyme regulated by Ca2+, indicating that compartmentalized stimulation of IP3-mediated Ca2+ signaling activates eNOS to provide a feedback mechanism to control vasoconstriction (63). The discovery of eNOS-derived NO signaling at the MEJ opened up the potential for a unique microdomain composed of redox switches.
Ca2+ signaling and redox events in VSMCs are followed by diffusion of Ca2+ and IP3 to ECs through MEJs. Three out of the four known GJ-forming vascular connexins (Cx37, Cx40, and Cx43) are found at MEJs to facilitate second messenger and electrical signaling (63, 64). Connexins at the MEJ are regulated by posttranslational modification, such as phosphorylation of specific serine (Ser) residues or through S-nitrosation/denitrosation. Modification of Cx43 at Cys271 controls GJ permeability through an eNOS/NO and S-nitrosoglutathione reductase (GSNOR)-dependent mechanism, highlighting an important redox switch controlling cross-communication between ECs and VSMCs (63) (Figure 2a,b).
Figure 2.
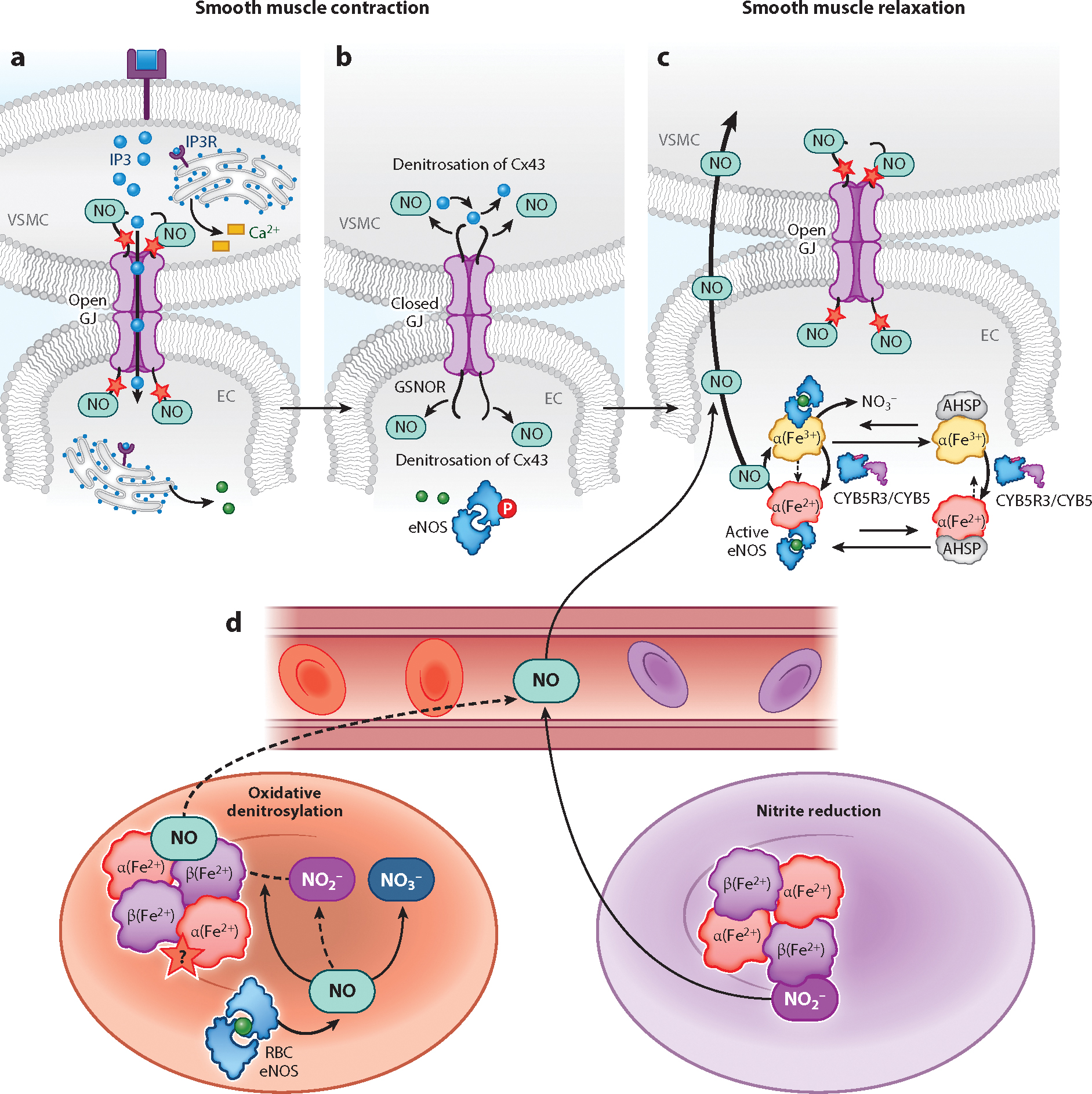
(a) S-Nitrosation of Cx43 facilitates the open state of GJs at the myoendothelial junctions to allow the intercellular exchange of signaling molecules. (b) IP3-induced Ca2+ release from the ER activates eNOS through phosphorylation (P circled in red). Denitrosation of Cx43 by GSNOR closes GJs and limits the diffusion of IP3. (c) NO generated by the activity of eNOS, which is mediated by the changes in the α-globin heme redox state. The NO generated during eNOS activity can diffuse from the EC to VSMC to cause vasodilatation and is important in maintaining the open probability of GJs through the S-nitrosation of Cx43. α-Globin is stabilized by AHSP before and complexes with eNOS to facilitate NO production. The redox state of the α-globin heme is regulated by CYB5R3 or eNOS. S-Nitrosation is marked with a red star. (d) In oxygenated RBCs (red), eNOS generates NO that can ultimately participate in vasorelaxation to regulate blood flow and blood pressure. Redox reactions of NO with oxygen, oxyhemoglobin, and deoxyhemoglobin result in nitrite NO2−, NO3−, and Fe2+-nitrosyl-hemoglobin. During gas exchange, the RBC partially deoxygenates (purple), and diffusible molecules that can signal for vessel relaxation are generated. NO2− undergoes a reductive reaction with deoxyhemoglobin, releasing diffusible NO as the oxygen tension approaches the point where 50% of the RBC hemoglobin has become deoxygenated. Reactive intermediates (NO2− and H2O2) formed in the process also facilitate the release of NO from Fe2+-nitrosyl-hemoglobin (oxidative denitrosylation) and participate in radical–radical interactions that generate the diffusible N2O3 for possible RBC export. The red star marks suspected S-nitrosation (S-nitrosothiol modification of hemoglobin), which is distinct from Fe2+-nitrosyl-hemoglobin formation. Abbreviations: AHSP, α-globin stabilizing protein; Cx43, gap junction–forming vascular connexin; CYB5, cytochrome B5; CYB5R3, cytochrome B5 reductase 3; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; GJ, gap junction; GSNOR, S-nitrosoglutathione reductase; H2O2, hydrogen peroxide; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; N2O3, dinitrogen trioxide; NO, nitric oxide; NO2−, nitrite; NO3−, nitrate; RBC, red blood cell; VMSC, vascular smooth muscle cell.
Transfer of IP3 through the MEJs induces IP3-mediated increases in endothelial [Ca2+]cyto through IP3Rs and TRPV4 channels, activating vasodilatory signaling by NO and endothelial-derived hyperpolarizing factor (EDHF). Interestingly, NO plays only a marginal role in agonist-induced vasodilatation compared to EDHF in resistance arteries (65). Vascular EDHF is thought to be a substance or electrical signal generated and released by healthy ECs for effecting VSMC hyperpolarization and relaxation. Although the exact identity of EDHF (e.g., K+, sulfur, eiconsanoids, C-type natriuretic peptide) and its mechanism of action still engender debate, the literature suggests that K+ is a common factor in EDHF signaling in the vasculature. EDHF signaling for vasodilatation increases as vessel diameters decrease—namely at sites of blood flow and blood pressure regulation—and relies on activation of BK channels. The switch from NO-to EDHF-based signaling cannot be explained by the lack of eNOS expression in the microcirculatory vessels (66). The initiation of EDHF signaling in ECs for VSMC hyperpolarization and relaxation relies on the activation of BK channels. The mechanism for differences in regulation of vasodilation in resistance arteries versus conduit arteries may be due to NO scavenging (67) (Figure 1e–f).
Recently α-globin was found to be expressed and concentrated at the MEJs in arteriolar ECs (68). In contrast to RBCs, where α-globin exists in a stable complex with β-globin, a recent study showed that EC α-globin undergoes stabilization when chaperoned by α-globin stabilizing protein (AHSP) prior to complexing with eNOS for regulation of NO diffusion from ECs to VSMCs (69) (Figure 2c). Disruption of the α-globin-eNOS complex, either by a peptide mimetic or genetic knockdown of either the α-globin or AHSP gene, has been shown to increase NO signaling and VSMC relaxation.
The redox state of the α-globin heme iron determines whether it scavenges NO or permits its diffusion. Reduced heme iron (Fe2+) promptly reacts with NO to form iron-nitrosyl-hemoglobin (Fe3+-NO), limiting its diffusion to VSMCs. However, oxidized heme iron (Fe3+) reacts very slowly with NO, allowing its intercellular diffusion to induce VSMC relaxation and regulation of vascular tone. The redox state of α-globin is controlled by cytochrome B5 reductase 3 (CYB5R3), which is a flavoprotein that complexes with eNOS and α-globin (70). In addition to CYB5R3, eNOS has also been shown to regulate α-globin reduction (69). Thus, the redox control of α-globin, with the reduced heme scavenging NO and the oxidized heme allowing NO diffusion, serves as a redox controlled gate for NO diffusion.
Other globins are expressed in nonerythroid cells and have been suggested to control NO and H2O2 bioavailability, thus controlling vascular tone. Neuroglobin (Ngb) and cytoglobin (Cygb) are different from hemoglobin and myoglobin, as they exhibit hexacoordinate heme binding, with both a proximal and distal heme pocket histidine binding to the heme iron. Interestingly, they possess two surface thiols that, when oxidized to form an intramolecular disulfide, allosterically promote a weakening of the distal histidine bond and increase ligand affinity and reactivity of the heme (71–73). For both Ngb and Cygb these cysteines thus serve as redox sensors that, when oxidized, increase ligand affinity and the reaction rate with molecules such as nitrite, which is reduced to form NO. In the case of Cygb there are subpopulations ranging from fully reduced thiols, single thiol oxidation, and intramolecular disulfide formation that determine heme-binding properties by modulating the histidine-heme affinity and ligand binding (71). The redox modulation of thiol oxidation state and heme-ligand binding is sensitive to physiological levels of H2O2, with a functional midpoint redox potential for the native Cygb intramolecular disulfide bond of −189 ± 4 mV, a value within the normal range of intracellular redox potentials (71). In the case of Ngb, mutation of either Cys55 or Cys46 to alanine stabilizes the six-coordinate structure and slows the reaction with nitrite (72). These results support the hypothesis that Cys38 and Cys83 on Cygb serve as sensitive redox sensors that modulate the Cygb distal heme pocket reactivity and ligand binding.
While the physiological function of both Ngb and Cygb remains uncertain, they both have lower expression levels than myoglobin and hemoglobin, suggesting they play a limited role in O2 regulation (74). A number of functional roles have been proposed, including (a) NO scavenging by the deoxygenation reaction to increase vascular tone (75), (b) nitrite reduction under deoxygenated conditions to promote NO signaling (76), superoxide scavenging (77), and (d) peroxidase activity that can catalyze the peroxidation of anionic phospholipids (including phosphatidylinositol phosphates), yielding mono-oxygenated molecular species (78). The surface thiol redox sensor–regulated control of these reactions most likely modulates vascular tone, responses to inflammatory and oxidative injury, and remodeling and cellular apoptosis (75, 79). Similar to hemoglobin intracellular α-globin and sGC, additional enzymatic redox control is determined by the oxidation and reduction of the heme by the cytochrome B5 reductase system (80, 81).
Phase 3: Control of Nitric Oxide in Smooth Muscle and Activation of Soluble Guanylyl Cyclase
The primary NO receptor in VSMCs is sGC. To reach optimal catalytic activity, sGC requires a heterodimeric structure formed from α and β subunits. The sGC protein has two types of α (α1 and α2) and two types of β (β1 and β2) subunits (82).The β2 subunit is expressed at the lowest level of all the subunits, resulting in the most common heterodimers being α1β1, followed by α2β1. It has been shown that VSMC-specific loss of sGCβ1 causes hypertension, and global deletion of sGCβ1 causes gut dysmotility and death (83).
The sGCβ subunit contains a heme region that can interact with NO, causing the heme-iron atom to be cleaved from the H105 residue of sGCβ (84). The binding of NO also facilitates conformational changes in the coiled-coil domains of both α and β subunits, resulting in the alignment of their catalytic sides to form a 5′-GTP binding motif. Heat shock protein 90 (HSP90) has been shown to drive heme into the heme-deficient sGCβ1 form during maturation, the dissociation of which is promoted by NO binding to support heterodimer formation with sGCα1 for an enzymatically active protein (85).
The redox state of the heme iron in sGC is central to NO signaling for cGMP production by the enzyme. When the sGC heme iron is reduced (Fe2+), it binds NO, and cGMP production occurs. When the sGC heme iron is oxidized (Fe3+), it only weakly binds NO, and cGMP production is blunted (86). Prior work has shown that CYB5R3 regulates the sGC heme redox state, particularly under oxidative stress conditions (87, 88). Oxidized sGC heme iron needs to be reduced back to its Fe2+ state to sense NO (70), which our work has shown is the role of CYB5R3 in VSMCs. It has yet to be determined whether CYB5R3 serves as a direct modulator of cysteine redox states during oxidative stress or affects S-nitrosation.
A key redox switch in regulating sGC function is disulfide bond formation between oxidation sensitive thiols, such as Cys489 and Cys571 of sGCβ1 (89). Treatment with thiol blockers (CdCl2, hydroxymercuribenzoate, N-ethylmaleimide, etc.) has been shown to decrease sGC responsiveness to NO (90). S-Nitrosation of specific cysteines in the β1 subunit has been shown to negatively regulate sGC activity in the presence of NO (91). In the presence of thioredoxin 1 (Trx1), S-nitrosated cysteines are reduced, allowing protein disulfide isomerase to form disulfide bonds between Trx1 Cys73 and sGCα1 Cys609. This interplay between Trx1 and sGC promotes the production of cGMP under high NO conditions, as shown by significantly decreased cGMP production with nitrosocysteine treatment during Trx1 inhibition and rescue of cGMP production during Trx1 overexpression (92).
Following the NO activation of sGC, protein kinase G1 (cGMP-activated protein kinase) is activated, eliciting relaxation of VSMCs through the inactivation of the myosin contractile machinery (93). There are two known splice variants of PKG1 (α and β) with different cGMP-binding affinity and phosphorylation substrate selectivity (94). PKG1α has redox-sensitive motifs that factor into the kinase’s function under pathological conditions. Disulfide bridge formation at the redox-sensitive Cys42 confers functionality on the kinase. Residues Cys117/Cys195 and Cys312/Cys518 within the high- and low-affinity cGMP-binding sites have been suggested to be sensitive to oxidative activation when enlisted for disulfide bridge formation (95). Studies with Cys42Ser mutant mice have revealed that interference with disulfide bond formation can yield diverging downstream effects for PKG1α. At baseline, Cys42Ser mutant mice display hypertension, less acetylcholine-dependent dilatation, decreased PKG activity after pro-oxidant exposure (H2O2), and impaired myocardial relaxation and contractility (96). However, when subjected to aortic constriction, Cys42Ser mutant mice were shown to be more resistant to severe pathology and death (97). These findings highlight the possible roles of cGMP signaling in certain pathological conditions.
ENDOTHELIAL TO SMOOTH MUSCLE CELL CROSS TALK AND CONTROL OF VASODILATION
Calcium Regulation of Nitric Oxide–Mediated Vasodilation
EC-mediated vasodilatation undergoes differential regulation determined by the size of the artery or arteriole. Whereas vasodilation in conduit arteries (i.e., aorta) occurs through eNOS-derived NO, vasodilation in systemic resistance arteries and arterioles can be driven by several distinct pathways: NO, EDHF, prostaglandins, H2O2, and hydrogen disulfide (H2S2). In contrast to VSMC [Ca2+]cyto, EC [Ca2+]cyto facilitates vasodilatation through eNOS-derived NO. Signaling molecules cross over from VSMCs to ECs, activating cellular Ca2+ signaling and NO generation by eNOS. NO synthesis by eNOS requires its precursor l-arginine, the cofactor tetrahydrobiopterin (BH4), flavin dinucleotide, flavin mononucleotide, CaM, and heme iron. For eNOS to produce NO, its oxygenase and reductase domains must be heterocomplexed into a dimer mediated by HSP90 (98). The eNOS heterodimer can become uncoupled in the absence of sufficient l-arginine or BH4, leading the oxygenase domain to preferentially produce superoxide (O2−) and contribute to oxidative stress in certain pathologies (98). Uncoupling of the eNOS enzyme also occurs with S-glutathionylation of Cys689 and Cys908 (99).
eNOS is tightly regulated, by both its location in caveolae and posttranslational modifications. The localization of eNOS in caveolae allows its interaction with the scaffold protein caveolin-1, which keeps it inactive. Posttranslational modifications also regulate eNOS activity in a manner that depends on the site and type of modification. eNOS is activated through phosphorylation of Ser1177 by PKA, PKB, AMPK, CaMK, and ERK1 and 2, and inhibited by the phosphorylation of tyrosine (Tyr) 495 by Rho kinase and PKC (5, 99, 100). In addition, phosphorylation of Tyr657 by tyrosine kinase lowers eNOS activity, and phosphorylation of Tyr81 by Src facilitates NO production (99). Redox switches are important for regulating eNOS activity; S-nitrosation of Cys94 and Cys99 or glycosylation of Ser1177 blunts enzyme activity, while acetylation of Lys610 and deacetylation of Lys497 and Lys507 increase enzyme activity (99).
In response to extracellular signals, EC cytosolic Ca2+ signaling is mostly initiated by Ca2+ release from intracellular stores through large,high-conductance Ca2+ channel IP3Rs and sustained by SOCE or TRP channels to induce eNOS activation or EDHF. The IP3Rs that participate in Ca2+ release come in three isoforms (IP3R1–3), all of which are present in ECs. IP3Rs facilitate Ca2+ mobilization from the SR/ER upon stimulation by GPCRs or tyrosine kinase receptors on the plasma membrane or by Ca2+ through CICR (101, 102). Endothelial GPCRs recruit phospholipase C (PLC) β2 and β3 to transform phosphatidylinositol 4,5-bisphosphate into DAG and IP3, while PLCγ1 couples with tyrosine kinase receptors to promote IP3 generation (103) (Figure 1d). Sensitization of IP3R channel activity is tightly regulated by oxidation of specific thiols. Mammalian IP3R1 has 60 thiol groups, a significant fraction of which are sensitive to oxidative posttranslational modifications (104). IP3R1 has been shown to have cytosolic (Cys292 and Cys1415) and ER lumen cysteines (Cys2496 and Cys2533) that are oxidized under basal conditions, and a handful of N-terminal cysteines (Cys206, Cys214, and Cys1397) that can be oxidized by H2O2 (Cys206, Cys767, and Cys1459) and thimerosal (104). S-Glutathionylation has been shown to increase IP3R activity in ECs after treatment with a thiol oxidizing agent (diamin) (105). Apart from IP3R1, the other two IP3R isoforms have undergone limited characterization in terms of redox regulation (104). IP3R1 and IP3R2 were shown to be more sensitive to redox modification to alter ligand sensitivity, while IP3R3 is relatively insensitive (106). H2O2 can also drive IP3-induced SR/ER Ca2+ release by directly acting on PLCγ1 and/or by stimulating IP3Rs. Indeed, H2O2 has been shown to promote IP3 production in aortic and mesenteric artery ECs (107). There is also evidence for exogenous H2O2 induction of a dose-dependent oscillatory [Ca2+]cyto increase in human aortic ECs (108). These oscillations were shown to be completely dependent on Ca2+ release from the ER, with no dependence on Ca2+ flux through the plasma membrane. By contrast, human umbilical vein ECs respond differently to H2O2, undergoing a marked reduction of ER Ca2+ concentration ([Ca2+]ER) (109). Of note, it has been speculated that H2O2 depolarization of mitochondria may inhibit IP3R-induced Ca2+ release in vascular ECs.
The SR/ER is the most important and largest intracellular Ca2+ store. Here, the SR/ER Ca2+ ATPase (SERCA) mainly regulates intracellular Ca2+ concentration and refills ER stores during SOCE (Figure 1d). SERCA is prone to increased activation by S-glutathionylation by peroxynitrite (ONOO−) generated from NO under conditions of oxidative stress (110). In atherosclerosis, the Cys674 site of SERCA was found to undergo irreversible oxidation to sulfonic acid, preventing NO-induced VSMC relaxation (111). Although modification of Cys674 by sulfonic acid has been shown in large vessels, it is unclear whether the same modification happens in the microcirculation.
After Ca2+ release, ER stores need to be refilled, and multiple mechanisms in the plasma membrane exist. STIM1 and STIM2, which sense ER Ca2+ levels, work together with ORAI (ORAI 1–3) Ca2+ channels to drive SOCE (Figure 1d). SOCE is primarily facilitated by changes in the ER Ca2+ content; however, it is also regulated through posttranslational modifications of STIM and ORAI proteins. Two STIM1 cysteines (Cys49 and Cys56) have been identified as ROS/RNS sensors that are modifiable by S-glutathionylation and S-nitrosation, affecting the stability of the EF-hand domains of calcium-binding proteins (112–114). S-Glutathionylation of Cys49 and Cys56 destabilizes the EF-hand domain, decreasing affinity for Ca2+ and causing STIM1 and ORAI1 activation (115). S-Nitrosation of Cys49 and Cys56 stabilizes the EF-hand domain, causing SOCE inhibition (113, 114). Little is known about the other three STIM1 cysteines; however, it has been suggested that the cytosolic Cys437 residue may serve as an oxidative stress sensor that drives conformational changes in ORAI1 before SOCE (112). For the STIM2 protein there are 15 cysteines, with four luminal cysteines corresponding to the ones found in STIM1, and 11 cytosolic cysteines that are unique (112). Cys313 was identified recently as a ROS sensor in STIM2. Oxidation of Cys313 interferes with STIM2 activation and inhibits SOCE, most likely through altering STIM2–STIM2 interactions (116). Redox modification of ORAI proteins can also occur (112). Oxidation of ORAI1 and ORAI2 at Cys195 has been shown to inhibit SOCE in various models. While ORAI3 does not contain a Cys195 site, it has other cysteine sites that may allow it to be oxidatively regulated. H2O2 treatment has been shown to prevent ORAI1–ORAI1 interactions, suggesting that Cys195 oxidization can prevent ORAI1 subunit interactions, causing a deficiency of pore-forming units. By contrast, H2O2 treatment increases STIM1–ORAI1 interactions (112). While the different redox modifications and their effect on SOCE have been extensively studied in the past, we lack an understanding of the role of different cellular oxidants and reductants in SOCE regulation.
Generation of [Ca2+]cyto transients in ECs can also occur via NCXrm. NCXrm is known to participate in vasodilatation through acetylcholine-stimulated activation of EC muscarinic receptor signaling (117). An association between NCX and eNOS has been shown to happen in caveolae. NCX-mediated increases in [Ca2+]cyto are coupled to both NO production and intermediate and small conductance Ca2+-activated K+ (IK/SK) channel activation in ECs. ROS-stimulated activation of NCX is suspected to occur via phosphorylation of the intracellular loop of NCX by Ser/Thr kinases (PKA or PKC) (118).
Endothelium-Derived Hyperpolarization
In the microcirculation EDHF is the main driver of vasodilatation. IK/SK channels play a crucial role in EC control of VSMC relaxation (Figure 1f). Endothelial IK/SK channels become activated upon interacting with Ca2+–CaM, resulting in hyperpolarization of the EC membrane and VSMC relaxation. Influx of extracellular Ca2+ for these signaling events is mediated through activation of various TRP channels.
TRPA1 is a crucial Ca2+ influx pathway in ECs. TRPA1 colocalizes with IK channels at the MEJs in cerebral arteries. TRPA1-mediated Ca2+ influx activates nearby IK channels to effect EC membrane hyperpolarization and subsequent vasoconstriction. TRPA1 channels also promote SR/ER Ca2+ release through IP3Rs (119) (Figure 1e, f). Interestingly, NOX2 has been found localized near TRPA1 channels in cerebral arteries. This suggests that NOX2-generated ROS, which are known to cause peroxidation of membrane lipids and 4-hydroxy-2-nonenal formation, can activate TRPA1 channels (120).
TRPV4 channel activity is regulated by numerous proteins and mechanisms, which can be important given the channel’s key role in mediating EC Ca2+ influx for blood pressure regulation. While TRPV4 channels can be activated by mechanical stimuli, it is suspected that they are not really mechanosensors and are mainly activated by signaling pathways that include cytochrome P450 activation or epoxygenases (121).
Under pathological conditions, such as pulmonary hypertension, it has been shown that TRPV4 signaling decreases with NOX1/inducible NOS-generated peroxynitrite in endothelial caveolae (122). In small pulmonary arteries, TRPV4-mediated Ca2+ sparks occur in response to ATP activation, promoting eNOS activity and NO release (Figure 1e). NO activates sGC-cGMP-PKG signaling, but the outcome is a lowering of Ca2+-dependent cooperative opening of TRPV4 channels in ECs. Hydrogen sulfide (H2S) is another gasotransmitter that can signal for vasodilatation, sometimes in collaboration with NO. EC-derived H2S has been shown to activate TRPV4 channels via sulfhydration, resulting in increased BK channel currents (123).
TRPC3 has been shown to localize close to IK/SK channels at the MEJs in rat mesenteric arteries (124) (Figure 1f). The TRPC3 channel is DAG sensitive and dependent on EC [Ca2+]cyto for controlling proliferation, migration, and tube formation linked to chemical (NO) and/or electrical stimulation during vasorelaxation. Activation of TRPC3 by tert-butyl hydroperoxide in pulmonary artery ECs has been shown to cause an NO-selective cation current (125). TRP channels may also form heterocomplexes with other TRP channels. TRPC1 has been found heterocomplexed with TRPV4 to activate eNOS in rabbit mesenteric arteries, causing vasodilatation (126) (Figure 1e). Heteromers of TRPC3 and TRPC4 have been detected in ECs, where they may undergo indirect activation by cellular ROS-induced PLC stimulation (127).
Lastly, the activation of P2X, PIEZO1, TRPP1, and TRPV4 channels can be stimulated to increase [Ca2+]cyto in a mechanosensitive manner, such as from blood flow–mediated shear stress on vascular endothelium (23). The result is eNOS activation and NO signaling with downstream activation of IK/SK channels, leading to EC-dependent hyperpolarization and VSMC relaxation.
THE IMPACT OF OXYGEN AND THE CONTRIBUTION OF RED BLOOD CELLS TO THE REGULATION OF VASCULAR TONE AND BLOOD FLOW
The role of RBCs in hypoxic vasodilatation illustrates the importance of O2 sensing and redox signaling to the maintenance of vascular homeostasis. At high O2 concentrations, RBC hemoglobin exists in a relaxed configuration (R-state), which has a higher O2 affinity, facilitating O2 binding from in the lung where O2 tension is high. Under low O2 conditions in the peripheral circulation in metabolically active tissues, RBC hemoglobin deoxygenates and assumes a tense configuration (T-state),which has a lower affinity for O2 and thus facilitates dissociation of O2 from hemoglobin for supply to less-oxygenated tissues. In addition to O2, the RBC dynamically responds to tissue metabolism via allosteric molecular interactions with proton, 2,3-Diphosphoglycerate (2,3-DPG) (modulated by intracellular glycolytic flux) and CO2 and in the intracellular domain of band 3. These interactions shift the hemoglobin P50, the partial pressure of O2 at which hemoglobin is 50% saturated with O2, allowing fine-tuning of O2 delivery in concert with tissue O2 consumption and metabolism. In addition to the canonical roles of hemoglobin in transporting O2, CO2, and buffering proton, it has been proposed that the hemoglobin and erythrocytes directly release ATP and NO under allosteric control to facilitate hypoxic vasodilation and signaling. While controversial, these models are elegant in presenting a pathway for direct coupling of hemoglobin-based hypoxic sensing to vasodilation.
Erythrocyte ATP Delivery Hypothesis
Three mechanisms have been proposed for RBC and hemoglobin-mediated hypoxic vasodilation. The first was proposed by Ellsworth, Sprague, and colleagues (128), who presented evidence that erythrocytes release ATP during deoxygenation, which activates purinergic receptors in endothelium to drive retrograde hypoxic vasodilation. Although this pathway has been understudied, a number of studies have continued to evaluate ATP release from RBCs to modulate vasodilation (129). It is our opinion that this pathway remains very promising, especially in light of recent clinical studies showing that the pharmacological modulation of the red cell isoform of pyruvate kinase (PK-R) can increase intracellular ATP levels (130, 131), potentially opening the door to metabolic therapies that enhance hypoxic vasodilation responses. Clearly, more work is required using modern methodologies to fully confirm this hypothesis.
S-Nitroso-Hemoglobin Hypothesis
The second proposed mechanism is the S-nitroso-hemoglobin hypothesis (132). This posits that NO binds to the heme of hemoglobin and then transfers in oxygenated blood the Cys93 residue forming a S-nitroso-thiol. The NO is then released from the thiol during hemoglobin deoxygenation, likely by transnitrosation to form a still unidentified intermediate X-NO species that has been proposed to facilitate vasodilation (132). A challenge is that the high concentrations of GSH in red cells favor transnitrosation to GSNO, which does not cross the RBC membrane (only l-cysteine can cross via the L-type neutral amino acid transporter) (133). S-Nitroso-hemoglobin can be synthesized by incubation of high concentrations of NO with RBCs and by treatment of RBCs with S-nitroso-cysteine. Once formed, these RBCs can promote vasodilation, but this does not appear to be O2-dependent (134). Furthermore, the chemical mechanism of NO binding to heme and transfer to cysteine has not been worked out: NO bound to the deoxygenated ferrous heme of hemoglobin will not directly transfer to a cysteine without the removal of an electron to form NO+ or a thiyl radical (a one-electron oxidation to form the covalent R-SNO). Indeed, no mechanism for this electron transfer step has been identified in the last two decades. From a physiological standpoint, the initial SNO-hemoglobin hypothesis was supported by the measurement of arterial-to-venous gradient that suggested artery-to-vein delivery of NO from Cys93 (132). However, these initial high levels and arterial-to-venous gradient of SNO-hemoglobin detected in vivo have not been reproduced, and the hypoxia-dependent release of NO has been difficult to assess and reproduce (133, 135–137). Perhaps most importantly, a Cys93Ala mutant mouse model was created and studied and appears to have intact physiological hypoxic vasodilation (138). One study suggesting an abnormal hypoxic response in this mouse model did not test canonical hypoxic vasodilation in vivo, only the microcirculatory vasodilatory response to ischemia (flow-mediated vasodilation) (139). A recent study used multiple assays to comprehensively test RBC-mediated NO signaling using red cells from the Cys93Ala mouse and found that signaling effects were evident with or without the Cys93 residue (140). From our analysis of the literature, it appears that SNO-hemoglobin can form under conditions of high NO flux, like sepsis, and when formed can contribute to vasodilation, but a dynamic hypoxia-redox response and a role in physiological NO signaling have not yet been established.
Nitrite Reductase Hypothesis
A third pathway proposed that links erythrocyte and hemoglobin deoxygenation to NO signaling is the nitrite reductase hypothesis (Figure 2d). This hypothesis posits that a classical electron and proton transfer reaction converts nitrite to NO, catalyzed by electron transfer from the deoxygenated ferrous heme of hemoglobin (137, 141, 142): nitrite (NO2−) + deoxyhemoglobin (Fe2+) + H+ − nitric oxide (NO) + methemoglobin (Fe3+) + OH−.
This hypothesis was advanced based on human translational studies showing that nitrite in plasma and RBCs has arterial-to-venous gradient, suggesting nitrite consumption across the circulation (137) and potent vasodilation during pharmacological nitrite infusions (137, 141). Unlike SNO-hemoglobin, the nitrite arterial-to-venous gradient and vasodilation by nitrite have been reproduced by numerous investigators (e.g., 135). In the presence of deoxygenated red cells in blood and in vitro, nitrite is converted to NO, which can be detected by NO formation in the RBC, where it binds to other deoxygenated hemoglobin molecules to form iron-nitrosyl-heme (Fe+2-NO) (141). NO formation from the reaction of nitrite with deoxyhemoglobin or deoxygenated erythrocytes has been shown to activate platelet-soluble guanylate cyclase, increase cGMP levels, inhibit platelet activation (137), and inhibit mitochondrial respiration via NO-dependent inhibition of complex IV (137, 143). The reaction of nitrite with deoxygenated hemoproteins has been broadly confirmed across all ferrous hemoproteins, including myoglobin, Cygb, Ngb, globin X, and nonhemoglobin redox active heme and molybdenum enzymes, suggesting broadly conserved pathways for hypoxic NO signaling via nitrite reduction (recently reviewed in 144).
The chemistry of the reaction of nitrite with hemoglobin has been extensively studied, and there is little controversy about the underlying mechanism. The reaction (shown above) is a second-order reaction, such that the rate of NO formation is dependent on the concentrations of nitrite and deoxygenated heme on hemoglobin (or heme in other globins and enzymes) (142). However, the rate of all second-order reactions also depends on the intrinsic reactivity with the heme, which is characterized by the bimolecular rate constant K. Interestingly, in hemoglobin this rate constant is in fact not constant and changes with the allosteric conformation of the R (oxy)-state and the T (deoxy)-state. The R-state rate constant is about 30 times faster than the T-state. This means that the rate of nitrite reduction and NO formation by hemoglobin is fastest during rapid deoxygenation of hemoglobin, when most of the hemoglobin is in the oxygenated R-state, but O2 molecules are released, freeing deoxygenated hemes for nitrite binding and conversion to NO. This rate is maximal around the P50 of hemoglobin, where 50% of the hemoglobin is deoxygenated. This chemistry is called R-state catalysis and provides a significant mechanism to link regional O2 utilization and O2 delivery to NO formation via nitrite reduction (142).
A major challenge for the nitrite reductase hypothesis has been how NO can escape from an RBC and avoid autocapture by vicinal deoxygenated hemoglobin, which has an extremely high NO affinity. One solution is that NO does, in fact, diffuse out from reactions at the erythrocyte membrane in an inefficient process. This is supported by studies that detect NO formation and release from nitrite-treated erythrocytes (137). The other possibility is that NO can be trapped on heme and released from heme, via a process called oxidative denitrosylation (137). NO2· and H2O2 are both intermediates that result from redox reactions between nitrite and oxygenated hemoglobin, which can both oxidize the heme–NO, facilitating the release of NO from ironnitrosyl-hemoglobin by oxidative denitrosylation (137, 145). This reaction also occurs around the P50 of hemoglobin (where auto-oxidation and nitrite oxidation are maximal). Finally, the reaction of nitrite and deoxyhemoglobin can generate a higher NO, N2O3, via reactions of NO with nitrogen dioxide (NO2·). Formation of N2O3 can result from radical–radical interactions between NO and NO2· and innersphere and outersphere reactions of NO and nitrite with ferric hemoglobin (145). The N2O3 molecule is more stable in hemoglobin solutions and could diffuse out of the RBC and homolyze to NO and NO2 or S-nitrosate plasma thiols, both pathways to the activation of sGC and vasodilation. This may explain how RBC eNOS-derived NO exits the RBC to modulate blood pressure and blood flow and limit cardiac ischemic injury, as reported by Wood et al. (146) and Leo et al. (147).
Thus, while the formation of NO and signaling of nitrite and deoxygenated hemoglobin, other globins, and deoxygenated RBCs have been extensively validated, the mediators and mechanisms that allow such signaling in the presence of high concentrations of scavenging globins are areas of active and continued investigation.
FUTURE DIRECTIONS
This review provides a generalized overview of blood pressure regulation through the interplay between RNS/ROS, redox switches, and Ca2+ signaling. Over the past few decades, studies using global antioxidant-focused approaches for fighting aging and diseases associated with oxidative stress (e.g., diabetes, cancer, and neurodegeneration) have largely failed, indicating that the global cellular redox state represents a poor therapeutic target. Meng et al. introduced the 5R principles of a precision redox approach: “Right species, Right place, Right time, Right level and Right target” (4, p. 1069). These principles focus on roles of redox switches in cellular compartments and confined spaces and the association of common gene polymorphisms to cardiovascular diseases. Importantly, the relative differences in redox signaling and reliance on Ca2+, NO, or EDHF as stimuli for vascular relaxation in large versus small vessels, detailed in this review, highlight the importance of distinguishing local from global ROS for improved understanding of cardiovascular pathologies.
The precise regulation of vascular tone is a chain of complex reactions involving signal transduction between different cell types to adapt to neurohumoral or mechanical stimuli. These pathways involve numerous enzymes regulated by signaling molecules (i.e.,ROS/RNS,K+,Ca2+), for which directional changes in activity may be detrimental to disease progression. Identifying the precise interplay between oxidants and reductants regulating Ca2+ signaling pathways or vice versa represents an interesting new direction in vascular biology. The recent availability of technologies that allow preclinical study of oxidation-reduction events at the subcellular level, such as the organelle or interorganellar interface with targeted GSH- and H2O2-sensing probes, may help with understanding the spatiotemporal redox events associated with cellular Ca2+ transients in ECs and VSMCs (148).
The key redox regulated players and pathways in EC and VSMC Ca2+-dependent vasoregulation were discussed in the first three sections of this review. Members of the voltage- and nonvoltage-gated channels and their regulators were shown to be under the direct or indirect regulation of redox switches. Nonetheless, there are other as-yet-uncharacterized targets and modulators. As an example, CYB5R3 was shown to regulate heme redox state in both ECs and VSMCs; however, whether it has a role in regulating other nonheme targets remains unstudied. There are four other members of the CYB5R family expressed in the vasculature, but their cellular localization and role in cellular processes remain undetermined. It is possible that different CYB5R isoforms may impact numerous Ca2+ channels and their regulators in vascular cells.
The study of common genetic variants impacting redox signaling and switches in cardiovascular disease and the functional consequences caused by these variants is also an emerging field. For example, the common polymorphism in SOD2 (SOD2V16A) in patients with sickle cell disease associates with increased cardiovascular consequences by curtailing mitochondrial complex IV activity and increasing ROS production (149, 150). Further identification of additional genetic biomarkers may lead to better precision medicine approaches for predicting the right therapy for the right patient.
ACKNOWLEDGMENTS
We thank Dr. Katherine C. Wood for the tremendous help in editing and Dr. Shuai Yuan and Robert J. Hall for their constructive comments on the text. Financial support for this work was provided by the US National Institutes of Health grants R35 HL 161177, R01 HL 149825, and R01 HL 153532 (A.C.S.); the US National Institutes of Health grants 5R01HL098032, 2R01HL125886, 5P01HL103455, and UH3HL143192 (M.T.G.); and the American Heart Association Established Investigator Award 19EIA34770095 (A.C.S.) and grant 5P01HL10345509 (M.T.G.).
Footnotes
DISCLOSURE STATEMENT
M.T.G. receives research support from Bayer HealthCare Pharmaceuticals and Globin Solutions, Inc.; is a co-inventor of patents and patent applications directed to the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning, which have been licensed by Globin Solutions, Inc.; is a shareholder, advisor, and director of Globin Solutions, Inc.; is also co-inventor on patents directed to the use of nitrite salts in cardiovascular diseases, which were previously licensed to United Therapeutics, and are now licensed to Globin Solutions, Inc. and Hope Pharmaceuticals; is a principal investigator in a research collaboration with Bayer HealthCare Pharmaceuticals to evaluate riociguat as a treatment for patients with sickle cell disease; is a textbook author and receives royalties from MedMaster Inc.; is a textbook editor and receives royalties from McGraw-Hill; actively serves as a scientific consultant for Forma Therapeutics (serving on the red blood cell scientific advisory board); has previously served as a consultant for Acceleron Pharma, Inc., Sujana Biotech, Epizyme, Inc., Catalyst Biosciences, Inc., Complexa, Novartis, United Therapeutics, Fulcrum, Actelion, Bayer HealthCare Pharmaceuticals, Pfizer, and Modus Therapeutics; and served on Bayer HealthCare LLC’s Heart and Vascular Disease Research Advisory Board. M.K. and A.C.S. are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Nehls V, Drenckhahn D. 1991. Heterogeneity of microvascular pericytes for smooth muscle type alphaactin. J. Cell Biol. 113:147–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Straub AC, Zeigler AC, Isakson BE. 2014. The myoendothelial junction: connections that deliver the message. Physiology 29:242–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhodin JA. 1967. The ultrastructure of mammalian arterioles and precapillary sphincters. J. Ultrastruct. Res. 18:181–223 [DOI] [PubMed] [Google Scholar]
- 4.Meng J, Lv Z, Zhang Y, Wang Y, Qiao X, et al. 2021. Precision redox: the key for antioxidant pharmacology. Antioxid. Redox Signal. 34:1069–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Förstermann U, Sessa WC. 2012. Nitric oxide synthases: regulation and function. Eur. Heart J. 33:829–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negri S, Faris P, Moccia F. 2021. Reactive oxygen species and endothelial Ca2+ signaling: Brothers in arms or partners in crime? Int. J. Mol. Sci. 22:9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snezhkina AV, Kudryavtseva AV, Kardymon OL, Savvateeva MV, Melnikova NV, et al. 2019. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2019:6175804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mailloux RJ. 2020. An update on mitochondrial reactive oxygen species production. Antioxidants 9:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Pagano PJ. 2017. Microvascular NADPH oxidase in health and disease. Free Radic. Biol. Med. 109:33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt HM, Kelley EE, Straub AC. 2019. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 21:101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, et al. 2014. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes 63:1381–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckman JS, Koppenol WH. 1996. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, andugly. Am. J. Physiol. Cell Physiol. 271:C1424–37 [DOI] [PubMed] [Google Scholar]
- 13.Hippeli S, Elstner EF. 1999. Transition metal ion-catalyzed oxygen activation during pathogenic processes. FEBS Lett. 443:1–7 [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Branicky R, Noë A, Hekimi S. 2018. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 217:1915–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George P 1947. Reaction between catalase and hydrogen peroxide. Nature 160:41–43 [DOI] [PubMed] [Google Scholar]
- 16.Meyer AJ, Hell R. 2005. Glutathione homeostasis and redox-regulation by sulfhydryl groups. Photosynth. Res. 86:435–57 [DOI] [PubMed] [Google Scholar]
- 17.Dringen R 2000. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62:649–71 [DOI] [PubMed] [Google Scholar]
- 18.Lubos E, Loscalzo J, Handy DE. 2011. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 15:1957–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. 2015. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40:435–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan L-J. 2014. Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxid. Med. Cell. Longev. 2014:343154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wani R, Nagata A, Murray BW. 2014. Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 5:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalinowski DS, Stefani C, Toyokuni S, Ganz T, Anderson GJ, et al.2016.Redox cycling metals: pedaling their roles in metabolism and their use in the development of novel therapeutics. Biochim. Biophys. Acta Mol. Cell Res. 1863:727–48 [DOI] [PubMed] [Google Scholar]
- 23.Ottolini M, Hong K, Sonkusare SK. 2019. Calcium signals that determine vascular resistance. Wiley Interdiscip. Rev. Syst. Biol. Med. 11:e1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bean BP, Sturek M, Puga A, Hermsmeyer K. 1986. Calcium channels in muscle cells isolated from rat mesenteric arteries: modulation by dihydropyridine drugs. Circ. Res. 59:229–35 [DOI] [PubMed] [Google Scholar]
- 25.Abd El-Rahman RR, Harraz OF, Brett SE, Anfinogenova Y, Mufti RE, et al. 2013. Identification of L-and T-type Ca2+ channels in rat cerebral arteries: role in myogenic tone development. Am. J. Physiol. Heart Circ. Physiol. 304:H58–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. 2005. The L-type calcium channel in the heart: the beat goes on. J. Clin. Investig. 115:3306–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moosmang S, Schulla V, Welling A, Feil R, Feil S, et al. 2003. Dominant role of smooth muscle L-type calcium channel Cav 1.2 for blood pressure regulation. EMBO J. 22:6027–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22:549–58 [DOI] [PubMed] [Google Scholar]
- 29.Blatter LA, Wier WG. 1994. Nitric oxide decreases [Ca2+]i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium 15:122–31 [DOI] [PubMed] [Google Scholar]
- 30.Jackson WF. 2017. Potassium channels in regulation of vascular smooth muscle contraction and growth. Adv. Pharmacol. 78:89–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muralidharan P, Cserne Szappanos H, Ingley E, Hool L. 2016. Evidence for redox sensing by a human cardiac calcium channel. Sci. Rep. 6:19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Catterall WA. 2011. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.VanBavel E, Sorop O, Andreasen D, Pfaffendorf M, Jensen BL. 2002. Role of T-type calcium channels in myogenic tone of skeletal muscle resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 283:H2239–43 [DOI] [PubMed] [Google Scholar]
- 34.Rossier MF. 2016. T-type calcium channel: a privileged gate for calcium entry and control of adrenal steroidogenesis. Front. Endocrinol. 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harraz OF, Brett SE, Welsh DG. 2014. Nitric oxide suppresses vascular voltage-gated T-type Ca2+ channels through cGMP/PKG signaling. Am. J. Physiol. Heart Circ. Physiol. 306:H279–85 [DOI] [PubMed] [Google Scholar]
- 36.Hashad AM, Harraz OF, Brett SE, Romero M, Kassmann M, et al. 2018. Caveolae link Cav3.2 channels to BKCa-mediated feedback in vascular smooth muscle. Arterioscler. Thromb. Vasc. Biol. 38:2371–81 [DOI] [PubMed] [Google Scholar]
- 37.Harraz OF, Welsh DG. 2013. Proteinkinase A regulation of T-type Ca2+ channels in rat cerebral arterial smooth muscle. J. Cell Sci. 126:2944–54 [DOI] [PubMed] [Google Scholar]
- 38.Ottolini M, Sonkusare SK. 2021. The calcium signaling mechanisms in arterial smooth muscle and endothelial cells. Compr. Physiol. 11:1831–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stojilkovic SS, Leiva-Salcedo E, Rokic MB, Coddou C. 2014. Regulation of ATP-gated P2X channels: from redox signaling to interactions with other proteins. Antioxid. Redox Signal. 21:953–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Billaud M, Chiu YH, Lohman AW, Parpaite T, Butcher JT, et al. 2015. A molecular signature in the pannexin1 intracellular loop confers channel activation by the α1 adrenoreceptor in smooth muscle cells. Sci. Signal. 8:ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Falck JR, Gopal VR, Inscho EW, Imig JD. 2004. P2X receptor-stimulated calcium responses in preglomerular vascular smooth muscle cells involves 20-hydroxyeicosatetraenoic acid. J. Pharmacol. Exp. Ther. 311:1211–17 [DOI] [PubMed] [Google Scholar]
- 42.Negri S, Faris P, Berra-Romani R, Guerra G, Moccia F. 2019. Endothelial transient receptor potential channels and vascular remodeling: extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 10:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramsey IS, Delling M, Clapham DE. 2006. An introduction to TRP channels. Annu. Rev. Physiol. 68:619–47 [DOI] [PubMed] [Google Scholar]
- 44.Planells-Cases R, Valente P, Ferrer-Montiel A, Qin F, Szallasi A. 2011. Complex regulation of TRPV1 and related thermo-TRPs: implications for therapeutic intervention. In Transient Receptor Potential Channels, ed. Islam MS, pp. 491–515. Dordrecht, Neth.: Springer; [DOI] [PubMed] [Google Scholar]
- 45.Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, et al. 2017. Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCα in arterial myocytes. J. Gen. Physiol. 149:639–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang XR, Lin AH, Hughes JM, Flavahan NA, Cao YN, et al. 2012. Upregulation of osmomechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 302:L555–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee EJ, Shin SH, Hyun S, Chun J, Kang SS. 2011. Mutation of a putative S-nitrosylation site of TRPV4 protein facilitates the channel activates. Anim. Cells Syst. 15:95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao S, Anishkin A, Zinkevich NS, Nishijima Y, Korishettar A, et al. 2018. Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. J. Biol. Chem. 293:5307–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Earley S 2013. TRPM4 channels in smooth muscle function. Pflügers Arch. 465:1223–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kovács ZM, Dienes C, Hézső T, Almássy J, Magyar J, et al. 2022. Pharmacological modulation and (patho)physiological roles of TRPM4 channel-part 1: modulation of TRPM4. Pharmaceuticals 15:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. 1999. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397:259–63 [DOI] [PubMed] [Google Scholar]
- 52.Adebiyi A, Thomas-Gatewood CM, Leo MD, Kidd MW, Neeb ZP, Jaggar JH. 2012. An elevation in physical coupling of type 1 IP3 receptors to TRPC3 channels constricts mesenteric arteries in genetic hypertension. Hypertension 60:1213–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, et al. 2009. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 23:2425–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, et al. 2007. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflügers Arch. 455:465–77 [DOI] [PubMed] [Google Scholar]
- 55.Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, et al. 2010. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 298:C993–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, et al. 2009. TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J. Physiol. 587:2429–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denniss A, Dulhunty AF, Beard NA. 2018. Ryanodine receptor Ca2+ release channel post-translational modification: central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 101:49–53 [DOI] [PubMed] [Google Scholar]
- 58.Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, et al. 2014. A PLCγ1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci. Signal. 7:ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dora KA, Doyle MP, Duling BR. 1997. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. PNAS 94:6529–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandow SL, Hill CE. 2000. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 86:341–46 [DOI] [PubMed] [Google Scholar]
- 61.Isakson BE, Duling BR. 2005. Heterocellular contact at the myoendothelial junction influences gap junction organization. Circ. Res. 97:44–51 [DOI] [PubMed] [Google Scholar]
- 62.Isakson BE. 2008. Localized expression of an Ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J. Cell Sci. 121:3664–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, et al. 2011. Compartmentalized connexin 43 S-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 31:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Straub AC, Johnstone SR, Heberlein KR, Rizzo MJ, Best AK, et al. 2010. Site-specific connexin phosphorylation is associated with reduced heterocellular communication between smooth muscle and endothelium. J. Vasc. Res. 47:277–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen G, Suzuki H, Weston AH. 1988. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 95:1165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakajima S, Ohashi J, Sawada A, Noda K, Fukumoto Y, Shimokawa H. 2012. Essential role of bone marrow for microvascular endothelial and metabolic functions in mice. Circ. Res. 111:87–96 [DOI] [PubMed] [Google Scholar]
- 67.Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, et al. 2012. Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature 491:473–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Straub AC, Gladwin MT. 2018. Escorting α-globin to eNOS: α-globin-stabilizing protein paves the way. J. Clin. Investig. 128:4755–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lechauve C, Butcher JT, Freiwan A, Biwer LA, Keith JM, et al. 2018. Endothelial cell α-globin and its molecular chaperone α-hemoglobin-stabilizing protein regulate arteriolar contractility. J. Clin. Investig. 128:5073–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rahaman MM, Nguyen AT, Miller MP, Hahn SA, Sparacino-Watkins C, et al. 2017. Cytochrome b5 reductase 3 modulates soluble guanylate cyclase redox state and cGMP signaling. Circ. Res. 121:137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeMartino AW, Amdahl MB, Bocian K, Rose JJ, Tejero J, Gladwin MT. 2021. Redox sensor properties of human cytoglobin allosterically regulate heme pocket reactivity. Free Radic Biol. Med. 162:423–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tiso M, Tejero J, Basu S, Azarov I, Wang X, et al. 2011. Human neuroglobin functions as a redox-regulated nitrite reductase. J. Biol. Chem. 286:18277–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reeder BJ, Ukeri J. 2018. Strong modulation of nitrite reductase activity of cytoglobin by disulfide bond oxidation: implications for nitric oxide homeostasis. Nitric Oxide 72:16–23 [DOI] [PubMed] [Google Scholar]
- 74.Burmester T, Hankeln T. 2014. Function and evolution of vertebrate globins. Acta Physiol. 211:501–14 [DOI] [PubMed] [Google Scholar]
- 75.Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, et al. 2017. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat. Commun. 8:14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Hemann C, Abdelghany TM, El-Mahdy MA, Zweier JL. 2012. Characterization of the mechanism and magnitude of cytoglobin-mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J. Biol. Chem. 287:36623–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zweier JL, Hemann C, Kundu T, Ewees MG, Khaleel SA, et al. 2021. Cytoglobin has potent superoxide dismutase function. PNAS 118:e2105053118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tejero J, Kapralov AA, Baumgartner MP, Sparacino-Watkins CE, Anthonymutu TS, et al. 2016. Peroxidase activation of cytoglobin by anionic phospholipids: mechanisms and consequences. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1861:391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fordel E, Thijs L, Martinet W, Schrijvers D, Moens L, Dewilde S. 2007. Anoxia or oxygen and glucose deprivation in SH-SY5Y cells: a step closer to the unraveling of neuroglobin and cytoglobin functions. Gene 398:114–22 [DOI] [PubMed] [Google Scholar]
- 80.Amdahl MB, DeMartino AW, Tejero J, Gladwin MT. 2017. Cytoglobin at the crossroads of vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 37:1803–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amdahl MB, Sparacino-Watkins CE, Corti P, Gladwin MT, Tejero J. 2017. Efficient reduction of vertebrate cytoglobins by the cytochrome b5/cytochrome b5 reductase/NADH system. Biochemistry 56:3993–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Budworth J, Meillerais S, Charles I, Powell K. 1999. Tissue distribution of the human soluble guanylate cyclases. Biochem. Biophys. Res. Commun. 263:696–701 [DOI] [PubMed] [Google Scholar]
- 83.Friebe A, Mergia E, Dangel O, Lange A, Koesling D. 2007. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. PNAS 104:7699–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, et al. 1994. Mutation of His-105 in the β1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. PNAS 91:2592–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghosh A, Stuehr DJ. 2017. Regulation of sGC via hsp90, cellular heme, sGC agonists, and NO: new pathways and clinical perspectives. Antioxid. Redox Signal. 26:182–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stasch J-P, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, HS AK, et al. 2006. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Investig. 116:2552–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wood KC, Durgin BG, Schmidt HM, Hahn SA, Baust JJ, et al. 2019. Smooth muscle cytochrome b5 reductase 3 deficiency accelerates pulmonary hypertension development in sickle cell mice. Blood Adv. 3:4104–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Durgin BG, Wood KC, Hahn SA, McMahon B, Baust JJ, Straub AC. 2022. Smooth muscle cell CYB5R3 preserves cardiac and vascular function under chronic hypoxic stress. J. Mol. Cell. Cardiol. 162:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alapa M, Cui C, Shu P, Li H, Kholodovych V, Beuve A. 2021. Selective cysteines oxidation in soluble guanylyl cyclase catalytic domain is involved in NO activation. Free Radic. Biol. Med. 162:450–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Craven PA, DeRubertis FR. 1978. Effects of thiol inhibitors on hepatic guanylate cyclase activity. Evidence for the involvement of vicinal dithiols in the expression of basal and agonist-stimulated activity. Biochim. Biophys. Acta Enzymol. 524:231–44 [DOI] [PubMed] [Google Scholar]
- 91.Fernhoff NB, Derbyshire ER, Underbakke ES, Marletta MA. 2012. Heme-assisted S-nitrosation desensitizes ferric soluble guanylate cyclase to nitric oxide. J. Biol. Chem. 287:43053–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang C, Alapa M, Shu P, Nagarajan N, Wu C, et al. 2017. Guanylyl cyclase sensitivity to nitric oxide is protected by a thiol oxidation-driven interaction with thioredoxin-1. J. Biol. Chem. 292:14362–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, et al. 1999. Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Iα. Science 286:1583–87 [DOI] [PubMed] [Google Scholar]
- 94.Francis SH, Busch JL, Corbin JD, Sibley D. 2010. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62:525–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Landgraf W, Regulla S, Meyer H, Hofmann F. 1991. Oxidation of cysteines activates cGMP-dependent protein kinase. J. Biol. Chem. 266:16305–11 [PubMed] [Google Scholar]
- 96.Prysyazhna O, Rudyk O, Eaton P. 2012. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 18:286–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakamura T, Ranek MJ, Lee DI, Hahn VS, Kim C, et al. 2015. Prevention of PKG1α oxidation augments cardioprotection in the stressed heart. J. Clin. Investig. 125:2468–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ghimire K, Altmann HM, Straub AC, Isenberg JS.2017.Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 312:C254–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heiss EH, Dirsch VM. 2014. Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 20:3503–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen F, Kumar S, Yu Y, Aggarwal S, Gross C, et al. 2014. PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+-toxins. PLOS ONE 9:e99823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moccia F, Berra-Romani R, Tanzi F. 2012. Update on vascular endothelial Ca2+ signalling: a tale of ion channels, pumps and transporters. World J. Biol. Chem. 3:127–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foskett JK, White C, Cheung K-H, Mak D-OD. 2007. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87:593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moccia F, Negri S, Shekha M, Faris P, Guerra G. 2019. Endothelial Ca2+ signaling, angiogenesis and vasculogenesis: just what it takes to make a blood vessel. Int. J. Mol. Sci. 20:3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joseph SK, Young MP, Alzayady K, Yule DI, Ali M, et al. 2018. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 293:17464–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lock JT, Sinkins WG, Schilling WP. 2012. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 590:3431–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, et al. 2014. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 289:8170–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun L, Yau HY, Lau OC, Huang Y, Yao X. 2011. Effect of hydrogen peroxide and superoxide anions oncytosolic Ca2+: comparison of endothelial cells from large-sized and small-sized arteries. PLOS ONE 6:e25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu Q, Corda S, Zweier JL, Capogrossi MC, Ziegelstein RC. 1998. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 97:268–75 [DOI] [PubMed] [Google Scholar]
- 109.Zheng Y, Shen X. 2005. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. 10:29–36 [DOI] [PubMed] [Google Scholar]
- 110.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, et al. 2004. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10:1200–7 [DOI] [PubMed] [Google Scholar]
- 111.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. 2008. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J. Mol. Cell. Cardiol. 44:361–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bhardwaj R, Hediger MA, Demaurex N. 2016. Redox modulation of STIM-ORAI signaling. Cell Calcium 60:142–52 [DOI] [PubMed] [Google Scholar]
- 113.Zhu J, Lu X, Feng Q, Stathopulos PB. 2018. A charge-sensing region in the stromal interaction molecule 1 luminal domain confers stabilization-mediated inhibition of SOCE in response to S-nitrosylation. J. Biol. Chem. 293:8900–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gui L, Zhu J, Lu X, Sims SM, Lu WY, et al. 2018. S-Nitrosylation of STIM1 by neuronal nitric oxide synthase inhibits store-operated Ca2+ entry. J. Mol. Biol. 430:1773–85 [DOI] [PubMed] [Google Scholar]
- 115.Prins D, Groenendyk J, Touret N, Michalak M. 2011. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 12:1182–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gibhardt CS, Cappello S, Bhardwaj R, Schober R, Kirsch SA, et al. 2020. Oxidative stress-induced STIM2 cysteine modifications suppress store-operated calcium entry. Cell Rep. 33:108292. [DOI] [PubMed] [Google Scholar]
- 117.Lillo MA, Gaete PS, Puebla M, Ardiles NM, Poblete I, et al. 2018. Critical contribution of Na+-Ca2+ exchanger to the Ca2+-mediated vasodilation activated in endothelial cells of resistance arteries. FASEB J. 32:2137–47 [DOI] [PubMed] [Google Scholar]
- 118.Wagner S, Rokita AG, Anderson ME,Maier LS. 2013. Redox regulation of sodium and calcium handling. Antioxid. Redox Signal. 18:1063–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qian X, Francis M, Solodushko V, Earley S, Taylor MS. 2013. Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 20:138–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, et al. 2015. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 8:ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. 2008. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 80:445–52 [DOI] [PubMed] [Google Scholar]
- 122.Daneva Z, Ottolini M, Chen YL, Klimentova E, Kuppusamy M, et al. 2021. Endothelial pannexin 1-TRPV4 channel signaling lowers pulmonary arterial pressure in mice. eLife 10:e67777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Naik JS, Osmond JM, Walker BR, Kanagy NL. 2016. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 311:H1437–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kochukov MY, Balasubramanian A, Abramowitz J, Birnbaumer L, Marrelli SP. 2014. Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium-activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J. Am. Heart Assoc. 3:e000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Balzer M, Lintschinger B, Groschner K. 1999. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc. Res. 42:543–49 [DOI] [PubMed] [Google Scholar]
- 126.Greenberg HZE, Carlton-Carew SRE, Khan DM, Zargaran AK, Jahan KS, et al. 2017. HeteromericTRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vascul. Pharmacol. 96–98:53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poteser M, Graziani A, Rosker C, Eder P, Derler I, et al. 2006. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 281:13588–95 [DOI] [PubMed] [Google Scholar]
- 128.Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. 2009. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology 24:107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McMahon TJ, Darrow CC, Hoehn BA, Zhu H. 2021. Generation and export of red blood cell ATP in health and disease. Front. Physiol. 12:754638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Al-Samkari H, van Beers EJ. 2021. Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther. Adv. Hematol. 12. 10.1177/20406207211066070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schroeder P, Fulzele K, Forsyth S, Ribadeneira MD, Guichard S, et al. 2022. Etavopivat, a pyruvate kinase activator in red blood cells, for the treatment of sickle cell disease. J. Pharmacol. Exp. Ther.380:210–19 [DOI] [PubMed] [Google Scholar]
- 132.McMahon TJ, Stone AE, Bonaventura J, Singel DJ, Stamler JS. 2000. Functional coupling of oxygen binding and vasoactivity in S-nitrosohemoglobin. J. Biol. Chem. 275:16738–45 [DOI] [PubMed] [Google Scholar]
- 133.Gladwin MT, Wang X, Reiter CD, Yang BK, Vivas EX, et al. 2002. S-Nitrosohemoglobin is unstable in the reductive erythrocyte environment and lacks O2/NO-linked allosteric function. J. Biol. Chem. 277:27818–28 [DOI] [PubMed] [Google Scholar]
- 134.Patel RP, Hogg N, Spencer NY, Kalyanaraman B, Matalon S, Darley-Usmar VM. 1999. Biochemical characterization of human S-nitrosohemoglobin. Effects on oxygen binding and transnitrosation. J.Biol. Chem. 274:15487–92 [DOI] [PubMed] [Google Scholar]
- 135.Bailey DM, Rasmussen P, Overgaard M, Evans KA, Bohm AM, et al. 2017. Nitrite and S-nitrosohemoglobin exchange across the human cerebral and femoral circulation: relationship to basal and exercise blood flow responses to hypoxia. Circulation 135:166–76 [DOI] [PubMed] [Google Scholar]
- 136.Gladwin MT, Ognibene FP, Pannell LK, Nichols JS, Pease-Fye ME, et al. 2000. Relative role of heme nitrosylation and β-cysteine 93 nitrosation in thetransport and metabolism of nitric oxide by hemoglobin in the human circulation. PNAS 97:9943–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.DeMartino AW, Kim-Shapiro DB, Patel RP, Gladwin MT. 2019. Nitrite and nitrate chemical biology and signalling. Br. J. Pharmacol. 176:228–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Isbell TS, Sun CW, Wu LC, Teng X, Vitturi DA, et al. 2008. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat. Med. 14:773–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang R, Hausladen A, Qian Z, Liao X, Premont RT, Stamler JS. 2022. Hypoxic vasodilatory defect and pulmonary hypertension in mice lacking hemoglobin β-cysteine93 S-nitrosylation. JCI Insight 7:e155234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun CW, Yang J, Kleschyov AL, Zhuge Z, Carlstrom M, et al. 2019. Hemoglobin β93 cysteine is not required for export of nitric oxide bioactivity from the red blood cell. Circulation 139:2654–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, et al. 2003. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9:1498–505 [DOI] [PubMed] [Google Scholar]
- 142.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, et al. 2005. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J. Clin. Investig. 115:2099–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shiva S 2013. Nitrite: a physiological store of nitric oxide and modulator of mitochondrial function. Redox Biol. 1:40–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dent MR, DeMartino AW, Tejero J, Gladwin MT. 2021. Endogenous hemoprotein-dependent signaling pathways of nitric oxide and nitrite. Inorg. Chem. 60:15918–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Basu S, Grubina R, Huang J, Conradie J, Huang Z, et al. 2007. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 3:785–94 [DOI] [PubMed] [Google Scholar]
- 146.Wood KC, Cortese-Krott MM, Kovacic JC, Noguchi A, Liu VB, et al. 2013. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler. Thromb. Vasc. Biol. 33:1861–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Leo F, Suvorava T, Heuser SK, Li J, LoBue A, et al. 2021. Red blood cell and endothelial eNOS independently regulate circulating nitric oxide metabolites and blood pressure. Circulation 144:870–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Booth DM, Várnai P, Joseph SK, Hajnóczky G. 2022. Fluorescence imaging detection of nanodomain redox signaling events at organellar contacts. STAR Protoc. 3:101119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dosunmu-Ogunbi AM, Wood KC, Novelli EM, Straub AC. 2019. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 3:2679–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dosunmu-Ogunbi A, Yuan S, Reynolds M, Giordano L, Sanker S, et al. 2022. SOD2 V16A amplifies vascular dysfunction in sickle cell patients by curtailing mitochondria complex IV activity. Blood 139:1760–65 [DOI] [PMC free article] [PubMed] [Google Scholar]