

Abstract
Acoustic tweezers have gained prominence as fundamental tools in life sciences and translational medical research for the manipulation of single cells in a high-throughput, precise, selective, and contact-free manner. However, protocols for the fabrication and experimental implementation of acoustic tweezers are lacking, thereby limiting the broad application of these powerful tools, which hold great potential for addressing challenges in the design of next-generation cellular assays and enhancing the understanding of complex biological systems. Herein, we describe a comprehensive protocol for applying acoustic tweezers to study single cells with an enhanced experimental throughput that is several orders of magnitude greater than traditional methods, including atomic force microscopy, micropipette aspiration, and optical tweezers. With this stepwise protocol, which includes device fabrication, instrumentation setup, and data acquisition, researchers can conduct versatile life sciences, biophysical studies, or clinical applications that require the trapping, patterning, pairing, and separation of single cells. In addition, this universal protocol can be adapted to study interdisciplinary topics, such as investigations into the spinning motion of colloids in material science or the development of acoustic-based quantum simulations in physics. Overall, the device fabrication requires ~12 h, the experimental setup of the acoustic tweezers takes 1-2 h, and the cell manipulation experiment takes ~30 min to complete.
Introduction
Advances in biophysical methodologies have fostered the understanding of many fundamental cellular processes, including cell proliferation1, differentiation2, migration3, apoptosis4, and immune recognition5. High-precision manipulation methods, such as atomic force microscopy6,7, micropipette aspiration8,9, and optical tweezers10,11, are applied to investigate these cellular processes, especially those significantly affected by dynamic mechanical perturbations. However, these state-of-the-art single cell manipulation techniques typically require direct physical contact with the cells, which can lead to detrimental effects12,13 such as those arising from cytoadherence between the cell and the external entity contacting the cell or from the introduction of chemical agents. Furthermore, these methods can only probe a single cell or one pair of cells simultaneously and require time-consuming and labour-intensive procedures6–11.
Recognizing the demand for a contact-free, high-throughput biophysical method for manipulating single cells14–17, we recently developed a new type of acoustic tweezers by applying harmonic acoustics for non-contact, dynamic, selective (HANDS) cell manipulation18. By leveraging the advantages of acoustic tweezers19-26, the HANDS platform can offer a contact-free and highly biocompatible method for single cell manipulation. More importantly, our HANDS platform can reversibly pair and separate single cells in a high-throughput, precise, repeatable, and programmable manner, all unique capabilities existing acoustic tweezers platforms21-26 cannot achieve. With our HANDS platform, we can pair and separate more than 100 pairs of cells simultaneously within suspension to quantify the cell-to-cell adhesion for rapid diagnostics. This time-efficient and parallel nature enables one researcher to generate reliable statistics for single-cell biophysical studies within 30 minutes by avoiding serial measurements of protracted experimental setup procedures from other established techniques6-11.
Although the development of our HANDS platform may enable new discoveries and inspire novel applications in life sciences, medicine, and biophysical studies6-11, detailed protocols including the fabrication and application of the acoustic tweezers for single cell manipulation, are currently lacking and have not been reported. Here, we present a comprehensive protocol including device fabrication, experimental setup, and data acquisition with acoustic tweezers. With this protocol, we anticipate that a trained researcher can conduct versatile studies that require trapping, patterning, pairing, and separation of single cells more efficiently than with other commonly applied techniques.
Overview of the technique
This protocol includes stepwise instructions (Table 1) for the device fabrication, experimental setup, data acquisition, and analysis for single cell manipulation with acoustic tweezers. A flow diagram of the detailed workflow is illustrated in Fig. 1. Specifically, we introduced the detailed fabrication processes (Fig. 1a and Supplementary Fig. 1) of the interdigital transducers, acoustic tweezers, and microfluidic chamber that have been used routinely in our laboratory. Due to the segmented transducer design, we can efficiently generate multi-harmonic surface acoustic waves (SAWs) and shape the acoustic waveforms to manipulate microparticles and cells dynamically. We also optimized the microfluidic chamber, which is enclosed with 150-μm-wide polydimethylsiloxane (PDMS) side walls separating the air and liquid sections (Supplementary Fig. 1c), to minimize the acoustic damping effect for efficient SAW generation. Furthermore, this protocol provides critical steps for the irreversible binding between the microfluidic chamber and the interdigital transducer substrate for acoustic tweezers fabrication. Following the fabrication processes, we discuss the experimental setup procedure that involves the equipment setup for programmable control of the acoustic tweezers, cell culture, and chemical perturbations (Fig. 1b), and the main processes of the device operation (Fig. 1c). Due to the fully programmable capability of the acoustic tweezers, we can achieve high-throughput, reversible cell-cell pairing and separation of single cells. Since we manipulate an array of single cells in suspension, the data acquisition can be conducted in parallel, acquiring time-efficient comprehensive statistics for rapid diagnostics.
TABLE 1 |.
Main processes of the protocol and their unique key features.
| Process | Key features | Steps | Figures |
|---|---|---|---|
| Microfluidic chamber fabrication | Optimized chamber design and fabrication process to minimize acoustic damping | 1-6 | Figure 1 |
| Interdigital transducer fabrication | Segmented transducers for efficient multi-harmonic SAWs generation | 7-12 | Figure 1 |
| Acoustic tweezers fabrication | Highly effective method for irreversible bonding | 13-18 | Figure 2a–c |
| Experimental setup | Full programmable control of acoustic fields | 19-36 | Figure 2d |
| Cell culture | Universal platform for manipulation of versatile cell types | 37-45 | Figure 3 |
| Single cell manipulation | High-throughput reversible cell-cell pairing and separation control | 46-64 | Figures 4a–c |
| Data acquisition and analysis | Time-efficient and parallel data acquisition | 65-72 | Figures 4d-f |
Fig. 1 |. Flow diagram of the workflow.
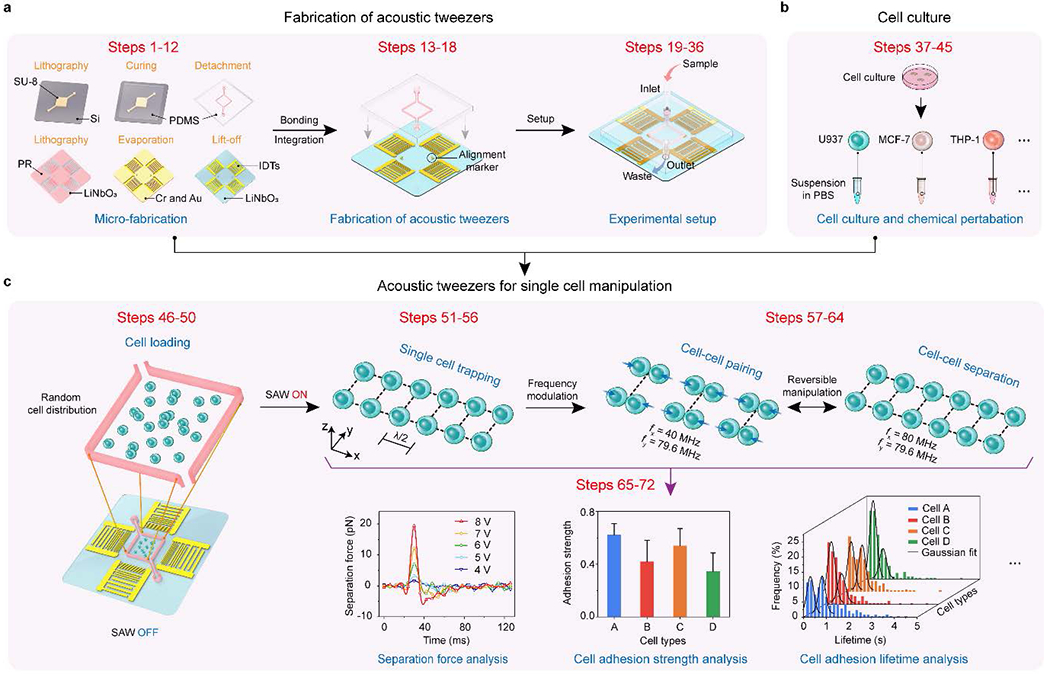
a, Fabrication steps of acoustic tweezers. b, Steps of cell culture. c, Steps of acoustic tweezers for single cell manipulation and data analysis18.
Applications and target audience
In the current implementation of acoustic tweezers, researchers in biology1-5, biophysics6-11, and medicine27-33 will find the protocol helpful in employing reversible cell-cell pairing for various research and drug development purposes. For example, by screening cancer cells and evaluating their potential metastatic differences through reversible cell-cell pairing studies18, the critical regulators in carcinogenesis could be uncovered from the associated complex signaling pathways. This protocol could also be applied by pairing two heterotypic cells with acoustic tweezers to provide a rapid screening method with dynamic control over heterogeneous intercellular interactions. In our previous work21, we have already quantitatively investigated the intercellular communication over a gap junction by pairing heterotypic cells. Other potential applications of this protocol range from immunotherapies, such as tumor-immune cell interactions6, to cell mechanosensing research (e.g., TCR-pMHC interactions9) at the single cell level.
Additionally, this protocol can be easily adapted and applied to enable new applications in acoustofluidics research34-42 by simply modifying the microfluidic chamber design. Engineers may find it extremely useful to use this protocol as a guideline when conducting experiments in various applications, including nanoparticle (extracellular vesicles, liposomes, and virus) isolation43, cell sorting and separation44, and even small organisms (C. elegans and zebrafish) manipulation45. More importantly, the SAW-programmable capability demonstrated in our recent paper18 also shows the potential application of this protocol in cross-disciplinary topics, such as investigations into the spinning motion of colloids18 in material science or the development of an acoustic-based quantum simulator46·47 and gauge field controller48 in physics. Thus, this protocol could be extended to become a universal protocol for the programmable manipulation of particles and cells49.
Advantages and limitations
Single cell manipulation techniques, such as atomic force microscopy6,7, micropipette aspiration8,9, and optical tweezers10,11, have been previously applied to study biophysics at the single cell level. However, in practical single cell assays, the currently available methods are extremely limited due to their reliance on physical contact with the cells and low experimental throughputs (Table 2). When using these contact-based methods, specific properties of cells, such as adhesion strength, are influenced by the molecules used to tether cells to a cantilever6 or bead10. As a result, it is challenging to isolate cell-cell interaction phenomena using contact-based methods. In addition, generating statistically significant results requires labor-intensive and lengthy experimental procedures.
TABLE 2 |.
Comparison of various single cell biophysical methods.
| Method | Biocompatibility | Contactless nature | Throughput |
|---|---|---|---|
| Optical tweezers | Low | No | Low |
| Atomic force microscopy | Moderate | No | Low |
| Micropipette aspiration | Moderate | No | Low |
| Acoustic tweezers | High | Yes | High |
To overcome these two main limitations, in this protocol, we introduce acoustic tweezers for single cell manipulation in a contact-free and parallel fashion with a throughput at least 100 times greater than existing single cell manipulation methods. Moreover, our tweezers offer several distinct advantages for studying single cells. The acoustic tweezers provide a rapid, repeatable (operating for more than 1,000 pairing cycles) and non-invasive method for directly performing the cell-cell pairing/separation process in aqueous solutions, which avoids time-consuming cell preparation protocols. In addition, the acoustic tweezers provide a self-aligning method for cellular manipulation, wherein the cells automatically form self-aligned pairs in the acoustic well traps without needing to fine-tune their positions with complex optical set-ups or expensive mechanical manipulators. Furthermore, the repeated cell-cell pairing/separation can be automized. All the cell-cell pairing parameters, such as cell-cell pairing/separation duration or force magnitudes, can also be programmed in advance. With these distinct advantages, our acoustic tweezers illustrated in this protocol enable a unique and high-throughput platform for controlling reversible cell-cell pairing for functional cellular assays.
In this protocol, the acoustic tweezers are optimized for manipulating single cells with size ranges from 10 μm to 15 μm. Currently, cells outside this size range may cause a loss in precision in the separation of paired cells in the tweezers. However, this issue can be addressed by modifying the design of the interdigital transducers18,26, and we guide how to change the design. For example, the acoustic manipulation of cell clusters or organisms (with sizes larger than 15 μm) can be achieved by decreasing the applied wavelength of SAWs by increasing the pitch of an electrode in the interdigital transducers. Furthermore, the current device operation in this protocol is conducted at room temperature (20 °C). Although our current system setup can be used for various temperatures, a temperature control system would be required to monitor the temperature changes.
Materials
Reagents
SU-8 25 photoresist (Kayaku Advanced Materials, cat. no. Y131263), cleanroom chemical
SU-8 developer (Kayaku Advanced Materials, cat. no. Y020100), cleanroom chemical
SPR-3012 photoresist (Kayaku Advanced Materials, cat. no. 10016863), cleanroom chemical
CD-26 developer (Kayaku Advanced Materials, cat. no. 10018050), cleanroom chemical
Acetone (VWR, cat. no. BDH1101-4LP)
Ethanol (VWR, cat. no. 89125-188)
PDMS pre-polymer and curing agent: SYLGARD184 (Ellsworth Adhesives, cat. no. 2065622)
(3-Aminopropyl) triethoxysilane (APTES), (Sigma-Aldrich, cat. no. 440140-500ML)
Chlorotrimethylsilane (TMSCI) (Sigma-Aldrich, cat. no. 386529)
Silver conductive epoxy (Amazon, cat. no. 8331D-14G)
Phosphate buffered saline (PBS) (Thermo Fisher Scientific, cat. no. 20012050)
Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, cat. no. 10564029)
RPMI 1640 (Thermo Fisher Scientific, cat. no. 72400120)
Fetal bovine serum (FBS) (Thermo Fisher Scientific, cat. no. 10437010)
Penicillin-Streptomycin (Thermo Fisher Scientific, cat. no. 15140122)
2-Mercaptoethanol (Sigma-Aldrich, cat. no. M6250-500ML)
Phorbol 12-myristate 13-acetate (PMA), (Sigma-Aldrich, cat. no. P8139-5MG)
TrypLE Express Enzyme (Thermo Fisher Scientific, cat. no. 12605028)
Cytochalasin D (Thermo Fisher Scientific, cat. no. PHZ1063)
SiR-actin kit (Cytoskeleton, cat. no. CY-SC001)
Equipment
Spin coater (Headway, model no. PWM32), cleanroom facility
Hot plate (CHROMALOX, model no. 1600), cleanroom facility
Mask aligner (Karl Suss, model no. MA6/BA6), cleanroom facility
E-beam evaporator (CHA Industries, model no. Solution E-Beam) for Cr and Au thin film deposition, cleanroom facility
Dual-channel function generator (Tektronix, model no. AFG3102C)
(Optional) Radio-frequency signal generator (Keysight, model no. N9310A)
(Optional) Radio-frequency signal generator (Keysight, model no. E4422B)
Power amplifier (Amplifier Research, model no. 30W1000B)
Oscilloscope (Keysight, model no. MSOX2024A)
Inverted microscope (Nikon, model no. Eclipse Ti)
Syringe pump (CETONI, model no. neMESYS)
Digital multimeter (FLUKE, model no. 115)
Coax BNC cable (50 ohms) (L-com, cat. no. CC58C-4)
Oscilloscope voltage probe (50 ohms) (TestEquity, cat. no. 25989.1)
Photomasks (Frontrange photomask)
Stainless steel tweezer (TED PELLA, cat. no. 532A)
4-inch silicon wafer (University Wafer, cat. no. 783)
4-inch Y-cut 128° LiNbO3 wafer (Precision Micro-Optics, cat. no. PWLN-431232)
150-mm-diameter petri dish (VWR, cat. no. 25384-326)
Polyethylene tubing (Warner Instruments, cat. no. PE-10)
Parafilm (VWR, cat. no. 52858-000)
Glass plier (Amazon, cat. no. B002YCDCQ6)
Diamond tipped glass cutter (Amazon, cat. no. B08L74YV3S)
Electric wire (Amazon, cat. no. B-30-1000)
Cell incubator (Thermo Fisher Scientific, model no. Heracell™ VIOS 160i)
Inverted confocal microscope (Zeiss, model no. Zeiss 880 Airyscan)
Water bath (Benchmark, model no. MyBlock)
Western blotting electrophoresis system (Bio-rad, model no. Mini-PROTEAN® Tetra)
Western blotting transfer system (Bio-rad, model no. Trans-Blot Turbo Transfer system)
Western blotting imaging system (Bio-rad, model no. ChemiDoc Touch Imaging System)
Cell culture dish (VWR, cat. no. 734-0006)
T-25 flask (Thermo Fisher Scientific, cat. no. 156367)
Ultra-low attachment culture dish (Sigma-Aldrich, cat. no. CLS3262)
Microscope control software (Nikon NIS-Elements)
MATLAB (MathWorks, MATLAB R2022a)
ImageJ (ImageJ 1.51w)
Procedure
Fabrication of microfluidic chamber ● TIMING 4 h
1| Spin-coat a 4-inch-diameter silicon wafer with SU-8 25 at 2420 r.p.m. for 30 s. Immediately bake the spin-coated wafer with a two-step contact hot plate process at 65 °C for 2.5 min and 95°C for 6 min to evaporate the solvent. Then, expose the baked wafer under the photolithography mask using a MA6/BA6 mask aligner system with the following parameters: expose dose = 180 mJ/cm2, UV power = 12 mJ/cm2.s, and exposure time =15 s.
2| Cure the exposed wafer with a two-step contact hot plate process at 65 °C for 1 min and 95 °C for 2.5 min to minimize resist cracking. Develop the cured wafer in SU-8 developer for 3 min and dry it with a nitrogen stream. Examine the developed SU-8 pattern under an upright microscope.
? TROUBLESHOOTING
■ PAUSE POINT The fabricated SU-8 mould can be stored in the wafer box at room temperature for years If it is handled with care.
3| To prevent the adhesion between SU-8 mould and PDMS, tape the fabricated mould in a 150-mm-diameter petri dish and expose the mould to 0.2 ml TMSCI for 30 min in the laminar flow hood.
! CAUTION TMSCI is toxic, corrosive, and combustible and should be handled in the laminar flow hood.
4| Pour degassed PDMS mixture with PDMS base and curing agent at a weight ratio of 10:1 into the petri dish. Cure the PDMS in an oven at 65 °C for 30 min.
5| Slowly peel the cured PDMS from the mould and attach both surfaces of the PDMS block with Parafilm to avoid potential contamination in the following procedures. Cut the PDMS into separated blocks, with each block containing an individual microfluidic chamber.
6| Punch both the inlet and outlet holes using a 0.75-mm hole puncher with the patterned microfluidic chamber facing up (Fig. 2a).
Fig. 2 |. Device fabrication and experimental setup of the acoustic tweezers.
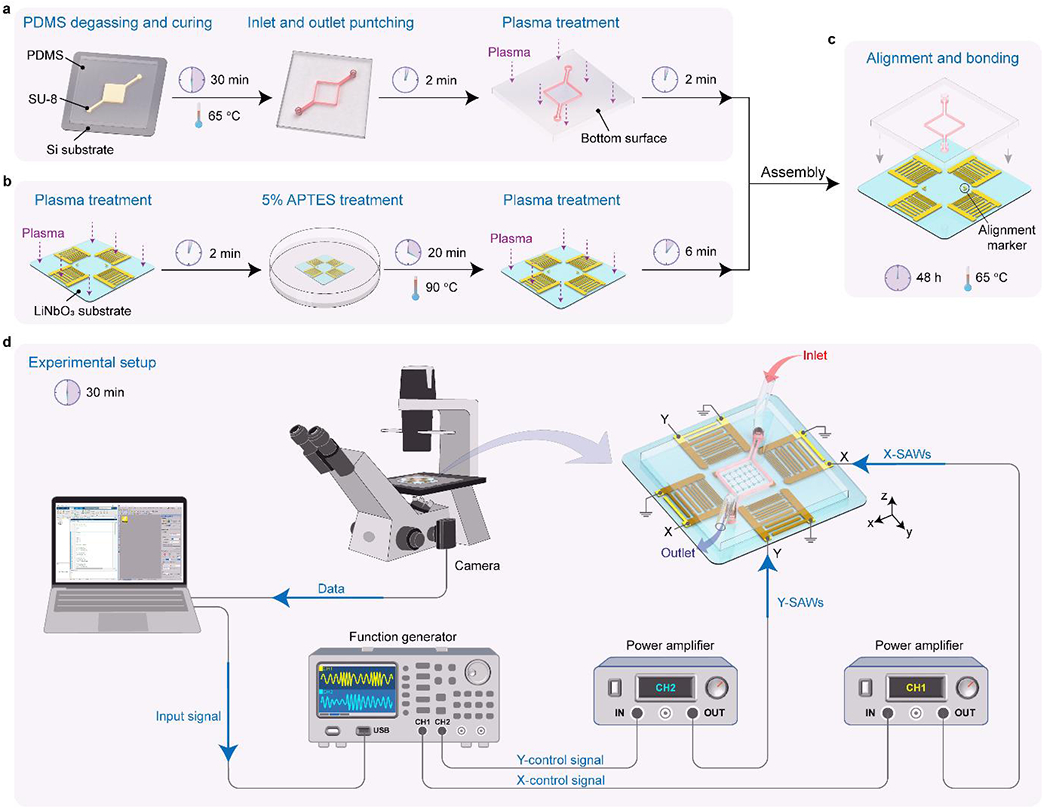
a, Fabrication steps of the microfluidic chamber. b, Fabrication steps of the interdigital transducers on the LiNbO3 substrate. c, Steps of alignment and bonding of the acoustic tweezers. d, Experimental setup of the acoustic tweezers to generate standing surface acoustic waves for single cell manipulation.
Fabrication of interdigital transducers ● TIMING 7 h
7| Spin-coat a 4-inch-diameter Y-cut 128° LiNbO3 wafer with SPR-3012 photoresist (PR) at 3000 r.p.m. for 45 s. Immediately bake the spin-coated wafer at 90°C for 1.5 min on a contact hot plate. Then, expose the baked wafer under a photolithography mask using the MA6/BA6 mask aligner system with the following parameters: UV power = 12 mJ/cm2.s, and exposure time =10 s.
8| Cure the exposed wafer at 115 °C for 1.5 min on a contact hot plate. Develop the cured wafer in CD-26 developer for 1.5 min. Rinse the wafer with deionized water and dry it with a nitrogen stream. Examine the developed SPR-3012 pattern under an upright microscope.
? TROUBLESHOOTING
9| Load the developed LiNbO3 wafers (4 or 5 wafers per batch) in the dome of an e-beam evaporator (CHA Industries, model no. Solution E-Beam) and conduct the metal deposition with a target thickness of 10 nm for chromium (Cr) as an adhesion layer and 80 nm for gold (Au), sequentially.
10| After evaporation, take the LiNbO3 wafers from the dome of the e-beam evaporator and clean any dust on the wafer with a dry nitrogen stream.
11| Place the LiNbO3 wafers in a 150-mm-diameter glass container and immerse them in acetone for the lift-off process (Fig. 2b). Seal the container with a cap and store it in the laminar flow hood for 2 h.
12| Sonicate the LiNbO3 wafers using an ultrasonic bath for 2 min. Clean the wafers with acetone and dry the wafer with an air stream.
■ PAUSE POINT The fabricated LiNbO3 wafers can be stored in the wafer box at room temperature indefinitely before proceeding.
Fabrication of acoustic tweezers ● TIMING 1 h
13| Dice the fabricated LiNbO3 wafers to individual chips with glass pliers and a diamond-tipped glass cutter. Clean the diced chips with acetone and dry the chips with an air stream.
14| Treat the LiNbO3 chips with plasma for 2 min. Then immerse the treated LiNbO3 chips in 5% APTES aqueous solution at 90 °C for 20 min. Rinse the APTES-treated chips with deionized water and dry the chips with an air stream.
▲ CRITICAL STEP The APTES-treatment is required to enhance the bonding strength between the PDMS chamber and the LiNbO3 substrate.
15| Clean any dust on the APTES-treated LiNbO3 substrate with an air stream. Peel the Parafilm off from both surfaces of the PDMS block fabricated from Step 6 and remove any dust by applying Scotch tape to the surface with the patterned microfluidic chamber.
16| Treat both the PDMS chamber and the APTES-treated LiNbO3 substrate with plasma for 3 min and 6 min, respectively.
17| Before bonding the PDMS chamber and the APTES-treated LiNbO3 substrate, perform the alignment with the help of alignment markers (Fig. 2c) under an inverted microscope. Gently apply the pressure on PDMS chamber to enhance the bonding strength between the PDMS chamber and the LiNbO3 substrate.
18| Connect each electrode on the bonded device to a 30-gauge electric wire using silver conductive epoxy. Store the fabricated device in an oven at 65 °C.
■ PAUSE POINT The devices can be taken from the oven after baking for 2 days. Then the fabricated devices can be stored at room temperature for months before proceeding.
Experimental setup ● TIMING 2 h
19| Test that the microfluidic chamber is firmly bonded on the LiNbO3 substrate by pressing all the side walls of the PDMS chamber and ensuring that there is no air cavity generated at the interference between the PDMS structure and the LiNbO3 substrate.
▲ CRITICAL STEP This bonding test is essential to ensure there is no leakage when injecting any fluidic sample.
? TROUBLESHOOTING
20| Test that the two electrodes of each IDT on the LiNbO3 substrate are not short-circuited with a digital multimeter.
▲ CRITICAL STEP This short circuit test is required to avoid electrical damage to the connected function generator, amplifier, and cables in the following steps.
? TROUBLESHOOTING
21 Cut two 25-cm pieces of polyethylene tubes (i.d. = 0.28 mm, o.d. = 0.61 mm) at a 45° angle and insert them into the inlet and outlet of the device with stainless steel tweezers.
22| Clean the bottom side of the device with ethanol to remove any dirt. Dry the device with an air stream and mount it onto the inverted microscope stage with Scotch tapes.
23| Inject 0.3 ml Pluronic F-127 aqueous solution (5% (wt/vol) in deionized water) into the microfluidic chamber through the polyethylene tube and coat the microfluidic chamber for 30 min.
▲ CRITICAL STEP The Pluronic-coating is required to prevent cell attachment on either PDMS chamber or the LiNbO3 substrate. During the Pluronic-coating, Steps 24–33 can be performed in parallel.
24| Connect the output port of a function generator (Tektronix, model no. AFG3102C) to the input port of a power amplifier (Amplifier Research, model no. 30W1000B) with a coax BNC cable (Fig. 2d). Connect the output port of the power amplifier to an alligator clip with the other coax BNC cable. Then connect the alligator clip to the electric wires (Step 18) of the acoustic tweezer device.
! CAUTION Make sure that all the function generators and amplifiers are turned off when connecting BNC cables. Please fasten all the BNC cables and connectors to avoid any electric surge.
25| Power up the function generator and set the output voltage to −20 dBm. Turn the frequency to 80 MHz to generate a sine signal and set the phase to 0°. Make sure the output signal indicator light is off.
26| Turn the power output knob of the amplifier to the lowest setting and turn on the amplifier.
! CAUTION The amplifier must be grounded with the supplied power cable to avoid any electric shock.
27| Connect the voltage probe (BNC terminator) with the oscilloscope (Keysight, model no. MSOX2024A) and set the probe to ×10 to protect the input port of the oscilloscope. Power up the oscilloscope and turn the attenuation to match the probe setting (x10) to monitor the RF signals accurately.
28| Connect the voltage probe to the electric wires of the acoustic tweezer device to monitor the RF signals that are applied to the IDTs.
! CAUTION If any electric surge is monitored by the oscilloscope, power off all the equipment, check the device, and repeat Step 20.
29| Turn the output signal of the function generator on to generate the RF sine signal. The output signal indicator light of the function generator should be on.
30| Monitor the detected sine signal with the oscilloscope, and the measured frequency should be 80 MHz. Monitor the detected output voltage with the oscilloscope when increasing it to the target value, such as 20 Vpp, by tuning the output voltage through either the function generator or the amplifier. After the setup of both frequency and output voltage, turn the output signal of the function generator off, and the output signal indicator light of the function generator should be off.
! CAUTION The output voltage of the function generator or the radiofrequency (RF) signal generators (Keysight, model no. N9310A and E4422B) cannot be set higher than 0 dBm to avoid any damage to the input port of the amplifier.
31| If a dual channel function generator or multiple signal generators and amplifiers are used, repeat Steps 24–30 to set up the other equipment.
32| Power up the white light of the inverted microscope and switch to the ×10 objective lens to image the microfluidic chamber. Turn on the CMOS camera or the fast camera for either long-term or high-frame-rate video recording, depending on the target experiment.
33| Rotate the polarizer chip until the double image effect caused by the light reflection from the LiNbO3 surface is eliminated.
34| Withdraw 5 ml PBS with a 5 ml syringe and insert the syringe into the hub of a 30-G needle. Insert the needle into the polyethylene tubing connected to the inlet of the device. Mount a 5 ml syringe with PBS solution on the syringe pump (CETONI, model no. neMESYS). Power up the syringe pump and set the syringe volume to 5 ml with an infusion speed of 100 μl min−1.
35| Run the infusion process and inject the PBS to wash away the Pluronic solution for 5 min.
36| Carefully check the microfluidic device under the microscope and wash away any dirt particles or bubbles with PBS.
? TROUBLESHOOTING
Cell culture and chemical perturbations ● TIMING 4 d
37| Culture the human myeloid leukemia cell line (U937 (ATCC-CRL1593.2)) in RPMI 1640 supplemented with 10% FBS and 1% penicillin-streptomycin with T-25 flasks (Fig. 3a). Culture the human monocytic leukemia cell line (THP-1 (ATCC-TIB202)) in RPMI 1640 supplemented with 0.05 mM 2-Mercaptoethanol, 10% FBS, and 1% penicillin-streptomycin with T-25 flasks (Fig. 3b). Culture the human breast cancer cell lines (MDA-MB-231 (ATCC-HTB26) and MCF-7 (ATCC-HTB22) and human embryonic kidney cell line (HEK 293T (ATCC-CRL11268)) in DMEM, containing 10% FBS and 1% penicillin-streptomycin in 100 mm tissue culture dishes (Fig. 3c).
Fig. 3 |. Cell culture and chemical perturbations.
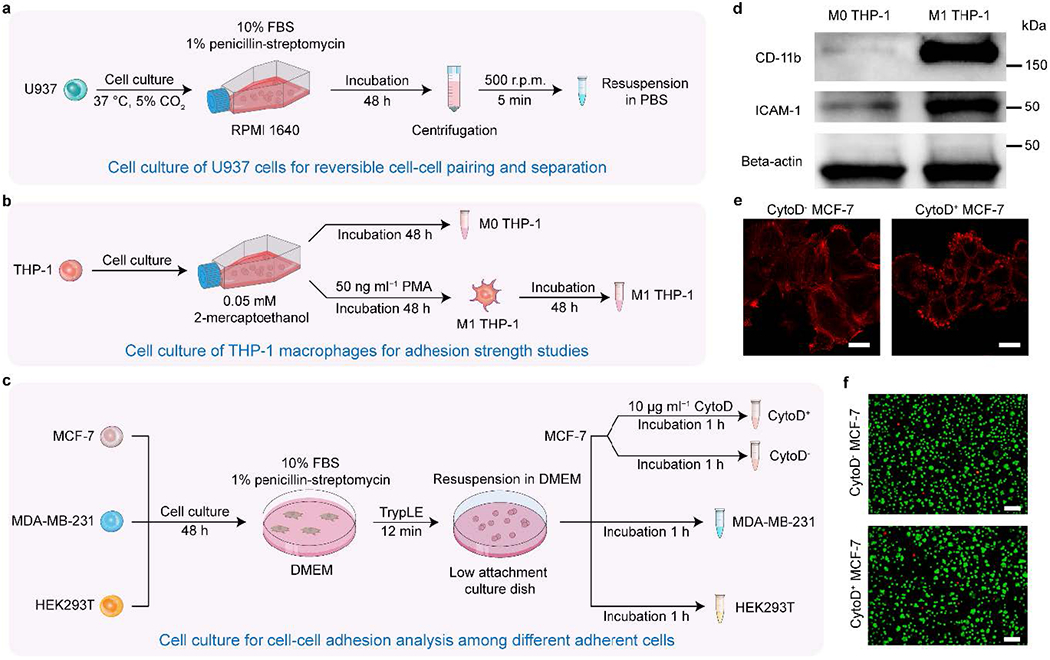
a, Cell culture of U937 cells for reversible cell-cell pairing and separation, b, Cell culture of THP-1 macrophages for adhesion strength studies, c, Cell culture for cell-cell adhesion analysis among different adherent cells, d, Western blot analysis of CD11b, ICAM-1, and Beta-actin for M0 and M1 THP-1 cells18. A high expression of CD11b in M1 THP-1 cells indicates successful cell differentiation from M0 THP-1 with PMA stimulation, e, Representative single z-layer images of F-actin from live MCF-7 cells, with and without CytoD stimulation, for both adherent and suspension cells (n=8 fields of view). Scale bar: 10 μm. f, Representative images of cell viability assays for MCF-7 cells with/without Cytochalasin D inhibition. For each condition, live/dead cell staining was conducted using Calcein-AM for live cells and SYTOX orange for dead cells. Scale bar: 100 μm.
38| Maintain all cultured cells in a cell incubator (Thermo Fisher Scientific, model no. Heracell™ VIOS 160i) at 37°C and a CO2 level of 5%. Replate all the cell lines with a concentration of 2 × 104 cells/ml every 2 d.
39| Differentiate the M0 phase THP-1 cells into macrophages (M1 phase THP-1) via incubation with 50 ng/ml PMA for 48 hours (Fig. 3b) and subsequent incubation in RPMI 1640 cell culture medium for another 48 h.
40| Conduct a Western blot analysis (Fig. 3d) to confirm the differentiation of M0 THP-1 cells into M1 THP-1 cells.
Count the suspended M0 THP-1 cells using a hemocytometer and count the adherent M1 THP-1 cells by assessing cell density under a microscope.
Prepare both M0 and M1 THP-1 cells with a concentration of 5 × 106 cells/ml and lyse the cells using 400 μl of radioimmunoprecipitation assay (RIPA) buffer added with a protease inhibitor cocktail. Incubate both samples on ice for 30 min.
Collect the lysis products and centrifuge them at 12,000 g for 15 min. Collect the supernatants (5 μl) and mix them with a tricine sample buffer at a 1:4 volume-to-volume ratio. Then, incubate the lysis products in a water bath (Benchmark, model no. MyBlock) at 95 °C for 6 min.
Load the samples into 12% acrylamide and conduct the two-step electrophoresis (Bio-rad, model no. Mini-PROTEAN® Tetra) for 10 min at 90 V and then 30 min at 110 V.
After electrophoresis, transfer the targeted proteins in the acrylamide gels to a PVDF membrane with a western blotting transfer system (Bio-rad, model no. Trans-Blot Turbo Transfer system).
Block the membrane with a blocking solution at room temperature for 1 h, then incubate the membranes with anti-CD11b, anti-ICAM-1, and anti-Beta-actin at the recommended dilution rates at 4 °C for 12 h.
After the first antibody incubation, incubate the membranes using horseradish peroxidase (HRP) conjugated secondary antibodies at room temperature for 1 h. Observe the targeted proteins’ bands on the membranes and obtain Western blot images (Bio-rad, model no. ChemiDoc Touch Imaging System).
41| Conduct cell staining and observe the actin structures with a confocal microscope to confirm the actin cytoskeleton inhibition effect due to the Cytochalasin D treatment.
Stain the endogenous F-actin with SiR-actin kit (Cytoskeleton, cat. no. CY-SC001) for MCF-7 cells previously diluted in a DMEM culture medium with 10% FBS. Incubate the cells at 37 °C and 5% CO2 for 3 h.
Discard the culture medium containing the dyes. Collect floating MCF-7 cells with TrypLE digestion at 37 °C for 12 min or collect adherent MCF-7 cells without TrypLE digestion.
Add 10 μg ml−1 Cytochalasin D to MCF-7 cells and incubate the treated cells in DMEM for 1 h to culture both the floating and adherent MCF-7 cells.
Use an inverted confocal microscope (Zeiss, model no. Zeiss 880 Airyscan) with a 63 × oil-immersion objective to observe actin structures in the Cytochalasin D treated and untreated groups for both adherent and floating MCF-7 cells (Fig. 3e).
42| Conduct cell staining to investigate the cell viability of Cytochalasin D treated and untreated cells (Fig. 3f). For both Cytochalasin D treated and untreated groups, conduct the cell staining with Calcein-AM and SYTOX orange for live and dead MCF-7 cells, respectively.
43| Detach adherent cells, such as MCF-7, MDA-MB-231, and HEK 293T cells, from 100 mm tissue culture dishes to study the adhesion strength among different cell lines. Digest the cells with TrypLE reagent treatment at 37 °C for 12 min.
44| Incubate all the detached cells in DMEM for 1 h with 100 mm low attachment tissue culture dishes to collect floating single cells.
45| Centrifuge the collected floating cells with a speed of 800 rpm for 5 min for all wash and resuspension steps.
Cell loading ● TIMING 10 min
46| Before proceeding with the single cell manipulation with acoustic tweezers, resuspend the cell pellet of the centrifuged cells in 1 ml PBS and withdraw the cell suspension with a 1 ml syringe. Insert the syringe into the hub of a 30-G needle and carefully remove air bubbles by tapping and pushing the syringe.
47| Carefully remove the 5 ml syringe (Step 34) from the inlet tube and push a drop of PBS out of the tube. Connect the needle of the 1 ml syringe (Step 46) to the tube in the PBS drop to avoid any air bubbles in the inlet tube.
48| Mount the 1 ml syringe on the syringe pump and inject the cell suspension into the microfluidic chamber with an infusion speed of 20 μl min−1 through the connected inlet tube (Fig. 4a). Monitor the microfluidic chamber through the CMOS camera and stop the infusion process of the syringe pump if any cell is observed.
Fig. 4 |. Anticipated results with the acoustic tweezers.
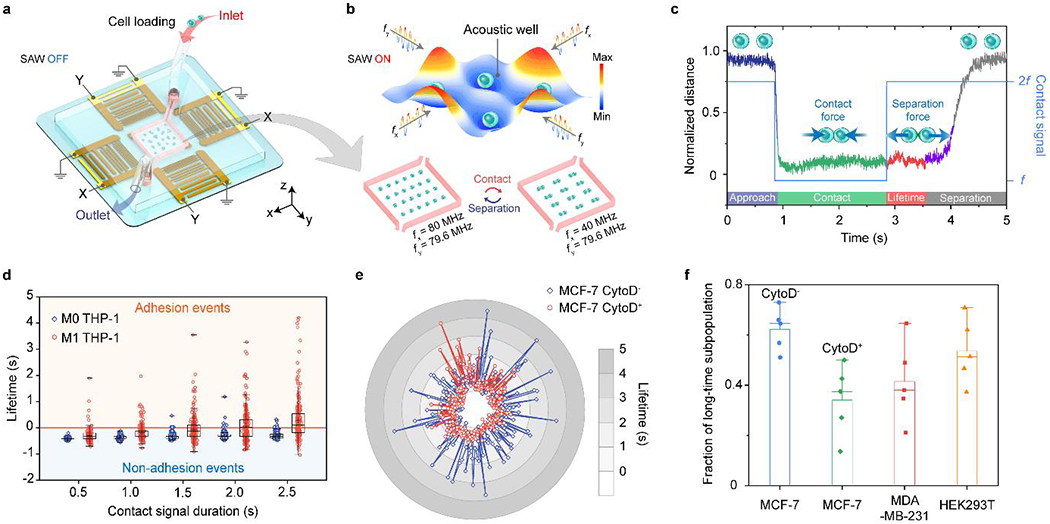
a, Setup of the acoustic tweezers for cell loading. b, Reversible cell-cell pairing and separation by cyclical switching of the applied harmonic standing surface acoustic waves. c, Representative normalized trace plot of cell-cell pairing and separation for adherent M1 THP-1 cells. d, Quantitative analysis of adhesion lifetime for M0 and M1 THP-1 cells (sample size n = 250) with respect to the contact signal duration from 0.5 s to 2.5 s with a 0.5 s interval. Data values of the minimum, lower quartile (25th percentile), median, upper quartile (75th percentile) and maximum are shown in the box plots. e, Representative adhesion lifetime plot for MCF-7 CytoD−and CytoD+ cells (n = 250). f, Fraction of long lifetime subpopulations for MCF-7 (CytoD−), MCF-7 (CytoD+), MDA-MB-231, and HEK293T cells18. The mean and s.d. of the fraction of long lifetime subpopulation are visualized. For each cell line in f, 5 cells pairs were tested repeatedly over 50 contact cycles per pair.
49| Tape both the inlet and outlet tubes onto the inverted microscope stage to minimize the cell perturbation caused by the vibration of tubing.
50| Collect waste samples from the outlet tube with a 1.5 ml polypropylene microcentrifuge tube.
Single-cell trapping ● TIMING 10 min
51| Set the frequencies to 80.0 MHz and 39.8 MHz for channel 1(CH1) and channel 2 (CH1) of the dual channel function generator (Fig. 2d).
52| Turn the output signal of the function generator on for both channels, and the output signal indicator lights should be on. Turn both amplifiers to apply standing surface acoustic waves to form a dot-like array of isolated acoustic wells.
53| Start the infusion process to inject the cells into the microfluidic chamber and monitor the cell trapping status with the CMOS camera.
54| Cyclically switch the frequency between 80 MHz and 40 MHz (Fig. 4b) for channel 1 of the dual channel function generator to increase the single cell trapping efficiency (Supplementary Fig. 2) by disassembling cell clusters.
55| After eight cycles of frequency switching, set the frequencies to 80.0 MHz for channel 1 of the dual channel function generator to form the single cell trapping per acoustic well.
56| Switch to the ×40 objective lens of the inverted microscope and tune the exposure light intensity to image single cells from the regions of interest (ROI) with high resolution.
Cell-cell pairing and separation ● TIMING 30 min
57| Set the frequency and duration for cell-cell pairing (40MHz, 2 s) and separation (80 MHz, 5 s) by programming the dual channel function generator with a customized MATLAB code or the signal generator with its built-in signal modulation function. The duration of cell-cell pairing can be tuned from 0.5 s to 2.5 s with a 0.5 s interval.
58| Record the ROI video with a duration of 6 min for 50 cycles manipulation with cell-cell pairing and separation.
59| Switch to the ×10 objective lens of the inverted microscope and decrease the exposure light intensity to image the microfluidic chamber. Start the infusion process to inject a new batch of cells into the microfluidic chamber. Repeat steps 54 to 58 to obtain more videos for cell-cell pairing and separation studies.
60| After the cell-cell pairing and separation experiment, turn the output signal of the function generator off. Close both the syringe pump and microscope software and turn all the equipment off.
61| Remove both polyethylene tubes from the inlet and outlet of the device.
62| Cut two 15-cm pieces of polyethylene tubes at a 45° angle and insert them into the inlet and outlet of the device with stainless steel tweezers.
63| Inject ethanol into the microfluidic chamber through the polyethylene tube and sonicate the device for 2 min to clean the microfluidic chamber.
64| Check the electrode and microfluidic chamber of the acoustic tweezer device under the microscope. Devices with no issues can be reused for subsequent experiments.
? TROUBLESHOOTING
■ PAUSE POINT The devices can be stored at room temperature for weeks.
Data analysis ● TIMING 5 d
65| Process the recorded ROI videos with ImageJ and measure the intercellular distance of a cell pair with cell-cell pairing and separation, as shown in Fig. 4c.
66| Calculate the separation force based on the measured displacement of particles (Supplementary Fig. 3a) and cells18 with known parameters, such as the radius, density, and compressibility.
67| For adherent cell lines, measure the contact duration tc from the duration between initial and final cell-cell contact based on the recorded ROI videos.
68| Measure the initial contact time, tic, which indicates the travelling time from the original single-cell trapping position to the cell-cell pairing position for each cell pairing cycle.
69| Calculate the adhesion lifetime, tl, based on the measured contact duration tc and the initial contact time tic. The adhesion lifetime is defined as the time difference between the deactivation of the contact signal and the beginning of retraction for the cell pair.
70| Analyse the adhesion lifetime difference with varied contact signal duration, from 0.5 to 2.5 s with a 0.5-s interval, for both M0 and M1 THP-1 cells, as shown in Fig. 4d.
71| Analyse the adhesion lifetime difference in MCF-7 cells with and without Cytochalasin D (CytoD) treatment, as shown in Fig. 3e and Fig. 4e.
72| Plot the lifetime distribution using a histogram plot (Supplementary Fig. 3b) and fit it using a double Gaussian kernel function with both short and long-lifetime subpopulations. Calculate the fraction of long lifetime subpopulations for indirectly measuring the differences in cell-cell adhesion strength among different cell lines (Fig. 4f).
Troubleshooting
Troubleshooting advice can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2 | White residue formed on the SU-8 pattern | The SU-8 photoresist is not fully developed | For mild residue issue, immerse the SU-8 patterned substrate into SU-8 developer for 30 s and dry the substrate to remove the white residue. For severe residue issue, immerse the SU-8 patterned substrate into acetone for 30 s and dry it with an air stream |
| 8 | Pattern distortion after development of the SPR-3012 photoresist | The photoresist is under-exposed or over-exposed | Measure the width of the developed pattern under the microscope. Increase or decrease the exposure time based on the measured results to achieve accurate pattern. Restart the process from step 7 |
| 19 | Defect in bonding between the LiNbO3 substrate and the microfluidic chamber | Mishandling in the APTES coating and plasma treatment process | Remove the defect PDMS chamber from the LiNbO3 substrate with a razor blade. Clean the PDMS residue and dry it with an air stream. Recycle the LiNbO3 substrate and restart the process from step 14 |
| 20 | Short circuit of the interdigital transducers | Defect in electrode fabrication or in silver epoxy process | Examine all the electrodes under an upright microscope. For the electrode damaged due to the fabrication process, the device cannot be used and recycled. For silver epoxy induced short circuit, scratch the epoxy to disconnect the shorted electrodes. |
| 36 | Air bubble trapped in the microfluidic chamber | Bubble trapped in the PBS, needle, or syringe | For bubble trapped in the needle and syringe, carefully remove air bubbles by tapping and pushing the syringe before injecting the PBS. For bubble already trapped in the microfluidic chamber, infuse the PBS with higher infusion speed (300 μl min−1) to squeeze out the air droplets. |
| 64 | Cell residue in the microfluidic chamber | Adherent cell residue on the PDMS walls or the LiNbO3 substrate | Inject ethanol with 5 ml syringe while sonicating the device to flush the microfluidic chamber for 2 min. Repeat this step if there is any residue detected under the microscope. |
Timing
Step 1-6, fabrication of microfluidic chamber: 4 h
Step 7-12, fabrication of microfluidic chamber: 7 h
Step 13-18, fabrication of acoustic tweezers: 1 h
Step 19-36, fabrication of acoustic tweezers: 2 h
Step 37-45, cell culture and chemical perturbations: 4 d
Step 46-50, cell loading: 10 min
Step 51-56, single cell trapping: 10 min
Step 57-64, cell-cell pairing and separation: 30 min
Step 65-72, data analysis: 5 d
Anticipated results
This protocol provides detailed procedures including device fabrication, experimental setup, and data acquisition with acoustic tweezers. We have highlighted the critical steps to fabricate successfully and setup the acoustic tweezers: (i) APTES-treatment to enhance the bonding strength between the PDMS chamber and the LiNbO3 substrate; (ii) essential bonding test to provide leakage-free sample handling; (iii) critical short circuit test to avoid device breakage and electrical equipment damages; (iv) adequate Pluronic-coating to prevent cell attachment to the LiNbO3 substrate. We also provide detailed guidance regarding how to conduct the cell loading, single cell trapping, and cell-cell pairing and separation with the acoustic tweezers. Since we can pair and separate an array of single cells simultaneously, we also demonstrate how to quantify the cell-to-cell adhesion strength in a time-efficient and parallel nature. As shown in Fig. 4b, high-throughput cell-cell pairing and separation manipulation can be achieved by cyclical switching the harmonic frequencies. In addition, we introduce how to obtain quantitative data easily from the recorded video to analyse the intercellular distance and calculate the adhesion lifetime (Fig. 4c). Based on the data analysis, a well-trained researcher may study the adhesion differences between adherent and non-adherent cells (Fig. 4d), quantify the variation of cell-cell adhesion strength caused by perturbations in the organization of the actin cytoskeleton (Fig. 4e), and investigate the variations in cell adhesion forces amongst different cell lines (Fig. 4f).
With this comprehensive protocol, we anticipate that researchers can duplicate our acoustic tweezers easily and conduct versatile biophysical studies that require trapping, patterning, pairing, and separation of single cells. We hope this protocol can enable the widespread adoption of acoustic tweezers for single cell studies in the life sciences, personalized medicine, and fundamental biophysical community, and even become a universal protocol for using acoustic tweezers to address interdisciplinary challenges in biology1-5, materials science50,51, and soft-matter physics52,53.
Supplementary Material
Acknowledgements
We acknowledge support from the Shared Materials Instrumentation Facility (SMIF) at Duke University. We acknowledge support from the National Institutes of Health (R01GM145960 (L.P.L), R01GM141055 (T.J.H.), R01GM132603 (T.J.H.), U18TR003778 (T.J.H.), and UH3TR002978 (T.J.H.)), the National Science Foundation (ECCS-1807601 (T.J.H.) and CMMI-2104295 (T.J.H.)), and the National Science Foundation Graduate Research Fellowship (1644868 (J.R.)).
Footnotes
Competing interests
T.J.H. has co-founded a start-up company, Ascent Bio-Nano Technologies Inc., to commercialize technologies involving acoustofluidics and acoustic tweezers. The remaining authors declare no competing interests.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Materials. Further information is available from the corresponding author upon reasonable request.
References
- 1.Barrow AD et al. Natural killer cells control tumor growth by sensing a growth factor. Cell 172, 534–548. e519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landsberg J. et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Galbraith CG, Yamada KM & Galbraith JA Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 315, 992–995 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Cunha LD et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175, 429–441. e416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T & Akira S The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology 11, 373 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Hosseini BH et al. Immune synapse formation determines interaction forces between T cells and antigen-presenting cells measured by atomic force microscopy. Proc. Natl. Acad. Sci 106, 17852–17857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrichs J, Helenius J & Muller DJ Quantifying cellular adhesion to extracellular matrix components by single-cell force spectroscopy. Nature Protocols 5, 1353–1361 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Chu YS et al. Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol 167, 1183–1194 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Chen W, Evavold BD & Zhu C Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 157, 357–368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dholakia K & Reece P Optical micromanipulation takes hold. Nano today 1, 18–27 (2006). [Google Scholar]
- 11.Feng Y. et al. Mechanosensing drives acuity of αβ T-cell recognition. Proc. Natl. Acad. Sci 114, E8204–E8213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guillaume-Gentil O. et al. Force-controlled manipulation of single cells: from AFM to FluidFM. Trends in biotechnology 32, 381–388 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Aimon S. et al. Membrane shape modulates transmembrane protein distribution. Developmental cell 28, 212–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skelley AM, Kirak O, Suh H, Jaenisch R & Voldman J Microfluidic control of cell pairing and fusion. Nat. Methods 6, 147–152 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cermak N. et al. High-throughput measurement of single-cell growth rates using serial microfluidic mass sensor arrays. Nat. Biotechnol 34, 1052–1059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S-Q et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol 36, 1156–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alapan Y. et al. Microrobotics and microorganisms: Biohybrid autonomous cellular robots. Annual Review of Control, Robotics, and Autonomous Systems 2, 205–230 (2019). [Google Scholar]
- 18.Yang S. et al. Harmonic acoustics for dynamic and selective particle manipulation. Nat. Mater 21, 540–546 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozcelik A. et al. Acoustic tweezers for the life sciences. Nat. Methods 15, 1021–1028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins DJ et al. Acoustic tweezers via sub–time-of-flight regime surface acoustic waves. Sci. Adv 2, e1600089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo F. et al. Controlling cell-cell interactions using surface acoustic waves. Proc. Natl. Acad. Sci 112, 43–48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins DJ et al. Two-dimensional single-cell patterning with one cell per well driven by surface acoustic waves. Nat. Commun 6, 8686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller PB, Barnkob R, Jensen MJH & Bruus H A numerical study of microparticle acoustophoresis driven by acoustic radiation forces and streaming-induced drag forces. Lab Chip 12, 4617–4627 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Shpak O. et al. Acoustic droplet vaporization is initiated by superharmonic focusing. Proc. Natl. Acad. Sci 111, 1697–1702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y. et al. A rapid and controllable acoustothermal microheater using thin film surface acoustic waves. Sens. Actuator A-Phys 318, 112508 (2021). [Google Scholar]
- 26.Tian Z. et al. Wave number–spiral acoustic tweezers for dynamic and reconfigurable manipulation of particles and cells. Sci. Adv 5, eaau6062 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulvana H, Cochran S & Hill M Ultrasound assisted particle and cell manipulation on-chip. Adv. Drug Deliv. Rev 65, 1600–1610 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Neužil P, Giselbrecht S, Länge K, Huang TJ & Manz A Revisiting lab-on-a-chip technology for drug discovery. Nat. Rev. Drug Discov 11, 620–632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke T, Braunmüller S, Schmid L, Wixforth A & Weitz D Surface acoustic wave actuated cell sorting (SAWACS). Lab Chip 10, 789–794 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Nilsson J, Evander M, Hammarström B & Laurell T Review of cell and particle trapping in microfluidic systems. Anal. Chim. Acta 649, 141–157 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Wiklund M & Hertz HM Ultrasonic enhancement of bead-based bioaffinity assays. Lab Chip 6, 1279–1292 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Reboud J. et al. Shaping acoustic fields as a toolset for microfluidic manipulations in diagnostic technologies. Proc. Natl. Acad. Sci 109, 15162–15167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garg N. et al. Whole-blood sorting, enrichment and in situ immunolabeling of cellular subsets using acoustic microstreaming. Microsystems & Nanoengineering 4, 1–9 (2018).31057891 [Google Scholar]
- 34.Baudoin M. et al. Folding a focalized acoustical vortex on a flat holographic transducer: miniaturized selective acoustical tweezers. Sci. Adv 5, eaav1967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeo LY & Friend JR Surface acoustic wave microfluidics. Annu. Rev. Fluid Mech 46, 379–406 (2014). [Google Scholar]
- 36.Huang PH et al. Acoustofluidic Synthesis of Particulate Nanomaterials. Adv. Sci 6, 1900913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu Y. et al. Acoustofluidic centrifuge for nanoparticle enrichment and separation. Sci. Adv 7, eabc0467 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melde K, Mark AG, Qiu T & Fischer P Holograms for acoustics. Nature 537, 518 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Ahmed D. et al. Neutrophil-inspired propulsion in a combined acoustic and magnetic field. Nat. Commun 8, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan X-D, Zou Z & Zhang L Acoustic vortices in inhomogeneous media. Phys. Rev. Res 1, 032014 (2019). [Google Scholar]
- 41.Glynne-Jones P, Boltryk RJ, Harris NR, Cranny AW & Hill M Mode-switching: A new technique for electronically varying the agglomeration position in an acoustic particle manipulator. Ultrasonics 50, 68–75 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Marzo A, Caleap M & Drinkwater BW Acoustic virtual vortices with tunable orbital angular momentum for trapping of mie particles. Phys. Rev. Lett 120, 044301 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Wang Z. et al. Acoustofluidic salivary exosome isolation: A liquid biopsy compatible approach for human papillomavirus–associated oropharyngeal cancer detection. The Journal of Molecular Diagnostics 22, 50–59 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren L et al. Standing surface acoustic wave (SSAW)-based fluorescence-activated cell sorter. Small 14, 1801996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J. et al. Surface acoustic waves enable rotational manipulation of Caenorhabditis elegans. Lab Chip 19, 984–992 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuetz MJ et al. Acoustic traps and lattices for electrons in semiconductors. Phys. Rev. X 7, 041019 (2017). [Google Scholar]
- 47.Schülein FJ et al. Fourier synthesis of radiofrequency nanomechanical pulses with different shapes. Nat. Nanotechnol 10, 512–516 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Zhao P. et al. Acoustically Induced Giant Synthetic Hall Voltages in Graphene. Phys. Rev. Lett 128, 256601 (2022). [DOI] [PubMed] [Google Scholar]
- 49.Krenner HJ & Westerhausen C Handy nanoquakes. Nat. Mater 21, 499–501 (2022). [DOI] [PubMed] [Google Scholar]
- 50.Li B, Zhou D & Han Y Assembly and phase transitions of colloidal crystals. Nat. Rev. Mater 1, 15011 (2016). [Google Scholar]
- 51.Hou J, Li M & Song Y Recent advances in colloidal photonic crystal sensors: Materials, structures and analysis methods. Nano Today 22, 132–144 (2018). [Google Scholar]
- 52.Lim MX, Souslov A, Vitelli V & Jaeger HM Cluster formation by acoustic forces and active fluctuations in levitated granular matter. Nat. Phys 15, 460–464 (2019). [Google Scholar]
- 53.Manoharan VN Colloidal matter: Packing, geometry, and entropy. Science 349,1253751 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Materials. Further information is available from the corresponding author upon reasonable request.