
Fig. 9. Schematic depicting MAF diversity and the pro-metastatic functions of pSTAT3+myMAFs.
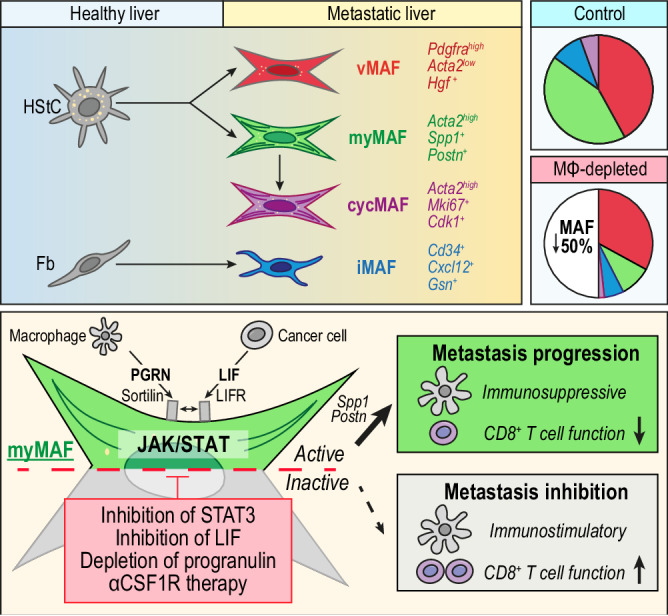
Illustration depicting the proposed cellular origin, subtyping, distribution, and mechanism of myMAF activation. In established liver metastatic PDAC, HStCs give rise to vMAFs, myMAFs, and cycMAFs, while portal fibroblasts give rise to iMAFs. In tumour bearing control mice, vMAFs and myMAFs are dominant, whereas iMAFs and cycMAFs are minor populations. In macrophage depleted mice (αCSF1R-treated) an overall reduction in fibrosis is primarily driven by the loss of myMAFs, resulting in an imbalance of vMAF/myMAF ratio. Mechanistically, progranulin and LIF, mainly derived from macrophages and cancer cells, respectively, co-opt to promote a myMAF phenotype via activation of JAK/STAT signalling. Progranulin binding to sortilin enhances the proximity of sortilin to LIFR, leading to JAK/STAT hyperactivation. Reciprocally, myMAF secreted periostin directly promotes cancer cell proliferation, whereas myMAF secreted osteopontin promotes an immunosuppressive macrophage phenotype. Pharmacological or genetic interference of cancer cell-macrophage-MAF crosstalk ablates the metastasis promoting functions of pSTAT3+myMAFs in metastatic PDAC.