
Figure 4. Data analysis pipeline and peak caller characteristics.
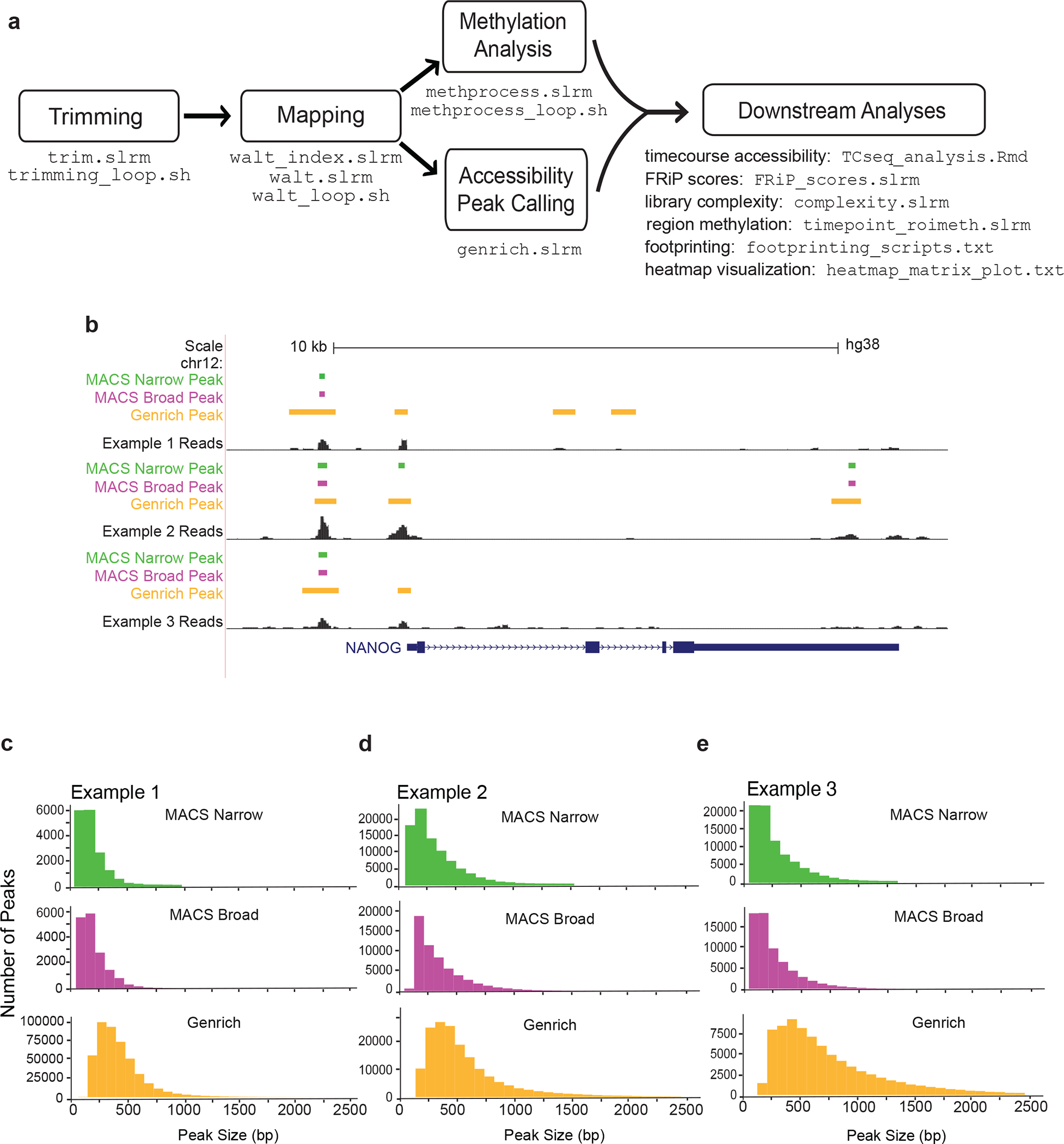
ATAC-Me data can be analyzed to determine both accessibility and methylation simultaneously. (a) Initial analysis steps are similar to other sequencing pipelines which begin with trimming of adaptors. These trimmed reads are then aligned to a reference index. As the fragments in ATAC-Me have been converted using sodium bisulfite they are aligned using a program that can accommodate T-rich reads. WALT49 is used to both create a reference genome index and map reads to this index. Following alignment, analysis branches to peak calling for accessibility using Genrich. Regions of increased chromatin accessibility have traditionally been identified through peak calling algorithms that determine genomic regions that contain a statistically significant enrichment of reads relative to background reads. Genrich incorporates biological replicates to create a set of consensus peaks. We provide additional details for MACS2, which can be used as an alternative to Genrich for calling peaks (See also Table 3). Parallel methylation analysis is performed using the MethPipe software48. A variety of downstream analyses are possible such as TF footprinting, differential accessibility, and regional methylation calculation. Scripts from this pipeline including example analyses are available at https://github.com/HodgesGenomicsLab/NatProtocols_ATACme56. (b) ATAC-Me signal tracks are displayed for MACS2 and Genrich peak calling methods, using a cutoff of p <0.05, to illustrate the differences between these two algorithms. We do not typically recommend calling peaks on samples individually, as there will be increased statistical strength and stringency when peak calling is performed on replicates and a consensus peak set is identified. (c-e) Histograms display the size distribution of regions called by Genrich and MACS2 (called using callpeak with and without the --broad argument, which composites broad regions from nearby highly enriched regions. At the lowest stringency p-value threshold (0.5), Genrich calls a larger number of peaks with wider size distributions.