
Fig. 3. Congenital aniridia workup.
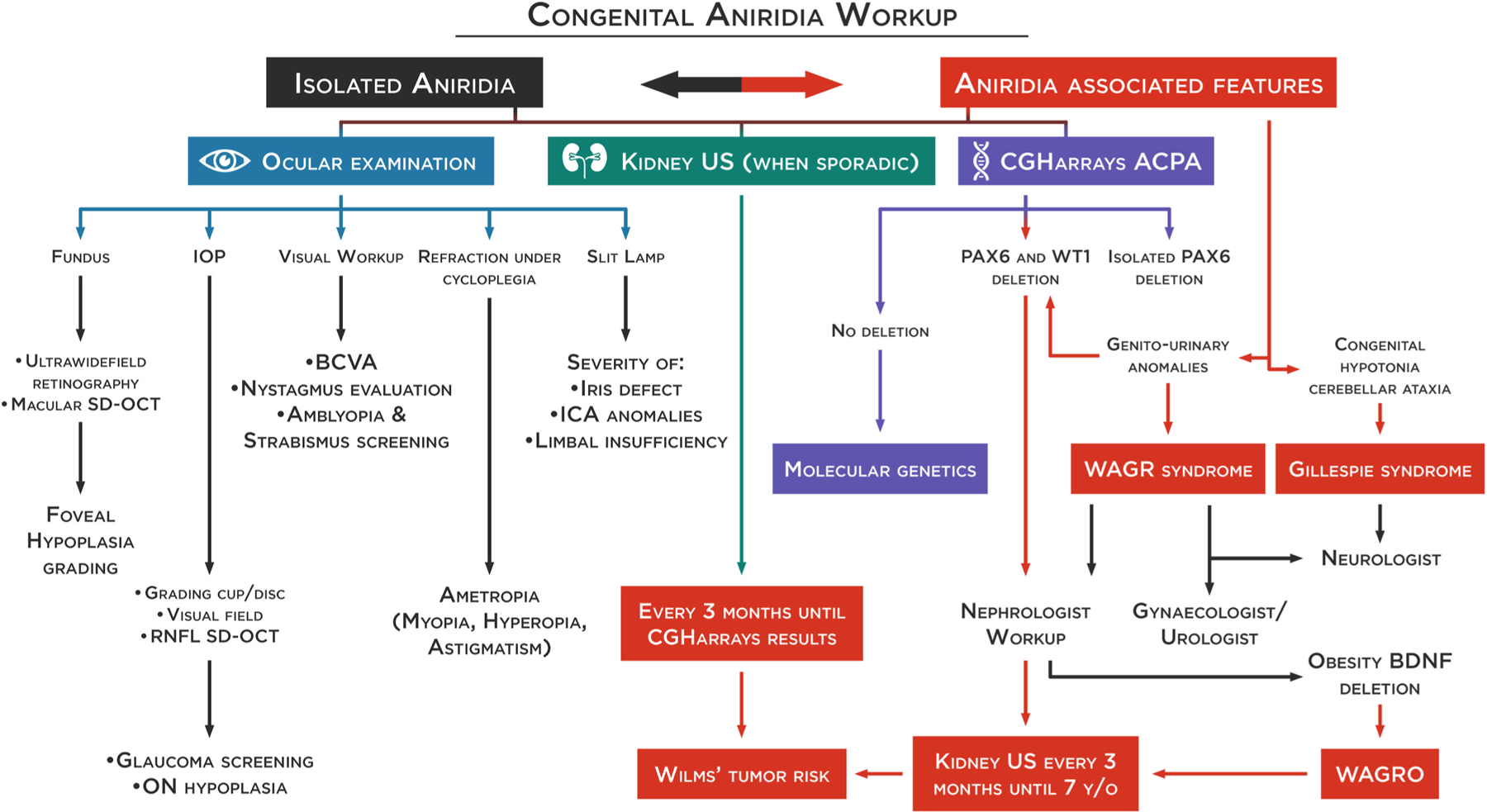
Differentiation of isolated or syndromic congenital aniridia is essential for appropriate management and follow-up. Prompt diagnosis of WAGR or WAGRO is critical to avoid vital risks. An extensive ophthalmological workup needs to be performed to characterize the ocular phenotype, especially foveal hypoplasia through spectral-domain optical coherence tomography (SD-OCT). A genetic workup should first include comparative genomic hybridization arrays to allow for the quick identification of isolated deletions on the PAX6 gene or deletions involving the WT1 gene. In sporadic cases, and until the results are from comparative genomic hybridization arrays, a kidney ultrasound should be performed every 3 months for early detection of a Wilms’ tumor. In case of WT1 deletion, a kidney ultrasound should be continued every 3 months until the age of 7. If no deletion is found, a molecular genetic test (NGS and/or WGS) will be performed to confirm the clinical diagnosis and identify the pathogenic variants allowing phenotype-genotype correlations. It is to be noted that NGS-based protocol has the potential of becoming the one-for-all test for molecular diagnosis of aniridia (see section 4.3 and 4.4), replacing the standard strategy represented here. BCVA: best-corrected visual acuity, ON: optic nerve, ICA: iridocorneal angle, RNFL: retinal nerve fiber layer.