
Figure 2.
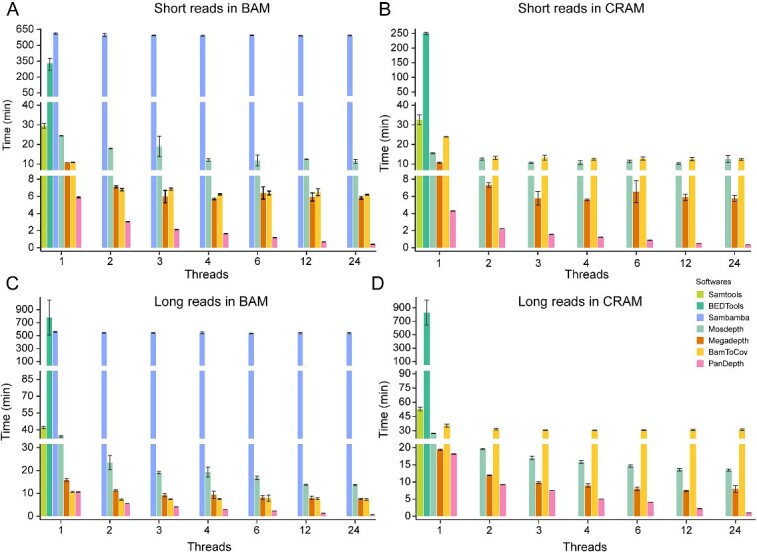
The computation time comparison of seven software tools using 150 GB sequencing reads in different numbers of threads for genome coverage calculations. (A) BAM file with short reads as input. (B) CRAM file with short reads as input. (C) BAM file with long reads as input. (D) CRAM file with long reads as input. The X-axis represents the number of threads and the Y-axis represents the computing time. The colored bars represent Samtools, BEDTools, Sambamba, MosDepth, MegaDepth, BamToCov and PanDepth, respectively. Error bars indicate standard deviation (n = 10).