

Abstract
Diffuse midline gliomas (DMGs) are devastating pediatric brain tumors recognized as the leading cause of cancer-related death in children. DMGs are high-grade gliomas (HGGs) diagnosed along the brain’s midline. Euchromatin is the hallmark feature of DMG, caused by global hypomethylation of H3K27 either through point mutations in histone H3 genes (H3K27M), or by overexpression of the enhancer of zeste homolog inhibitory protein. In a clinical trial for adults with progressive HGGs, a 22-year-old patient with a thalamic DMG, H3 K27-altered, showed a remarkable clinical and radiological response to dordaviprone (ONC201). This response in an H3 K27-altered HGG patient, coupled with the lack of response of patients harboring wildtype-H3 tumors, has increased the clinical interest in dordaviprone for the treatment of DMG. Additional reports of clinical benefit have emerged, but research defining mechanisms of action (MOA) fall behind dordaviprone’s clinical use, with biomarkers of response unresolved. Here, we summarize dordaviprone’s safety, interrogate its preclinical MOA identifying the mitochondrial protease “ClpP” as a biomarker of response, and discuss other ClpP agonists, expanding the arsenal of potential weapons in the fight against DMG. Finally, we discuss combination strategies including ClpP agonists, and their immunomodulatory effects suggestive of a role for the tumor microenvironment in DMG patient response.
Keywords: CLPP, CLPP agonist, DMG, dordaviprone, ONC201
Key Points.
Dordaviprone shows emerging clinical benefits for the treatment of diffuse midline glioma (DMG).
In DMG, dordaviprone is an agonist of the mitochondrial protease ClpP.
CLPP is overexpressed in DMG patients, highlighting the therapeutic potential of ClpP agonists.
Diffuse midline glioma (DMG), including those that arise in the pontine region of the brainstem (referred to as diffuse intrinsic pontine glioma [DIPG]), is the most aggressive and lethal childhood cancer, harboring a median overall survival (OS) of 9–11 months, with a 2-year OS of <10%.1,2 Over 60 years of clinical trials testing cytotoxic chemotherapies, chemoradiation, precision- and immuno-therapies, surgical resection, and local therapeutic delivery in the treatment of DMG have not improved patient outcomes.3
In 2021, The World Health Organization (WHO) published the fifth classification of central nervous system (CNS) tumors and reclassified DIPG as “diffuse midline glioma, H3 K27-altered,” representing high-grade gliomas (HGG) located along the midline structures of the brain.4 The new classification recognized global hypomethylation of lysine 27 of histone H3 (H3K27) as a ubiquitous feature of DMG, driving epigenetic signatures that promote promiscuous expression of oncogenes resulting in rapid tumor growth,3,5 as well as imparting immune system privilege.6
Remarkably, 80% of DMG cases harbor missense mutations at lysine 27 in histone H3 in either HIST1H3B/C (H3.1K27M) or H3F3A (H3.3K27M) genes, where the amino acid lysine is substituted to a methionine.7,8 Patients carrying wildtype-H3 overexpress the enhancer of zeste homolog inhibitory protein (EZHIP) which harbors biochemical properties similar to the H3K27M mutation, both of which drive hypomethylation of H3K27.2,9
There are subtle differences in the median OS of individual DMG patients harboring different H3-alterations and tumor localizations, that is, pons compared with thalamus.2 Indeed, patients with H3.1K27M mutations are most frequently diagnosed with tumors in the pontine region of the brainstem at a median age of 4 years old and harbor a median OS of 12–15 months.2 Whereas, H3.3K27M alterations are frequently diagnosed in the pons, but are also seen in the spine, midbrain, and thalamus with patients diagnosed at a median age of 6–7 years of again with OS of 9 months.2,10 Lastly, DMG patients carrying wildtype H3K27 but overexpression of EZHIP, are diagnosed with tumors evenly distributed along the midline and diagnosed at a median age of 4.6 years of age, with slightly increased OS of 15 months compared with H3.1K27M and H3.3K27M patients.2
During development, the functional organization of chromatin is regulated by the polycomb repressive complex 2 (PRC2), particularly, by the catalytic enzyme of the lysine methyltransferase EZH2 responsible for the deposition of trimethylated marks (me3) on histone tails, specifically at H3K27.3 EZH2 directly regulates chromatin accessibility and the transcriptional efficiency/activity of the developing midline.3 Although H3 mutations are heterozygous and only contribute to a fraction of the total pool of H3 protein (3%–17%), its effects are global,7 promoting dominant negative H3K27 trimethylation (H3K27me3).7,11,12 H3 proteins carrying the mutant methionine (H3K27M) sequester the EZH2 methyltransferases through interactions with its C-terminal SET domain13 to reduce the global activity of the PRC2 complex.10,14 This, coupled with the co-enrichment of elevated H3K36me2 deposited by the nuclear receptor binding SET domain protein 1/2 (NSD1/2),15,16 inhibit spreading of repressive H3K27me3 marks that are preferentially retained at unmethylated CpG islands, affecting lowly expressed genes influencing neurogenesis.8,11,17–19 Global loss of H3K27me3 promotes H3K27 acetylation by p300/CBP (histone acetyltransferase p300-EP300 bound to CREB-binding protein CBP-CREBBP) of non-H3K27M mutated H3 protein.7 Acetylated H3K27 then preferentially localizes to gene promoters and/or enhancers to activate transcription.
Unfortunately, there are no therapies available that target the H3-alterations that give rise to DMG. However, dordaviprone (also referred to as ONC201 and TIC10) is a first in class, brain penetrant, small molecule of the imipridone family, with reported case study benefits for the treatment of H3K27M mutant DMG,20 and in clinical trials following radiation and at disease progression (NCT02525692).21 This has encouraged off-patent development,22,23 and clinical controversy.24 Here, we outline the discovery of dordaviprone for the treatment of DMGs and provide a summary of the known mechanisms of action (MOA) and touch on other therapies that exploit similar MOAs for the potential improved treatment of DMG.
Discovery of Dordaviprone (ONC201) for the Treatment of DMG
The first report of dordaviprone was by a patent filed in 1973 outlining its chemical structure. While no MOA or biologics were reported, there was speculation of potential clinical utility for the treatment of CNS disorders.25 Dordaviprone's potential as an anticancer agent began to be uncovered in 2013, when it was discovered in a small molecule screen of p53-independent inducers of TNF-related apoptosis-inducing ligand (TRAIL) mediated cell death in colorectal cancer cell lines in vitro.26 This stimulated interest in its clinical potential due to a lack of therapeutics that are effective against TP53-mutant tumors and its potential pan-cancer utility.
During a Phase II clinical trial of dordaviprone in adult glioblastoma patients, using 625 mg administered orally every 3 weeks (NCT02525692), a 22-year-old female patient harboring an H3K27M positive thalamic HGG, confirmed via biopsy, experienced a near complete objective response including regression of the primary thalamic lesion, durable 3-years later.27 This led to significant clinical interest in the potential use of dordaviprone in pediatric H3K27M mutant DMG, where H3K27M is the defining feature. Hence, an expanded access program was launched to further examine its therapeutic potential. A preliminary clinical study using dordaviprone in patients harboring DMG or DIPG, post-radiation therapy (RT) and with confirmed signs of radiological progression commenced,employing dordaviprone orally 625 mg per week.21 Among the 4 pediatric patients who started dordaviprone post-RT, 2 patients remained in second progression-free survival (2PFS) for 53 and 81 weeks, with several others experiencing radiographic regression after commencement, including 1 patient with a complete response. These findings encouraged numerous other clinical trials to examine dordaviprone in multiple tumor types as a single agent and in combination (Tables 1 and 2).Recently, the combined results of two DMG clinical trials testing dordaviprone (NCT03134131 and NCT03416530) following initial RT but prior to recurrence, demonstrated an increased median OS of 21.7 months compared to historical, while those treated after recurrence had a median OS of 9.3 months (Table 1).* Remarkably, DMG patients who experienced extended benefits from dordaviprone treatment, showed reversed hallmark loss of H3K27me3, identified by IHC upon tissue autopsy, with in vitro data suggesting the mechanism to be through metabolic inhibition of the Jumonji C domain (Jmj C) family of histone lysine demethylases.
Table 1.
Dordaviprone Clinical Trials in Central Nervous System Cancers
| Identifier | Conditions | Date | Phase | Intervention | Description | Published Results |
|---|---|---|---|---|---|---|
| NCT02038699 |
|
Jan 2014 to Dec 2016. | Phase 1/2 |
|
Testing ONC201 in patients with advanced cancer. The Phase 1 portion will inform the dosing and safety profile, while the Phase 2 portion will determine efficacy. | Withdrawn. |
| NCT02525692 |
|
Jan 2016 to Dec 2023. | Phase 2 |
|
Testing the efficacy of ONC201 in patients with recurrent glioblastoma or WHO Grade IV gliomas with the H3 K27M mutation. | Among the 14 patients with recurrent disease prior to initiation of ONC201 treatment, median PFS is 14 weeks, and the median OS is 17 weeks. Among the 4 pediatric patients enrolled, 2 DIPG patients remain progression-free for at least 53 and 81 weeks. |
| NCT03134131 |
|
28 Apr 2017 to 14 Mar 2022. | Expanded access |
|
Expanding access to ONC201 in eligible patients with previously treated glioma harboring the H3 K27M mutation and/or in midline high-grade gliomas. | Eleven of 41 patients were enrolled following radiation but prior to recurrence; 30 of 41 patients were enrolled with recurrent disease. Patient survival data was combined with data from NCT03416530, reporting a median OS of non-recurrent H3K27M-DMG patients treated with ONC201 (n=35) of 21.7 months from diagnosis and 9.3 months at recurrence. |
| NCT03295396 |
|
31 Oct 2017 to 15 May 2023. | Phase 2 |
|
To determine the efficacy and safety of ONC201 in adult patients with recurrent H3 K27M high-grade glioma. | Active, not recruiting. No results posted. |
| NCT03416530 |
|
29 Jan 2018 to 31 May 2023. | Phase 1 |
|
Multicenter, open-lab, 7 arms, dose escalation study of ONC201 for pediatric patients with newly diagnosed DIPG and recurrent/refractory H3 K27M gliomas. | Twenty-four of 30 patients were enrolled following radiation but prior to recurrence; 6 of 30 patients were enrolled with recurrent disease. Patient survival data was combined with data from NCT03134131, reporting a median OS of patients with non-recurrent H3K27M-DMG treated with ONC201 (n=35) of 21.7 months from diagnosis and 9.3 months at recurrence. The utility of cf-tDNA in both plasma and CSF of DMG patients was shown to be feasible and to harbor clinical utility. H3K27M VAF values in cf-tDNA were a strong biomarker of response. |
| NCT04617002 |
|
5 Nov 2020 to Feb 2023. | Expanded access |
|
An intermediate-size expanded access protocol to provide ONC201 to patients harboring H3 K27M-mutant and/or midline gliomas who cannot access ONC201. | Available. No results posted. |
| NCT04629209 |
|
15 Feb 2021 to 30 June 2023. | Phase 2 |
|
Open-label study testing intra-tumoral concentrations and radiographic efficacy in adults with EGFR-low glioblastoma. Excluding patients with midline gliomas, H3K27M mutations, or IDH mutations. | Withdrawn (change in approach to study). No results posted. |
| NCT04854044 |
|
1 May 2021 to 1 July 2026. | Phase 1 |
|
Combines ONC201 with standard-of-care radiotherapy before tumor resection in recurrent glioblastoma (GBM) patients. | Withdrawn (The P.I. is not prepared to move forward at this time). |
| NCT05009992 |
|
20 Oct 2021 to 30 June 2027. | Phase 2 |
|
A Phase 2 trial that determines if the combination of ONC201 and paxalisib is effective for treating patients with diffuse midline gliomas (DMGs). | Recruiting. No results posted. |
| NCT05392374 |
|
26 May 2022. | Expanded access |
|
Intermediate study to provide access for patients with diffuse intrinsic pontine gliomas who cannot access ONC201 through clinical trials. | No longer available, no results posted. |
| NCT05476939 |
|
29 Sept 2022 to Sept 2031. | Phase 3 |
|
Also known as BIOMEDE 2.0, it is the second stage of a multi-arm, adaptive platform trial. It is a randomized open-label Phase-3 controlled trial evaluating the efficacy of ONC201 in comparison with everolimus and subsequently historical controls. | Recruiting. No results posted. |
| NCT05580562 |
|
23 Jan 2023 to Aug 2026. | Phase 3 |
|
Randomized double-blind, placebo-controlled, parallel-group, international Phase 3 study for patients with newly diagnosed H3 K27M-mutant diffuse midline glioma, combined with frontline radiotherapy. Excludes patients diagnosed with DIPG. | Recruiting. No results posted. |
Table 2.
Dordaviprone Clinical Trials across Non-CNS Cancers
| Identifier | Conditions | Date | Phase | Intervention | Description | Published Results |
|---|---|---|---|---|---|---|
| NCT02250781 |
|
12 Jan 2015 to 25 Oct 2018. | Phase 1 |
|
Testing the side effects and best dosing for ONC201 in treating patients with advanced solid tumors. | Completed. No results posted. |
| NCT02324621 |
|
20 Feb 2015 to 23 Oct 2018. | Phase 1 |
|
To evaluate long-term efficacy of ONC201 in patients with solid tumors that have metastasized for patients who have previously benefitted from the drug. | Completed. Prolonged stable disease for >6 months was observed in 23.8% (5/21) of patients. |
| NCT02420795 |
|
3 Nov 2015 to 16 Nov 2020. | Phase 1/2 |
|
Testing safety and efficacy of ONC201 in treating patients with non-Hodgkin’s lymphoma that is recurrent or refractory. | Terminated (Per PI request). Median progression-free survival on 625 mg ONC201 was a median of 5 weeks. |
| NCT02392572 |
|
3 Nov 2015 to 30 Nov 2023. | Phase 1/2 |
|
Testing the safety and dosing of ONC201 in patients with relapsed/refractory acute leukemia or high-risk myelodysplastic syndrome. | Recruiting. No results posted. |
| NCT02609230 |
|
5 Nov 2015 to 26 Mar 2020. | Phase 1 |
|
Testing the safety and MTD of ONC201 in patients with advanced solid tumors or multiple myeloma. | Completed. No results posted. |
| NCT02863991 |
|
1 Jan 2016 to 31 Dec 2022. | Phase 1/2 |
|
Open-label study of ONC201 in combination with dexamethasone for patients with relapsed/refractory multiple myeloma. | Active, not recruiting. |
| NCT03099499 |
|
8 June 2017 to Sept 2023. | Phase 2 |
|
Testing the clinical benefit of ONC201 in women with recurrent or metastatic endometrial cancers, especially those with alterations in the PI3K/mTOR pathway. | Suspended due to slow accrual. |
| NCT03034200 |
|
2 Aug 2017 to 30 Nov 2022. | Phase 2 |
|
Testing whether ONC201 has efficacy in tumor and metastases reduction in PC–PG (pheochromocytoma–paraganglioma) and other neuroendocrine tumors. | Completed. The trial was separated into 3 arms; cohorts A (PC-PG) and B (other neuroendocrine tumors) were treated once a week with ONC201, while dosage was twice a week in cohort C. Of the 10 PC-PG patients in cohort A, 5 exhibited a partial response, with 2 additional patients having stable disease for more than 3 months. In arm B, there was 1 partial responder and 2 with stable disease for more than 3 months, from 12 total patients. Patients enrolled in arm C with PC-PG (n=8) showed 1 partial responder and 7 with stable disease at 3 months. |
| NCT03394027 |
|
17 Jan 2018 to 7 Oct 2021. | Phase 2 |
|
Testing ONC201 in each disease cohort to see if there is lasting efficacy in shrinking tumors. | No objective responses were achieved, however, 18% (4/22) of patients had prolonged stable disease (>9 cycles). |
| NCT03485729 |
|
21 Mar 2018 to 31 Dec 2022. | Phase 2 |
|
Two-stage, non-randomized, open label, 2-arm trial of ONC201 in women with metastatic or recurrent Type II endometrial cancer, who have already failed at least 1 prior chemotherapy regimen. | Active, not recruiting. No results posted. |
| NCT03492138 |
|
26 Mar 2018 to 13 Jan 2020. | Phase 1/2 |
|
Single arm, open-label study for patients with relapsed/refractory multiple myeloma testing ONC201, ixazomib, and dexamethasone. | Terminated due to low enrollment. No results posted. |
| NCT03733119 |
|
13 Nov 2018 to 13 Feb 2021. | Phase 2 |
|
Combining ONC201 and a methionine-restricted diet for patients with metastatic or unresectable triple-negative breast cancer (TNBC) | Terminated. The trial was terminated due to slow accrual of patients and statistical analysis was not completed. |
| NCT03932643 |
|
30 July 2019 to July 2025. | Phase 1 |
|
Pilot study of 20 patients with AML/MDS to determine the safety and preliminary efficacy of ONC201 as a maintenance therapy. | Recruiting. No results posted. |
| NCT03791398 |
|
15 Nov 2019 to 5 Aug 2021. | Phase 1b/2 |
|
Single arm, open label, pharmacokinetic, pharmacodynamics, and efficacy study of ONC201 in combination with Opdivo (Nivolumab) in adult patients with metastatic colorectal cancer. | Terminated. 13 patients were enrolled, and all died of disease progression. Median extension of survival was 58 days. The study was terminated due to lack of efficacy. |
| NCT04055649 |
|
21 Jan 2020 to 27 April 2024. | Phase 2 |
|
An efficacy trial for combining ONC201 and paclitaxel for patients with platinum-resistant epithelial ovarian, fallopian tube, and recurrent/refractory primary peritoneal cancer. | Recruiting. No results posted. |
| NCT05542407 |
|
15 Apr 2023 to 15 Jan 2025. | Phase 1 |
|
Testing the combination of ONC201 and atezolizumab in patients in obese and non-obese metastatic/recurrent endometrial cancer. | Not yet recruiting. |
| NCT05630794 |
|
13 May 2023 to 1 Feb 2025. | Phase 1 |
|
Testing ONC201 in preventing colorectal cancer in preventing colorectal cancer in patients with familial adenomatous polyposis (FAP) or multiple polyps. | Not yet recruiting. No results posted. |
Access to Dordaviprone through Unconventional Means
Dordaviprone is characterized by a unique heterocyclic pharmacophore that produces no stereoisomers, is highly stable, produced via facile reactions, is aqueous, and passively penetrates the blood–brain barrier (BBB). However, the 1973 patented structure of dordaviprone was mistakenly reported as a [4,3-d] linear structure, rather than the [3,4-e] angular structure identified by recent NMR studies.22,28 Wagner et al., re-synthesized both the angular and linear isomers and definitively demonstrated the structures of each, confirming that only the angular isomer exhibited the anti-cancer biology associated with dordaviprone.28 The rumored clinical response by DMG patients, and the limited opportunities for patients to receive it worldwide, led to ONC201 becoming available via prescription from a German oncologist and dispensed by a local pharmacy (called GsONC201). Capitalizing on “individual healing attempt” (individueller Heilversuch), GsONC201 became a medical treatment option in Germany as it deviates from the medical standard, aimed at treating a specific patient.24 However, conjecture remained whether this synthesized version of dordaviprone was the reported inactive [4,3-d] linear isomer of the originating compound,28 or the active angular isomer. Duchatel et al., investigated GsONC201, donated from families that had purchased it from Germany, using nuclear magnetic resonance (NMR) spectroscopy, it was identified that the compound was the same active [3,4-e] structural isomer used in clinical trials,22 and that the German drug showed analogous in vitro and in vivo anti-DMG effects to the genuine dordaviprone provided under a material transfer agreement. Although hundreds of patients have now received GsONC201, data provided by the clinical teams of 28 pontine H3K27M+ DMG patients starting 7.5 months following their diagnosis (median duration), showed a median OS of 18 months, with patients who received re-irradiation in combination with German dordaviprone survived 22 months.22 A recent follow-on study compared the survival of 27 DMG patients with confirmed H3-alterations who purchased and received GsONC201 (n = 18) compared with patients who did not (n = 9), reporting a median (OS) of 19.9 versus 10.9 months, respectively.23
Safety and Tolerability of Dordaviprone
Dordaviprone is widely considered a safe compound. Its safety profile was first published by Allen et al., in 2013, where it was shown that dordaviprone did not alter cell cycle profiles or decrease clonogenic survival of normal fibroblasts at the same doses that are toxic to colorectal cancer cell lines HCT116 (TP53−/−).26 Reversibly, dordaviprone does, however, decrease the proliferation of fibroblasts, but proliferation is restored 24 h after drug removal.29 Subsequently, dordaviprone has been tested on normal bone marrow mononuclear cells, lung epithelial cells, astrocytes, and macrophages without any effect on cell viability at relevant doses.30–32
The safety profile in vitro is mirrored in vivo, where Sprague–Dawley rats treated with a single dose of dordaviprone, ranging from 0 to 225 mg/kg, showed no mortalities or dose-limiting toxicities (DLTs).29 Only the highest dose resulted in signs of toxicity, including decreased activity, body weight gain, and food consumption as well as abnormal gait. However, both decreased activity and abnormal gait quickly resolved. Furthermore, both blood and urine tests as well as tissue examination suggested no significant toxicities nor long-lasting affects due to dordaviprone. Similar results were obtained when treating beagle dogs with equivalent doses of dordaviprone. Beagles treated with high doses (42 and 120 mg/kg) showed symptoms including salivation, vomitus, loose feces as well as decreased activity on day of administration with the symptoms resolving within a day. Both these preliminary studies suggest no observed adverse event level (NOAEL) at the corresponding human dose of approximately 1.25 g.29
The first-in-human clinical trial of dordaviprone set out to determine the recommended Phase II dose (RP2D) as well as conducted safety appraisal for dordaviprone delivered using dose escalation regimen starting at 125 mg, and ranging up to 625 mg.33 Dordaviprone was given orally every 21 days to patients with advanced solid tumors. No adverse events (AE) > Grade 1 were observed at any dose, with Grade 1 AEs including fever, nausea, and emesis in 1 patient, and increased serum amylase in 2 patients. The mean half-life of dordaviprone was determined to be 11.3 h, with a Cmax (maximal concentration) of 6.3 µg/mL and Tmax (the time it takes for a drug to reach Cmax) at 1.8 h after administration.33 The mean AUC observed was 37.7 h × µg/mL and the mean CL/F was 25.2 l/h.
Following the optimistic safety profile using 625 mg dordaviprone once every 3 weeks, a Phase I study investigated the safety of 375 or 625 mg dordaviprone orally once per week.34 Likewise, no AE >Grade 1 or DLTs were reported. Pharmacokinetic (PK) evaluation identified a mean half-life of 9.4 h, Cmax of 4.3 µg/mL, and AUC of 34.3 h × µg/mL, with similar results after the first and fourth dose. More recently, administration of dordaviprone twice a week on 2 consecutive days has been investigated. Similarly, preliminary studies showed no DLTs or serious AE associated with dordaviprone used twice a week.35 These data were built upon by Gardner et al., studies, where a Phase 1 trial of pediatric DMG patients administered 625 mg (weight adjusted), identified similar Cmax (2.3 µg/mL),36 however, within the therapeutic ranges reported by Stein et al.33 The maximum-tolerated dose (MTD) was not identified, calling for further clinical investigations. To date, the most common drug-related AEs are fatigue, decreased lymphocyte count, nausea, and vomiting.
Dordaviprone Mechanism of Action: DRD2 or ClpP?
For the most part, DMG clinical trials examined the dordaviprone safety and efficacy profile without the establishment of robust preclinical data for its use, including no in vivo DMG orthotopic xenograft studies, or understanding of its cellular target. The MOA of dordaviprone has been subject to rigorous debate over recent time. Indeed, the initial MOA reported by Allen et al., in colon cancer suggested dordaviprone promotes TRAIL-mediated apoptosis independent of p53, leading to inhibition MAPK/ERK and PI3K/Akt signaling.26
Dordaviprone as a Selective DRD2 Antagonist
Using a Bayesian machine-learning approach, dopamine receptors, specifically dopamine receptor D2 (DRD2), was identified as a targets of dordaviprone in non-DMG cancer cell lines,37 with in silico modeling confirming dordaviprone binds to the active site of DRD2.22 Further characterization revealed dordaviprone treatment modulated cyclic adenosine monophosphate (cAMP), a measure of DRD2 activation.37 DRD2 is a member of the 5 G-protein coupled dopamine receptor family, split into 2 groups: D1-like (DRD1 and DRD5), which induce cAMP production through an α stimulatory subunit (Gαs-coupled), and D2-like (DRD2, DRD3, and DRD4), which inhibit cAMP production through an α inhibitory subunit (Gαi-coupled).38 DRD2 has been reported as highly expressed across brain tumor types, promoting tumor growth, and has emerged as a therapeutic target for gliomas.21,39 DRD2 signaling has previously been targeted in glioblastomas using anti-psychotic therapies, such as haloperidol, decreasing downstream ERK signaling and subsequent tumor growth,40 however, clinical trial results in non-DMG HGGs have not supported DRD2 antagonism as an MOA in this disease setting.
Neurotransmitter research in the context of psychiatric disorders such as schizophrenia shows that patients using antipsychotics that antagonize DRD2 experience lower incidences of cancer, even though this patient population is usually associated with social behaviors that increase cancer incidences (excessive smoking and alcohol usage).41 This provides some support for dordaviprone’s proposed MOA through DRD2 antagonism, as neurotransmitters such as dopamine and serotonin contribute to tumor initiation.42 Notably, using a G protein-coupled receptor (GPCR) screen, dordaviprone did not antagonize any other GPCRs/or dopamine receptors and is shown to harbor more potent anti-tumor effects than more traditional DRD2 antagonists, while maintaining a wider therapeutic index.43 DRD2 receptors harbor specific binding pockets containing both orthosteric and allosteric residues, both of which dordaviprone can bind to, allowing for tight molecular docking.22,44 Initially, dordaviprone received attention due to promising preclinical data showing upregulation of TRAIL, a potent apoptosis-promoting pathway in multiple cancers.26 Antagonism of DRD2 has also been shown to reduce cell viability in triple-negative breast cancer (TNBC),38 however, in a TRAIL-independent manner.45 Intriguingly, evidence in colon cancer cell lines suggest that DRD5 may influence the sensitivity of tumor cells to dordaviprone, as it is a negative regulator of DRD2 antagonism and is associated with reduced sensitivity.43
Although there is very little preclinical evidence correlating DRD2 antagonism and response in DMG, there is in silico and in vitro modeling providing evidence that dordaviprone binds to GPCR, in a selective way.44 Additionally, there is little information into the interactions of other dopamine receptors, and how these may contribute to its anti-tumor effects. However, mRNA expression does not tend to correlate with dordaviprone sensitivity, in fact, tumor cells with no DRD2 expression have shown sensitivity to dordaviprone (Figure 1A),46 suggesting alternative MOAs.47,48
Figure 1.

Expression of dordaviprone cellular targets across pediatric cancers, compared with diffuse gliomas. (A) DRD2 and (B) CLPP mRNA expression in diffuse glioma (including diffuse midline glioma, diffuse pontine glioma, and other high-grade gliomas), low-grade glioma, medulloblastoma, ependymoma, neuroblastoma, osteosarcoma, kidney, adrenal gland, AML (acute myeloid leukemia), ALL (acute lymphoid leukemia), and NBM (normal bone marrow). Diseases are grouped into BT (brain tumor), ST (solid tumor), and HM (hematological malignancies). RNA expression data were analyzed from publicly available data (normalized fragments per kilobase of transcript per million mapped reads [FPKM]).46 (C) CLPP protein expression (z-score) across pediatric central nervous system cancers analyzed from publicly available data.49 The dotted line is the median expression level for diffuse glioma, while turquoise dots are less than the dotted line and orange have the median above the dotted line. Statistical significance determined compared only to diffuse gliomas (one-way ANOVA, *P < .05,**P < .01,***P < .001,****P < .0001).
Dordaviprone is an Agonist of the Mitochondria Protease ClpP
More recent studies show dordaviprone is a potent agonist of the ATP-dependent Clp protease proteolytic subunit (ClpP), a mitochondrial protease that degrades mitochondrial respiratory chain proteins to disrupt energy homeostasis.50,51 Huge energy demands are required for rapidly proliferating DMG cells located alone the midline, and are primarily filled by the production of adenosine triphosphate (ATP) by the respiratory chain complexes. Substrates generated from glucose are produced during glycolysis and converted into coenzyme A (acetyl-CoA) which enters the tricarboxylic acid cycle (TCA cycle), to produce nicotinamide adenine dinucleotide + hydrogen (NADH) to generate a proton gradient across the inner mitochondrial membrane (Figure 2).52,53
Figure 2.

Normal mitochondrial function in cells. ATP production in mitochondria is produced by a cascade of reaction including glycolysis, tricarboxylic acid (TCA) cycle, or electron transport chain (ETC). The ClpXP complex, comprised of ClpP and ClpX, degrades misfolded proteins in the mitochondria, to maintain its functional integrity.
Mitochondria harbor intricate protein quality control systems, regulated by molecular chaperones which repair misfolded proteins, while proteases, such as the ClpXP complex, degrade misfolded proteins that are beyond repair.52 The ClpXP complex is made of 2 subunits; ClpP and ClpX. The ClpP proteins form a chamber-like structure of 2, stacked heptameric rings, creating a hollow core, known as axial pores (Figure 3).48 Hydrophobic binding sites (H sites) then act as attachment points for the ClpX protein, activating the complex.48,52 ClpX is part of a AAA+ (ATPase Associated with diverse cellular Activities) chaperone class of proteins, a well-known ATPase used in a range of protein machines.54 ClpX acts as a protein quality control system, ensuring that only misfolded proteins are proteolyzed due to the recognition of aromatic residues and specific sequences only visible in misfolded proteins.50,55
Figure 3.

Hyperactivation of ClpP by dordaviprone. During respiration, misfolded proteins are identified by a degradation tag and degraded by the ClpXP complex, passing through the axial pore. Dordaviprone replaces ClpX and binds ClpP, in H sites, removing protein selectivity and hyperactivating its protease activity. Additionally, dordaviprone increases axial pore size, resulting in more efficient degradation of mitochondrial respiratory complex proteins, regardless of their folding.
Given the high demands on energy production to fuel the malignant proliferation of DMG; and the observation that ClpP is overexpressed across diffuse gliomas at an mRNA and protein level (Figure 1B and C),46,49,56 degradation of the mitochondria matrix heralds huge anti-tumor potential.51 Dordaviprone is able to selectively bind to the H sites of ClpP, displacing ClpX, leading to its degradation as dordaviprone concentration increases.51 Upon dordaviprone attachment to the ClpP H sites, axial pores enlarge, allowing for increased protein influx into the complex (Figure 3),22 causing ClpP to lose the chaperoning of ClpX, and the ability to identify misfolded proteins and translocate them to the ClpXP matrix for degradation.51
Duchatel et al., confirmed ClpP target of dordaviprone using in silico binding of dordaviprone to ClpP, and robust structural stability leading to the degradation of mitochondrial proteins, such as SDHA and SDHB; components of Complex II of the ETC, in DMG cell lines in vitro and in vivo.22 This was further supported by Przystal et al., also showing ClpP as a key mediator of the response to dordaviprone in DMG.51 These findings were then validated through the use of CRIPSR/Cas-9 mediated knockdown of ClpP in DMG cell lines, where dordaviprone efficacy was lost in cell lines that were previously sensitive.57
Broadly, the process whereby dordaviprone binds to ClpP destabilizes the homeostatic levels of proteins in the mitochondria, increasing reactive oxygen species (ROS) and catalyzing the integrated stress response (ISR), an adaptational signaling pathway that allows cells to shutdown protein synthesis.51,58 In DMG, dordaviprone has been shown to dramatically increase mitochondrial ROS levels in a time- and dose-dependent manner, leading to mitochondrial structural deformities.51 Indeed, studies have reported increased ATF4 (activating transcription factor 4) and CHOP (C/EBP homology protein); known biomarkers for ROS and ISR, to be increased following dordaviprone exposure.31 This has been further examined in DMG revealing ATF4, and CHOP to be significantly upregulated following treatment.22,51
ATF4 is a regulator of the mitochondrial stress response, driving the translation of cytoprotective genes through activation of the ISR to protect the mitochondria.59 ATF4 mediates CHOP response, which is involved in the regulation of genes responsible for proliferation as well as energy metabolism, and has been shown to be an essential factor in the regulation of endoplasmic reticulum (ER) stress-induced apoptosis.59–61 Thus, treatments that activate the ISR, such as dordaviprone, drive increased expression of these proteins.61 Consequently, tumor cells will undergo mitochondrial degradation-dependent apoptosis, leading to the inability of tumor cells to proliferate and survive.
ClpP is the Prominent Biomarker of Response to Dordaviprone in DMG
Cancer cells harboring limited expression of DRD2 are also sensitive to dordaviprone, suggestive of alternative MOAs.50,62 Bonner et al., conducted a meta-analysis using the publicly available Cancer Dependency Map (DepMap) data,63 to show that CLPP mRNA expression levels correlated with dordaviprone sensitivity, across non-DMG cancer cell lines.47 Furthermore, recent DMG cell line data corroborated this analysis showing cells with high ClpP protein expression had increased sensitivity to dordaviprone in vitro.56 DepMap did not correlate DRD2 mRNA expression with dordaviprone sensitivity,47 nor did it at a transcript level in DMG cell lines.56 Although, DRD2 protein expression levels in DMG cell lines in vitro correlated with dordaviprone sensitivity, knockout of DRD2 was lethal to all DMG cell lines in vitro, with some of these DMG cells lines refractory to dordaviprone exposure.56 Additionally, Kline et al., showed that transient knockdown of DRD2 in colorectal cancer cells minimally impacts response to dordaviprone, suggesting DRD2 may not be critical for its anticancer MOA.64
Here, we performed analysis of DRD2 and CLPP mRNA expression levels obtained from publicly available pediatric cancer transcriptomic data sets, established by St. Jude Hospital, to show limited DRD2 expression across pediatric cancers (Figure 1A). Neuroblastoma (median = 3.099 normalized fragments per kilobase of transcript per million mapped reads [FPKM]) harbored significantly increased expression compared with diffuse gliomas (median = 0.4941, P ≤ .001), while there was no difference across brain tumors; and as expected, expression in hematological malignancies was significantly lower than diffuse gliomas (AML median = 0.00331, P = .0005, ALL median = 0.002202, P = .0004).56 Given the very low level mRNA expression reported herein (Figure 1A), it is intriguing that molecular inhibition of DRD2 blocked proliferation of DMG cell lines in vitro, including DMG cell lines that lacked dordaviprone sensitivity.56DRD2 expression levels in DMG and other non-brainstem pediatric HGGs does not appear to be different,56 with recent studies in glioblastoma cell lines showing molecular inhibition of DRD2 to not effect neurosphere formation or proliferation in vitro.65 Potentially, the co-occurrence of high protein expression levels of both DRD2 and CLPP correlates with sensitivity to dordaviprone in vitro, whereas the mRNA levels of DRD2 or CLPP do not. Currently, we are unaware whether high-level protein expression of DRD2 and CLPP correlates or co-occurs with dordaviprone sensitivity in non-brainstem pediatric HGG cell lines.
CLPP mRNA is highly expressed in diffuse gliomas compared with lower-grade brain tumors, while solid tumors and hematological malignancies were either significantly increased or showed no difference (Figure 1B). More aggressive cancer subtypes tended to express more CLPP mRNA than low-grade cancers. Analysis of CLPP protein expression across pediatric brain tumors,49 also identified increased expression in diffuse gliomas compared with all other tumor types of the CNS, potentially highlighting its utility as a precision medicine for diffuse gliomas, solid tumors, as well as acute myeloid and lymphoblastic leukemias and hence it is currently in clinical studies in hematological malignancies (Table 1, Figure 1C). Taken together, these data indicate that ClpP is a more robust biomarker of response to dordaviprone rather than DRD2 in diffuse gliomas,47,56 with more work required to determine the role of DRD2 in response to dordaviprone in DMG.56
Potential for Combination Treatment Strategies Including Dordaviprone in DMG
To date, there have been 3 different preclinical combination strategies using dordaviprone in DMG; (1) therapies targeting enzymes responsible for epigenetic posttranslational modifications, (2) a kinase inhibitor, and (3) standard of care radiotherapy (RT). The altered chromatin landscape of DMGs are, for the most part, reversible (except for the 3%–17% of H3 peptides harboring the H3K27M mutation). Therefore, drugs targeting epigenetic enzymes have been combined with dordaviprone and have shown to hold some synergistic potential, at least in vitro. Histone deacetylases (HDACs), enzymatically remove acetyl groups, regulating gene expression across many different cancer types. Nguyen et al., showed HDAC inhibition with either panobinostat and romidepsin-induced synthetic lethality when combined with imipridones which suppressed tumor cell metabolism in glioblastoma cells.66 Unfortunately, most HDAC inhibitors have poor BBB permeability,67 reducing their potential as a treatment for DMG. An additional epigenetic targeting strategy is to inhibit the histone methyltransferase catalytic enzyme EZH2, of the PRC2 complex, which is retained at genes responsible for neurogenesis and differentiation, processes inhibited in DMG.17,68 The rationale for their use is to sensitize DMG cells further to dordaviprone as they mimic the H3K27M mutation that has been associated with patient response to dordaviprone.21 EZH2 inhibitors EPZ-6438 (tazemetostat), FDA approved for the treatment of follicular lymphoma and epithelial sarcoma, and PF-06821497, currently in clinical trials for multiple cancers (NCT05767905 and NCT03460977) have been combined with dordaviprone in DMG with some in vitro synergistic potential.48 This synergy has been shown across several tumor cell lines, most potently in the glioblastoma cell line, U251, with some efficacy in the highly aggressive DMG cell line, SF8628.48 Due to the limited in vivo testing of this triple therapy, there is still some way to go before we determine the clinical potential of these therapies for the treatment of DMG.
Dordaviprone targets mitochondrial energy homeostasis in DMG, however, in DMG cell lines harboring reduced sensitivity, redox-activated PI3K/Akt signaling promotes metabolic adaptation.56 Using 13 patient-derived cell lines, representative of the molecular landscape of the DMG patient population (ie, 15% EZHIP, 31% H3.1K27M, 54% H3.3K27M), varying levels of sensitivity to dordaviprone were identified, mimicking the patient experience, where patients often fail upfront treatment. Employing a proteogenomic profiling approach, exposure to dordaviprone in DMG cells increased mitochondrial oxidative stress that was seen to promote redox activation of PI3K/Akt signaling, resulting in metabolic adaption and cell survival. Using paxalisib, a brain penetrant PI3K/Akt inhibitor, additive, and synergistic combinations were shown both in vitro and in vivo, with limited toxicities seen in orthotopic models, or in healthy, human peripheral blood mononuclear cell controls. These studies also correlated phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutations (frequently altered DMG gene5) with increased sensitivity to dordaviprone. This is likely due to constitutive activation caused by mutations in the catalytic subunit of PI3K/Akt/mTOR signaling; resulting in cells not being able to further upregulate PI3K/Akt signaling in response to treatment, and hence reducing the metabolic adaption necessary for additional growth following ClpP-agonism. Whereas, TP53 mutations (second most frequently mutated gene in DMGs5) were correlated with decreased sensitivity.56 Importantly, 2 DMG case studies experienced increased progression-free survival (PFS) using the combination of dordaviprone and paxalisib, with a patient starting the combination soon after the completion of RT demonstrating a dramatic tumor regression and continued PFS, 24 months following diagnosis (continuing). This study provided the impetus to test RT in combination with dordaviprone or paxalisib at diagnosis or progression, or the combination of dordaviprone and paxalisib within 14 weeks of the completion or RT, with each arm supported by a consolidation strategy including dordaviprone and paxalisib in a Phase II clinical trial for DMG/DIPG—NCT05009992 (Table 1).
Many clinical trials combine dordaviprone with RT in patients diagnosed with DMG (Table 1). Focal RT has the potential to reduce tumor volume, extending survival by 2–3 months, while temporarily improving neurological symptoms (~80%).69 Once tumors become refractory or throughout disease course, an increasing number of patients receive re-irradiation, supporting a clinical benefit for some.70 Gardner et al., examined the tolerability of ONC201 in combination with RT in the Phase 1 clinical trial (NCT03416530) and did not see AE when administered concurrently.36 Duchatel et al., correlated the use of re-irradiation in conjunction with dordaviprone, showing that the combination significantly increased median OS compared with patient receiving dordaviprone alone.22 Together these data suggest that dordaviprone may be rationally combined with upfront RT.
Dordaviprone Modulates the Immune Microenvironment
Emerging data suggests that dordaviprone might not solely work by targeting cancer cells directly, but also by initiating an immune response (Figure 4).71 Indeed, several types of immune cells express DRD2 and TRAIL.72,73 Preclinical studies using immune-competent mice engrafted with the colorectal cancer lines MC38 and CT126 identified increased numbers of both T cells (CD4 and CD8) and Natural Killer (NK) cells following dordaviprone treatment, within the tumor microenvironment (TME) and spleen as well as in the peripheral circulation.74 T cells and NK cells were upregulated in the blood of tumor naïve mice as well, suggesting dordaviprone to directly influence the immune cell populations, and not exclusively through a tumor-immune interaction. Furthermore, tumor cells resistant to dordaviprone in vitro became sensitized in vivo, hypothesizing that dordaviprone may work by activating an immune response against cancer cells. NK cell tumor infiltration was also observed in the breast cancer murine model, E0771, following dordaviprone treatment.75 Furthermore, pretreatment of breast cancer cells with dordaviprone prior to the addition of NK cells in vitro, resulted in more efficient killing, suggesting that dordaviprone sensitizes cancer cells to an immune response. Supporting these preclinical results, a tumor tissue biopsy from a patient with metastatic prostate cancer post dordaviprone treatment, showed tumor infiltration of granzyme B+ NK cells.34 Additionally, prostate cancer patients showed increased levels of NK cells in the peripheral blood up to 3 days after dordaviprone treatment.74
Figure 4.

Dordaviprone influences the immune microenvironment. (A) Dordaviprone increases levels of circulating inflammatory cytokines and effector molecules. (B) Proposed mechanism of dordaviprone-induced release of sequestered T cells in the bone marrow. Dordaviprone antagonizes DRD2 signaling, preventing β-arrestin from internalizing S1PR1. S1PR1 surface localization promotes T-cell trafficking. (C) Dordaviprone increases CD4+ and CD8+ T cell levels, as well as NK cells both in the peripheral blood and in the tumor microenvironment of solid tumors, (D) to reprogram macrophage metabolism driving the proinflammatory M1 phenotype.
Increased levels of CD45+ immune cells as well as CD8+ T cells were identified in a patient with mantle cell lymphoma 6 months after cessation of dordaviprone treatment (7 cycles of 125 mg every 3 weeks).76 However, no NK cells were detected, with authors hypothesizing that this might be due to NK cells being part of the rapid innate immune response and thus no longer detectable after 6 months. However, more definitive studies are required to confirm this finding. Further evidence for an immunomodulatory role for dordaviprone, showed that treatment increased serum levels of both cytokines and effector molecules, including IL-17A, TNF-α, IL-6, IL-10, Granzyme A and B, and perforin in adult patients with solid tumors refractory to standard treatments compared with pretreatment, especially in those who experienced >12 weeks PFS.
Macrophages have also been shown to be modulated by dordaviprone treatment. Primary human monocyte-derived macrophages were shown to switch from respiration to glycolytic ATP production after dordaviprone treatment, in the absence of tumor cells, likely due to mitochondrial stress.30 Glycolysis in macrophages has been linked to the proinflammatory subtype M1,77 suggesting that dordaviprone drives proinflammatory reprogramming of macrophages. Indeed, macrophages significantly upregulated production and secretion of IL-1β, as well as exhibited a dose-dependent trend of increased TNF although not significant.30 However, when co-culturing macrophages with glioblastoma cells in the presence of dordaviprone, no additional decrease in cell viability was observed compared with dordaviprone alone. Only minor changes in expression of inflammatory markers including IL-β1 was seen. This highlights the importance of immune and cancer cell interactions, a field becoming more important as we progress, raising the question as to whether the lack of effect in a co-culture system was due to the immunosuppressive crosstalk between the glioblastoma cells and the immune cells. Indeed, further studies are required to determine these cell-to-cell interactions. It remains to be determined if this could be a result of dosing and/or timing of treatment, macrophages being isolated from other immune cells, including T cells, or the lack of parenchyma/stroma, and how the lack of interactions affects response and will play out in the pons of patients with DMG.
Thus far, no published studies have investigated the immunomodulatory effects of dordaviprone in DMG, however, the potential proinflammatory effects of dordaviprone are promising. DMGs harbor a “cold” TME, characterized by few numbers of tumor-infiltrating lymphocytes as well as inflammatory factors.6,68,78,79 Due to a lack of lymphocytes as well as immune checkpoint proteins, immunotherapies including immune checkpoint inhibitors (ICIs) have thus far failed to increase survival in DMG patients, highlighting the need for therapeutics that help to recruit immune cells into the TME.
Dordaviprone’s potential role in the promotion of immune cell infiltration into the TME suggests it might be effectively combined with ICIs. Importantly, dordaviprone increases levels of both NK cells and T cells, both expressing PD-1, making the blockade of PD-1 an attractive approach. Indeed, combining 100 mg/kg dordaviprone with PD-1 inhibitors decreased tumor volume of mice orthotopically engrafted with the undifferentiated colon carcinoma cell line CT26, compared with PD-1 inhibitors alone.74 However, the effect was not seen with lower doses of dordaviprone or in mice with chemically induced murine colon adenocarcinoma cells MC38 pressing the need for DMG studies investigating the efficacy of combining dordaviprone with ICIs.
Even though there is evidence for dordaviprone-induced immune cell recruitment, the mechanism underpinning dordaviprone’s immunomodulatory effects remains to be determined. Like DMG, glioblastoma patients harbor a cold TME. Chongsathidkiet et al., identified that both glioblastoma patients as well as glioblastoma immune competent murine glioma models suffer from T cell lymphopenia, specifically CD4+ cells that likely contribute to the cold TME.80 The T cells were shown to be sequestered in the bone marrow due to loss of surface expression of the sphingosine 1 phosphate receptor 1 (S1PR1), required for T cells to travel between tissues. This loss of S1PR1 is likely mediated through β-arrestin, downstream of DRD2, suggesting a potential mechanism of dordaviprone-induced immune cell modulation, the systemic antagonism of DRD2. Intriguingly, the sequestration of T cells seems to be due to tumor location. When engrafting the same tumor cells either intracranially or subcutaneously, only the intracranial tumors resulted in T cell sequestration. Furthermore, systemic immune suppression has also been observed in patients with traumatic brain injury, identifying a strong-communication link between the CNS and the immune system.81 The mechanism underpinning these observation remains to be elucidated; however, β-arrestin/S1PR1 mediated T-cell sequestration and dordaviprone’s role in immunomodulation may suggest dopaminergic signaling might play a yet to be determined but significant role.
Next-generation ClpP Agonists
The promising clinical benefits testing dordaviprone in clinical trials, expanded access schemes and through unorthodox routes, coupled with the identification of CLPP overexpression in aggressive cancers including DMG, has stimulated the development of new ClpP agonists called “TR” analogs. These compounds do not cross-react with, or bind to DRD2, and hence are pure ClpP-agonists.52,57,62,82
TR compounds differ from imipridones by substitutions of small lipophilic moieties, combined with optimized benzyl residues (Table 3),83 resulting in increased specificity for ClpP and hence amplified potency.62 Further refinement of the compound by the removal of the carbonyl oxygen and methyl residue, increased shape complementarity, and optimized noncovalent interactions, resulting in the latest of the TR ClpP-agonists, called “TR-107”; shown to promote ClpP activity and drive anticancer responses in the low nanomolar range.52,57 Cells harboring CLPP knockout, treated with TR chemicals did not show sensitivity or increased ATF4 activity, demonstrating the ClpP selectivity of these compounds.82 Treatment of TNBC cell lines with TR-107 promoted ClpP-mediated degradation of mitochondrial TCA cycle proteins in a dose-dependent manner, however, did not promote a TRAIL-mediated apoptotic response, rather caused cancer cell cytostasis.57,82 This suggests that TR compounds may need to be combined with RT, or targeted therapies like paxalisib to show a response in DMG.56
Table 3.
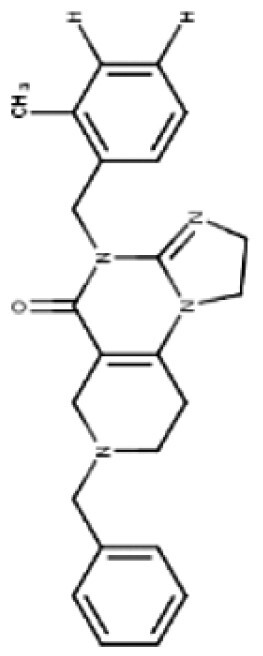
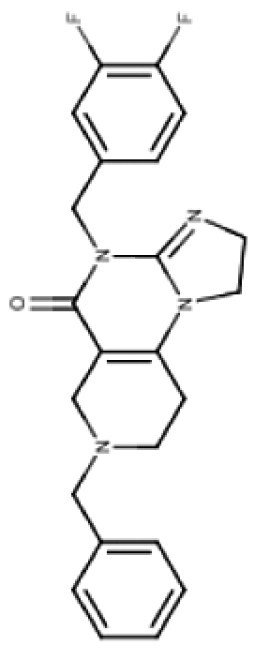
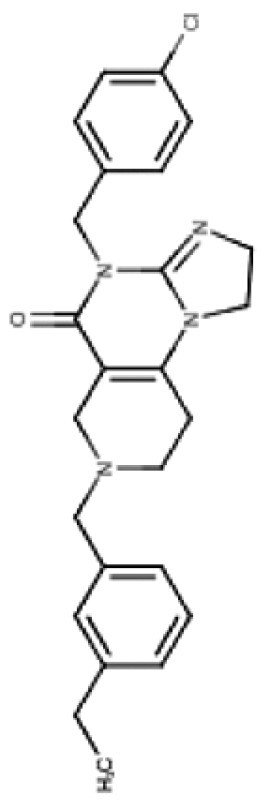
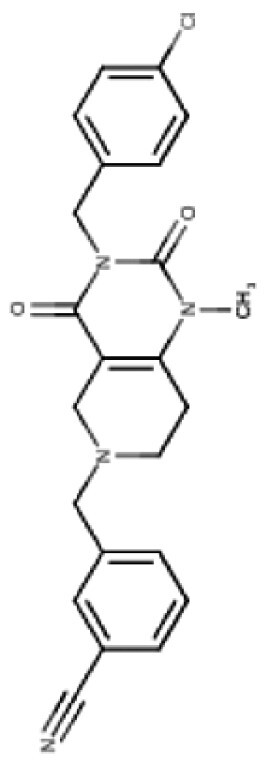
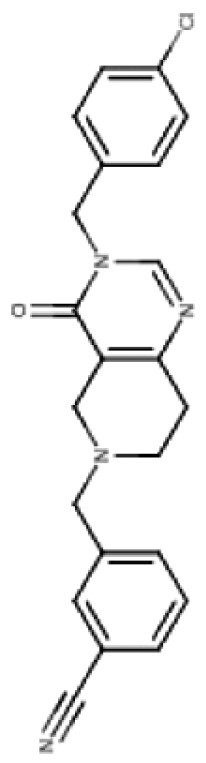
Predicted Brain Penetration Potential of ClpP-agonists as Determined by CNS-MPO.
| Name | ONC201 | ONC206 | TR-27 | TR-57 | TR-65 | TR-107 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chemical structure |
|
|
|
|
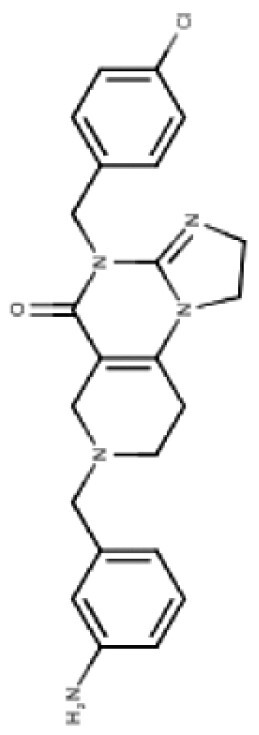
|
|
||||||
| CNS-MPO Parameter | Actual | Score | Actual | Score | Actual | Score | Actual | Score | Actual | Score | Actual | Score |
| LOGP | 3.05 | 0.97 | 2.83 | 1.00 | 4.10 | 0.45 | 3.06 | 0.97 | 2.32 | 1.00 | 3.13 | 0.93 |
| LOGD | 2.24 | 0.88 | 2.03 | 0.98 | 3.27 | 0.36 | 3.68 | 0.66 | 1.42 | 1.00 | 2.78 | 0.61 |
| MW | 386.50 | 0.81 | 408.45 | 0.65 | 434.97 | 0.46 | 420.90 | 0.57 | 421.93 | 0.56 | 390.87 | 0.78 |
| TPSA | 39.15 | 0.96 | 39.15 | 0.96 | 39.15 | 0.96 | 67.65 | 1.00 | 65.17 | 1.00 | 59.70 | 1.00 |
| HBD | 0 | 1.00 | 0.00 | 1.00 | 0.00 | 1.00 | 0.00 | 1.00 | 2.00 | 0.50 | 0.00 | 1.00 |
| PKA | 8.13 | 0.93 | 8.12 | 0.94 | 8.16 | 0.92 | 7.54 | 1.00 | 8.23 | 0.88 | 7.49 | 1.00 |
| CNS-MPO Score | 5.55/6 | 5.54/6 | 4.16/6 | 5.19/6 | 4.94/6 | 5.32/6 | ||||||
Subjecting imipridones and TR-compounds to analysis using the central nervous system multi-parameter optimization (CNS-MPO) computation tool,84 predicted all ClpP-agonists as good candidates to cross the BBB and enter the brain/tumor when delivered systemically (Table 3). CNS-MPO predicts ONC201 and ONC206 to have the best CNS penetration, followed by TR-107 and TR-57, however, at this time, there are no reports of preclinical or clinical examination of TR-compounds in DMG.
Discussion
Emerging clinical data suggests that some DMG patients treated with dordaviprone experience tumor regression and neurological improvement, and in some cases, increased median OS.20–23 These early phase trial results and alternative access opportunities have encouraged phase III efficacy testing in patients diagnosed with DMG, H3 K27-altered located along the midline structures of the brain, but excluding patients harboring tumors located in the pontine region of the brainstem (NCT05580562, Table 1). Potentially, patients with midline/thalamic DMGs show an improved response to dordaviprone, compared with those patients diagnosed with pontine tumors. This is likely due to differing immune cell populations in the TME of DIPG compared with thalamic DMG, where monocyte-derived macrophages are the dominant immune cell population, while DIPGs are enriched in brain-resident microglia.68 So not only does this promote different immune responses to the drug, but these immune cell populations promote a mesenchymal cell state for patients with thalamic lesions, whereas DIPGs exhibit stemness properties including oligodendrocyte precursor cell (OPC)-like capable of self-renewal.68,85
The brain’s high oxygen demand,86,87 pinpoints oxidative phosphorylation, and hence the mitochondria, as an essential factor in the energy supply needed for the rapid cellular proliferation of DMG. Thus, DMGs require abundant respiratory chain proteins to meet these high energy demands. It is therefore clear that this underpins a metabolic dependency in DMG, and hence a therapeutic vulnerability that dordaviprone may exploit. Assessment of CLPP gene and protein show high expression in diffuse gliomas and other aggressive pediatric cancers (Figure 1B and C), highlighting the clinical potential of ClpP agonists.51,56 Development of new selective ClpP agonists, including the TR compounds, may add additional weapons to the barren armory of options we currently have for the treatment of DMG.
Dordaviprone’s excellent safety profile makes it an ideal candidate for the combination strategies that are necessary if we are to achieve long-term positive outcomes, particularly for children diagnosed with DIPG. Hence, dordaviprone is currently being tested in combination with RT and paxalisib in the international clinical trial “Combination Therapy for the Treatment of Diffuse Midline Gliomas” (NCT05009992), underpinned by extensive preclinical and case study results established across 8 international laboratories and coordinated by the Pacific Neuro-Oncology Consortium (PNOC022).56 Although we are still some way from achieving long-term survival, dordaviprone appears to be one of the options needed for the future development of complex multimodal combination strategies that will improve outcomes for children, adolescents, and young adults diagnosed with DMG, H3 K27-altered.
Acknowledgments
We acknowledge all children and their families tragically affected by DIPG/DMG. Figures 2–4 were created with BioRender.com.
Contributor Information
Evangeline R Jackson, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia.
Mika L Persson, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia.
Cameron J Fish, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia.
Izac J Findlay, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia.
Sabine Mueller, DIPG/DMG Center Zurich, University Children’s Hospital Zürich, Zurich, Switzerland; Department of Neurology, Neurosurgery and Pediatric, UCSF, San Francisco, California, USA.
Javad Nazarian, DIPG/DMG Center Zurich, University Children’s Hospital Zürich, Zurich, Switzerland; Center for Genetic Medicine Research, Children’s National Hospital, Washington, District of Columbia, USA; The George Washington University, School of Medicine and Health Sciences, Washington, District of Columbia, USA.
Esther Hulleman, Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands, Utrecht, Netherlands.
Jasper van der Lugt, Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands, Utrecht, Netherlands.
Ryan J Duchatel, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia.
Matthew D Dun, Cancer Signalling Research Group, School of Biomedical Sciences and Pharmacy, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia; Precision Medicine Research Program, Hunter Medical Research Institute, New Lambton Heights, New South Wales , Australia; Paediatric Program, Mark Hughes Foundation Centre for Brain Cancer Research, College of Health, Medicine and Wellbeing, University of Newcastle, Callaghan, New South Wales, Australia.
Funding
M.D.D. was supported by a Cancer Institute NSW Fellowship. M.D.D. holds an NHMRC Investigator Grant—GNT1173892. The contents of the published material are solely the responsibility of the research institutions involved or individual authors and do not reflect the views of NHMRC. M.D.D. was also supported by a ChadTough Defeat DIPG New Investigator grant. R.J.D. is supported by a ChadTough Defeat DIPG fellowship grant. E.R.J. is supported by the Hunter Cancer Research Alliance and the Josephine Dun Scholarship from the Isabella and Marcus Foundation and Miette Skiller Scholarship Fund a sub-fund of the Australian Communities Foundation. M.L.P. is recipient of the RUN DIPG International HDR Scholarship. I.J.F. is recipient of the RUN DIPG “Moving Towards a Cure” HDR Scholarship and CureCell ExCELLerate Award. This project was supported by the DIPG/DMG Collaborative: The Cure Starts Now Foundation, The Cure Starts Now Foundation Australia, Brooke Healey Foundation, Wayland Villars Foundation, ChadTough Foundation, Aidan’s Avengers, Austin Strong, Cure Brain Cancer, Jeffrey Thomas Hayden Foundation, Laurie’s Love Foundation, Love Chloe Foundation, Musella Foundation, Pray Hope Believe, Reflections of Grace, Storm the Heavens Fund, Aubreigh’s Army, Whitley’s Wishes, Ryan’s Hope, Benny’s World, The Isabella and Marcus Foundation, Lauren’s Fight for Cure, Robert Connor Dawes Foundation, The Gold Hope Project, Julia Barbara Foundation, Lily Larue Foundation, American Childhood Cancer Organization, RUN DIPG, Gabriella’s Smile Foundation, and Snapgrant.com. Additional funding was also received from Strategic Group, McDonald Jones Foundation, VINVAGroup, Kiriwina Investments, Australian Lions Childhood Cancer Research Foundation, Pacific Pediatric Neuro-Oncology Consortium Foundation, Yuvaan Tiwari Foundation, Charlie Teo Foundation, Little Legs Foundation, The Kids' Cancer Project, Tour De Cure, Liv Like a Unicorn, Edie’s Kindness Project, Maitland Cancer Appeal Limited, Blackjack Foundation, Hunter Medical Research Institute, and the Evie Poolman Research Gift via the Kinghorn Foundation.
Conflict of Interest Statement
M.D.D. is a parent to a child lost to diffuse intrinsic pontine glioma (DIPG), and the Founder and a Director of the not-for-profit charity RUN DIPG Pty Ltd.
Supplement sponsorship
This article appears as part of the supplement “H3 K27M-mutant Glioma: Disease State Overview,” sponsored by Chimerix, Inc.
Authorship Statement
M.D.D. and R.J.D. conceived and designed the study. E.R.J., M.L.P., C.F., I.J.F., S.M, J.N, E. H., J.V.L., R.J.D., and M.D.D. designed the figures, wrote the manuscript, and edited and approved the final version of the manuscript.
References
- 1. Jackson S, Patay Z, Howarth R, et al. Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neurooncol. 2013;114(3):339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A collaborative report from the international and European society for pediatric oncology DIPG registries. J Clin Oncol. 2018;36(19):1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duchatel RJ, Jackson ER, Alvaro F, et al. Signal transduction in diffuse intrinsic pontine glioma. Proteomics. 2019;19(21–22):e1800479. [DOI] [PubMed] [Google Scholar]
- 4. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Findlay IJ, De Iuliis GN, Duchatel RJ, et al. Pharmaco-proteogenomic profiling of pediatric diffuse midline glioma to inform future treatment strategies. Oncogene. 2022;41(4):461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Persson ML, Douglas AM, Alvaro F, et al. The intrinsic and microenvironmental features of diffuse midline glioma: Implications for the development of effective immunotherapeutic treatment strategies. Neuro Oncol. 2022;24(9):1408–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27(9):985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rushing EJ. WHO classification of tumors of the nervous system: Preview of the upcoming 5th edition. Memo - Mag Eur Med Oncol. 2021;14(2):188–191. [Google Scholar]
- 10. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124(3):439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24(5):660–672. [DOI] [PubMed] [Google Scholar]
- 12. Ajuyah P, Mayoh C, Lau LMS, et al. Histone H3-wild type diffuse midline gliomas with H3K27me3 loss are a distinct entity with exclusive EGFR or ACVR1 mutation and differential methylation of homeobox genes. Sci Rep. 2023;13(1):3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7(1):11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. [DOI] [PubMed] [Google Scholar]
- 15. Yu JR, LeRoy G, Bready D, et al. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci Adv. 2021;7(29):eabg7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stafford JM, Lee CH, Voigt P, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv. 2018;4(10):eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harutyunyan AS, Krug B, Chen H, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun. 2019;10(1):1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23(4):483–492. [DOI] [PubMed] [Google Scholar]
- 19. Funato K, Major T, Lewis PW, Allis CD, Tabar V.. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (New York, N.Y.). 2014;346(6216):1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hall MD, Odia Y, Allen JE, et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: A case report. J Neurosurg Pediatr. 2019;23(6):719–725. [DOI] [PubMed] [Google Scholar]
- 21. Chi AS, Tarapore RS, Hall MD, et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J Neurooncol. 2019;145(1):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duchatel RJ, Mannan A, Woldu AS, et al. Pre-clinical and clinical evaluation of German-sourced ONC201 for the treatment of H3K27M mutant diffuse intrinsic pontine glioma. Neuro Oncol Adv. 2021;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanrikulu B, Yasar AH, Canpolat C, et al. Preliminary findings of German-sourced ONC201 treatment in H3K27 altered pediatric pontine diffuse midline gliomas. J Neurooncol. 2023;163(3):565–575. [DOI] [PubMed] [Google Scholar]
- 24. André N, Buyens G, Bouffet E, Walker D, Dun MD.. Access to new drugs in paediatric oncology: Can we learn from the ongoing ONC201 saga? Lancet Oncol. 2023;24(3):209–212. [DOI] [PubMed] [Google Scholar]
- 25. Allen JE, Kline CLB, Prabhu VV, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7(45):74380–74392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Allen JE, Krigsfeld G, Mayes PA, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5(171):171ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arrillaga-Romany I, Chi AS, Allen JE, et al. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget. 2017;8(45):79298–79304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagner J, Kline CL, Pottorf RS, et al. The angular structure of ONC201, a TRAIL pathway-inducing compound, determines its potent anti-cancer activity. Oncotarget. 2014;5(24):12728–12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen JE, Crowder RN, El-Deiry WS.. First-in-class small molecule ONC201 induces DR5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS One. 2015;10(11):e0143082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Geiss C, Witzler C, Poschet G, Ruf W, Regnier-Vigouroux A.. Metabolic and inflammatory reprogramming of macrophages by ONC201 translates in a pro-inflammatory environment even in presence of glioblastoma cells. Eur J Immunol. 2021;51(5):1246–1261. [DOI] [PubMed] [Google Scholar]
- 31. Ishizawa J, Kojima K, Chachad D, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liguori NR, Sanchez Sevilla Uruchurtu A, Zhang L, et al. Preclinical studies with ONC201/TIC10 and lurbinectedin as a novel combination therapy in small cell lung cancer (SCLC). Am J Cancer Res. 2022;12(2):729–743. [PMC free article] [PubMed] [Google Scholar]
- 33. Stein MN, Bertino JR, Kaufman HL, et al. First-in-human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res. 2017;23(15):4163–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stein MN, Malhotra J, Tarapore RS, et al. Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J ImmunoTher Cancer. 2019;7(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gardner S, Koschmann C, Tarapore R, et al. Safety of ONC201 administered two consecutive days per week in pediatric h3 k27m-mutant glioma patients. Neuro Oncol. 2021;23.(Supplement_6).):vi67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gardner SL, Tarapore RS, Allen J, et al. Phase I dose escalation and expansion trial of single agent ONC201 in pediatric diffuse midline gliomas following radiotherapy. Neurooncol Adv. 2022;4(1):vdac143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Madhukar NS, Khade PK, Huang L, et al. A Bayesian machine learning approach for drug target identification using diverse data types. Nat Commun. 2019;10(1):5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tegowski M, Fan C, Baldwin AS.. Thioridazine inhibits self-renewal in breast cancer cells via DRD2-dependent STAT3 inhibition, but induces a G1 arrest independent of DRD2. J Biol Chem. 2018;293(41):15977–15990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Staley A, Tucker K, Yin Y, et al. Highly potent dopamine receptor D2 antagonist ONC206 demonstrates anti-tumorigenic activity in endometrial cancer. Am J Cancer Res. 2021;11(11):5374–5387. [PMC free article] [PubMed] [Google Scholar]
- 40. Li J, Zhu S, Kozono D, et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget. 2014;5(4):882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dalton SO, Johansen C, Poulsen AH, et al. Cancer risk among users of neuroleptic medication: A population-based cohort study. Br J Cancer. 2006;95(7):934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caragher SP, Hall RR, Ahsan R, Ahmed AU.. Monoamines in glioblastoma: Complex biology with therapeutic potential. Neuro Oncol. 2017;20(8):1014–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prabhu VV, Madhukar NS, Gilvary C, et al. Dopamine receptor d5 is a modulator of tumor response to dopamine receptor D2 antagonism. Clin Cancer Res. 2019;25(7):2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prabhu VV, Morrow S, Rahman Kawakibi A, et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia. 2020;22(12):725–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yuan X, Kho D, Xu J, et al. ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells. Oncotarget. 2017;8(13):21626–21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McLeod C, Gout AM, Zhou X, et al. St. Jude cloud: A pediatric cancer genomic data-sharing ecosystem. Cancer Discov. 2021;11(5):1082–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bonner ER, Waszak SM, Grotzer MA, Mueller S, Nazarian J.. Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro Oncol. 2020;23(4):542–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wedam R, Greer YE, Wisniewski DJ, et al. Targeting mitochondria with ClpP agonists as a novel therapeutic opportunity in breast cancer. Cancers (Basel). 2023;15(7):1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petralia F, Tignor N, Reva B, et al. ; Children’s Brain Tumor Network. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. 2020;183(7):1962–1985.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ishizawa J, Zarabi SF, Davis RE, et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell. 2019;35(5):721–737.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Przystal JM, Cosentino CC, Yadavilli S, et al. Imipridones affect tumor bioenergetics and promote cell lineage differentiation in diffuse midline gliomas. Neuro Oncol. 2022;24.(9).):1438–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mabanglo MF, Wong KS, Barghash MM, et al. Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome. Structure. 2023;31(2):185–200.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonora M, Patergnani S, Rimessi A, et al. ATP synthesis and storage. Purinergic Signal. 2012;8(3):343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang SG, Maurizi MR, Thompson M, Mueser T, Ahvazi B.. Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J Struct Biol. 2004;148(3):338–352. [DOI] [PubMed] [Google Scholar]
- 55. Liu K, Ologbenla A, Houry WA.. Dynamics of the ClpP serine protease: A model for self-compartmentalized proteases. Crit Rev Biochem Mol Biol. 2014;49(5):400–412. [DOI] [PubMed] [Google Scholar]
- 56. Jackson ER, Duchatel RJ, Staudt DE, et al. ONC201 in combination with paxalisib is a therapeutic strategy for diffuse midline glioma. Cancer Res. 2023;83(14): 2421–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fennell EMJ, Aponte-Collazo LJ, Wynn JD, et al. Characterization of TR-107, a novel chemical activator of the human mitochondrial protease ClpP. Pharmacol Res Perspect. 2022;10(4):e00993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Choo JAMY, Schlösser D, Manzini V, Magerhans A, Dobbelstein M.. The integrated stress response induces R-loops and hinders replication fork progression. Cell Death Dis. 2020;11(7):538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. [DOI] [PubMed] [Google Scholar]
- 60. Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S.. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 1996;395(2–3):143–147. [DOI] [PubMed] [Google Scholar]
- 61. Wang S, Chen XA, Hu J, et al. ATF4 gene network mediates cellular response to the anticancer PAD inhibitor YW3-56 in triple-negative breast cancer cells. Mol Cancer Ther. 2015;14(4):877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Graves PR, Aponte-Collazo LJ, Fennell EMJ, et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem Biol. 2019;14(5):1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell. 2017;170(3):564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kline CLB, Ralff MD, Lulla AR, et al. Role of dopamine receptors in the anticancer activity of ONC201. Neoplasia. 2018;20(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jeon HM, Oh YT, Shin YJ, et al. Dopamine receptor D2 regulates glioblastoma survival and death through MET and death receptor 4/5. Neoplasia. 2023;39:100894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nguyen TTT, Shang E, Schiffgens S, et al. Induction of synthetic lethality by activation of mitochondrial ClpP and inhibition of HDAC1/2 in glioblastoma. Clin Cancer Res. 2022;28(9):1881–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vitanza NA, Biery MC, Myers C, et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naïve diffuse midline glioma models. Neuro Oncol. 2021;23(3):376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu I, Jiang L, Samuelsson ER, et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat Genet. 2022;54(12):1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tinkle CL, Campbell K, Han Y, et al. Radiation dose response of neurologic symptoms during conformal radiotherapy for diffuse intrinsic pontine glioma. J Neurooncol. 2020;147(1):195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fontanilla HP, Pinnix CC, Ketonen LM, et al. Palliative reirradiation for progressive diffuse intrinsic pontine glioma. Am J Clin Oncol. 2012;35(1):51–57. [DOI] [PubMed] [Google Scholar]
- 71. Foster JB, Alonso MM, Sayour E, et al. Translational considerations for immunotherapy clinical trials in pediatric neuro-oncology. Neoplasia. 2023;42:100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu Z, Zhai XR, Du ZS, et al. Dopamine receptor D2 on CD4(+) T cells is protective against neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain Behav Immun. 2021;98:110–121. [DOI] [PubMed] [Google Scholar]
- 73. Kayagaki N, Yamaguchi N, Nakayama M, et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol. 1999;163(4):1906–1913. [PubMed] [Google Scholar]
- 74. Wagner J, Kline CL, Zhou L, et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest. 2018;128(6):2325–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ralff MD, Jhaveri A, Ray JE, et al. TRAIL receptor agonists convert the response of breast cancer cells to ONC201 from anti-proliferative to apoptotic. Oncotarget. 2020;11(42):3753–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Romaguera JE, Lee HJ, Tarapore R, et al. Integrated stress response and immune cell infiltration in an ibrutinib-refractory mantle cell lymphoma patient following ONC201 treatment. Br J Haematol. 2019;185(1):133–136. [DOI] [PubMed] [Google Scholar]
- 77. Liu Y, Xu R, Gu H, et al. Metabolic reprogramming in macrophage responses. Biomark Res. 2021;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lin GL, Nagaraja S, Filbin MG, et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018;6(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lieberman NAP, DeGolier K, Kovar HM, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chongsathidkiet P, Jackson C, Koyama S, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hazeldine J, Lord JM, Belli A.. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front Neurol. 2015;6:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fennell EMJ, Aponte-Collazo LJ, Pathmasiri W, et al. Multi-omics analyses reveal ClpP activators disrupt essential mitochondrial pathways in triple-negative breast cancer. Front Pharmacol. 2023;14:1136317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Xu R, Liu Y.. Imidazo-pyrimidone compounds, and preparation method and application thereof. Google Patents. 2020. [Google Scholar]
- 84. Wager TT, Hou X, Verhoest PR, Villalobos A.. Central nervous system multiparameter optimization desirability: Application in drug discovery. ACS Chem Neurosci. 2016;7(6):767–775. [DOI] [PubMed] [Google Scholar]
- 85. Filbin MG, Tirosh I, Hovestadt V, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science (New York, N.Y.). 2018;360(6386):331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Salim S. Oxidative stress and the central nervous system. J Pharmacol Exp Ther. 2017;360(1):201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Venneti S, Kawakibi AR, Ji S, et al. Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discov. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]