

Abstract
In congenital diaphragmatic hernia (CDH), abdominal organs are displaced into the chest, compress the lungs, and cause mediastinal shift. This contributes to development of pulmonary hypoplasia and hypertension, which is the primary determinant of morbidity and mortality for affected newborns. The severity is determined using prenatal imaging as early as the first trimester and is related to the laterality of the defect, extent of lung compression, and degree of liver herniation. Comprehensive evaluation of fetal CDH includes imaging-based severity assessment, severity assessment, and evaluation for structural or genetic abnormalities to differentiate isolated from complex cases. Prenatal management involves multispecialty counseling, consideration for fetal therapy with fetoscopic endoluminal tracheal occlusion (FETO) for severe cases, monitoring and intervention for associated polyhydramnios or signs of preterm labor if indicated, administration of antenatal corticosteroids in the appropriate setting, and planned delivery to optimize the fetal condition at birth. Integrated programs that provide a smooth transition from prenatal to postnatal care produce better outcomes. Neonatal care involves gentle ventilation to avoid hyperinflation and must account for transitional physiology to avoid exacerbating cardiac dysfunction and decompensation. Infants who have undergone and responded to FETO have greater pulmonary capacity than expected, but cardiac dysfunction seems unaffected. In about 25–30% of CDH neonates extracorporeal life support is utilized, and this provides a survival benefit for patients with the highest predicted mortality, including those who underwent FETO. Surgical repair after initial medical management for the first 24–48 hours of life is preferred since later repair is associated with delayed oral feeding, increased need for tube feeds, and increased post-repair ventilation requirement and supplemental oxygen at discharge. With overall survival rates >70%, contemporary care involves management of chronic morbidities in the context of a multidisciplinary clinic setting.
Keywords: Congenital diaphragmatic hernia (CDH), extracorporeal life support, fetoscopic endoluminal tracheal occlusion (FETO), gentle ventilation, surgical repair
Introduction
Background
Congenital diaphragmatic hernia (CDH) is the consequence of a partial or complete defect in the diaphragm that allows displacement of abdominal contents into the thoracic cavity during fetal development. Intrathoracic crowding and compression of the fetal lungs by abdominal organs predispose the neonate to intrinsic bilateral pulmonary hypoplasia and hypertension, which manifest after birth (1,2). CDH is present in approximately 1 in 3,000 births, with over 80% being on the left side, and is associated with significant neonatal and infant morbidity and mortality (1-4). Up to 70% of CDHs are detected prenatally. The degree of lung compression is related to prenatally acquired pulmonary hypoplasia and is a significant determinant of postnatal care complexity and ultimately, infant survival (5-8). In the last decade, significant developments in CDH care have transformed the management approach to one that begins before delivery. This strategy harnesses the benefits of improved prenatal risk stratification, offers the option for fetal therapy for selected cases, and allows for coordinated delivery and care transition to a pediatric care setting that optimizes lung function, decreases the risk of infant mortality and morbidity, and improves overall outcome.
Rationale and knowledge gap
Advancements in pre- and postnatal care have shown potential benefits for CDH management resulting in improved outcomes in integrated care programs (9). However, knowledge gaps can arise when developments within the various specialties that provide CDH care may not be universally recognized leading to potential delays in optimizing care.
Objective
This review aims to summarize advances in pre- and postnatal management of infants with CDH and highlight opportunities for integrating care.
Risk stratification of CDH
Recognizing the importance of standardized risk adjustment to evaluate outcomes for children with CDH, the Congenital Diaphragmatic Hernia Study Group agreed on a standardized classification scheme for defect size as observed during hernia repair. Defects are coded as “A” defects if entirely surrounded by diaphragmatic muscle, “B” defects if less than 50% of the chest wall is devoid of diaphragm, “C” defects if more than 50% of the chest wall lacks a diaphragm and “D” defects if the diaphragm is mostly absent (Figure 1) (10). Defect size is the primary determinant of respiratory, gastrointestinal, and neurologic morbidity, and together with a coexisting major cardiac anomaly, it is the overarching determinant of survival (Figure 2). The association between larger defect size and adverse outcome is due to the greater magnitude of intrathoracic organ herniation earlier in gestation, ultimately resulting in more severe pulmonary hypoplasia at birth (5,11). The ability to diagnose CDH by prenatal ultrasound and quantify factors that correlate with infant outcome forms the basis of prenatal risk stratification and informs management decisions including the option to undergo fetal treatment when indicated.
Figure 1.

Schematic representation of diaphragmatic hernia size at surgery. Visual representation of a standardized classification scheme for defect size as observed during hernia repair. “A” defects are entirely surrounded by diaphragmatic muscle, “B” defects have less than 50% of the chest wall devoid of diaphragm, “C” defects have more than 50% of the chest wall lacking diaphragm and in “D” defects the diaphragm is mostly absent.
Figure 2.
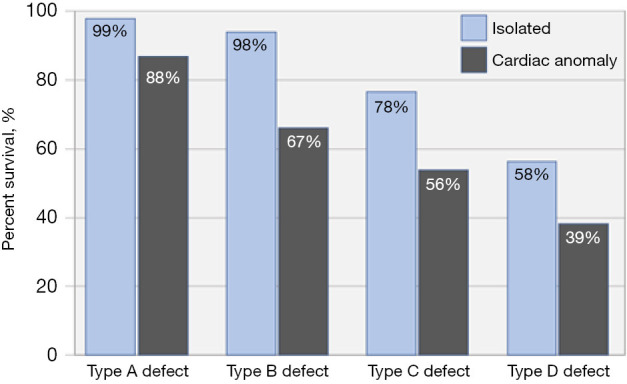
Survival in relationship to hernia size and presence of a cardiac defect. The bar graphs represent the observed survival in four types of left diaphragmatic hernia, stratified by the presence of an isolated defect or a coexisting cardiac anomaly [based on ref. (11)].
Prenatal diagnosis and identification of isolated and complex CDH
CDH is suspected when prenatal ultrasound demonstrates mediastinal shift to the contralateral side of the hernia and evidence of intrathoracic herniation of abdominal contents, which may be observed as early as the first trimester (Figure 3). When the diaphragmatic defect is on the left side bowel is most frequently seen in the thorax, and the stomach bubble is at the level of the heart in 80% (12). The likelihood for liver, spleen and even kidney herniation increases with the size of the defect. In right-sided defects the liver is the primary abdominal organ that herniates upward due to its normal intraabdominal position. The similar ultrasound echotexture of compressed lung and liver make it difficult to distinguish these two organs and contributes to lower prenatal detection of right CDH (6). Overall, up to 74% of CDH is diagnosed prenatally by these ultrasound findings (Figure 4). The likelihood and timing of diagnosis is related to the defect size. Larger defects can be detected earlier in gestation. Accordingly, while first trimester diagnosis is only made in 30% of CDH (13), these are more likely to be larger defects that carry a worse prognosis after birth.
Figure 3.

Prenatal ultrasound appearance of diaphragmatic hernia in early gestation. Prenatal ultrasound images obtained in two patients with a left diaphragmatic hernia at 13 weeks’ gestation (A) and 16 weeks’ gestation (B). The first trimester defect was suspected due to the rotation of the cardiac axis to the left and the intrathoracic retrocardiac position of the stomach. The second patient was diagnosed based on the marked mediastinal shift of the heart towards the right of the fetus and the intrathoracic location of the stomach.
Figure 4.

Ultrasound appearance of left and right diaphragmatic hernia in the second trimester. Prenatal ultrasound images obtained in a patient with left diaphragmatic hernia at 26 weeks’ gestation (A) and a right diaphragmatic hernia at 22 weeks’ gestation (B). The left diaphragmatic hernia is characterized by a right mediastinal shift of the heart and intrathoracic bowel and stomach herniation. The contralateral right lung is posterior to the right atrium and there is reduction of the observed to estimated lung to head ratio to a moderate hernia severity. In right diaphragmatic hernia the cardiac axis is rotated to the left and the mediastinal shift is less marked despite intrathoracic herniation of the liver. Both lungs measure smaller in size and there is a pericardial effusion (*) which is common in this setting.
Once the diagnosis of CDH is suspected and the laterality has been determined the next step is evaluation for associated physical, genetic or syndromic conditions. The presence of any of these findings distinguishes isolated from complex CDH. In almost 50% of fetuses with CDH additional structural abnormalities are identified on prenatal imaging (14). The most common structural abnormalities in non-syndromic and syndromic conditions are cardiac defects identified in approximately 30% of patients (15) (Tables 1,2). In addition to structural anomalies, detailed survey for dysmorphic features is essential as it may point to a range of genetic conditions that are identified in 2–33% of patients with CDH dependent on the testing strategy employed (16-19). Compared to traditional karyotype, microarray testing has a higher diagnostic yield for genetic abnormalities extending beyond the common aneuploidies and is considered most appropriate if parents desire genetic evaluation (19).
Table 1. Non-syndromic structural anomalies associated with congenital diaphragmatic hernia.
| Location | Anomalies | Frequency |
|---|---|---|
| Cardiovascular | Ventricular septum defect | 25–30% |
| Atrial septum defect | ||
| Tetralogy of Fallot | ||
| Double outlet right ventricle | ||
| Transposition of the great arteries | ||
| Shone complex | ||
| Hypoplastic left heart | ||
| Coarctation of the aorta | ||
| Thoracic | Bronchopulmonary sequestration | 2–10% |
| Congenital pulmonary airway malformation | ||
| Abdominal | Bowel malrotation | 2–10% |
| Omphalocele | ||
| Horseshoe kidney | ||
| Pelvic kidney | ||
| Musculoskeletal | Polydactyly or syndactyly | 1–15% |
| Limb reduction defects | ||
| Nervous system | Neural tube defects | 1–10% |
| Hydrocephaly | ||
| Others | Undescended testes | 5% |
Table 2. Common genetic abnormalities associated with congenital diaphragmatic hernia.
| Category | Genetic abnormalities or syndromes | Additional prenatal findings |
|---|---|---|
| Aneuploidy | Trisomy 21, 18, 13, 22, 16 | Cardiac, CNS, craniofacial, shortened limbs, nuchal edema, hydrops, polyhydramnios |
| Tetrasomy 12p (Pallister-Killian) | Nuchal thickening, shortened long bones, polyhydramnios | |
| Translocation der (22) t(11:22) (q23:q11) | Nuchal thickening, craniofacial, FGR | |
| Monosomy 15q | Cardiac, facial, FGR, talipes, SUA | |
| Monosomy 4p (Wolf-Hirshhorn) | Cardiac, facial, digital, talipes, FGR | |
| 8p.23.1 deletion | Cardiac, FGR | |
| 1q41-1q42 deletion | Cardiac, craniofacial, cleft palate, talipes | |
| Xpter-Xp22 | Variable prenatal phenotype | |
| 16p11.2 microdeletion | Nonspecific prenatal phenotype | |
| 15q24 microdeletion | Craniofacial, digital, genital, FGR | |
| Mendelian disorders | Xq26 (Simpson-Golabi-Behmel) | Macrosomia, organomegaly, omphalocele, macroglossia, polydactyly, polyhydramnios |
| 11p15.5 (Beckwith-Wiedemann) | Macrosomia, omphalocele, macroglossia | |
| Xp | Cardiomyopathy, microphthalmia | |
| Xp22 (Goltz) | Syndactyly | |
| Xp22 (Craniofrontonasal) | Coronal synostosis, hypertelorism, digital | |
| 11p13 (Denys-Drash) | Nephromegaly, ambiguous genitalia | |
| Syndromes with unidentified genetic etiology | Fryns | Cardiac, facial, cleft lip/palate, neuronal heterotopias, limb, genitourinary |
| Gershoni-Baruch | Omphalocele, radial ray abnormalities | |
| Goldenhar | Cardiac, absent ears, cleft lip palate, vertebral | |
| Pentalogy of Cantrell | Ectopia cordis, bifid sternum, omphalocele |
CNS, central nervous system; FGR, fetal growth restriction; SUA, single umbilical artery.
Other than extrapulmonary abnormalities there may be ultrasound findings associated with CDH that are not considered anomalies but may impact clinical management (20,21). Intrafetal fluid effusions are seen in 5% of left, and 29% of right CDH without any adverse effect (22). Approximately 15% of fetuses have ineffective gastrointestinal passage of swallowed amniotic fluid which can predispose to polyhydramnios and precipitation of preterm labor due to uterine overdistention. Overall, over 20% of CDH deliver prematurely with the severity of the hernia as a primary determinant of polyhydramnios and preterm birth (23).
A small left ventricle is commonly seen in left CDH and its decrease in dimensions is proportional to the degree of mediastinal shift (Figure 5). Normal fetal cardiovascular development requires preferential ductus venosus shunting of oxygenated umbilical venous blood to the left side of the heart ensuring adequate nutrient and oxygen supply of the coronary and cerebral circulations (24). Prenatal distortion of these flow dynamics and the associated decrease in oxygen and nutritional content of left ventricular preload as well as decrease in stroke volume from external compression has been linked to abnormal cardiac growth and myocardial dysfunction (25,26). The spectrum of left-sided heart disease may range from very mild dysfunction to aortic coarctation or even hypoplastic left heart (27).
Figure 5.

Third trimester ultrasound findings in left congenital diaphragmatic hernia. Prenatal ultrasound images obtained in a patient with left diaphragmatic hernia at 37 weeks’ gestation demonstrate a left diaphragmatic hernia with compression and relative hypoplasia of the left ventricle. RV, right ventricle; LV, left ventricle.
Prenatal stratification of CDH severity
Approximately 40% of prenatally diagnosed CDH cases are associated with other anomalies while 60% are isolated (7,28-31). Mortality of complex CDH is determined by the combined impact of pulmonary hypoplasia and associated physical or genetic abnormalities with expected survival as low as 15% (29). In isolated CDH determination of defect laterality, prenatal quantification of lung size, degree of liver herniation, and overall mediastinal displacement refines prediction of survival and morbidity. A number of prenatal imaging parameters have been described utilizing ultrasound as well as magnetic resonance imaging (MRI) to quantify mediastinal displacement and a standardized prenatal imaging protocol to capture these key variables has been proposed (8).
Postmortem pulmonary hypoplasia is defined as a lung weight to body weight ratio ≤0.015 before 28 weeks’ gestation and ≤0.012 thereafter (32,33). Recognizing the relationship between prenatal lung size and postnatal pulmonary hypoplasia, quantification of lung size is a crucial factor in prenatal imaging for patients with isolated CDH. In 1996, Metkus and coworkers described the ultrasound-derived two-dimensional lung area measurement of the contralateral lung at the level of the cardiac four-chamber view in left CDH (34). Using the head circumference as a reference value for gestational age, they determined that the lung-to-head ratio (LHR) in the presence of intrathoracic liver herniation predicted survival most reliably between 22–26 weeks’ gestation. However, since the head and lungs grow at different rates, predictive reliability of the LHR varies across gestation and predictive cutoffs must be adjusted for gestational epochs (35). This may also be further accentuated in the presence of fetal growth restriction leading to smaller head measurements. Jani and colleagues developed the observed to expected LHR (o/e LHR) where the lung area is expressed as a percentage of the expected measurement for gestational age (36,37). The o/e LHR predicts survival reliably across gestation and lung area measurement is most reproducible when measured using the trace method (38,39). Another technique that uses the ultrasound traced lung to thorax area ratio in the transverse plane (40). Since these ratios do not reflect all three lung dimensions ultrasound and MRI techniques were developed to estimate volumes of both the contralateral and ipsilateral lungs. Ultrasound and MRI derived measurements correlate more closely for the contralateral lung although mean volume estimated by ultrasound can be up to 25% smaller than those measured by MRI (41). MRI derived lung volumes have been generated from 17 weeks onward using dedicated postprocessing software (42) and correlate with postmortem lung volumes (43,44). These reference ranges can be utilized to interpret total fetal lung volume (TFLV), observed to expected TFLV (o/e TFLV), the percentage of predicted lung volume (45) (PPLV) or the lung volume to body weight ratio (46) obtained from patients with CDH (Table 3). Ultrasound visualization of the ipsilateral lung is inferior to MRI and therefore measurements correlate better for the contralateral lung. Inconsistencies between the two imaging modalities are further attributable to variable contribution of the ipsilateral lung to the total lung volume (47).
Table 3. Prenatal imaging parameters for prognostic assessment in congenital diaphragmatic hernia.
| Imaging parameter | Acronym | Imaging modality | Technique |
|---|---|---|---|
| Lung to head ratio | LHR | Ultrasound | The ratio of the contralateral lung area (at the level of the cardiac four chamber view) to the head circumference in millimeters. Three lung measurement techniques are described: |
| • Longest diameter method: product of longest diameter of the lung by its longest perpendicular diameter | |||
| • AP method: product of AP lung diameter at mid-clavicular line by perpendicular diameter at AP diameter midpoint | |||
| • Trace method: manual tracing of the lung circumference | |||
| Observed to expected lung to head ratio | o/e LHR | Ultrasound | Lung to head ratio of the contralateral lung is normalized for expected size for gestational age (38) |
| Total fetal lung volume | TFLV | MRI | Sum of three dimensionally measured lung volumes for both lungs |
| Observed to expected total fetal lung volume | o/e TFLV | MRI | The total fetal lung volume is expressed as a percentage of the expected total lung volume using gestational age references (45,46) |
| Percent of predicted lung volume | PPLV | MRI | Expressed as the measured thoracic volume minus measured mediastinal volume (47) |
| Lung volume to body weight ratio | LVBWR | Ultrasound | Lung volume measured by 3-dimensional ultrasound divided by fetal weight estimated by the Hadlock formula |
| Lung to thorax ratio | L/T | Ultrasound | Transverse lung and thoracic area measured at the level of the four-chamber view using the trace method |
| Liver herniation | Liver-up | Ultrasound | Qualitative determination of any liver portion above the diaphragm |
| Percent liver herniation | LiTR; %HL | MRI | Determination of the amount of intrathoracic liver herniation above the plane of the diaphragm expressed in two ways: |
| • LiTR: ratio of intrathoracic liver to total thoracic volume | |||
| • %HL: ratio of intrathoracic liver to total liver volume | |||
| Stomach grading | Cordier | Ultrasound | Position of the stomach assessed on the four‐chamber view of the heart |
| • Grade I: stomach not seen | |||
| • Grade II: next to the apex of the heart, with no structure in between the stomach and the sternum | |||
| • Grade III: stomach visualized along from the apex of the heart and abdominal structures anteriorly | |||
| • Grade IV: stomach with its larger portion posterior to the level of the AV heart valves | |||
| Basta | Ultrasound | In the true axial plane at the level of the four-chamber view of the heart | |
| • Grade I: stomach intraabdominal | |||
| • Grade II: anterior left chest contacting the anterior chest wall | |||
| • Grade III: mid-to posterior chest possibly contacting posterior chest wall | |||
| • Grade IV: retrocardiac with at least a portion of the stomach posterior to the left atrium of the heart within the right chest | |||
| Kitano | MRI | Grade I: stomach intraabdominal | |
| Grade II: left chest intrathoracic | |||
| Grade III: less than half of the stomach herniated into right chest | |||
| Grade IV: more than half of the stomach herniated into the right chest | |||
| Mediastinal shift angle | MSA | MRI | The angle between a sagittal reference line between the posterior surfaces of the vertebral body and the sternum with a line between the vertebral body and the right atrium |
LHR, lung-to-head ratio; AP, antero-posterior; o/e, observed to expected; TFLV, total fetal lung volume; MRI, magnetic resonance imaging; MSA, mediastinal shift angle; PPLV, percentage of predicted lung volume; LVBWR, lung volume to body weight ratio; L/T, lung to thorax; LiTR, liver in thorax ratio; %HL, percent liver herniation; AV, atrioventricular.
Liver herniation can be classified qualitatively as intrathoracic or intraabdominal or quantified by measuring the fraction of the total liver length extending above the diaphragm (48). A degree of operator dependence in ultrasound detection of liver herniation led to the development of stomach grading in left CDH. Because intrathoracic stomach herniation and position are dependent on the edge of the left diaphragmatic defect, presence of the stomach in the posterior or mid-thorax is an excellent predictor of liver herniation (Table 3) (49). Particularly grade 3 and 4 stomach herniation have a 100% positive predictive value for intrathoracic liver herniation and associated postnatal risk for adverse outcome (50-52). Accordingly, intrathoracic stomach position is considered an indirect outcome predictor for increased mortality which in left-CDH is related to the degree of liver herniation likely to be present under these circumstances (53). Another imaging technique to evaluate the magnitude of intrathoracic herniation of abdominal organs is mediastinal shift angle (MSA), which has been described for left and right CDH and can be performed using ultrasound or MRI (54-57) (Table 3). The prognostic use of the MSA is based on the hypothesis that contralateral lung volume in CDH is not only influenced by the presence of abdominal organs but also by displacement of the mediastinal axis in the opposite direction. The normal mediastinal angle of 15–20 degrees is increased significantly in fetuses with left and right CDH. An increased mediastinal angle correlates with the decrease in lung volume and predicted postnatal pulmonary hypertension, extracorporeal life support (ECLS) by membrane oxygenation need and length of hospital stay similarly to o/e LHR or o/e TFLV (54-57).
Research about prenatal risk stratification of CDH has established several important principles. Prenatally acquired pulmonary hypoplasia, which is proportional to the decrease in lung dimension and liver herniation, are the primary factors responsible for neonatal pulmonary morbidity and early mortality. The predictive accuracy of prenatal imaging is not interchangeable between left and right CDH, and the latter has to be considered a distinct disease entity requiring a dedicated prognostic approach. This may be due to several factors, including obligatory liver herniation in right-sided defects, the physiologically smaller volume of the contralateral lung which shares the left thoracic cavity with the heart, and less robust statistics available for this rare condition (58). Accordingly, right CDH should not be simply considered a variant of left-sided defects but a distinct disease entity requiring specific prenatal risk stratification models to direct management.
In left-sided isolated CDH, the observed to expected contralateral lung size and liver position are reliable predictors of neonatal respiratory function, anticipated care complexity, defect size, and the associated mortality and morbidity in survivors. With stringent adherence to reliable imaging standards, ultrasound measured o/e LHR, MRI measured o/e TFLV, and qualitative or quantitative liver herniation perform comparably in identifying patients with severe, moderate, and mild diaphragmatic hernia prenatally (59-63). The ultrasound-based assessment of left CDH the o/e LHR and liver position has been used to identify the highest risk patients to determine the potential benefits of fetal therapy.
Fetal therapy for CDH by fetoscopic tracheal occlusion
The potential value of fetal therapy for patients with left-sided isolated CDH was established following several key observations. The accuracy of prenatal assessment was able to identify patients with a high likelihood of lethal pulmonary hypoplasia as the primary contributor to early mortality (36,37). Compared to milder forms, survival of severe CDH did not significantly improve over a decade (11,64). There was experimental evidence that temporary prenatal tracheal occlusion partly reversed pulmonary hypoplasia (65). At the same time prenatal CDH repair, or open surgical techniques for tracheal occlusion did not improve outcomes and were associated with significant infant morbidity and mortality (66,67). In 2004, a new minimally invasive fetoscopic endoluminal tracheal occlusion (FETO) technique with the off-label use of a latex vascular occlusion balloon became available that circumvented the significant risks demonstrated for prior fetal surgical approaches (68).
Establishing the feasibility and safety of this technique led to two randomized trials (Tracheal Occlusion to Accelerate Lung Growth: TOTAL) evaluating survival for severe left CDH (o/e LHR <25% and liver up) and 6-month O2 dependence for moderate left CDH (o/e LHR 25–34.9% and 35–44.9% with liver herniation) (69-71) (Figure 6). In these trials, FETO was performed in up to 14 fetal therapy centers at 27+0 to 29+6 weeks for severe CDH and between 30+0 to 31+6 weeks for moderate CDH with planned balloon removal at 34+0 to 34+6 weeks for both groups (Video 1). Following balloon removal neonates delivered in over 40 tertiary care facilities with standardized neonatal care according to international guidelines (72). For severe left CDH, survival to discharge was significantly higher after FETO {40% compared to 15% with expectant management [relative risk (RR) =2.67; 95% confidence interval (CI): 1.22–6.11; P=0.009]}. In moderate left CDH, 6-month oxygen dependence was not significantly decreased by FETO. Other neonatal outcomes were comparable for severe- and moderate-risk CDH. The primary risk associated with FETO was preterm delivery which was highest with earlier balloon insertion (Table 4).
Figure 6.
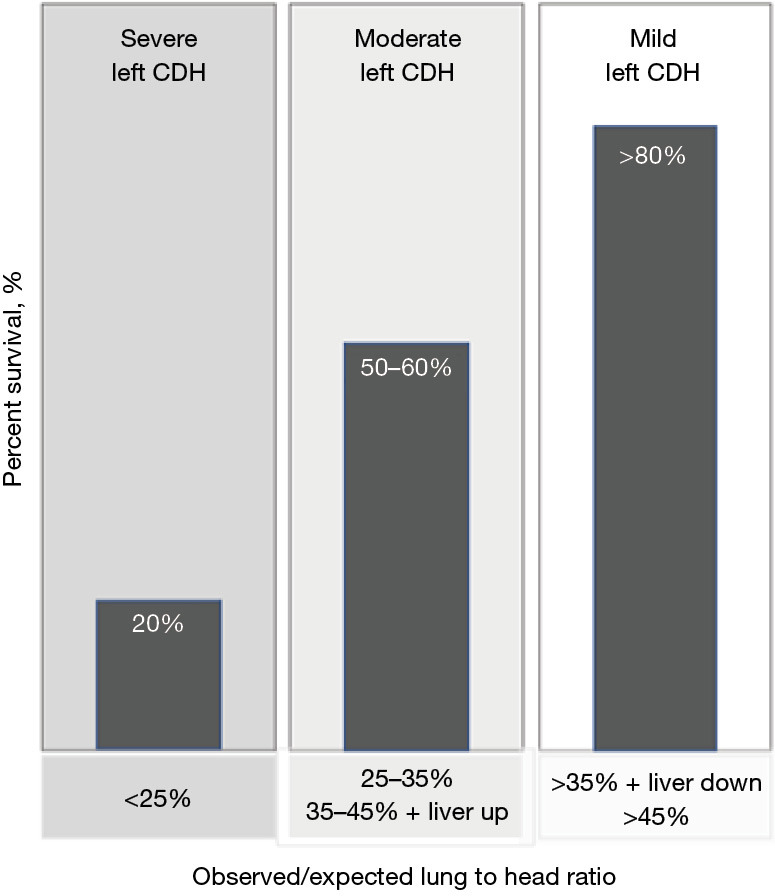
Left CDH severity assessed by observed to expected lung to head ratio and liver position. The figure illustrates the prenatal severity assessment of isolated left congenital diaphragmatic hernia based on the ultrasound measured observed lung to head ratio and liver position and the anticipated postnatal survival (37,38). This assessment was utilized to determine eligibility for fetoscopic tracheal occlusion in the randomized TOTAL trials (70,71). CDH, congenital diaphragmatic hernia.
Video 1.
Fetoscopic balloon insertion and removal.
Table 4. Pooled and individual trial outcomes in the TOTAL trials.
| Parameter | Severe CDH | Moderate CDH | Pooled data | |||||
|---|---|---|---|---|---|---|---|---|
| FETO (n=40) | Expectant (n=40) | FETO (n=98) | Expectant (n=98) | FETO (n=145) | Expectant (n=142) | |||
| Median o/e LHR | 21.0 (19.6, 23.3) | 21.0 (18.0, 23.0) | 30.9 (28.0, 34.0) | 31.0 (28.0, 34.5) | 28.0 (24.0, 33.0) | 28.0 (23.2, 33.4) | ||
| Liver herniation | 36 [90] | 35 [88] | 79 [81] | 78 [80] | 121 [83] | 116 [82] | ||
| Survival until NICU discharge | 16 [40] | 6 [15] | 62 [63] | 49 [50] | 79 [54] | 55 [39] | ||
| Without supplemental O2 at 6 months | 9 [22] | 3 [8] | 53 [54] | 43 [44] | 63 [43] | 46 [32] | ||
| Gestational age at delivery | 34.6 (32.2, 36.6) | 38.4 (36.5, 39.1) | 35.9 (34.3, 37.9) | 38.1 (37.0, 38.9) | 35.6 (33.8, 37.3) | 38.3 (37.0, 39.0) | ||
| Delivery <34 weeks | 16/40 [40] | 0/38 [0] | 19/97 [20] | 7/94 [7] | 35/137 [26] | 7/132 [5] | ||
| Neonatal repair | 20/38 [53] | 14/38 [37] | 81/97 [84] | 70/94 [74] | 91/135 [67] | 84/108 [78] | ||
| Patch repair | 18/20 [90] | 11/14 [79] | 67/81 [83] | 50/70 [71] | 85/101 [84] | 61/84 [73] | ||
| Repair day of life | 2 (2, 5) | 7 (4, 9) | 3 (2, 6) | 3 (2, 5) | – | – | ||
| ECLS use | 2/38 [5] | 11/38 [29] | 20/97 [21] | 19/94 [20] | 22/135 [16] | 30/132 [23] | ||
Data are n [%], n/N [%] or median (interquartile range). CDH, congenital diaphragmatic hernia; FETO, fetoscopic tracheal occlusion; o/e LHR, observed to expected lung to head ratio; NICU, neonatal intensive care unit; ECLS, extracorporeal life support.
A secondary analysis of pooled data from the severe and moderate TOTAL trials suggested a survival benefit for severe and moderate CDH with FETO associated prematurity as the primary factor limiting this benefit (73). However, low rates of neonatal repair, ECLS utilization and survival with expectant management in the TOTAL trials raised questions about the generalizability of FETO benefits to pediatric CDH programs with better outcomes with traditional management (4,74,75). Other investigators have also reported repair of all neonates with up to 50% ECLS utilization achieving over 80% survival at discharge without fetal therapy (76-78).
A recent meta-analysis found that the survival benefit of FETO is most apparent in care settings where the fetal and neonatal therapy programs operate in an integrated institutional care setting (9). Given the higher background survival rates for CDH achieved at pediatric high volume centers the benefit of FETO may extend beyond a measurable impact on survival but also decrease respiratory (79), gastrointestinal as well as neurodevelopmental morbidity (80). Based on the current level I evidence FETO should be strongly considered for isolated severe left sided CDH, recognizing that the anticipated survival benefit may vary based on care setting and integration with the pediatric management expertise. In the United States, FETO can currently only be offered under an investigational device exemption with the oversight of the Food and Drug Administration. While immediate benefits of FETO may become more apparent when associated prematurity risks can be mitigated, potential long-term benefits require further study (79).
Prenatal and delivery management
Optimal CDH care is initiated prenatally. As part of the prenatal severity assessment, parents have the opportunity to gather in-depth information on postnatal management strategies and expected outcomes from a range of pediatric experts that form the CDH management team. Prenatal surveillance of fetal growth and development is important. For patients that experience polyhydramnios and preterm labor, amnioreduction and medical management can prevent premature delivery (81). Obstetric management strategies that decrease the risk for membrane rupture and preterm birth after FETO have been described, thereby increasing the likelihood for a planned term delivery (78).
In preparation for delivery maternal administration of a course of betamethasone or dexamethasone to enhance fetal lung maturity should be considered in alignment with current society recommendations (82). Because multiple maternal steroid courses are associated with fetal and neonatal adverse effects, prenatal administration is limited to two courses in the presence of continued risks for preterm birth. Patients undergoing FETO and subsequent balloon removal are at risk for preterm birth in the context of these procedures. Accordingly, two courses of maternal betamethasone should be considered concurrently with these two procedures (70,71,78,79). In patients managed expectantly, the potential risks and benefits of steroids in the setting of anticipated neonatal respiratory insufficiency should be discussed with the parents. Based on the benefits of steroids in normal infants at increased risk for pulmonary immaturity (83), a single course of maternal steroid administration can be considered in the week prior to delivery irrespective of gestational age.
Planned delivery at 39 weeks is optimal because it offers control over the delivery setting. Delivery route (i.e., vaginal versus cesarean delivery) should be guided by obstetric factors and ideally be scheduled to allow an early week daytime delivery. This facilitates the availability of dedicated staffing and the ability for an early scheduled CDH repair within a one-week window. Obstetric management should aim for delivery of a neonate with a normal cord arterial pH value to minimize the depth of respiratory acidemia during neonatal transition. Key elements of a CDH delivery are the swift establishment of a reliable airway to maximize the likelihood for control of ventilation pressures after birth, and gastric decompression to maximize lung excursion. This is best accomplished by pre-delivery coordination planning with the pediatric management team.
Principles of neonatal management of CDH
Neonatal CDH care focuses on three overlapping domains: pulmonary hypoplasia, pulmonary hypertension, and biventricular cardiac dysfunction (27,72,84). These patients present with issues inherent to their underlying disease as well as those attributable to fetal-to-neonatal transition and when applicable, prematurity. Therefore, the neonatologist must attempt to identify which proportion of the patient’s symptoms are transitional and temporary, versus those inherent to the underlying structural disease. Initial management of CDH patients must begin with ventilator practices that optimize ventilation and V/Q matching, allowing postnatal pulmonary vascular resistance (PVR) to drop as naturally as possible. Without optimal ventilation, these infants suffer from acidosis, high PVR, and exacerbated cardiac dysfunction, increasing the chances that their care may require ECLS. Oxygen should be offered judiciously, as excessive exposure may attenuate the subsequent efficacy of nitric oxide, should it become necessary later in the infant’s care (85,86).
Ventilation strategies and care of asymmetrically hypoplastic lungs in CDH
Caring for asymmetric lungs requires clinicians to find the delicate balance between atelectasis of the larger lung and hyperinflation of the smaller lung. Hyperinflation is likely a more frequent issue than atelectasis, prevented by the use of lower mean airway pressures that better approximate the functional residual capacity (FRC) of hypoplastic lungs. Concerns for hyperinflation naturally include parenchymal injury and air leak, but the most common finding is mechanical compression of the surrounding alveolar vasculature, with cumulative compression leading to an iatrogenic increase in PVR. Furthermore, severe hyperinflation and the resulting increased intrathoracic pressure is well known to reduce pulmonary venous return, in this case to an already vulnerable heart. To further evaluate inflation with regards to ventilator choice, the VICI trial compared conventional ventilation with high-frequency oscillation as the initial mode of ventilation, finding that infants who started their course on an oscillator remained intubated longer and were more likely to use inhaled nitric oxide and ECLS (87). Therefore, it is generally recommended that infants start their journey on conventional ventilation.
Pulmonary hypoplasia is variable and prenatally predicted using o/e LHR, which is helpful in suggesting postnatal pulmonary expectations but not prescriptive for outcome. Postnatal pre-operative management should focus on ventilating the larger lung well, to an expansion of 8–9 ribs on chest radiograph. The smaller lung ipsilateral to the hernia should not be visible on pre-operative radiograph. Visible inflation of the ipsilateral lung indicates that the contralateral lung is no longer the path of least resistance for inspiratory airflow, having already exceeded maximum inflation capacity. Low positive end-expiratory pressures respect the low FRC inherent to hypoplastic lungs (27,72) and a volume-based inspiratory strategy allows instantaneous pressure titration to dynamic postnatal compliance. Infants who undergo successful FETO treatment with uneventful balloon removal have greater pulmonary capacity than otherwise expected, allowing ease with ventilation (88). Most centers do not alter their a priori ventilator strategy specific to a history of FETO, but rather to the patient’s presentation and need for support.
The multifactorial pulmonary hypertension of CDH
The etiology of pulmonary hypertension is multifactorial and dynamic in infants with CDH, with the overall cumulative pulmonary vascular resistance declining with time during the neonatal period (89,90). The first contributor derives from normal transitional physiology. Pulmonary vascular pressures are high in all infants at the time of birth and remain high until appropriately ventilated. In normal infants, a cry is usually sufficient, and V/Q matching leads to a drop in PVR. This process is delayed in specific situations such as prematurity, cesarean birth, maternal or fetal illness, or cardiopulmonary disease like CDH. Transitional pulmonary hypertension should decline with time once adequate ventilation is established. The second contributor in infants with CDH is pulmonary vascular hypoplasia, potentially limiting the amount of pulmonary blood flow even after pressures have significantly declined. Third, infants with cardiopulmonary disease undergo vascular remodeling that starts antenatally in an effort to compensate for the physiologic inadequacies of their structural disease (91,92). Signal pathway alterations lead to extensive muscularization that may reduce responsiveness to pulmonary vasodilators (93). PVR also may be actively increased by iatrogenic circumstances, such as hyperinflation, atelectasis, acidosis, hypothermia, inadequate sedation, or certain medications (94).
Active management of PVR essentially revolves around allowing fetal-to-neonatal transition to occur as smoothly as possible, while avoiding iatrogenic factors that increase PVR. Keeping the primary focus on gentle ventilation will allow PVR to drop smoothly and naturally, utilizing the infant’s own pulmonary potential. Until pulmonary vascular pressures have dropped enough to welcome the entirety of right ventricular output, prostaglandin E1 may be used to keep the ductus arteriosus open and detour some deoxygenated blood to the descending aorta (95,96). Clinicians must be willing to accept low post-ductal PO2 or oxygen saturations, as these are necessary consequences of using the ductus arteriosus as a pop-off for the right ventricle. Saturations, PO2, A-a gradients, and lactate levels used for judging progress and adequate tissue perfusion should all come from pre-ductal sources.
When ventilation is not enough to reduce pulmonary hypertension, clinicians may consider using inhaled pulmonary vasodilators such as nitric oxide for further vasodilation (89). Drug delivery is contingent on optimal ventilation; however, efficacy does not necessarily reduce the incidence of ECLS or mortality (97,98). Clinicians must also be wary of the adverse effects of pulmonary vasodilators, as efforts to increase pulmonary blood flow may overwhelm the underdeveloped left heart. Signs of such pathology include pulmonary edema exacerbated cardiac dysfunction (99). Providers may choose to proactively treat with inotropes and/or diuretics to preempt these side effects.
Management of cardiac dysfunction in CDH
The anatomic distortion of CDH has critical effects on the fetal heart, leading to maldevelopment and dysfunction, which appears to be proportional to defect size (89,100-102). Infants with CDH who do not undergo FETO tend to have pulmonary and cardiac disease that are parallel in prenatal risk severity, whereas those who undergo FETO often demonstrate lung disease that is much milder than their cardiac findings.
Cardiac dysfunction in CDH is bilateral, consisting of an overworked right ventricle and an underdeveloped left ventricle. Medical therapy consists of relieving unnecessary afterload to the right side, and unnecessary preload to the left side. Prostaglandin E1, as mentioned before, allows fetal circulation to continue (95,96). Inotropic drugs such as epinephrine or milrinone may also be indicated. Early echocardiograms should focus on function and direction of ductus and foramen ovale shunting to help guide therapy.
Extracorporeal life support
Despite advances in prenatal fetal interventions and improved understanding of transition physiology and early postnatal therapies, ECLS is employed in 25–30% of CDH babies as a rescue means of support (103). Prior to the availability of fetal therapy, ECLS showed the greatest survival benefit in patients with the highest predicted mortality (104). Various predictive models exist encompassing prenatal and postnatal factors estimating the fetus’ or neonate’s risk for need of ECLS (105,106). Contemporary data reveal excess mortality with ECLS use in neonates with low and moderate risk CDH, but continued survival benefit in those with high-risk disease (107). The ECLS service is a valuable and important member of the multidisciplinary CDH team, and prenatal counseling discussions in higher risk cases can provide valuable information regarding the specifics of ECLS and outcomes. Additionally, as a resource intensive technology, awareness of impending CDH deliveries is helpful for ECLS resource planning. Higher volume centers with a well-integrated approach seamlessly encompassing the fetal, neonatal, surgical, and ECLS services; volume and experience portend a survival benefit (107).
Standardized criteria for ECLS cannulation should be used with multidisciplinary agreement on maximum and time-limited trials of medical support prior to transitioning to extracorporeal support. Both veno-venous (VV) support via a dual lumen cannula and veno-arterial (VA) support via two separate cannulas are options for ECLS cannulation of the neonate with CDH and are associated with similar outcomes (108). For the neonate with pure respiratory failure or respiratory failure with borderline cardiac failure, VV-ECLS may be preferred since it preserves the carotid artery and may have inotropic effects secondary to the delivery of oxygenated blood to the coronary vessels. Conversion to VA-ECLS is usually an option should the cardiac failure progress. However, primary VA cannulation remains the most popular approach for initiating ECLS support in CDH (>80% per the Extracorporeal Life Support Organization registry) due to a variety of factors, including institutional preference, more reliable cannula flows, and the ability to directly support cardiac dysfunction until the patient can be medically optimized. VV-ECLS is also not possible in lower birthweight neonates due to the small size of the right internal jugular vein.
All CDH patients on ECLS support require systemic anticoagulation to maintain the circuit and to prevent thromboembolic complications. Nevertheless, cannulation onto ECLS need not delay surgical repair of the hernia defect once the neonate is adequately supported. Registry data from the CDH Study Group suggested that early repair on ECLS (usually defined as repair within 24–48 hours of cannulation) is superior to both delayed repair on ECLS and repair after decannulation from ECLS (109). While major bleeding was historically a major barrier to safe operative repair on ECLS, the risk can now be substantially mitigated through peri-operative protocols that emphasize meticulous surgical technique and anti-fibrinolytic infusions, such as aminocaproic acid or tranexamic acid (110-112).
A standardized approach to ECLS care, including ongoing use of gentle, lung protective ventilation both during the duration of the ECLS run and in the transition from weaning to decannulating from ECLS support, is paramount for the care of CDH neonates. The balance lies in providing adequate support while limiting complications of ECLS such as neurologic sequelae, bleeding, and infection and avoiding iatrogenic barotrauma, such as aggressive ventilator use in an attempt to shorten the duration of ECLS. Based on recent CDH Study Group data, the mean duration on ECLS in CDH neonates is 11 days (103).
In CDH fetuses undergoing FETO, there was a higher usage of ECLS in integrated pre- and postnatal care programs associated with improved overall survival (9). In addition to the same institution care, integrated programs are more likely to have protocolized postnatal care algorithms. Integrated programs had higher rates of hospital survival at 70.7% (versus 45.7% at nonintegrated institutions); in multilevel regression analysis, access to an ECLS program was a survival predictor, although this may have been a surrogate for a more advanced level of neonatal care. Single center data reveal that in fetuses with poor response to FETO, defined as post FETO o/e TLV increase <10%, there was higher ECLS utilization and lower survival when compared with fetuses that had a more robust response to fetal therapy after controlling for gestational age at delivery and defect severity (113). Even with more widespread experience with FETO, ECLS may still be required in those with the more severe disease phenotype, and robust involvement of the ECLS team ensures readiness to support these babies.
Intraoperative and postoperative management
Although emergent repair of the CDH defect at birth was initially advocated to reduce mediastinal mass effect, this practice has been abandoned to allow for physiologic stability under medical management during the initial 24–48 hours of life (114,115). CDH neonates without evidence of liver herniation and who can be adequately supported on minimal ventilator settings are excellent candidates for minimally invasive thoracoscopic repair using 3 mm ports. In those with moderate or severe cardiopulmonary disease, transabdominal repair through a subcostal incision continues to be the favored surgical approach (85% of cases) due to lower rates of recurrent hernia, among other reasons (116). For larger hernia defects (some type B and all type Cs and Ds), a synthetic permanent mesh [(polytetrafluoroethylene (PTFE)] is the most popular material used to close the defect. While the mean age at repair is 7 days based on recent data from the CDH Study Group, it was day 4 for infants where ECLS was not utilized (117,118). In this second group, later repair was associated with delayed full oral feeding and increased need for tube feeds as well as increased post-repair ventilation requirement and supplemental oxygen at discharge (118).
After surgical repair of the diaphragmatic defect, care focuses on weaning off cardiorespiratory support and optimizing nutrition. Pulmonary hypertension crises, often triggered by inadequate pain control and sedation, can also manifest in the post-operative period. Therefore, coordination of sedation and weaning of narcotics are important in facilitating ongoing stability on ventilatory support. The transition to non-invasive modes of ventilation (such as continuous positive airway pressure and high-flow oxygen) is often required. Based on recent CDH Study Group data, mean ventilator duration was 17 days, and approximately half required some supplemental oxygen support at 30 days of life. Fortunately, tracheostomy is relatively uncommon (4%) and is employed in the most severe cases in which there were multiple failures to extubate or to wean from positive pressure ventilation (119).
Infants with severe CDH are at increased risk for high nutritional morbidity. Some patients have a prolonged ileus and are hypermetabolic, requiring parenteral nutrition supplementation to meet the higher basal metabolic demands suggested by normative references for age in critical illness (120). The prevalence of significant malnutrition at discharge has been estimated to be 26% (121). More than 30% of infants are discharged on supplemental tube feeds, and over 10% received surgical feeding tube procedures (e.g., gastrostomy tubes) to ensure caloric needs owing to poor oral feeding (11). Patients at high risk for severe gastroesophageal reflux disease (GERD) include those with large hernia defects, FETO survivors, and those with liver herniation or substantial gastric distortion on prenatal imaging (122). CDH survivors with ongoing severe GERD that is refractory to standard medical therapy might be candidates for anti-reflux procedures. The operative intervention rate for fundoplication in CDH has been reported to be as high as 20–33% (123). Although overall postnatal survival rates have only modestly increased from 69% to 74% over the past two decades, this represents a statistically significant improvement when risk-adjusted for disease severity in a recent multicenter study (11).
Long-term care
Increased survivorship of CDH infants has prompted a shift in focus toward the management of chronic morbidity affecting many organ systems in a unique cohort of patients with complex medical and surgical needs that can extend into adolescence and beyond. For these reasons, structured and long-term health surveillance of CDH patients, including those who had fetal therapy, within a multidisciplinary clinic setting can be helpful for affected children and their families (80,124). Surveillance guidelines developed in collaboration with the American Academy of Pediatrics tend to be more useful for CDH patients with a moderate or severe disease phenotype (125).
Nutritional status affects approximately 25% of survivors but tends to improve during the first year of life (126). Hernia recurrence is a concern in CDH, particularly in those with larger defects within the first three years of life. Symptomatic recurrence often manifests with bowel obstruction or respiratory distress. However, many recurrent cases are asymptomatic, prompting the need for serial surveillance by chest radiography (127). Postnatal risk factors for long-term pulmonary morbidity include the need for respiratory support at 30 days, which predicts ongoing morbidity at 1 and 5 years, the need for supplemental oxygen, and a subsequent diagnosis of asthma (128). More recently, FETO-related tracheal complications, including tracheomalacia and tracheomegaly, have been reported in a small fraction of children (129). Though CDH is inherently associated with pulmonary hypertension related to vascular remodeling, only a subset will proceed to experience clinically significant chronic pulmonary vascular disease. Infants who require pulmonary vasodilator therapy during hospitalization, are discharged on pulmonary vasodilators, or have significant echo findings have been found more likely to have chronic disease (130,131). Evidence of pulmonary hypertension is observed in 10–20% of patients at discharge, but most survivors tend to experience clinical and echocardiographic resolution over time (132). Prenatal risk factors for persistent pulmonary hypertension include liver herniation and low o/e LHR or TFLV. At least one-quarter of CDH survivors will have some form of neurodevelopmental impairment, which ranges from motor or sensory deficits to cognitive, language, and behavioral challenges (133). In those who undergo fetal therapy, there is now emerging data to suggest that neurodevelopmental outcomes at 24 months of age are improved compared to those with severe CDH who were managed expectantly in the prenatal period.
Conclusions
Advances in fetal and pediatric care for patients with CDH can optimize outcome, especially if applied in an integrated care model utilizing the expertise of all involved specialties. Early prenatal diagnosis and prognostic assessment provides parents the opportunity to consider the spectrum of management options. For patients eligible for FETO, higher infant survival can be anticipated, particularly if risks of preterm birth can be mitigated. For all pregnancies with fetal CDH, carefully planned delivery timing and circumstances facilitates transition to early neonatal intensive care. Use of lung-protective ventilation strategies and a balanced cardiovascular management approach that harnesses the benefits of physiologic neonatal transition and minimizes iatrogenic exacerbation of pulmonary hypertension and cardiac dysfunction. For patients with severe disease, ECLS can be lifesaving and tailored to address the primary contributor to deterioration. ECLS is of greatest benefit when lung protective ventilation strategy and principles of cardiovascular management are maintained. Timing of surgical correction is directed by the severity of the condition but is typically possible in the first week of life. Early surgical repair after initial neonatal stabilization confers benefits for respiratory function and achievement of feeding goals. As more infants with CDH survive, a long-term multidisciplinary care model is essential to address ongoing cardiorespiratory, gastrointestinal, and developmental care needs. For the subset of infants eligible for fetal therapy, management in such a multidisciplinary care model suggests they may have additional benefits that extend beyond increased survival for patients with severe disease.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Peer Review File: Available at https://tp.amegroups.com/article/view/10.21037/tp-23-602/prf
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://tp.amegroups.com/article/view/10.21037/tp-23-602/coif). A.A.B. receives royalties from UpToDate and in-kind donation of fetoscopy sheath equipment from Karl Storz. J.L.M. is supported by Janssen Research and Development, LLC; serves as a Data Safety Monitoring Board member for FETO, and on the Executive Board of the North American Fetal Therapy Network. The other authors have no conflicts of interest to declare.
References
- 1.Lally KP. Congenital diaphragmatic hernia. Curr Opin Pediatr 2002;14:486-90. 10.1097/00008480-200208000-00022 [DOI] [PubMed] [Google Scholar]
- 2.Sola JE, Bronson SN, Cheung MC, et al. Survival disparities in newborns with congenital diaphragmatic hernia: a national perspective. J Pediatr Surg 2010;45:1336-42. 10.1016/j.jpedsurg.2010.02.105 [DOI] [PubMed] [Google Scholar]
- 3.Gray BW, Fifer CG, Hirsch JC, et al. Contemporary outcomes in infants with congenital heart disease and bochdalek diaphragmatic hernia. Ann Thorac Surg 2013;95:929-34. 10.1016/j.athoracsur.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 4.Snoek KG, Greenough A, van Rosmalen J, et al. Congenital Diaphragmatic Hernia: 10-Year Evaluation of Survival, Extracorporeal Membrane Oxygenation, and Foetoscopic Endotracheal Occlusion in Four High-Volume Centres. Neonatology 2018;113:63-8. 10.1159/000480451 [DOI] [PubMed] [Google Scholar]
- 5.Harting MT, Lally KP. The Congenital Diaphragmatic Hernia Study Group registry update. Semin Fetal Neonatal Med 2014;19:370-5. 10.1016/j.siny.2014.09.004 [DOI] [PubMed] [Google Scholar]
- 6.Burgos CM, Frenckner B, Luco M, et al. Prenatally versus postnatally diagnosed congenital diaphragmatic hernia – Side, stage, and outcome. J Pediatr Surg 2019;54:651-5. 10.1016/j.jpedsurg.2018.04.008 [DOI] [PubMed] [Google Scholar]
- 7.Gallot D, Boda C, Ughetto S, et al. Prenatal detection and outcome of congenital diaphragmatic hernia: a French registry-based study. Ultrasound Obstet Gynecol 2007;29:276-83. 10.1002/uog.3863 [DOI] [PubMed] [Google Scholar]
- 8.Russo FM, Cordier AG, De Catte L, et al. Proposal for standardized prenatal ultrasound assessment of the fetus with congenital diaphragmatic hernia by the European reference network on rare inherited and congenital anomalies (ERNICA). Prenat Diagn 2018;38:629-37. 10.1002/pd.5297 [DOI] [PubMed] [Google Scholar]
- 9.Sferra SR, Miller JL, Cortes M S, et al. Postnatal care setting and survival after fetoscopic tracheal occlusion for severe congenital diaphragmatic hernia: A systematic review and meta-analysis. J Pediatr Surg 2022;57:819-25. 10.1016/j.jpedsurg.2022.05.011 [DOI] [PubMed] [Google Scholar]
- 10.Lally KP, Lasky RE, Lally PA, et al. Standardized reporting for congenital diaphragmatic hernia–an international consensus. J Pediatr Surg 2013;48:2408-15. 10.1016/j.jpedsurg.2013.08.014 [DOI] [PubMed] [Google Scholar]
- 11.Putnam LR, Harting MT, Tsao K, et al. Congenital Diaphragmatic Hernia Defect Size and Infant Morbidity at Discharge. Pediatrics 2016;138:e20162043. 10.1542/peds.2016-2043 [DOI] [PubMed] [Google Scholar]
- 12.Cordier AG, Cannie MM, Guilbaud L, et al. Stomach position versus liver-to-thoracic volume ratio in left-sided congenital diaphragmatic hernia. J Matern Fetal Neonatal Med 2015;28:190-5. 10.3109/14767058.2014.906576 [DOI] [PubMed] [Google Scholar]
- 13.Syngelaki A, Hammami A, Bower S, et al. Diagnosis of fetal non-chromosomal abnormalities on routine ultrasound examination at 11-13 weeks’ gestation. Ultrasound Obstet Gynecol 2019;54:468-76. 10.1002/uog.20844 [DOI] [PubMed] [Google Scholar]
- 14.Sweed Y, Puri P. Congenital diaphragmatic hernia: influence of associated malformations on survival. Arch Dis Child 1993;69:68-70. 10.1136/adc.69.1_Spec_No.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin AE, Pober BR, Adatia I. Congenital diaphragmatic hernia and associated cardiovascular malformations: type, frequency, and impact on management. Am J Med Genet C Semin Med Genet 2007;145C:201-16. 10.1002/ajmg.c.30131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slavotinek AM. The genetics of common disorders – congenital diaphragmatic hernia. Eur J Med Genet 2014;57:418-23. 10.1016/j.ejmg.2014.04.012 [DOI] [PubMed] [Google Scholar]
- 17.Holder AM, Klaassens M, Tibboel D, et al. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet 2007;80:825-45. 10.1086/513442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lurie IW. Where to look for the genes related to diaphragmatic hernia? Genet Couns 2003;14:75-93. [PubMed] [Google Scholar]
- 19.Yu L, Wynn J, Ma L, et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet 2012;49:650-9. 10.1136/jmedgenet-2012-101135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thébaud B, Azancot A, de Lagausie P, et al. Congenital diaphragmatic hernia: antenatal prognostic factors. Does cardiac ventricular disproportion in utero predict outcome and pulmonary hypoplasia? Intensive Care Med 1997;23:10062-9. 10.1007/s001340050457 [DOI] [PubMed] [Google Scholar]
- 21.Byrne FA, Keller RL, Meadows J, et al. Severe left diaphragmatic hernia limits size of fetal left heart more than does right diaphragmatic hernia. Ultrasound Obstet Gynecol 2015;46:688-94. 10.1002/uog.14790 [DOI] [PubMed] [Google Scholar]
- 22.Van Mieghem T, Cruz-Martinez R, Allegaert K, et al. Outcome of fetuses with congenital diaphragmatic hernia and associated intrafetal fluid effusions managed in the era of fetal surgery. Ultrasound Obstet Gynecol 2012;39:50-5. 10.1002/uog.10097 [DOI] [PubMed] [Google Scholar]
- 23.Barbosa BML, Rodrigues AS, Carvalho MHB, et al. Spontaneous prematurity in fetuses with congenital diaphragmatic hernia: a retrospective cohort study about prenatal predictive factors. BMC Pregnancy Childbirth 2018;18:27. 10.1186/s12884-017-1652-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baschat AA. The fetal circulation and essential organs-a new twist to an old tale. Ultrasound Obstet Gynecol 2006;27:349-54. 10.1002/uog.2762 [DOI] [PubMed] [Google Scholar]
- 25.Gupta VS, Popp EC, Ebanks AH, et al. Isolated aortic arch anomalies are associated with defect severity and outcome in patients with congenital diaphragmatic hernia. Pediatr Surg Int 2022;39:69. 10.1007/s00383-022-05354-1 [DOI] [PubMed] [Google Scholar]
- 26.Allan LD, Sharland G, Tynan MJ. The natural history of the hypoplastic left heart syndrome. Int J Cardiol 1989;25:341-3. 10.1016/0167-5273(89)90226-X [DOI] [PubMed] [Google Scholar]
- 27.Lakshminrusimha S, Keszler M, Yoder BA. Care of the infant with congenital diaphragmatic hernia. In: Keszler M, Gautham KS. editors. Goldsmith’s Assisted Ventilation of the Neonate. Seventh Edition. Elsevier; 2022: 446-57.e2. [Google Scholar]
- 28.Skari H, Bjornland K, Haugen G, et al. Congenital diaphragmatic hernia: a meta-analysis of mortality factors. J Pediatr Surg 2000;35:1187-97. 10.1053/jpsu.2000.8725 [DOI] [PubMed] [Google Scholar]
- 29.Witters I, Legius E, Moerman P, et al. Associated malformations and chromosomal anomalies in 42 cases of prenatally diagnosed diaphragmatic hernia. Am J Med Genet 2001;103:278-82. 10.1002/ajmg.1564 [DOI] [PubMed] [Google Scholar]
- 30.Stege G, Fenton A, Jaffray B. Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics 2003;112:532-5. 10.1542/peds.112.3.532 [DOI] [PubMed] [Google Scholar]
- 31.Colvin J, Bower C, Dickinson JE, et al. Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics 2005;116:e356-63. 10.1542/peds.2004-2845 [DOI] [PubMed] [Google Scholar]
- 32.Askenazi SS, Perlman M. Pulmonary hypoplasia: lung weight and radial alveolar count as criteria of diagnosis. Arch Dis Child 1979;54:614-8. 10.1136/adc.54.8.614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wigglesworth JS, Desai R, Guerrini P. Fetal lung hypoplasia: biochemical and structural variations and their possible significance. Arch Dis Child 1981;56:606-15. 10.1136/adc.56.8.606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metkus AP, Filly RA, Stringer MD, et al. Sonographic predictors of survival in fetal diaphragmatic hernia. J Pediatr Surg 1996;31:148-51; discussion 151-2. 10.1016/S0022-3468(96)90338-3 [DOI] [PubMed] [Google Scholar]
- 35.Yang SH, Nobuhara KK, Keller RL, et al. Reliability of the lung-to-head ratio as a predictor of outcome in fetuses with isolated left congenital diaphragmatic hernia at gestation outside 24-26 weeks. Am J Obstet Gynecol 2007;197:30.e1-7. 10.1016/j.ajog.2007.01.016 [DOI] [PubMed] [Google Scholar]
- 36.Peralta CF, Cavoretto P, Csapo B, et al. Assessment of lung area in normal fetuses at 12-32 weeks. Ultrasound Obstet Gynecol 2005;26:718-24. 10.1002/uog.2651 [DOI] [PubMed] [Google Scholar]
- 37.Jani J, Nicolaides KH, Keller RL, et al. Observed to expected lung area to head circumference ratio in the prediction of survival in fetuses with isolated diaphragmatic hernia. Ultrasound Obstet Gynecol 2007;30:67-71. 10.1002/uog.4052 [DOI] [PubMed] [Google Scholar]
- 38.Jani J, Nicolaides KH, Benachi A, et al. Timing of lung size assessment in the prediction of survival in fetuses with diaphragmatic hernia. Ultrasound Obstet Gynecol 2008;31:37-40. 10.1002/uog.5198 [DOI] [PubMed] [Google Scholar]
- 39.Abbasi N, Ryan G, Johnson A, et al. Reproducibility of fetal lung-to-head ratio in left diaphragmatic hernia across the North American Fetal Therapy Network (NAFTNet). Prenat Diagn 2019;39:188-94. 10.1002/pd.5413 [DOI] [PubMed] [Google Scholar]
- 40.Usui N, Kitano Y, Okuyama H, et al. Reliability of the lung to thorax transverse area ratio as a predictive parameter in fetuses with congenital diaphragmatic hernia. Pediatr Surg Int 2011;27:39-45. 10.1007/s00383-010-2725-z [DOI] [PubMed] [Google Scholar]
- 41.Jani JC, Cannie M, Peralta CF, et al. Lung volumes in fetuses with congenital diaphragmatic hernia: comparison of 3D US and MR imaging assessments. Radiology 2007;244:575-82. 10.1148/radiol.2442061158 [DOI] [PubMed] [Google Scholar]
- 42.Deshmukh S, Rubesova E, Barth R. MR assessment of normal fetal lung volumes: a literature review. AJR Am J Roentgenol 2010;194:W212-7. 10.2214/AJR.09.2469 [DOI] [PubMed] [Google Scholar]
- 43.Rypens F, Metens T, Rocourt N, et al. Fetal lung volume: estimation at MR imaging-initial results. Radiology 2001;219:236-41. 10.1148/radiology.219.1.r01ap18236 [DOI] [PubMed] [Google Scholar]
- 44.Ward VL, Nishino M, Hatabu H, et al. Fetal lung volume measurements: determination with MR imaging–effect of various factors. Radiology 2006;240:187-93. 10.1148/radiol.2393050583 [DOI] [PubMed] [Google Scholar]
- 45.Barnewolt CE, Kunisaki SM, Fauza DO, et al. Percent predicted lung volumes as measured on fetal magnetic resonance imaging: a useful biometric parameter for risk stratification in congenital diaphragmatic hernia. J Pediatr Surg 2007;42:193-7. 10.1016/j.jpedsurg.2006.09.018 [DOI] [PubMed] [Google Scholar]
- 46.Ruano R, Aubry MC, Dumez Y, et al. Predicting neonatal deaths and pulmonary hypoplasia in isolated congenital diaphragmatic hernia using the sonographic fetal lung volume-body weight ratio. AJR Am J Roentgenol 2008;190:1216-9. 10.2214/AJR.07.3078 [DOI] [PubMed] [Google Scholar]
- 47.Jani J, Cannie M, Done E, et al. Relationship between lung area at ultrasound examination and lung volume assessment with magnetic resonance imaging in isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 2007;30:855-60. 10.1002/uog.5168 [DOI] [PubMed] [Google Scholar]
- 48.Victoria T, Bebbington MW, Danzer E, et al. Use of magnetic resonance imaging in prenatal prognosis of the fetus with isolated left congenital diaphragmatic hernia. Prenat Diagn 2012;32:715-23. 10.1002/pd.3890 [DOI] [PubMed] [Google Scholar]
- 49.Bootstaylor BS, Filly RA, Harrison MR, et al. Prenatal sonographic predictors of liver herniation in congenital diaphragmatic hernia. J Ultrasound Med 1995;14:515-20. 10.7863/jum.1995.14.7.515 [DOI] [PubMed] [Google Scholar]
- 50.Kitano Y, Okuyama H, Saito M, et al. Re-evaluation of stomach position as a simple prognostic factor in fetal left congenital diaphragmatic hernia: a multicenter survey in Japan. Ultrasound Obstet Gynecol 2011;37:277-82. 10.1002/uog.8892 [DOI] [PubMed] [Google Scholar]
- 51.Cordier AG, Jani JC, Cannie MM, et al. Stomach position in prediction of survival in left-sided congenital diaphragmatic hernia with or without fetoscopic endoluminal tracheal occlusion. Ultrasound Obstet Gynecol 2015;46:155-61. 10.1002/uog.14759 [DOI] [PubMed] [Google Scholar]
- 52.Basta AM, Lusk LA, Keller RL, et al. Fetal Stomach Position Predicts Neonatal Outcomes in Isolated Left-Sided Congenital Diaphragmatic Hernia. Fetal Diagn Ther 2016;39:248-55. 10.1159/000440649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou C, Wang J, Song S, et al. The grading of stomach position for postnatal outcomes in isolated left-sided congenital diaphragmatic hernia: A systematic review and meta-analysis. Prenat Diagn 2023;43:1008-17. 10.1002/pd.6383 [DOI] [PubMed] [Google Scholar]
- 54.Savelli S, Bascetta S, Carducci C, et al. Fetal MRI assessment of mediastinal shift angle in isolated left congenital diaphragmatic hernia: A new postnatal survival predictive tool? Prenat Diagn 2020;40:136-41. 10.1002/pd.5619 [DOI] [PubMed] [Google Scholar]
- 55.Romiti A, Viggiano M, Conforti A, et al. Ultrasonographic assessment of mediastinal shift angle (MSA) in isolated left congenital diaphragmatic hernia for the prediction of postnatal survival. J Matern Fetal Neonatal Med 2020;33:1330-5. [DOI] [PubMed] [Google Scholar]
- 56.Ding W, Gu Y, Wang H, et al. Prenatal MRI assessment of mediastinal shift angle as a feasible and effective risk stratification tool in isolated right-sided congenital diaphragmatic hernia. Eur Radiol 2024;34:1524-33. 10.1007/s00330-023-10178-1 [DOI] [PubMed] [Google Scholar]
- 57.Romiti A, Viggiano M, Savelli S, et al. Comparison of mediastinal shift angles obtained with ultrasound and magnetic resonance imaging in fetuses with isolated left sided congenital diaphragmatic hernia. J Matern Fetal Neonatal Med 2022;35:269-74. 10.1080/14767058.2020.1716714 [DOI] [PubMed] [Google Scholar]
- 58.Spaggiari E, Stirnemann JJ, Sonigo P, et al. Prenatal prediction of pulmonary arterial hypertension in congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 2015;45:572-7. 10.1002/uog.13450 [DOI] [PubMed] [Google Scholar]
- 59.Jani JC, Cordier AG, Martinovic J, et al. Antenatal ultrasound prediction of pulmonary hypoplasia in congenital diaphragmatic hernia: correlation with pathology. Ultrasound Obstet Gynecol 2011;38:344-9. 10.1002/uog.9031 [DOI] [PubMed] [Google Scholar]
- 60.Jani JC, Benachi A, Nicolaides KH, et al. Prenatal prediction of neonatal morbidity in survivors with congenital diaphragmatic hernia: a multicenter study. Ultrasound Obstet Gynecol 2009;33:64-9. 10.1002/uog.6141 [DOI] [PubMed] [Google Scholar]
- 61.Basurto D, Russo FM, Papastefanou I, et al. Pulmonary hypertension in congenital diaphragmatic hernia: Antenatal prediction and impact on neonatal mortality. Prenat Diagn 2022;42:1303-11. 10.1002/pd.6207 [DOI] [PubMed] [Google Scholar]
- 62.Bebbington M, Victoria T, Danzer E, et al. Comparison of ultrasound and magnetic resonance imaging parameters in predicting survival in isolated left-sided congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 2014;43:670-4. 10.1002/uog.13271 [DOI] [PubMed] [Google Scholar]
- 63.Oluyomi-Obi T, Kuret V, Puligandla P, et al. Antenatal predictors of outcome in prenatally diagnosed congenital diaphragmatic hernia (CDH). J Pediatr Surg 2017;52:881-8. 10.1016/j.jpedsurg.2016.12.008 [DOI] [PubMed] [Google Scholar]
- 64.van den Hout L, Schaible T, Cohen-Overbeek TE, et al. Actual outcome in infants with congenital diaphragmatic hernia: the role of a standardized postnatal treatment protocol. Fetal Diagn Ther 2011;29:55-63. 10.1159/000322694 [DOI] [PubMed] [Google Scholar]
- 65.Flageole H, Evrard VA, Piedboeuf B, et al. The plug-unplug sequence: an important step to achieve type II pneumocyte maturation in the fetal lamb model. J Pediatr Surg 1998;33:299-303. 10.1016/S0022-3468(98)90451-1 [DOI] [PubMed] [Google Scholar]
- 66.Skarsgard ED, Meuli M, VanderWall KJ, et al. Fetal endoscopic tracheal occlusion (‘Fetendo-PLUG’) for congenital diaphragmatic hernia. J Pediatr Surg 1996;31:1335-8. 10.1016/S0022-3468(96)90823-4 [DOI] [PubMed] [Google Scholar]
- 67.Harrison MR, Adzick NS, Flake AW, et al. Correction of congenital diaphragmatic hernia in utero: VI. Hard-earned lessons. J Pediatr Surg 1993;28:1411-7; discussion 1417-8. 10.1016/S0022-3468(05)80338-0 [DOI] [PubMed] [Google Scholar]
- 68.Deprest J, Gratacos E, Nicolaides KH, et al. Fetoscopic tracheal occlusion (FETO) for severe congenital diaphragmatic hernia: evolution of a technique and preliminary results. Ultrasound Obstet Gynecol 2004;24:121-6. 10.1002/uog.1711 [DOI] [PubMed] [Google Scholar]
- 69.Jani JC, Nicolaides KH, Gratacós E, et al. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol 2009;34:304-10. 10.1002/uog.6450 [DOI] [PubMed] [Google Scholar]
- 70.Deprest JA, Nicolaides KH, Benachi A, et al. Randomized Trial of Fetal Surgery for Severe Left Diaphragmatic Hernia. N Engl J Med 2021;385:107-18. 10.1056/NEJMoa2027030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deprest JA, Benachi A, Gratacos E, et al. Randomized Trial of Fetal Surgery for Moderate Left Diaphragmatic Hernia. N Engl J Med 2021;385:119-29. 10.1056/NEJMoa2026983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Snoek KG, Reiss IK, Greenough A, et al. Standardized Postnatal Management of Infants with Congenital Diaphragmatic Hernia in Europe: The CDH EURO Consortium Consensus – 2015 Update. Neonatology 2016;110:66-74. 10.1159/000444210 [DOI] [PubMed] [Google Scholar]
- 73.Van Calster B, Benachi A, Nicolaides KH, et al. The randomized Tracheal Occlusion To Accelerate Lung growth (TOTAL)-trials on fetal surgery for congenital diaphragmatic hernia: reanalysis using pooled data. Am J Obstet Gynecol 2022;226:560.e1-560.e24. 10.1016/j.ajog.2021.11.1351 [DOI] [PubMed] [Google Scholar]
- 74.Stolar CJH, Flake AW, Losty PD. Fetal Surgery for Severe Left Diaphragmatic Hernia. N Engl J Med 2021;385:2111-2. 10.1056/NEJMc2115673 [DOI] [PubMed] [Google Scholar]
- 75.Baschat AA, Miller JL, Kunisaki SM. Increased survival following fetoscopic endoluminal tracheal occlusion for diaphragmatic hernia. J Pediatr 2021;238:338-42. 10.1016/j.jpeds.2021.08.061 [DOI] [PubMed] [Google Scholar]
- 76.Belfort MA, Olutoye OO, Cass DL, et al. Feasibility and Outcomes of Fetoscopic Tracheal Occlusion for Severe Left Diaphragmatic Hernia. Obstet Gynecol 2017;129:20-9. 10.1097/AOG.0000000000001749 [DOI] [PubMed] [Google Scholar]
- 77.Style CC, Olutoye OO, Belfort MA, et al. Fetal endoscopic tracheal occlusion reduces pulmonary hypertension in severe congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 2019;54:752-8. 10.1002/uog.20216 [DOI] [PubMed] [Google Scholar]
- 78.Baschat AA, Rosner M, Millard SE, et al. Single-Center Outcome of Fetoscopic Tracheal Balloon Occlusion for Severe Congenital Diaphragmatic Hernia. Obstet Gynecol 2020;135:511-21. 10.1097/AOG.0000000000003692 [DOI] [PubMed] [Google Scholar]
- 79.Bergh E, Baschat AA, Cortes MS, et al. Fetoscopic Endoluminal Tracheal Occlusion for Severe, Left-Sided Congenital Diaphragmatic Hernia: The North American Fetal Therapy Network Fetoscopic Endoluminal Tracheal Occlusion Consortium Experience. Obstet Gynecol 2024;143:440-8. 10.1097/AOG.0000000000005491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sferra SR, Nies MK, Miller JL, et al. Morbidity in children after fetoscopic endoluminal tracheal occlusion for severe congenital diaphragmatic hernia: Results from a multidisciplinary clinic. J Pediatr Surg 2023;58:14-9. 10.1016/j.jpedsurg.2022.09.042 [DOI] [PubMed] [Google Scholar]
- 81.Deprest J, Brady P, Nicolaides K, et al. Prenatal management of the fetus with isolated congenital diaphragmatic hernia in the era of the TOTAL trial. Semin Fetal Neonatal Med 2014;19:338-48. 10.1016/j.siny.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 82.Society for Maternal-Fetal Medicine (SMFM) . Electronic address: pubs@smfm.org, Reddy UM, Deshmukh U, et al. Society for Maternal-Fetal Medicine Consult Series #58: Use of antenatal corticosteroids for individuals at risk for late preterm delivery: Replaces SMFM Statement #4, Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery, August 2016. Am J Obstet Gynecol 2021;225:B36-42. 10.1016/j.ajog.2021.07.023 [DOI] [PubMed] [Google Scholar]
- 83.Stutchfield P, Whitaker R, Russell I, et al. Antenatal betamethasone and incidence of neonatal respiratory distress after elective caesarean section: pragmatic randomised trial. BMJ 2005;331:662. 10.1136/bmj.38547.416493.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kinsella JP, Steinhorn RH, Mullen MP, et al. The Left Ventricle in Congenital Diaphragmatic Hernia: Implications for the Management of Pulmonary Hypertension. J Pediatr 2018;197:17-22. 10.1016/j.jpeds.2018.02.040 [DOI] [PubMed] [Google Scholar]
- 85.DeKoninck PLJ, Horn-Oudshoorn EJJ, Knol R, et al. Knowledge Gaps in the Fetal to Neonatal Transition of Infants With a Congenital Diaphragmatic Hernia. Front Pediatr 2021;9:784810. 10.3389/fped.2021.784810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lakshminrusimha S, Russell JA, Steinhorn RH, et al. Pulmonary hemodynamics in neonatal lambs resuscitated with 21%, 50%, and 100% oxygen. Pediatr Res 2007;62:313-8. 10.1203/PDR.0b013e3180db29fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Snoek KG, Capolupo I, van Rosmalen J, et al. Conventional Mechanical Ventilation Versus High-frequency Oscillatory Ventilation for Congenital Diaphragmatic Hernia: A Randomized Clinical Trial (The VICI-trial). Ann Surg 2016;263:867-74. 10.1097/SLA.0000000000001533 [DOI] [PubMed] [Google Scholar]
- 88.Gien J, Kinsella JP, Behrendt NJ, et al. Improved survival for infants with severe congenital diaphragmatic hernia. J Perinatol 2022;42:1189-94. 10.1038/s41372-022-01397-3 [DOI] [PubMed] [Google Scholar]
- 89.Gien J, Kinsella JP. Management of pulmonary hypertension in infants with congenital diaphragmatic hernia. J Perinatol 2016;36 Suppl 2:S28-31. 10.1038/jp.2016.46 [DOI] [PubMed] [Google Scholar]
- 90.Gien J, Palmer C, Liechty K, et al. Early Abnormalities in Gas Exchange in Infants with Congenital Diaphragmatic Hernia. J Pediatr 2022;243:188-92. 10.1016/j.jpeds.2021.12.009 [DOI] [PubMed] [Google Scholar]
- 91.Kool H, Mous D, Tibboel D, et al. Pulmonary vascular development goes awry in congenital lung abnormalities. Birth Defects Res C Embryo Today 2014;102:343-58. 10.1002/bdrc.21085 [DOI] [PubMed] [Google Scholar]
- 92.Mous DS, Kool HM, Wijnen R, et al. Pulmonary vascular development in congenital diaphragmatic hernia. Eur Respir Rev 2018;27:170104. 10.1183/16000617.0104-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kumar VHS, Dadiz R, Koumoundouros J, et al. Response to pulmonary vasodilators in infants with congenital diaphragmatic hernia. Pediatr Surg Int 2018;34:735-42. 10.1007/s00383-018-4286-5 [DOI] [PubMed] [Google Scholar]
- 94.McNamara PJ, Giesinger RE, Lakshminrusimha S. Dopamine and Neonatal Pulmonary Hypertension-Pressing Need for a Better Pressor? J Pediatr 2022;246:242-50. 10.1016/j.jpeds.2022.03.022 [DOI] [PubMed] [Google Scholar]
- 95.Hari Gopal S, Patel N, Fernandes CJ. Use of Prostaglandin E1 in the Management of Congenital Diaphragmatic Hernia-A Review. Front Pediatr 2022;10:911588. 10.3389/fped.2022.911588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lawrence KM, Berger K, Herkert L, et al. Use of prostaglandin E1 to treat pulmonary hypertension in congenital diaphragmatic hernia. J Pediatr Surg 2019;54:55-9. 10.1016/j.jpedsurg.2018.10.039 [DOI] [PubMed] [Google Scholar]
- 97.Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Pediatrics 1997;99:838-45. 10.1542/peds.99.6.838 [DOI] [PubMed] [Google Scholar]
- 98.Noh CY, Chock VY, Bhombal S, et al. Early nitric oxide is not associated with improved outcomes in congenital diaphragmatic hernia. Pediatr Res 2023;93:1899-906. 10.1038/s41390-023-02491-8 [DOI] [PubMed] [Google Scholar]
- 99.Moore SS, Keller RL, Altit G. Congenital Diaphragmatic Hernia: Pulmonary Hypertension and Pulmonary Vascular Disease. Clin Perinatol 2024;51:151-70. 10.1016/j.clp.2023.10.001 [DOI] [PubMed] [Google Scholar]
- 100.Le LS, Kinsella JP, Gien J, et al. Failure to Normalize Biventricular Function Is Associated with Extracorporeal Membrane Oxygenation Use in Neonates with Congenital Diaphragmatic Hernia. J Pediatr 2023;260:113490. 10.1016/j.jpeds.2023.113490 [DOI] [PubMed] [Google Scholar]
- 101.Massolo AC, Paria A, Hunter L, et al. Ventricular Dysfunction, Interdependence, and Mechanical Dispersion in Newborn Infants with Congenital Diaphragmatic Hernia. Neonatology 2019;116:68-75. 10.1159/000499347 [DOI] [PubMed] [Google Scholar]
- 102.Patel N, Lally PA, Kipfmueller F, et al. Ventricular Dysfunction Is a Critical Determinant of Mortality in Congenital Diaphragmatic Hernia. Am J Respir Crit Care Med 2019;200:1522-30. 10.1164/rccm.201904-0731OC [DOI] [PubMed] [Google Scholar]
- 103.Sferra SR, Guo M, Gonzalez Salazar AJ, et al. Sex-Specific Differences in Congenital Diaphragmatic Hernia Mortality. J Pediatr 2023;259:113481. 10.1016/j.jpeds.2023.113481 [DOI] [PubMed] [Google Scholar]
- 104.Does extracorporeal membrane oxygenation improve survival in neonates with congenital diaphragmatic hernia? The Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg 1999;34:720-5. 10.1016/S0022-3468(99)90363-9 [DOI] [PubMed] [Google Scholar]
- 105.Kays DW, Talbert JL, Islam S, et al. Improved Survival in Left Liver-Up Congenital Diaphragmatic Hernia by Early Repair Before Extracorporeal Membrane Oxygenation: Optimization of Patient Selection by Multivariate Risk Modeling. J Am Coll Surg 2016;222:459-70. 10.1016/j.jamcollsurg.2015.12.059 [DOI] [PubMed] [Google Scholar]
- 106.Jancelewicz T, Brindle ME, Harting MT, et al. Extracorporeal Membrane Oxygenation (ECMO) Risk Stratification in Newborns with Congenital Diaphragmatic Hernia (CDH). J Pediatr Surg 2018;53:1890-5. 10.1016/j.jpedsurg.2018.04.014 [DOI] [PubMed] [Google Scholar]
- 107.Jancelewicz T, Langham MR, Jr, Brindle ME, et al. Survival Benefit Associated With the Use of Extracorporeal Life Support for Neonates With Congenital Diaphragmatic Hernia. Ann Surg 2022;275:e256-63. 10.1097/SLA.0000000000003928 [DOI] [PubMed] [Google Scholar]
- 108.Guner YS, Harting MT, Fairbairn K, et al. Outcomes of infants with congenital diaphragmatic hernia treated with venovenous versus venoarterial extracorporeal membrane oxygenation: A propensity score approach. J Pediatr Surg 2018;53:2092-9. 10.1016/j.jpedsurg.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dao DT, Burgos CM, Harting MT, et al. Surgical Repair of Congenital Diaphragmatic Hernia After Extracorporeal Membrane Oxygenation Cannulation: Early Repair Improves Survival. Ann Surg 2021;274:186-94. 10.1097/SLA.0000000000003386 [DOI] [PubMed] [Google Scholar]
- 110.Low ZK, Tan ASM, Nakao M, et al. Congenital diaphragmatic hernia repair in patients requiring extracorporeal membrane oxygenation: are outcomes better with repair on ECMO or after decannulation? Interact Cardiovasc Thorac Surg 2021;32:632-7. 10.1093/icvts/ivaa303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu PT, Jen HC, Rice-Townsend S, et al. The role of ECMO in the management of congenital diaphragmatic hernia. Semin Perinatol 2020;44:151166. 10.1053/j.semperi.2019.07.005 [DOI] [PubMed] [Google Scholar]
- 112.Smithers CJ, Zalieckas JM, Rice-Townsend SE, et al. The Timing of Congenital Diaphragmatic Hernia Repair on Extracorporeal Membrane Oxygenation Impacts Surgical Bleeding Risk. J Pediatr Surg 2023;58:1656-62. 10.1016/j.jpedsurg.2022.12.030 [DOI] [PubMed] [Google Scholar]
- 113.Espinoza J, King A, Shamshirsaz AA, et al. Characterization of Suboptimal Responses to Fetoscopic Endoluminal Tracheal Occlusion in Congenital Diaphragmatic Hernia. Fetal Diagn Ther 2023;50:128-35. 10.1159/000530549 [DOI] [PubMed] [Google Scholar]
- 114.Langer JC, Filler RM, Bohn DJ, et al. Timing of surgery for congenital diaphragmatic hernia: is emergency operation necessary? J Pediatr Surg 1988;23:731-4. 10.1016/S0022-3468(88)80413-5 [DOI] [PubMed] [Google Scholar]
- 115.West KW, Bengston K, Rescorla FJ, et al. Delayed surgical repair and ECMO improves survival in congenital diaphragmatic hernia. Ann Surg 1992;216:454-60; discussion 460-2. 10.1097/00000658-199210000-00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Putnam LR, Tsao K, Lally KP, et al. Minimally Invasive vs Open Congenital Diaphragmatic Hernia Repair: Is There a Superior Approach? J Am Coll Surg 2017;224:416-22. 10.1016/j.jamcollsurg.2016.12.050 [DOI] [PubMed] [Google Scholar]
- 117.Gupta VS, Harting MT, Lally PA, et al. Mortality in Congenital Diaphragmatic Hernia: A Multicenter Registry Study of Over 5000 Patients Over 25 Years. Ann Surg 2023;277:520-7. 10.1097/SLA.0000000000005113 [DOI] [PubMed] [Google Scholar]
- 118.Gupta VS, Shepherd ST, Ebanks AH, et al. Association of timing of congenital diaphragmatic hernia repair with survival and morbidity for patients not requiring extra-corporeal life support. J Neonatal Perinatal Med 2022;15:759-65. 10.3233/NPM-221072 [DOI] [PubMed] [Google Scholar]
- 119.Al Baroudi S, Collaco JM, Lally PA, et al. Clinical features and outcomes associated with tracheostomy in congenital diaphragmatic hernia. Pediatr Pulmonol 2020;55:90-101. 10.1002/ppul.24516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Haliburton B, Chiang M, Marcon M, et al. Nutritional Intake, Energy Expenditure, and Growth of Infants Following Congenital Diaphragmatic Hernia Repair. J Pediatr Gastroenterol Nutr 2016;62:474-8. 10.1097/MPG.0000000000001000 [DOI] [PubMed] [Google Scholar]
- 121.Bairdain S, Khan FA, Fisher J, et al. Nutritional outcomes in survivors of congenital diaphragmatic hernia (CDH)-factors associated with growth at one year. J Pediatr Surg 2015;50:74-7. 10.1016/j.jpedsurg.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 122.Verla MA, Style CC, Mehollin-Ray AR, et al. Prenatal Imaging Features and Postnatal Factors Associated with Gastrointestinal Morbidity in Congenital Diaphragmatic Hernia. Fetal Diagn Ther 2020;47:252-60. 10.1159/000501555 [DOI] [PubMed] [Google Scholar]
- 123.Diamond IR, Mah K, Kim PC, et al. Predicting the need for fundoplication at the time of congenital diaphragmatic hernia repair. J Pediatr Surg 2007;42:1066-70. 10.1016/j.jpedsurg.2007.01.046 [DOI] [PubMed] [Google Scholar]
- 124.Safavi A, Synnes AR, O’Brien K, et al. Multi-institutional follow-up of patients with congenital diaphragmatic hernia reveals severe disability and variations in practice. J Pediatr Surg 2012;47:836-41. 10.1016/j.jpedsurg.2012.01.032 [DOI] [PubMed] [Google Scholar]
- 125.American Academy of Pediatrics Section on Surgery. American Academy of Pediatrics Committee on Fetus and Newborn ; Lally KP, et al. Postdischarge follow-up of infants with congenital diaphragmatic hernia. Pediatrics 2008;121:627-32. 10.1542/peds.2007-3282 [DOI] [PubMed] [Google Scholar]
- 126.Haliburton B, Mouzaki M, Chiang M, et al. Pulmonary function and nutritional morbidity in children and adolescents with congenital diaphragmatic hernia. J Pediatr Surg 2017;52:252-6. 10.1016/j.jpedsurg.2016.11.020 [DOI] [PubMed] [Google Scholar]
- 127.Jancelewicz T, Chiang M, Oliveira C, et al. Late surgical outcomes among congenital diaphragmatic hernia (CDH) patients: why long-term follow-up with surgeons is recommended. J Pediatr Surg 2013;48:935-41. 10.1016/j.jpedsurg.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 128.Cauley RP, Potanos K, Fullington N, et al. Pulmonary support on day of life 30 is a strong predictor of increased 1 and 5-year morbidity in survivors of congenital diaphragmatic hernia. J Pediatr Surg 2015;50:849-55. 10.1016/j.jpedsurg.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tho ALW, Rath CP, Tan JKG, et al. Prevalence of symptomatic tracheal morbidities after fetoscopic endoluminal tracheal occlusion: a systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed 2023;109:52-8. 10.1136/archdischild-2023-325525 [DOI] [PubMed] [Google Scholar]
- 130.Lusk LA, Wai KC, Moon-Grady AJ, et al. Persistence of pulmonary hypertension by echocardiography predicts short-term outcomes in congenital diaphragmatic hernia. J Pediatr 2015;166:251-6.e1. 10.1016/j.jpeds.2014.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mahmood B, Murthy K, Rintoul N, et al. Predicting treatment of pulmonary hypertension at discharge in infants with congenital diaphragmatic hernia. J Perinatol 2022;42:45-52. 10.1038/s41372-021-01249-6 [DOI] [PubMed] [Google Scholar]
- 132.Kraemer US, Leeuwen L, Krasemann TB, et al. Characteristics of Infants With Congenital Diaphragmatic Hernia Who Need Follow-Up of Pulmonary Hypertension. Pediatr Crit Care Med 2018;19:e219-26. 10.1097/PCC.0000000000001464 [DOI] [PubMed] [Google Scholar]
- 133.Montalva L, Raffler G, Riccio A, et al. Neurodevelopmental impairment in children with congenital diaphragmatic hernia: Not an uncommon complication for survivors. J Pediatr Surg 2020;55:625-34. 10.1016/j.jpedsurg.2019.05.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as