


Abstract
The MutS family of DNA repair proteins recognizes base pair mismatches and insertion/deletion mismatches and targets them for repair in a strand-specific manner. Photocrosslinking and mutational studies previously identified a highly conserved Phe residue at the N-terminus of Thermus aquaticus MutS protein that is critical for mismatch recognition in vitro. Here, a mutant Escherichia coli MutS protein harboring a substitution of Ala for the corresponding Phe36 residue is assessed for proficiency in mismatch repair in vivo and DNA binding and ATP hydrolysis in vitro. The F36A protein is unable to restore mismatch repair proficiency to a mutS strain as judged by mutation to rifampicin or reversion of a specific point mutation in lacZ. The F36A protein is also severely deficient for binding to heteroduplexes containing an unpaired thymidine or a G:T mismatch although its intrinsic ATPase activity and subunit oligomerization are very similar to that of the wild-type MutS protein. Thus, the F36A mutation appears to confer a defect specific for recognition of insertion/deletion and base pair mismatches.
INTRODUCTION
DNA mismatch repair plays a critical role in mutation avoidance in virtually all organisms (reviewed in 1–3). The repair of DNA biosynthetic errors by this mismatch repair pathway increases the overall fidelity of DNA replication by as much as 1000-fold. In addition to its role in post-replication mismatch repair, components of the mismatch repair machinery exhibit an inhibitory effect on homologous recombination involving exchange between evolutionarily divergent sequences (4). In eukaryotes, mismatch repair proteins are also required for meiotic recombination where they function to ensure the proper segregation of chromosomes (reviewed in 5). The discovery that defects in mismatch repair genes cause hereditary non-polyposis colon cancer highlights the importance of mismatch repair in maintaining genome stability (reviewed in 1,6,7).
In Escherichia coli, the correction of mismatches arising from biosynthetic errors is carried out by the methyl-directed mismatch repair system. Genetic and biochemical studies have identified at least 10 gene products involved in mismatch repair: MutS, MutL, MutH, DNA heliase II, single-stranded DNA-binding protein, single-strand exonucleases Exo I, Exo VII or RecJ, DNA polymerase III holoenzyme and DNA ligase (reviewed in 1). MutS recognizes seven of eight base pair mismatches (only C:C mismatches are not corrected by this pathway) as well as small insertion/deletion mismatches of one to three unpaired bases. Together, MutS and MutL then activate MutH in an ATP-dependent fashion resulting in incision by MutH of the transiently unmethylated daughter strand at hemimethylated GATC sites, a step that confers strand specificity (8–11). Excision repair is initiated by DNA helicase II (12) which, like MutH, is activated by MutS and MutL (13,14). Excision of the daughter strand is carried out by exonucleases having either a 5′→3′ or 3′→5′ polarity (15,16) facilitating bidirectional repair in which a GATC site can be on either side of a mismatch (17).
The highly conserved MutS family of DNA mismatch repair proteins recognizes mismatched bases that arise through replication errors, homologous recombination and physical damage to bases. Whereas prokaryotic genomes generally encode a single mutS gene, in Saccharomyces cerevisiae, three mutS homologs, MSH2, MSH3 and MSH6, have been implicated in nuclear post-replicative repair, and a similar number have been identified in humans. Most MutS proteins appear to function as dimers with prokaryotic MutS proteins being homodimeric and eukaryotic complexes being heterodimers of MSH2 and MSH6 (MutSα) or MSH2 and MSH3 (MutSβ) (reviewed in 2). In addition to their mismatch binding activity, all MutS proteins have an intrinsic ATPase activity and share four highly conserved nucleotide binding motifs. Two of these correspond to the Walker A and Walker B motifs (18) while the remaining two motifs are unique to the ABC ATPase superfamily (19).
Previous photocrosslinking and mutagenesis experiments identified a highly conserved Phe residue at the N-terminus of Taq MutS protein that appeared to be critical for mismatch recognition in vitro (20). Phe39 of Taq MutS was specifically crosslinked to an insertion/deletion heteroduplex DNA in which 5-iodo-dU was present as an unpaired base. When Phe39 was mutated to Ala, the resulting F39A protein failed to bind an insertion/deletion heteroduplex DNA although it dimerized like wild-type MutS, was thermostable like the wild-type protein and had a fully functioning ATPase activity. Mutation of the corresponding Phe residue of yeast MSH6 to Ala, F337A, or serine, F337S, resulted in a dominant negative phenotype in mutator assays (21,22). Mutant MSH2–MSH6 (F337A) complexes were shown to be deficient for binding to insertion/deletion heteroduplexes in vitro although residual DNA binding activity was detected as judged by the stimulation of the ATP hydrolysis activity of MSH2–MSH6 by heteroduplex DNA (21).
Unresolved issues in mismatch repair include the role of mismatch recognition in signaling downstream mismatch repair steps such as the activation of MutH for DNA cleavage and the role of mismatch recognition by MutS in the regulation of homologous recombination by mismatch repair proteins. As discussed above, E.coli MutS serves as a paradigm for the MutS family of proteins, facilitating both genetic and biochemical approaches to the study of mismatch repair. Here, we report the substitution of Ala for Phe36 of the E.coli MutS protein. The mutant F36A protein appears to have a specific defect in mismatch binding, failing to recognize either an insertion/deletion mismatch or a G:T base pair mismatch and is unable to complement a mutS strain in two mutator assays.
MATERIALS AND METHODS
Site-directed mutagenesis
Escherichia coli TX2929 and pTX412 were provided by Malcolm Winkler (University of Texas, Houston, TX). The F36A MutS mutation was constructed using a two-step PCR method (23). The T193TT codon (Phe) was altered to GCT (Ala) (nucleotide position corresponds to GenBank accession no. U69873). The PCR product was gel purified and cloned into pET17xb (Novagen, Madison, WI) as an XbaI–EcoRV fragment. The remaining mutS coding sequence was subsequently cloned in as an EcoRV–EcoRV fragment to yield pAY02 containing the F36A mutation. The corresponding sequence of wild-type mutS was also cloned into pET17xb to yield pAY01. The amplified coding regions were verified by DNA sequencing.
Complementation assay
For rifampicin resistance assays, TX2929 (CC106 mutS201::Tn5 Kmr) was transformed with pET17xb, pAY01 or pAY02. Transformants were plated on LB–agar plates containing 100 µg/ml carbenicillin (Sigma, St Louis, MO) and 30 µg/ml kanamycin sulfate (Sigma) and incubated at 37°C. Two independent clones from each of two different transformations were inoculated to 5 ml LB containing 25 µg/ml carbenicillin and 12.5 µg/ml kanamycin and grown overnight with shaking. Cultured cells were collected by centrifugation and washed once with Minimal A medium (24). Appropriate dilutions were plated in duplicate on Minimal A–agar supplemented with 25 µg/ml carbenicillin, 35 µg/ml kanamycin sulfate, 1 mM IPTG (Gold Biotechnologies, St Louis, MO) and 2% glucose in the presence or absence of 35 µg/ml rifampicin (Sigma). Colonies were scored after incubation at 37°C for 2 days.
For lac+ reversion assays, transformants grown in Minimal A medium as described above were plated in duplicate at appropriate dilutions on Minimal A–agar supplemented with 25 µg/ml carbenicillin, 35 µg/ml kanamycin sulfate, 2% lactose, 1 mM IPTG, 500 µg/ml phenyl-β-d-galactoside (Sigma) and 40 µg/ml Xgal (Gold Biotechnologies). Viability was determined by plating on Minimal A plates supplemented with antibiotics, IPTG and 2% glucose. Colonies were scored after incubation at 37°C for 2 days.
Protein expression and purification
HMS174(DE3)pLysS strain (Novagen) was transformed with pAY01 or pAY02 and plated onto Tryptone Broth plates supplemented with 1% glucose, 100 µg/ml carbenicillin and 35 µg/ml chloramphenicol (Sigma). Transformants were inoculated into Tryptone Broth supplemented with 1% glucose, 100 µg/ml carbenecillin, 35 µg/ml chloramphenicol and grown overnight at 37°C with shaking. The overnight cultures were centrifuged, resuspended and used to inoculate to LB (Quality Biological Inc., Gaithersburg, MD) supplemented with 1% glucose, 100 µg/ml carbenicillin and 35 µg/ml chloramphenicol. The cultures were grown at 30°C and protein expression was induced at OD600 = 0.5 by the addition of IPTG to a final concentration of 0.3 mM. Cells were harvested 2 h after induction with centrifugation and resuspended into 80 ml of binding buffer (20 mM Tris–HCl, pH 7.6, 5 mM KHPO4, 500 mM NaCl and 10% glycerol) supplemented with Complete EDTA-free protease inhibitor (Roche, Indianapolis, IN). Cell suspensions were stored at –20°C.
All purification steps were performed at 4°C. The thawed cell suspension from 1 l of culture was cleared by centrifugation at 36 000 g for 20 min at 4°C. The cleared lysate (~35 ml) was mixed for 10 min with gentle shaking with 5 ml Ni-NTA Agarose (Qiagen, Valencia, CA) previously equilibrated with binding buffer. Purification was performed in a batch-wise manner. The resin was washed four times with 30 ml binding buffer followed by four washes with 30 ml wash buffer [20 mM Tris–HCl, pH 7.6, 5 mM KHPO4, pH 7.6, 500 mM NaCl, 50 mM imidazole, pH 7.6 (Sigma) and 10% glycerol]. The proteins were eluted twice with 8 ml elution buffer [20 mM Tris–HCl, pH 7.6, 5 mM KHPO4, pH 7.6, 500 mM NaCl, 400 mM imidazole, pH 7.6 (Sigma), 10% glycerol]. Proteins eluted from the nickel resin were exchanged into buffer K (20 mM Tris–HCl, pH 7.6, 200 mM KCl, 0.1 mM dithiothreitol, 0.01 mM EDTA and 20% glycerol) by passage over a pre-equilibrated NAP-25 column (Amersham Pharmacia, Piscataway, NJ). The concentration of protein was determined by the modified Bradford assay (Bio-Rad, Hercules, CA) with BSA standard.
Analytical gel filtration
Protein (10 µg) was loaded onto a Superdex S-200 PC 3.2/30 column (Amersham Pharmacia) equilibrated with buffer K at room temperature at a flow rate of 20 µl/min using the SMART system (Amersham Pharmacia). The elution profile was monitored by OD280.
Steady-state ATPase
Assays were performed in 20 mM Tris–HCl, pH 7.6, 100 mM NaCl, 5 mM MgCl2, 0.5 mM CaCl2, 0.1 mM dithiothreitol, 100 µg/ml BSA, 100 nCi/µl [α-32P]ATP (3000 Ci/mmol, NEN, Boston, MA). Cold ATP was added to a final concentration of 5, 8, 10, 12, 20 and 50 µM for F36A protein and 5, 7, 10, 15, 20, 40 and 50 µM for wild-type protein. All assays were performed in duplicate immediately after the final protein purification step. Each reaction mixture was preheated to 37°C for 3 min and reactions were initiated by the addition of protein to a concentration of 100 nM. Three microliters was removed at 0, 0.5, 1, 1.5, 2, 3, 4 and 5 min after the initiation of reactions and quenched by the addition of 2 µl stop solution containing 33 mM each of AMP, ADP and ATP. Reactions (0.5 µl) were spotted on prewashed PEI–cellulose thin-layer chromatography plates (Polygram CEL300 PEI, Machery-Nagel, Easton, PA), and developed in 0.4 M LiCl (Sigma) and 1 M formic acid (Sigma). The results were analyzed on a Fuji phosphorimager, and quantified with MacBAS v2.52.
Heteroduplex DNA binding assays
For gel mobility shift assays, varying amounts of His-tagged wild-type or F36A protein were incubated with 100 pM 32P-labeled heteroduplex DNA at 20°C for 10 min in 20 mM HEPES, pH 7.8, 5 mM MgCl2, 50 mM NaCl, 100 µg/ml BSA and 1 mM dithiothreitol. Samples were brought to 3% w/v Ficoll and electrophoresed on 5% non-denaturing polyacrylamide gels in Tris–glycine buffer pH 8.3 (25 mM Tris–HCl, 250 mM glycine) at 4°C for 2–3 h at 130 V with buffer recirculation. The extent of binding was quantified on a Fuji phosphoimager using MacBAS software. The fraction bound (radioactivity of the bound complex/total radioactivity) was calculated and plotted as a function of the protein concentration. Data were fitted to an equation for a simple two-state binding process (25).
For filter binding assays, varying amounts of wild-type or F36A protein were incubated with 0.25 pmol 32P-labeled 37 bp DNA having an unpaired thymidine, a G:T mismatch or an A:T perfect base pair (see legend to Fig. 3 for sequence) in 20 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 1 mM CaCl2, 1 mM β-mercaptoethanol and 0.1 mM EDTA in a total volume of 50 µl. After 30 min at 4°C, samples were filtered through a double membrane system consisting of BA85 nitrocellulose and NA45 DEAE (Schleicher and Schuell, Keene, NH) as previously described (26).
Figure 3.
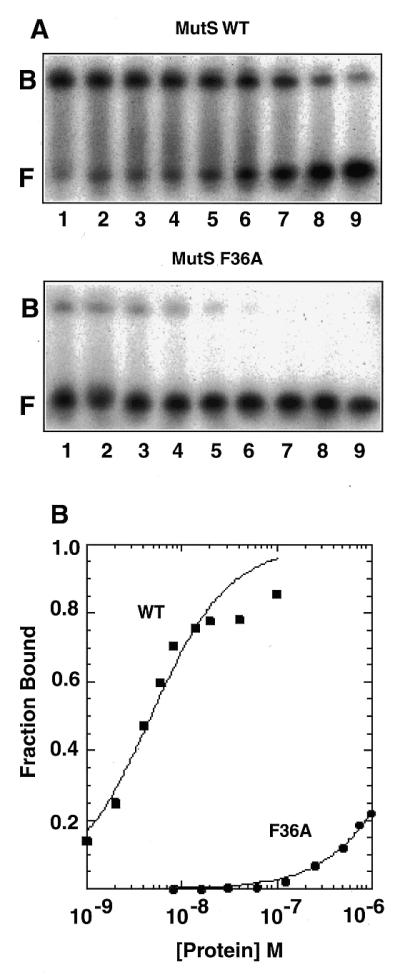
Binding of wild-type and F36A MutS proteins to a heteroduplex DNA containing an unpaired thymidine. (A) His6-tagged wild-type (WT) (top) and F36A (bottom) proteins were incubated with a 32P-labeled 37 bp heteroduplex DNA containing an unpaired thymidine and were electrophoresed on native polyacrylamide gels as described in Materials and Methods. The sequence of the top strand of the heteroduplex DNA substrate is 5′-ATA CCG TCG ACG CTA GCG T GCG GCT CGT CGT CGA CCT (unpaired T is in bold). Protein concentrations for the wild-type protein were: lane 1, 100 nM; lane 2, 40 nM; lane 3, 20 nM; lane 4, 14 nM; lane 5, 8 nM; lane 6, 6 nM; lane 7, 4 nM; lane 8, 2 nM; lane 9, 1 nM. For F36A: lane 1, 1000 nM; lane 2, 750 nM; lane 3, 500 nM; lane 4, 250 nM; lane 5, 125 nM; lane 6, 63 nM; lane 7, 32 nM; lane 8, 16 nM; lane 9, 8 nM. F, free DNA; B, bound MutS–DNA complexes. (B) Quantitation of the binding data.
RESULTS
Function of F36A MutS in vivo
Site-specific mutagenesis of the coding region of E.coli MutS protein was carried out as described in Materials and Methods, resulting in a single amino acid substitution of Ala for Phe36, a highly conserved residue among prokaryotic and eukaryotic MutS proteins (Fig. 1). Phe36 of E.coli MutS resides at the same relative position as Phe39 of Taq MutS which was identified in previous crosslinking and mutagenesis studies as being critical for mismatch binding (20).
Figure 1.

Sequence alignment of the region containing a conserved Phe residue. The F39 residue of Taq MutS identified in crosslinking studies is denoted by an asterisk. Corresponding Phe residues in other MutS proteins are highlighted and the residue number indicated on the right.
The biological activity of the mutant E.coli F36A MutS protein was determined by complementation in TX2929 (27), a mutS derivative of the Cupples–Miller mutator tester strain CC106 (28). Complementation was measured by the frequency of spontaneous mutation to rifampicin resistance or by reversion of the F′–lacZ mutation in TX2929 that occurs as a result of a specific A:T→G:C transition. TX2929 was transformed with pAY01 encoding wild-type His6-MutS protein, pAY02 encoding F36A His6-MutS protein or vector alone, pET17xb. Transformants expressing F36A protein had a mutator phenotype as judged by the relative frequency of spontaneous mutation which was 72-fold higher than that of transformants expressing wild-type protein (pAY01) in the rifampicin resistance assay and 10 000-fold higher than pAY01 transformants in the lacZ reversion assay (Table 1). The frequencies of rifampicin resistance and reversion to lac+ of F36A transformants were very similar to those of transformants harboring the pET17xb vector alone. These results indicated that replacement of Phe36 of MutS with Ala resulted in a significant loss of mismatch repair capability in vivo.
Table 1. Complementation by wild-type and F36A MutS.
| Plasmid | Mutation frequency | |
|---|---|---|
| Rifampicin resistance (× 10–7) | lacZ+ (× 10–9) | |
| pAY01 (MutS) |
1.0 ± 0.5 |
1.0 ± 2.2 |
| pAY02 (F36A) |
160 ± 33 |
12 000 ± 1900 |
| pET17xb | 570 ± 100 | 9700 ± 1900 |
Four independent clones from two transformations of TX2929 (CC106 mutS201::Tn5;Kmr) harboring pAY01 (His-tagged wild-type MutS), pAY02 (His-tagged F36A) or pET17xb (vector) were assessed for their ability to complement the mutS strain by scoring for mutation to rifampicin resistance or reversion to lacZ+ of a specific A:T→G:C transition in the lacZ gene.
Oligomerization of wild-type and F36A MutS
His-tagged versions of wild-type and F36A MutS proteins were purified from overexpressing E.coli HMS174 (DE3) pLysS cells as described in Materials and Methods. Judging from Coomassie stained SDS polyacrylamide gels, the proteins were in excess of 95% pure (Fig. 2A).
Figure 2.

Oligomerization of the F36A and wild-type MutS proteins. (A) His6-tagged wild-type and F36A proteins were purified on nickel–agarose columns as described in Materials and Methods and visualized on 4–20% denaturing SDS–polyacrylamide gels. Protein standard (left lane) is Mark12 Wide range (Novex, San Diego, CA). (B) S-200 Superdex analytical gel filtration chromatography. Purified wild-type and F36A proteins were chromatographed on S-200 PC 3.2/30 column as described in Materials and Methods and detected by absorbance at 280 nm. (C) Plot of Kav, the partition coefficient, versus the logarithm of molecular weight of wild-type (WT) and F36A proteins eluting in the major early peak from S-200 columns. Molecular weight standards are thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa) and BSA (66 kDa).
Wild-type E.coli MutS protein whose monomer subunit is 97 kDa has been shown to oligomerize to form dimers and higher order oligomers in solution (29). The oligomerization of the purified His-tagged wild-type and F36A proteins were investigated by gel filtration chromatography on an analytical S-200 Superdex column (Fig. 2B and C). The elution profiles of the His-tagged wild-type and F36A proteins were virtually identical. The major peak eluted at a position corresponding to 500–600 kDa with broad tailing of the eluted proteins to a position corresponding to dimer. No monomeric species were observed. Protein samples stored at –20°C for 3 days, 1 week or 2 weeks were similarly analyzed by gel filtration, but did not show significant changes (data not shown). This size distribution of oligomers is very similar to that observed previously for E.coli MutS (29). We conclude that the F36A mutation does not interfere with oligomerization of MutS.
ATP hydrolysis
Bacterial and eukaryotic MutS proteins have an ATP hydrolysis activity that yields ADP and inorganic phosphate (reviewed in 1,2,30). The steady-state ATPase activities of His-tagged wild-type and F36A E.coli MutS protein were assessed in parallel. The ATPase activity of the F36A mutant protein was very similar to that of the wild-type protein at 37°C (Table 2). Both obeyed Michaelis–Menten kinetics with a Km of 20 and 17 µM for the wild-type and F36A proteins, respectively. Maximal hydrolysis rates (kcat) were 11 and 12 min–1, respectively. These values are in qualitative agreement with previously determined values for Salmonella typhimurium MutS, kcat = 0.3 min–1 and Km = 7 µM (31), E.coli MutS, kcat = 1 min–1 and Km = 36 µM (29), and His-tagged E.coli MutS, kcat = 7 min–1 and Km = 15 µM (32). From this data, we conclude that the F36A mutation has no significant effect on the rate of ATP hydrolysis by MutS.
Table 2. Steady-state ATP hydrolysis of His-tagged wild-type and F36A MutS.
| Km (µM) | Vmax (µM/min) | kcat (µM ADP/min/µM MutS) | |
|---|---|---|---|
| Lineweaver–Burk | |||
| MutS wild-type | 23.3 ± 2.0 | 1.15 ± 0.08 | 12 |
| F36A | 9.9 ± 3.7 | 0.89 ± 0.16 | 9 |
| Eadie–Hofstee | |||
| MutS wild-type | 20.5 ± 1.8 | 1.06 ± 0.04 | 11 |
| F36A | 16.6 ± 8.6 | 1.21 ± 0.40 | 12 |
ATP hydrolysis was determined as described in Materials and Methods at 37°C.
DNA binding
The ability of the F36A mutant protein to bind a 37 bp heteroduplex DNA containing an unpaired thymidine was assessed in gel mobility shift assays (Fig. 3). The wild-type MutS protein readily bound to the insertion/deletion heteroduplex DNA. In contrast, the F36A mutant protein bound only weakly to the same heteroduplex. Quantitation of the data, shown in Figure 3B, reveals that substitution of Ala for Phe36 reduced the relative affinity for heteroduplex DNA by some three orders of magnitude. The best fit of the data assuming one heteroduplex DNA bound per dimer yields apparent Kd values of 1.1 × 10–8 and 1.4 × 10–5 M for the wild-type and F36A proteins, respectively. The apparent Kd values are only approximations given the heterogeneous population of oligomers and evidence for non-equivalence of subunits in higher order MutS oligomers with respect to DNA binding (29). Nonetheless, these data demonstrate that a single amino acid substitution at Phe36 has a profound effect on heteroduplex DNA binding by E.coli MutS. These results are similar to previous observations for the Taq MutS protein where substitution of Ala for Phe39 also abolished mismatch binding (20).
The ability of the F36A mutant protein to bind to a 37 bp heteroduplex DNA containing a G:T mismatch was investigated using a filter binding assay. As is evident in the results shown in Figure 4, the F36A protein failed to bind to either a Δ1 insertion/deletion substrate (unpaired thymidine) or a G:T mismatch present in the identical sequence context even at a protein concentration of 200 nM (20 pmol). Although the filter binding experiments carried out at 4°C cannot be directly compared with gel mobility shift assays, there is excellent qualitative agreement between the two assays. These results demonstrate that substitution of Ala for Phe36 abolishes binding to both an insertion/deletion type mismatch and a base pair mismatch. Additional filter binding experiments with a perfectly paired homoduplex showed that although the wild-type E.coli MutS binds weakly to the homoduplex, the extent of homoduplex binding by the F36A mutant protein was undetectable over background (data not shown).
Figure 4.

Binding of wild-type and F36A MutS proteins to a heteroduplex DNA containing a G:T mismatch. (Top) His-tagged wild-type (WT) and F36A proteins were incubated at 4°C with a heteroduplex containing an unpaired thymidine (Δ1, see Fig. 3), and analyzed by filter binding as described in Materials and Methods. (Bottom) Filter binding experiments were carried out as above, but with a 37 bp heteroduplex containing a G:T mismatch.
DISCUSSION
We have shown that substitution of Ala for Phe36 in E.coli MutS protein confers a specific defect in DNA mismatch binding. Binding to both an insertion/deletion heteroduplex containing an unpaired thymidine and a G:T base pair mismatch are significantly impaired. The F36A protein is also defective for mismatch repair in vivo, failing to complement a mutS strain in two different mutator assays measuring the frequency of reversion of a lacZ point substitution and mutation to rifampicin resistance. Support for the idea that the mutation affects DNA binding directly rather than through some indirect means such as protein destabilization stems from the observations that the F36A mutant protein oligomerizes in an identical fashion to wild-type protein and has an ATPase activity whose steady-state parameters are essentially unchanged from that of wild-type protein.
The data presented here, together with other studies, suggest that the highly conserved Phe residue present in the N-terminal region of MutS proteins plays a critical role in mismatch binding in many MutS proteins. Previous crosslinking and mutational studies identified a Phe residue at the N-terminus of Taq MutS as being critical for DNA binding (20). Substitution of Ala for Phe39 of Taq MutS reduced the affinity of the mutant protein for heteroduplex containing an unpaired thymidine by three orders of magnitude, a finding that we have reproduced here for the corresponding F36A E.coli mutant. Alani and colleagues have characterized a MSH2–MSH6 complex in yeast in which the MSH6 subunit harbors a substitution of Ala for Phe337, the residue that corresponds to Taq Phe39 and E.coli Phe36. In the case of the mutant yeast complex, binding to a heteroduplex containing an insertion/deletion mismatch was disrupted, and the overexpression of the F337A protein in yeast cells resulted in a dominant negative phenotype (21). Similarly, mutation of msh6 at the chromosomal locus to yield yMSH2–MSH6 (F337S) complexes in which the Phe was mutated to Ser conferred a dominant mutator phenotype in S.cerevisiae (22). Finally, the corresponding Phe432→Ala mutation has been introduced in the human MSH6 subunit resulting in loss of mismatch binding by the human MSH2–MSH6 (MSHα) complex (J.Jiricny, personal communication).
Recently, we have determined the crystal structure of Taq MutS and its complex with a heteroduplex DNA containing an insertion/deletion mismatch to 3.2 and 2.2 Å resolution, respectively (33). In the co-crystal structure, the DNA is sharply kinked at the mismatch towards the major groove by ~60°, and a novel protein–DNA interaction is revealed in which the unpaired thymidine is neither stacked in the duplex nor flipped out, but displaced towards the minor groove by ~2 Å. Phe39 approaches the DNA from the minor-groove side, stacks onto the unpaired thymidine and contacts the 3′ adjacent base with the tip of its phenyl ring. Thus, the Phe residue figures prominently in the stabilization of the unpaired thymidine. The data presented here suggests that a similar mechanism of induced fit is utilized by E.coli MutS protein.
An unresolved question is the role of the Phe residue in binding to base pair mismatches. The data presented here suggest that the Phe residue is also important for binding to base pair mismatches since the E.coli F36A protein is deficient for binding to a G:T mismatch as well as an insertion/deletion mismatch. Additional support for the notion that binding to insertion/deletion and base pair mismatches follows a similar scheme stems from previous chemical footprinting studies of Taq MutS–heteroduplex complexes. In that study, dimethyl sulfate protection, a probe of the major groove, and 1,10 copper phenanthroline modification, a probe of the minor groove, suggested that protein–DNA contacts and DNA distortion were similar for heteroduplexes containing an unpaired thymidine or a G:T mismatch (34).
Interestingly, in Taq (20), E.coli (this study), yeast (21) and human (J.Jiriciny, personal communication) MutS proteins, substitution of Ala for the conserved Phe residue disrupts not only mismatch binding, but binding to homoduplex DNA as well. These findings suggest that the Phe residue participates in some way in the binding to homoduplex DNA by MutS that must occur during the search for a mismatch.
The identification of a mutation in E.coli MutS protein that confers a defect specific for mismatch recognition is potentially useful for addressing a number of important problems. For example, one can investigate the role of mismatch recognition in the activation of MutH for cleavage by MutS and MutL protein and the role of mismatch recognition by MutS in the regulation of homologous recombination. The data presented here suggest that Phe36 is critical for recognition of both insertion/deletion and base pair mismatches. A better understanding of the structural basis for the broad substrate specificity of MutS proteins awaits additional structural and biochemical studies.
NOTE ADDED IN PROOF
The role of Phe36 in mismatch binding by E.coli MutS has been confirmed in a crystal structure of an E.coli MutS-G:T mismatch complex (35).
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Joe Jiricny for communicating results prior to publication, Malcolm Winkler for providing TX412 and TX2929, Galya Obmolova for advice on protein purification, Alison Herr for DNA binding studies and Dan Camerini-Otero for reading the manuscript. We thank George Poy for oligonucleotide syntheses and automated DNA sequencing and Linda Robinson and Arlene Jenkins for their help. A.Y. was supported in part by a fellowship from The Japan Society for the Promotion of Science.
REFERENCES
- 1.Modrich P. and Lahue,R. (1996) Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 2.Jiricny J. (1998) EMBO J., 17, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horst J.-P., Wu,T.-H. and Marinus,M.G. (1999) Trends Microbiol., 7, 29–36. [DOI] [PubMed] [Google Scholar]
- 4.Rayssiguier C., Thaler,D.S. and Radman,M. (1989) Nature, 342, 396–401. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa T., Datta,A. and Kolodner,R.D. (1999) Proc. Natl Acad. Sci. USA, 96, 14186–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishel R. and Kolodner,R.D. (1995) Curr. Opin. Genet. Dev., 5, 382–395. [DOI] [PubMed] [Google Scholar]
- 7.Umar A. and Kunkel,T.A. (1996) Eur. J. Biochem., 238, 297–307. [DOI] [PubMed] [Google Scholar]
- 8.Welsh K.M., Lu,A.L., Clark,S. and Modrich,P. (1987) J. Biol. Chem., 262, 15624–15629. [PubMed] [Google Scholar]
- 9.Lahue R.S., Au,K.G. and Modrich,P. (1989) Science, 245, 160–164. [DOI] [PubMed] [Google Scholar]
- 10.Ban C. and Yang,W. (1998) EMBO J., 17, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall M.C. and Matson,S.W. (1999) J. Biol. Chem., 274, 1306–1312. [DOI] [PubMed] [Google Scholar]
- 12.Dao V. and Modrich,P. (1998) J. Biol. Chem., 273, 9202–9207. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi M., Dao,V. and Modrich,P. (1998) J. Biol. Chem., 273, 9197–9201. [DOI] [PubMed] [Google Scholar]
- 14.Hall M.C., Jordan,J.R. and Matson,S.W. (1998) EMBO J., 17, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viswanathan M. and Lovett,S.T. (1998) Genetics, 149, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris R.S., Ross,K.J., Lombardo,M.-J. and Rosenberg,S.M. (1998) J. Bacteriol., 180, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper D.L., Lahue,R.S. and Modrich,P. (1993) J. Biol. Chem., 268, 11823–11829. [PubMed] [Google Scholar]
- 18.Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorbalenya A.E. and Koonin,E.V. (1990) J. Mol. Biol., 213, 583–591. [DOI] [PubMed] [Google Scholar]
- 20.Malkov V.A., Biswas,I., Camerini-Otero,R.D. and Hsieh,P. (1997) J. Biol. Chem., 272, 23811–23817. [DOI] [PubMed] [Google Scholar]
- 21.Bowers J., Sokolsky,T., Quach,T. and Alani,E. (1999) J. Biol. Chem., 274, 16115–16125. [DOI] [PubMed] [Google Scholar]
- 22.Das Gupta R. and Kolodner,R.D. (2000) Nature Genet., 24, 53–56. [DOI] [PubMed] [Google Scholar]
- 23.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 24.Miller J.H. (1992) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Giraud-Panis M.-J. and Lilley,D.M.J. (1998) J. Mol. Biol., 278, 117–133. [DOI] [PubMed] [Google Scholar]
- 26.Biswas I. and Hsieh,P. (1996) J. Biol. Chem., 271, 5040–5048. [DOI] [PubMed] [Google Scholar]
- 27.Feng G. and Winkler,E.W. (1995) Biotechniques, 19, 956–965. [PubMed] [Google Scholar]
- 28.Cupples C.G. and Miller,J.H. (1989) Proc. Natl Acad. Sci. USA, 86, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjornson K.P., Allen,D.J. and Modrich,P. (2000) Biochemistry, 39, 3176–3183. [DOI] [PubMed] [Google Scholar]
- 30.Kolodner R.D. and Marsischky,G.T. (1999) Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 31.Haber L.T. and Walker,G.C. (1991) EMBO J., 9, 2707–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worth L.W., Bader,T., Yang,J. and Clark,S. (1998) J. Biol. Chem., 273, 23176–23182. [DOI] [PubMed] [Google Scholar]
- 33.Obmolova G., Ban,C., Hsieh,P. and Yang,W. (2000) Nature, in press. [Google Scholar]
- 34.Biswas I. and Hsieh,P. (1997) J. Biol. Chem., 272, 13355–13364. [DOI] [PubMed] [Google Scholar]
- 35.Lamers M.H., Perrakis,A., Enzlin,J.H., Winterwerp,H.H.K., de Wind,N. and Sixma,T.K. (2000) Nature., in press. [DOI] [PubMed] [Google Scholar]