

Abstract
Chronic inflammation associated with lung cancers contributes to immunosuppressive tumor microenvironments, reducing CD8+ T-cell function and leading to poor patient outcomes. A disintegrin and metalloprotease domain 9 (ADAM9) promotes cancer progression. Here, we aim to elucidate the role of ADAM9 in the immunosuppressive tumor microenvironment. A bioinformatic analysis of TIMER2.0 was used to investigate the correlation of ADAM9 and to infiltrate immune cells in the human lung cancer database and mouse lung tumor samples. Flow cytometry, immunohistochemistry, and RNA sequencing (RNA-seq) were performed to investigate the ADAM9-mediated immunosuppressive microenvironment. The coculture system of lung cancer cells with immune cells, cytokine array assays, and proteomic approach was used to investigate the mechanism. By analyzing the human LUAD database and the mouse lung cancer models, we showed that ADAM9 was associated with the immunosuppressive microenvironment. Additionally, ADAM9 released IL6 protein from cancer cells to inhibit IL12p40 secretion from dendritic cells, therefore leading to dendritic cell dysfunction and further affecting T-cell functions. Proteomic analysis indicated that ADAM9 promoted cholesterol biosynthesis and increased IL6-STAT3 signaling. Mechanistically, ADAM9 reduced the protein stability of LDLR, resulting in reduced cholesterol uptake and induced cholesterol biosynthesis. Moreover, LDLR reduction enhanced IL6-STAT3 activation. We reveal that ADAM9 has a novel biological function that drives the immunosuppressive tumor microenvironment by linking lung cancer’s metabolic and signaling axes. Thus, by targeting ADAM9 an innovative and promising therapeutic opportunity was indicated for regulating the immunosuppression of lung cancer.
Keywords: ADAM9, STAT3, IL6, cholesterol, immune suppression
Introduction
Generally, the immune system is considered a well-trained network of cells and organs working to detect and attack pathogens and abnormal microbes in hosts. In the tumor environment, the constant stimulation of cell debris and unrepaired tissue damage usually cause chronic inflammation during cancer development. Chronic inflammation can potentially weaken the therapeutic effect of cancer immunotherapy [1]. Interleukin 6 (IL6) is a pleiotropic cytokine involved in inflammation, metabolism, and many fundamental processes. Through activation of signal transducer, transcription 3 (STAT3), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), IL6 promotes cancer cell survival, metastasis, angiogenesis, and immunosuppression [2]. IL6 stimulates STAT3 activation to reduce the expression of interleukin 12 (IL12) and major histocompatibility complex (MHC) in dendritic cells. Therefore, dendritic cells fail to initiate a T-cell response against cancer cells. Since STAT3 disrupts the anticancer immune response, blockade of IL6-STAT3 signaling pathway has been initiated to improve the efficacy of immunotherapy [3]. AZD9150 is an antisense oligonucleotide inhibitor of STAT3 that has been shown to exhibit antitumor activity in non-small cell lung cancer (NSCLC) patients [4]. These studies collectively highlight the vital role of STAT3 in cancer progression. However, the unidentified cellular factor(s) derived from IL6-STAT3 signaling pathway in cancer cells needs to be investigated.
Reprogramming of cellular metabolism is one of the emerging hallmarks in cancers [5]. Substantially, the induction of cholesterol biosynthesis contributes to lung cancer progression [6]. NSCLC tumors with low infiltration of immune cells presented a significant increase in cholesterol biosynthesis [7]. Cholesterol-depleting drugs, such as statins, have been demonstrated to improve the efficacy of immunotherapy. Notably, cholesterol-depleting drugs could also decrease IL6 secretion and reduce IL6-mediated inflammation [8,9]. However, the molecular basis of the cholesterol-depleting drugs functioning in IL6 is still unclear.
ADAM9 has been shown to regulate cell-cell and cell-matrix interactions and promote cancer cell metastasis [10]. Our previous study provided evidence that ADAM9 promoted lung cancer metastasis to the brain by upregulating CUB-domain-containing protein 1 (CDCP1), a regulator of anoikis resistance [11]. Additionally, ADAM9 was associated with vascular remodeling and tumor angiogenesis by increasing vascular endothelial growth factor-A (VEGF-A) and angiopoietin-2 (ANGPT2) expression. Clinically, ADAM9 expression was correlated with poor outcomes in lung adenocarcinoma patients [12]. Moreover, ADAM9 has been shown to affect the cancer cells of the immune system. For example, ADAM9 sheds MHC class I polypeptide-related sequence A (MICA), a stress-induced ligand for the natural killer Group 2 member D (NKG2D) receptor, leading to the escape of a cancer cell from natural killer (NK) cells [13]. Whether ADAM9 influences other immune cells in the tumor microenvironment is unclear.
In this study, we reveal that ADAM9 stimulates the immunosuppressive tumor environment in lung cancer through activation of IL6-STAT3 signaling and inhibition of IL12p40 secretion and MHC class II expression in dendritic cells. Notably, ADAM9 reduces low-density lipoprotein receptor (LDLR) expression to further induce intracellular cholesterol biosynthesis in lung cancer cells and led to the activation of IL6-STAT3 signaling.
Materials and methods
Cell culture and reagents
Human lung adenocarcinoma cell lines Bm7 (RRID: CVCL_A5BS) and Brm were used as previously described [11]. The mouse lung adenocarcinoma cell line TC-1 was used as previously described [14]. Bm7 and Brm cells were cultured in DF12 with 10% FBS, penicillin, and streptomycin. Human lung adenocarcinoma A549 cells (RRID: CVCL_A549; ATCC, CCL-185), TC1 cells and mouse macrophage Raw264.7 cells (ATCC, TIB-71) were cultured in RPMI 1640 with 10% FBS, penicillin, and streptomycin. All cell lines were free of mycoplasma contamination and have been authenticated by short tandem repeat genotyping. Human recombinant IL6 and M-CSF were obtained from PeproTech (Rosemont Illinois, USA). Mouse recombinant IFNγ, IL6, M-CSF, and GM-CSF were purchased from PeproTech. Human recombinant GM-CSF and IL4 were obtained from SinoBiological (Beijing, China). Fatostatin, Stattic, chloroquine (CQ), cycloheximide (CHX), MG-132, LPS and simvastatin were purchased from Sigma-Aldrich (St. Louis, USA). Purified NA/LE Rat IgG isotype control and GolgiStop were obtained from BD Bioscience (California, USA). Bodipy-cholesterol was purchased from Cayman (Michigan, USA). Anti-mouse CD3 antibody, ultra-LEAF-purified anti-mouse CD28 antibody, and ultra-LEAF-purified anti-mouse IL-6 antibody were purchased from Biolegend (California, USA).
Gene knockdown and knockout
Gene knockdown of human ADAM9 was established using two lentiviral vectors (clone E: TRCN0000046982; clone H: TRCN0000290528). Gene knockdown of human LDLR was established using three lentiviral vectors (clone A: TRCN0000262146; clone C: TRCN0000282124). These plasmids were obtained from the National RNAi Core Facility, Academia Sinica (Taipei, Taiwan). The CRISPR RNA-guided Cas9 nuclease gene targeting system was used to knock out the ADAM9 gene in mouse TC1 and human Brm lung cancer cells with a ToolGen kit (ToolGen Inc., City and Country).
Animal studies
For subcutaneous tumor models, TC1 cells (2.5 × 105) were injected into the right and left flanks of six-week-old C57BL/6 mice (National Laboratory Animal Center, Taipei, Taiwan). Tumor volume was calculated using the following equation: tumor volume = length × width × width/2. All animal experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee of China Medical University (CMUIACUC-2018-001 and CMUIACUC-2022-125).
RNA sequencing and analysis
RNA sequencing was performed by Genomics Biotech Co., Ltd. (Taipei, Taiwan) as previously described [15]. Briefly, total RNA was extracted by TRIzol Reagent (Invitrogen, Massachusetts, USA), and the quality was measured using an RNA6000 Nano kit on an Agilent 2100 Bioanalyzer (Agilent, California, USA) for further library construction for next-generation sequencing. The functional enrichment analysis of the differentially expressed genes was performed as previously described [16].
Real-time quantitative PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen), and cDNA synthesis was performed using the SuperScript™ III Reverse Transcriptase Kit (Thermo Fisher Scientific). Quantitative PCR was performed by ChamQ Universal SYBR qPCR Master Mix (Vazyme, Jiangsu, China). The sequences of the gene-specific primers are listed in Table S1.
Immunohistochemistry (IHC) staining
IHC staining was performed with a CD3 antibody (Cat. No. ab16669; Abcam, Cambridge, UK). The horseradish peroxidase-conjugated avidin-biotin complex (ABC) was obtained from the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) and AEC chromogen (Vector Laboratories). The sections were counterstained with hematoxylin/mounted, and the positively stained cells were measured with ImageJ software (version 1.52a, Wayne Rasband, National Institutes of Health, USA).
Quantification of cytokines by ELISA
Human IL6 was measured using a human IL6 ELISA Kit (OptEIA™ Set for human IL-6; BD Biosciences). Human IL12p40 was measured using a human IL12p40 ELISA Kit (OptEIA™ Set for human IL-12 (p40); BD Biosciences). Murine IFNγ was measured using a murine IFNγ ELISA Kit (OptEIA™ Mouse IFN-γ (AN-18) ELISA Set; BD Biosciences). Murine IL6 was measured using a murine IL6 ELISA Kit (OptEIA™ Set for mouse IL-6; BD Biosciences). Murine IL12p40 was measured using a murine IL12p40 ELISA Kit (OptEIA™ Set for mouse IL12 (p40); BD Biosciences). All experiments were performed according to the manufacturer’s instructions.
Analyses of in vitro dendritic cell and macrophage differentiation
Mouse bone marrow cells were collected from six-week-old C57BL/6 mice. Mouse bone marrow cells (1 × 106 cells/ml) were subsequently treated with 10 ng/ml mouse GM-CSF on Day 1, 4, and 6. The cells were differentiated into dendritic cells on Day 8 [17]. Additionally, mouse bone marrow cells were treated with 10 ng/ml mouse M-CSF on Day 1, Day 4, and Day 6. The cells were then differentiated into macrophages on Day 8 [18]. Peripheral blood mononuclear cells were collected from healthy human donors. The study methodologies were approved by the Institutional Review Board of China Medical University Hospital (CMUH111-REC3-153 (AR-1)). CD14+ cells were enriched using BD IMag™ Anti-human CD14 Magnetic Particles according to the manufacturer’s instructions. CD14+ cells (1 × 106 cells/ml) were treated with 50 ng/ml human M-CSF on Day 1, 4, and 6. Subsequently, CD14+ cells were differentiated into macrophages on Day 8 [19]. In addition, CD14+ cells were treated with 800 U/ml human GM-CSF and 500 U/ml human IL4 on Day 1 and 500 U/ml human GM-CSF and 300 U/ml human IL4 on Day 4 and 6. CD14+ cells were then differentiated into dendritic cells on Day 8 [20].
Antibodies for Western blotting
The following antibodies were used for Western blotting: anti-ADAM9 antibody (GTX130081, GeneTex, California, USA), anti-STAT3 antibody (GTX104616, GeneTex), anti-phospho-Tyr705-STAT3 antibody (GTX118000, GeneTex), anti-SRC antibody (#2108, Cell signaling, Massachusetts, USA), anti-phospho-Tyr416-SRC antibody (#6943, Cell signaling), anti-phospho-Tyr418-SRC antibody (44-660G, Thermo Fisher Scientific, Massachusetts, USA), anti-AKT antibody (#2920, Cell signaling), anti-phospho-Ser473-AKT antibody (#9271, Cell signaling), anti-phospho-Thr308-AKT antibody (#9275, Cell signaling), anti-phospho-(Tyr1022/1023) JAK1 antibody (44-422, Invitrogen), anti-phospho-(Tyr1007/1008) JAK2 antibody (#3771, Cell signaling), anti-NF-kB p65 antibody (ab16502, Abcam), anti-phospho-Ser536-NF-kB p65 (#3033, Cell signaling), anti-LDLR antibody (10785-1-AP, ProteinTech, Chicago, USA), anti-PCSK9 antibody (sc-515082, Santa Cruz, Texas, USA), anti-SREBP2 (557037, BD Biosciences), anti-GADPH antibody (10494-1-AP, ProteinTech), anti-β-actin antibody (ab8226, Abcam).
Flow cytometry
The antibodies used for flow cytometry were anti-mouse CD45 (Cat. No. 561037; BD Biosciences), anti-mouse I-A+I-E/MHC-II (Cat. No. 107630; BioLegend), anti-mouse CD8a (Cat. No. 553033; BD Biosciences), anti-mouse CD11c (Cat. No. 553802; BD Biosciences), anti-mouse CD40 (Cat. No. 124612; BioLegend), anti-human/mouse Granzyme B (Cat. No. 515406; BioLegend), and anti-human HLA-DR (Cat. No. 559866; BD Biosciences). Cells were incubated with individual antibodies and placed on ice for 30 min. Subsequently, intracellular staining was performed by Cytofix/Cytoperm and Perm/Wash buffer (BD Biosciences) according to the manufacturer’s instructions. All flow cytometry data were acquired on BD FACSVerse™ instruments (BD Biosciences).
Isolation of membrane proteins and proteomics
TC1 cell membrane proteins were isolated by a Compartmental Protein Extraction Kit (K3012010, BioChain, California, USA) according to the manufacturer’s instructions. The protein samples were separated using 10% SDS-PAGE. After suitable cutting (<1 mm3), the gel pieces were digested with produce tryptic peptides and further identified as previously described [21]. Briefly, an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) equipped with an Ultimate 3000 RSLC system (Dionex, California, USA) and a nanoelectrospray ion source (New Objective, Massachusetts, USA) were applied for MS analysis. The proteins that exhibited at least a 0.66-fold decrease and ratio Adj P Value <0.01 in control and ADAM9 KO TC1 cells (Ctr v.s A9KO) were identified.
Statistical analysis
Student’s t test was applied for at least three independent biological replicates to calculate significance. McNemar’s test and Fisher’s exact test were applied for IHC analysis.
Results
ADAM9 is associated with the immunosuppressive environment
To investigate whether ADAM9 was associated with tumor immunity, we first analyzed the correlation between ADAM9 and tumor-infiltrated immune cells using the TIMER2.0 database [22]. As shown in Figure 1A, ADAM9 expression was significantly correlated with reduced CD8+ T-cell infiltration in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), which was demonstrated in patient specimens (Figure 1A). Subsequently, we established syngeneic lung tumor models by subcutaneously injecting mouse TC1 cells into immune-competent C57BL/6 mice. Our results demonstrated a reduced tumor growth rate in the ADAM9 knockout (A9KO) group compared to the control group (Figure 1B). Consistent with the clinical relevance, ADAM9 expression reduced tumor-infiltrated CD3+ T cells compared to the A9KO group (Figure S1A). Furthermore, ADAM9 reduced the infiltration of CD8+ T cells, specifically cytotoxic CD8+ T cells (GranzB+CD8+), as determined by flow cytometry analysis aimed at characterizing T cell subtypes within tumors (Figure 1C). To further define the molecular mechanisms by which ADAM9 expression mediates immune regulation, we performed RNA sequencing and then analyzed the expression profile of associated genes in subcutaneous tumors from mice bearing WT or A9KO TC1 cells. Using TIMER2.0 to determine the abundance of immune cell types in tumors, the results showed elevated CD4+ and CD8+ T cell, B cell, and NK cell infiltration in A9KO TC1 tumors compared to control WT tumors (Figure S1B). This result indicated that ADAM9 was related to the immune suppressive tumor microenvironment (TME). Moreover, to explore the ADAM9-mediated genes involved in regulating immunity in the tumor microenvironment, named TME-modulated genes, we compared the transcriptomes of WT tumors with those of A9KO tumors. Specifically, we focused on the differential genes from the compared tumor groups that showed no change in cultured cancer cell lines to represent the TME-modulated genes. The enriched pathway analysis of TME-modulated genes revealed that they were involved in the immune response, including the regulation of cytokine production, lymphocyte activation, and IFNγ production (Figure 1D). Interestingly, IL12B (IL12p40), a subunit of IL12, was detected in the enriched biological functions of TME-modulated genes (Table 1). IL12p40 is produced by immune cells, including dendritic cells [23], and is a pleiotropic cytokine that can regulate dendritic cell migration and naïve T-cell activation [24]. Elevated IL12p40 proteins were detected in A9KO TC1 tumors compared to control tumors from C57BL/6 mice (Figure 1E). Moreover, significantly increased dendritic cell (CD45+CD11c+) infiltration was detected in A9KO TC1 tumors (Figure S1C). Altogether, ADAM9 is associated with the immune suppressive cells and mediates reduced IL12p40 proteins in the tumor microenvironment.
Figure 1.
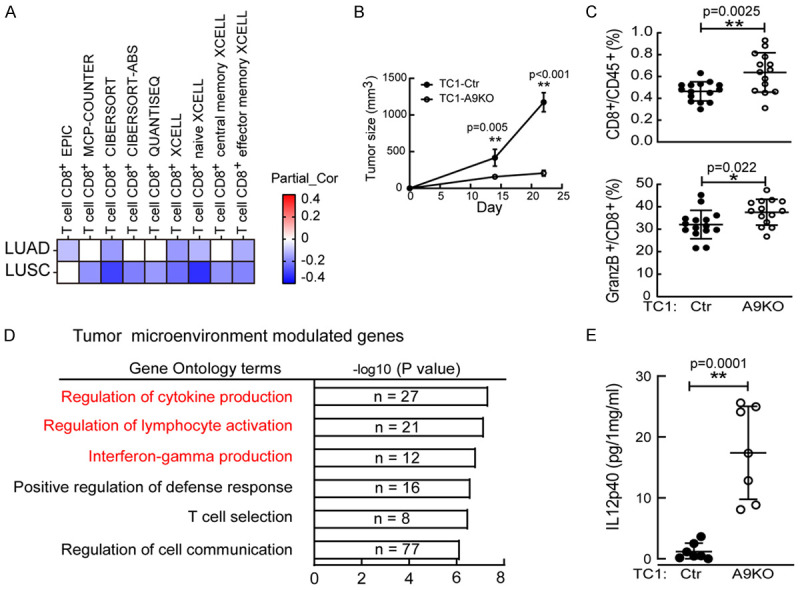
ADAM9 is associated with the immunosuppressive environment. A. The heatmap depicts the Spearman correlation analysis of CD8+ T-cell infiltration and ADAM9 gene expression in lung adenocarcinoma (LUAD, n = 515) and lung squamous cell carcinoma (LUSC, n = 501) by multiple algorithms. B. Tumor size analysis. Wild-type (Ctr; n = 6) or ADAM9 knockout (A9KO; n = 6) TC1 cells were injected subcutaneously into C57BL/6 mice. C. Flow cytometry staining of CD8+ T cells and GranzB+CD8+ T cells in Ctr and A9KO TC1 tumors. D. Functional analysis of 3522 ADAM9-mediated genes influenced only in tumors but not in cultured cells. E. IL12p40 detection in lung tumors harvested from Ctr and A9KO TC1 C57BL/6 mice by ELISA. *P<0.05 and **P<0.01.
Table 1.
ADAM9-mediated genes involved in regulating immunity in tumor microenvironment (TME)
| GO.ID | Term | log10 (p-value) | Genes |
|---|---|---|---|
| GO:0001817 | Regulation of cytokine production | 7.37 | Ubash3a, Il17f, Cd209d, Irf4, Cd2, Il12b, Cd27, Gbp4, Cd3e, Card11, Cxcl5, Clec2i, Il6, Txk, Ccr7, Clec9a, Clec7a, Ffar2, Il12rb1, Klre1, Klrk1, Inhbb, Il27, Crtam, Tlr11, Nlrp12, Ltf |
| GO:0051249 | Regulation of lymphocyte activation | 7.19 | Il12b, Cd27, Ikzf3, Cd3e, Card11, Samsn1, Clec2i, Il6, H2-Oa, Dpp4, Ccr7, Slamf7, Ctla4, Tbx21, Il12rb1, Slfn1, Btla, Flt3, Il27, Cd5, Tnfsf11 |
| GO:0032609 | Interferon-gamma production | 6.85 | Itk, Cd2, Il12b, Cd27, Cd3e, Txk, Ccr7, Il12rb1, Klre1, Klrk1, Avpr2, Il27 |
| GO:0031349 | Positive regulation of defense response | 6.62 | Irf4, Il12b, Klrb1c, Sh2d1a, Il6, Ccr7, Gfi1, Clec7a, Ffar2, Klre1, Klrk1, Crtam, Tlr11, S100a9, Ltf, Tnfsf11 |
| GO:0045058 | T cell selection | 6.52 | Themis, Bcl11b, Irf4, Cd3d, Cd3e, Card11, Il6, Ccr7 |
| GO:0010646 | Regulation of cell communication | 6.17 | Ubash3a, Il17f, Epha7, Rgs9bp, Rasd2, Fasl, Adamts3, Adora1, Eps8l1, Irf4, Bmp7, Il12b, Cd27, Grm7, Bmpr1b, Tbx18, Npnt, Cd3e, Card11, Cxcl5, Gata1, Csf3, Srpx, Stap1, Cxxc4, Gsc, Nrk, Cthrc1, Il6, Txk, Ccr7, Cacna1e, Esr1, Fgf23, Dennd2d, Gfi1, Bcl2a1d, Rgl3, Smad6, Ffar2, Sh2d2a, Gpr158, Cd8a, Acap1, Ngfr, Arhgap15, Sla2, Rgs18, Rgs17, Bcl2l14, Ubd, Igfbp5, Bmp5, Inhbb, P2ry1, Ucn2, Axin2, Ncam1, Lama2, Nlrp12, Ppargc1a, Ephb1, Cacng7, Sectm1a, Cdh13, S100a9, Cspg5, Ltf, Cplx4, Chga, Bik, Foxa1, Blk, Cysltr2, Sh3tc2, Adamts12, Tnfsf11 |
| GO:0016337 | Single organismal cell-cell adhesion | 6.09 | Epha7, Cd2, Mycn, Acan, Bmp7, Bmpr1b, Tbx18, Npnt, Gata1, Lama3, Dpp4, Podxl2, Clec7a, Col11a1, Ttyh1, Slc7a11, Ncam1, S100a9, Rnase10, Tnfsf11 |
| GO:0042127 | Regulation of cell proliferation | 5.96 | Fasl, Adora1, Bcl11b, Tnmd, Mycn, Bmp7, Il12b, Tbx18, Ikzf3, Cd3e, Card11, Gata1, Csf3, Amica1, Srpx, Nrk, Cthrc1, Clec2i, Aldh3a1, Il6, Kcna5, Dpp4, Ccr7, Esr1, Ctla4, Smad6, Il12rb1, Tesc, Avpr2, Slfn1, Ngfr, Plk5, Tbx2, Btla, Igfbp5, Bmp5, Axin2, Flt3, Il27, Ncam1, Foxo6, Ppargc1a, Cdh13, Ltf, Npr3 |
ADAM9 inhibits IL12p40 secretion from dendritic cells
Both T-cell receptor and CD40 ligand (CD40L) signaling are crucial for T-cell activation, and their engagement on MHC class II and CD40 enhances dendritic cells to secrete IL12p40 [25]. First, we measured the surface protein levels of CD40 and MHC class II (IA/IE) on tumor-infiltrated dendritic cells which showed increased levels in the A9KO group (Figure S2A). Next, we examined if ADAM9 in cancer cells inhibited IL12p40 secretion from dendritic cells. To address this question, bone marrow-derived dendritic cells (BMDCs) were cocultured with WT or A9KO TC1 cells. Increases in IL12p40 secretion and MHC class II expression in BMDCs were observed in the A9KO group (Figure 2A). Additionally, the conditioned medium of cultured A9KO TC1 cells also increased IL12p40 and MHC class II expression in BMDCs (Figure 2B). This finding suggests that ADAM9 expression in cancer cells regulates the function of dendritic cells through secreted factors. Consistent with mouse TC1 cells, conditioned medium collected from A9KO human lung cancer Brm cells induced IL12p40 secretion from human monocyte-derived dendritic cells (MDDCs) (Figure 2C). To investigate whether coculture of A9KO lung cancer cells with BMDCs can activate BMDC function for T-cell activation, we established a lymphocyte-BMDC coculture model. Consistent with the observations from the in vivo animal model, A9KO TC1 cells stimulated BMDCs to increase the proportion of cytotoxic GramzB+CD8+ T cells (Figure 2D). In addition to affecting dendritic cells, ADAM9 also manipulated IL12p40 production in macrophages (Figure S2B and S2C). Altogether, through inhibiting IL12p40 secretion from dendritic cells and macrophages by unidentified secreted factors from lung cancer cells, ADAM9 can further affect CD8+ T-cell activation.
Figure 2.
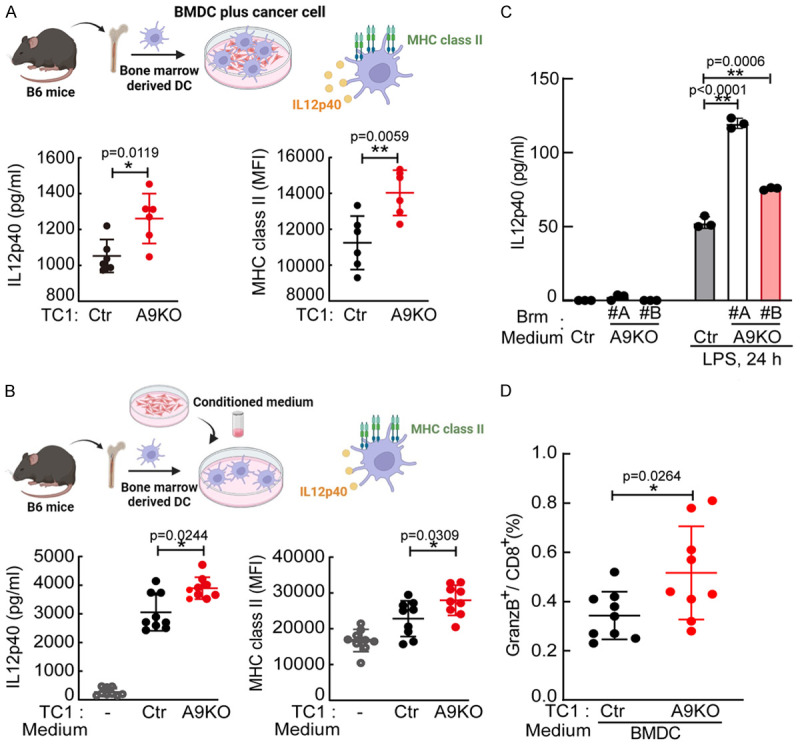
ADAM9 inhibits IL12p40 secretion from dendritic cells. A. Mouse bone marrow-derived dendritic cells (BMDCs) were cultured with Ctr or A9KO TC1 cells (1 × 105 cells). Conditioned medium was collected to detect the IL12p40 concentration by ELISA. BMDCs were harvested to detect MHC class II expression by flow cytometry staining. B. BMDCs were treated with conditioned medium collected from Ctr or A9KO TC1 cells. After replacing the medium, the supernatant of TC1-educated BMDCs cultured for 48 h was collected to detect IL12p40. BMDCs were harvested to detect MHC class II expression by flow cytometry staining. C. Monocyte-derived dendritic cells (MDDCs) were treated with conditioned medium collected from Ctr or A9KO Brm cells for 24 h. After replacing the medium, MDDCs were stimulated with 100 ng/ml LPS for 24 h. Conditioned medium was collected to detect the IL12p40 concentration by ELISA. D. BMDCs were treated with conditioned medium from Ctr or A9KO TC1 cells for 24 h. Lymphocytes from tumor-free mouse inguinal lymph nodes were pretreated with anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) for 48 hr. TC1-educated BMDCs were cocultured with active lymphocytes (5:1) for 48 h. Cells were harvested to detect the percentage of GranzB+CD8+ T cells. *P<0.05 and **P<0.01.
ADAM9-mediated IL6 secretion from lung cancer cells inhibits IL12p40 secretion from dendritic cells
Considering that secreted factors from lung cancer cells could regulate the function of antigen-presenting cells, we subsequently used a cytokine array assay to identify potential composites between conditioned media from control and ADAM9 KO TC1 cells. IL6 was the top differential cytokine between control and ADAM9 KO TC1 cells (Figure S3A), and the phenomenon of ADAM9 promoting IL6 secretion was confirmed in both normal and starvation culture conditions (Figure 3A). Ectopic expression of ADAM9 wild type and catalytic mutant (E348A) in lung cancer cells increased IL6 secretion, despite slightly lower IL6 in catalytic mutant compared with wild type (Figures 3B and S3B), suggesting that ADAM9-induced IL6 production was independent of its catalytic activity. In particular, a weakly positive correlation (R = 0.15, P = 0.00066) between the expression of ADAM9 and IL6 in the LUAD patient database by GEPIA was found (http://gepia.cancer-pku.cn/) (Figure 3C). To further examine whether IL6 could control BMDC function, BMDCs were treated with recombinant IL6. Our findings revealed that IL6 inhibited IL12p40 secretion from BMDCs (Figure S3C) and downregulated MHC class II expression in BMDCs (Figure S3D). Furthermore, IL6 blockade using a neutralizing antibody significantly augmented IL12p40 secretion from TC1-educated BMDCs (Figure 3D) and moderately enhanced MHC class II expression (Figure S3E). IL6 has been shown to activate STAT3 signaling by increasing phospho-STAT3 (pSTAT3), a transcription factor turning on downstream gene expression [26]. In addition, pSTAT3 contributes to IL6 production under starvation conditions [27]. We found that Stattic, a STAT3 inhibitor, increased IL12p40 secretion from TC1-educated BMDCs (Figure S3C), but showed no change in MHC class II expression (Figure S3D). A previous paper reported that IL6 promoted arginase 1 (ARG1) expression and inhibited the antigen presenting ability of dendritic cells [28]. Interestingly, conditioned medium collected from ADAM9 KO TC1 cells exhibited a reduction in ARG1 expression in BMDCs (Figure 3E). Furthermore, the expression of several immunosuppressive molecules, including CD274 (PD-L1) and cholesterol 25-hydroxylase (CH25H), was also decreased in ADAM9 KO educated BMDCs. Moreover, these immunosuppressive gene levels were reduced in Stattic treatment. Collectively, ADAM9-mediated IL6 from lung cancer cells reduces IL12p40 secretion and MHC class II expression in dendritic cells.
Figure 3.
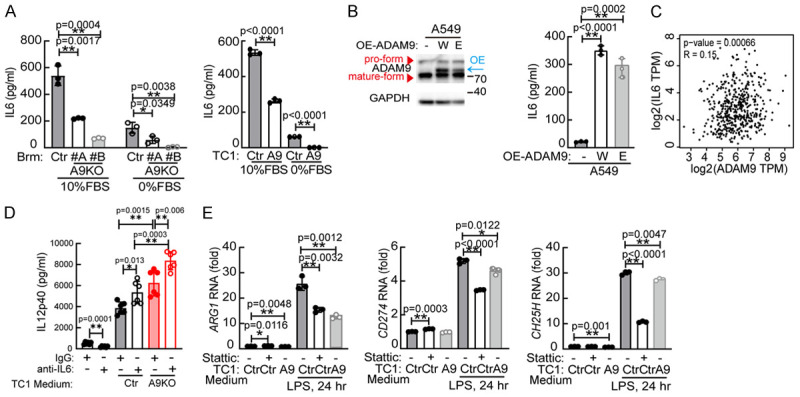
ADAM9-mediated IL6 secretion from lung cancer cells inhibits IL12p40 secretion from dendritic cells. A. IL6 detection in conditioned medium from Ctr or A9KO lung cancer cells. B. A549 cells were transfected with wild-type (W) and E348A mutant (E) ADAM9 plasmids for 24 h, and then the supernatant was collected to detect the IL6 concentration. C. Correlation between ADAM9 and IL6 was determined by GEPIA from lung adenocarcinoma (LUAD, n = 515). D. BMDCs were treated with conditioned medium from TC1 cells in the presence of anti-IL6 (100 ng/ml) blocking antibody or control IgG antibody for 24 h. After replacing the medium, the supernatant of TC1-educated BMDCs cultured for 48 h was collected to detect IL12p40. E. BMDCs were pretreated with 2 μM Stattic for 1 h. Subsequently, the cells were treated with conditioned medium from Ctr or A9 (ADAM9 knockout) TC1 cells for 24 h. After replacing the medium, TC1-educated BMDCs were stimulated with 100 ng/ml LPS for 24 h. Cells were harvested to detect RNA expression. *P<0.05 and **P<0.01.
ADAM9 promotes IL6-STAT3 signaling in lung cancer cells
Next, we explored whether ADAM9 mediated IL6-STAT3 signaling in lung cancer cells. We found a positive correlation (R = 0.39, P<0.01) between ADAM9 and STAT3 in LUAD tumors from the GEPIA database (http://gepia.cancer-pku.cn/) (Figure 4A). IL6-related genes, including IL6, STAT3, CCAAT-enhancer-binding proteins (C/EBP) [29], and IL6Rβ (IL6ST), were downregulated in ADAM9 KO cells upon IL6 stimulation (Figure 4B). Moreover, ADAM9 silencing inhibited IL6Rβ RNA (IL6ST) and IL6Rβ protein (GP130) expression in lung cancer cells (Figures 4C and S4A). Furthermore, the protein levels of STAT3 and pSTAT3 were elevated in control TC1 tumor tissues compared to ADAM9 KO tumor tissues from mice models (Figure 4D). These results demonstrate that ADAM9 is related to IL6-mediated STAT3 expression and activation. ADAM9 proteins were undetectable in ADAM9 knockout TC1 cells compared to control cells (Figure 4F), confirming the successful maintenance of the ADAM9 knockout condition. ADAM9 demonstrates widespread expression in human tissues and is observed in various cell types, including monocytes, macrophages, neutrophils, keratinocytes, and fibroblasts [30]. In the ADAM9 KO tumor group, low levels of ADAM9 proteins were still detected throughout the entire tumor, encompassing both cancerous and non-cancerous cells (Figure 4D). Therefore, it may originate from non-cancerous cells within the tumors. Fetal bovine serum (FBS) was reported to induce STAT3 phosphorylation/activation and nuclear accumulation for cell proliferation [31]. Indeed, we found STAT3 phosphorylation is higher in 10% FBS culture condition than 0% FBS culture condition after IL6 stimulation (Figure 4E). Moreover, pSTAT3 proteins were suppressed in ADAM9-silenced cells compared to control cells after IL6 stimulation under starvation conditions (Figure 4E and 4F). Of note, IL6 promoted ADAM9 protein expression in lung cancer cells, suggesting an amplification loop between ADAM9 and IL6. However, the phenomenon of positive correlation of ADAM9 and STAT3 is obviously detected in IL6 stimulation under starvation. In addition, we found that ADAM9 knockdown reduced STAT3 phosphorylation but not suppressed the STAT3 proteins in Bm7 cells (Figure S4B), suggesting unclear mechanism may contribute to ADAM9 regulating STAT3 proteins, particularly under starvation. Overall, we reveal that ADAM9 increases IL6-STAT3 signaling and forms a signaling loop between ADAM9 and IL6 in lung cancer cells.
Figure 4.
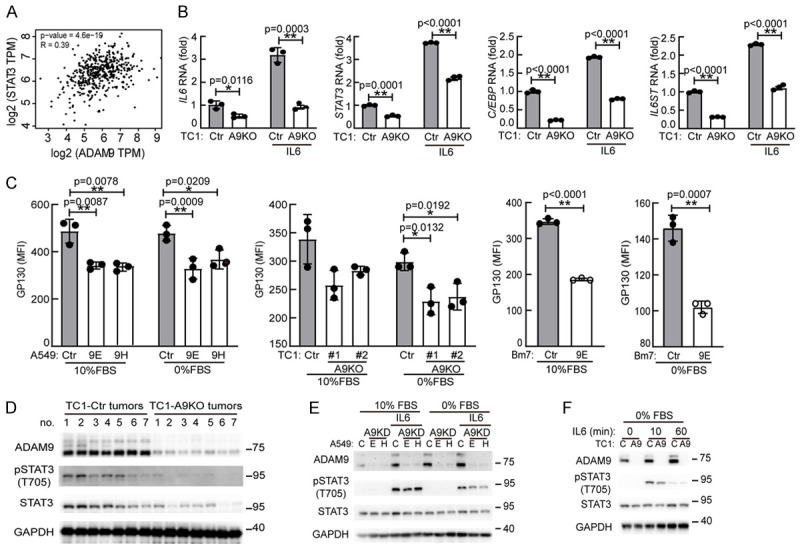
ADAM9 promotes IL6-STAT3 signaling in lung cancer cells. A. Correlation between ADAM9 and STAT3 expression was determined by GEPIA from lung adenocarcinoma (LUAD, n = 515). B. RT-qPCR of IL6 response genes. Ctr and A9KO TC1 cells were cultured in 0% FBS and treated with 50 ng/ml IL6 for 2 h. C. Ctr- and ADAM9-silenced cells were cultured in 10% and 0% FBS for 24 h. Cells were harvested to detect IL6 receptor β (GP130) proteins by flow cytometry staining. *P<0.05 and **P<0.01. D. Subcutaneous tumors from C57BL/6 (B6) mice were harvested. Western blot analysis was utilized to determine the indicated proteins. E. Western blot analysis of the indicated proteins. Ctr and ADAM9 knockdown (A9KD) A549 cells were cultured in 10% and 0% FBS with 50 ng/ml IL6 for 15 min. F. Western blot analysis of the indicated proteins. Wild-type (Ctr) and ADAM9 knockout (A9) TC1 cells were cultured in 0% FBS and treated with 50 ng/ml IL6.
ADAM9 upregulates cholesterol biosynthesis pathways for increasing IL6-STAT3 signaling
To further investigate the mechanism of ADAM9-mediated IL-6-STAT3 signaling under starvation, we identified the differential expression of membrane proteins from control and ADAM9-silenced cells under normal and starvation conditions by a comprehensive LC-MS/MS method. Remarkably, cholesterol biosynthesis-related proteins were significantly reduced in ADAM9-silenced cells, especially under starvation conditions (Figure 5A). Using a functional annotation tool, Database for Annotation Visualization and Integrated Discovery (DAVID), we identified a group of proteins involved in cholesterol biosynthesis, including squalene epoxidase (SQLE), NAD(P)-dependent steroid dehydrogenase-like (NSDHL) and lanosterol synthase (LSS), that were suppressed in ADAM9-silenced TC1 cells under starved condition (Figure 5B). We further validated and confirmed the RNA levels of these identified candidates, which were reduced in ADAM9-silenced lung cancer cells (Figure 5C and 5D). Additionally, the expression of these cholesterol biosynthesis-related genes was significantly elevated in ADAM9 KO lung cancer cells with ectopic expression of ADAM9 (Figure S5A). Hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is a rate-limiting enzyme catalyzing cholesterol biosynthesis [32], and sterol regulatory element-binding protein 2 (SREBP2) is a key transcription factor for cholesterol biosynthesis [33]. We detected lower RNA levels of HMGCR and SREBP2 genes in ADAM9-silenced cells under starvation (Figure S5B). Next, two selective inhibitors for cholesterol biosynthesis, simvastatin and fatostatin, were used to block HMGCR activity and SREBP2 maturation, respectively. Fatostatin inhibited SREBP2 more actively than simvastatin treatment, and reduced the IL6-stimulated pSTAT3 under starvation conditions (Figure 5E). Additionally, fatostatin reduced the transcriptional levels of NSDHL, SQLE, and LSS (Figure S5C) and STAT3 (Figure S5D). Overall, we demonstrated that ADAM9 enhanced cholesterol biosynthesis and led to an increase in IL6-STAT3 signaling.
Figure 5.
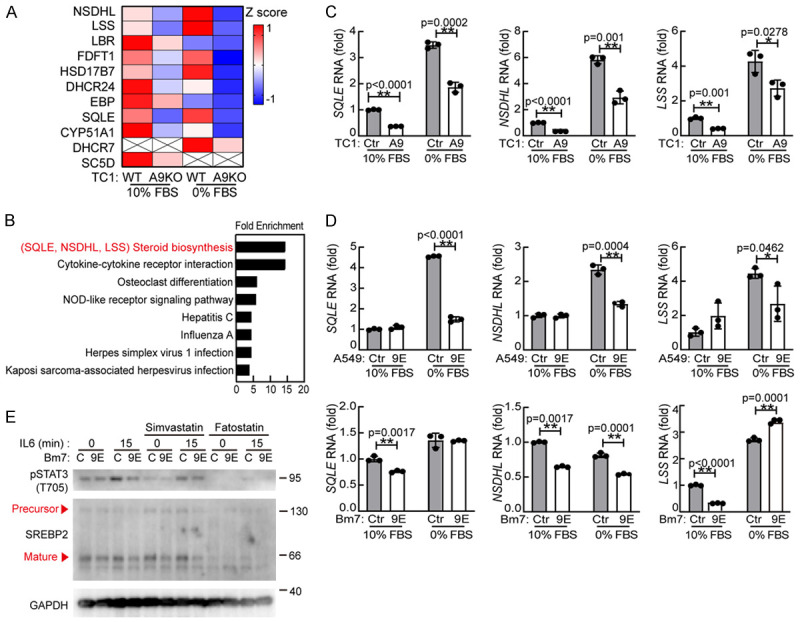
The cholesterol biosynthesis pathway mediates IL6-STAT3 signaling. A. LC-MS/MS analysis of the membrane proteins in WT or A9KO TC1 cells cultured in 10% or 0% FBS for 24 hr. B. The Database for Annotation Visualization and Integrated Discovery (DAVID) was used to analyze enriched functionally related protein groups under starvation conditions. C. Ctr and ADAM9 knockout (A9) TC1 cells were cultured in 10% FBS or 0% FBS for 24 h. RT-qPCR of cholesterol biosynthesis-related genes. D. Ctr and ADAM9 knockdown (9E) A549 and Bm7 cells were cultured in 10% FBS or 0% FBS for 24 hr. RT-qPCR of cholesterol biosynthesis-related genes. E. Wild-type (C) and ADAM9 knockdown (9E) Bm7 cells were cultured in 0% FBS and treated with simvastatin (5 μM) or fatostatin (20 μM). After 12 h, the cells were treated with 50 ng/ml IL6 for 15 min. *P<0.05 and **P<0.01.
LDLR inhibits cholesterol biosynthesis and IL6-STAT3 signaling
Cholesterol homeostasis is precisely controlled by de novo cholesterol biosynthesis and cholesterol transport [34]. LDLR is a critical cholesterol transporter that mediates cholesterol uptake and countervails cholesterol biosynthesis. Downregulation of LDLR has been shown to promote cholesterol biosynthesis-related gene expression in hepatocellular carcinoma [35]. We found ADAM9 KO increased LDLR protein levels while ectopic expression of ADAM9 reduced LDLR expression (Figure 6A). Next, we delineated whether LDLR regulates IL6 secretion in lung cancer cells. We found that starvation stimulated LDLR expression and ADAM9 KD increased LDLR proteins (Figure S6A). Notably, effective knock down of LDLR by shLDLR#A increased IL6 secretion in ADAM9 KD Bm7 lung cancer cells, while the IL6 levels remained high with no alteration in control Bm7 cells (Figure S6B). Similarly, downregulation of LDLR in ADAM9-silenced Brm lung cancer cells stimulated IL6 secretion (Figure 6B). Furthermore, the gene expression of the cholesterol biosynthesis pathway was increased in lung cancer cells under downregulation of LDLR (Figure 6C). Subsequently, we tested whether LDLR manipulates IL6-STAT3 signaling. The downregulation of LDLR enhanced IL6-stimulated STAT3 protein expression and activation, particularly in starvation (Figures 6D and S6C). Altogether, ADAM9 downregulated LDLR to induce IL6-STAT3 activation in lung cancer cells.
Figure 6.
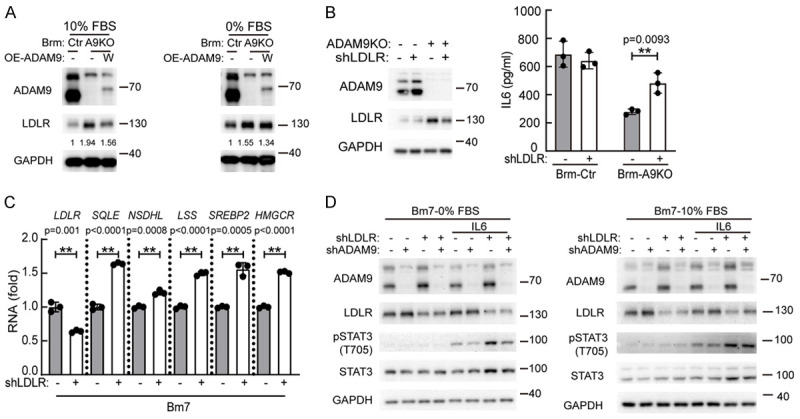
ADAM9-mediated LDLR reduction increases cholesterol biosynthesis and IL6-STAT3 signaling. A. Brm cells were transfected with ADAM9 plasmids. After 24 h, the cells were cultured in 10% FBS and 0% FBS. Western blot analysis of the indicated proteins. B. Brm cells were cultured in 10% FBS for 24 h and the medium was collected to detect the IL6 concentration. Cell lysates were analyzed by Western blot analysis. C. Bm7 cells were cultured in 10% FBS for 24 hr. RT-qPCR of cholesterol biosynthesis-related genes. D. Bm7 cells were cultured with 50 ng/ml IL6 for 15 min. Western blot analysis of the indicated proteins. *P<0.05 and **P<0.01.
ADAM9 reduces LDLR stability
Considering LDLR-mediated cholesterol uptake in cells, we subsequently used bodipy-cholesterol to trace cholesterol transport affected by ADAM9. ADAM9-silenced cells displayed higher cholesterol uptake levels than control cells by immunofluorescence staining (Figure 7A) and flow cytometry (Figure S7A). Conversely, the cholesterol efflux capacity was reduced in ADAM9-silenced cells or showed no significant change in different lung cancer cells (Figure S7B). This data indicated that ADAM9 silencing mainly promoted cholesterol uptake. Compared with control cells, ADAM9-silenced cells showed increased LDLR proteins, but the RNA levels of LDLR were decreased or unchanged (Figure S7C). Therefore, we speculated that this phenomenon could occur through posttranslational regulation. To address this question, a cycloheximide (CHX) chase assay was performed to detect LDLR protein degradation. As expected, LDLR proteins were reduced faster in control cells than in ADAM9-silenced cells (Figure 7B). Proprotein convertase subtilisin/kexin type-9 (PCSK9) is a downstream gene of SREBP2 and binds to LDLR for lysosomal degradation [36]. We found that PCSK9 proteins were reduced in ADAM9-silenced cells compared to control cells and that blockade of lysosomal degradation by chloroquine rescued LDLR proteins (Figure 7C). This result indicated that ADAM9 silencing in lung cancer cells diminished PCSK9-mediated LDLR degradation and maintained high levels of LDLR proteins in cells. Overall, we demonstrate that ADAM9 reduces LDLR protein stability through PCSK9-mediated LDLR in the lysosomal degradation pathway to promote cholesterol biosynthesis in lung cancer cells.
Figure 7.
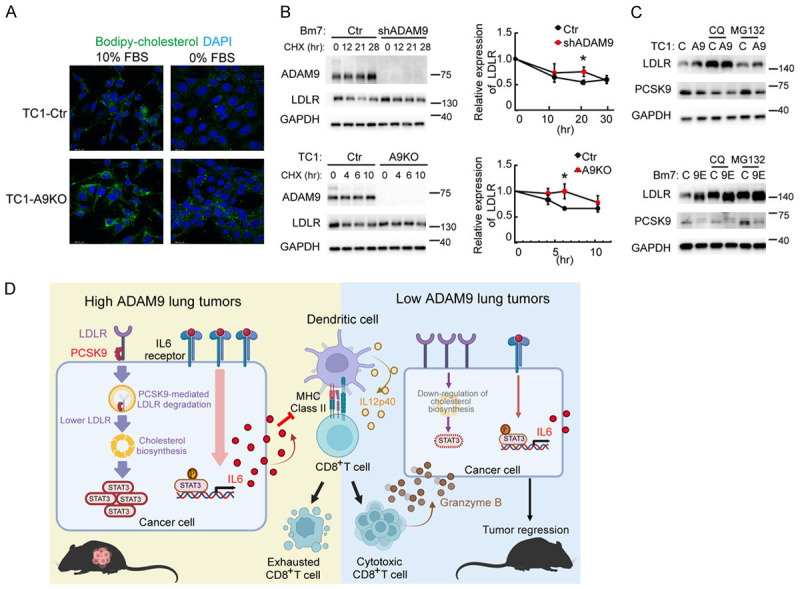
ADAM9 reduces LDLR stability. A. Immunofluorescence staining of Bodipy-cholesterol in Ctr and A9KO TC1 cells cultured in 0% or 10% FBS for 24 h. B. Ctr- or ADAM9-silenced Bm7 and TC1 cells were cultured in 10% FBS. Cells were treated with 50 μg/ml cycloheximide (CHX) at different times. *P<0.05 and **P<0.01, when comparing all data with the control group. C. Western blot analysis of the LDLR and PCSK9 proteins. Ctr- or ADAM9-silenced Bm7 and TC1 cells were cultured in 10% FBS and treated with 10 μM chloroquine (CQ) or 10 μg/ml MG132 for 24 h. D. A diagram depicts that ADAM9 abundant lung cancer inhibits LDLR protein expression to trigger the cholesterol biosynthesis pathway for STAT3 expression. Moreover, ADAM9 increases IL6-STAT3 signaling for IL6 secretion. Cancer cell-mediated IL6 inhibited IL12p40 secretion and antigen presentation from dendritic cells. Therefore, ADAM9-abundant lung cancer-educated dendritic cells failed to activate cytotoxic CD8+ T cells.
Discussion
Aggressive tumor cells usually enhance IL6-STAT3 signaling and impair the functional maturation of dendritic cells to activate T cells and elicit antitumor immunity [37]. In this study, we reveal for the first time that ADAM9 enhances the IL6-STAT3 signaling pathway for driving the immunosuppressive tumor environment (Figure 7D). Silencing ADAM9 in lung cancer cells drives dendritic cell activation, thereby increasing the capability of dendritic cells to activate CD8+ T cells. Notably, ADAM9 increases IL6-STAT3 signaling through the degradation of LDLR, leading to the induction of cholesterol biosynthesis to maintain cholesterol homeostasis and STAT3 expression. We provide new insight into the contribution of ADAM9 correlation with immune suppression by linking cholesterol homeostasis and IL6/STAT3 signaling pathways.
The major roles of ADAM9 in tumorigenesis have been previously defined to facilitate the release tumor cells growth factors [38] as well as angiogenesis [12]. Research articles reported that ADAM9 cleaved the extracellular matrix, leading to chronic lung inflammation [39], and that ADAM9 correlated with lower CD8+ infiltration [40]. However, whether ADAM9 can stimulate lung cancer progression by increasing inflammation remains unclear. We demonstrated that ADAM9 activated IL6-STAT3 signaling in lung cancer cells. Furthermore, ADAM9 induced IL6 production, causing inefficient dendritic cells and thereby inhibiting T-cell activation. Consistent with a recent finding, it is indicated that ADAM9 reduced T-cell activation in advanced renal cell carcinoma [40].
Using comprehensive LC-MS/MS analysis, we found that ADAM9-silenced lung cancer cells exhibited lower cholesterol biosynthesis-related protein expression, especially under starvation conditions. Likewise, regulating cholesterol biosynthesis-related gene expression was concomitant with STAT3 phosphorylation and IL6 signaling. This result suggested that cholesterol biosynthesis activated IL6-STAT3 signaling. Consistent with a recent report that serum starvation conditions enhanced cholesterol biosynthesis [41], we expounded on the function of starvation in the manipulation of IL6 signaling by ADAM9. Rapidly proliferating cancer cells require sufficient cholesterol metabolites to support and maintain growth, survival, energy consumption, and signal transduction [42]. Therefore, the efficacy of cholesterol biosynthesis is correlated with tumor progression, and cholesterol-lowering agents have been applied for cancer therapy [43]. For example, fatostatin, a specific inhibitor of SREBP activation, was originally developed to inhibit insulin-induced adipogenesis and reduce the amounts of body fats [44]. It has also been reported to possess antitumor properties through reduction of lipid/cholesterol biosynthesis [45]. Moreover, our results showed that fatostatin inhibited IL6-STAT3 activation in lung cancer cells, suggesting that cholesterol biosynthesis could stimulate inflammatory and immune suppression.
Our findings showed that LDLR proteins were reduced in ADAM9-abundant lung cancer cells due to PCSK9-mediated LDLR lysosomal degradation. PCSK9 has been shown to induce the expression of proinflammatory cytokines, including IL1β, TNFα, IL6, and MCP-1, in macrophages [46]. Whether PCSK9 also mediates the activation of IL6-STAT3 signaling in lung cancer cells needs further research. Instead of regulating cholesterol homeostasis, PCSK9 also promoted Toll-like receptor 4 (TLR 4) and proinflammatory cytokine expression. Moreover, agents developed for PCSK9 inhibition were employed to treat vascular inflammation [47], indicating the interaction between cholesterol and inflammation. Although we found that ADAM9 can inhibit cholesterol uptake and increase cholesterol synthesis, further research is needed to determine if ADAM9 regulates the levels of cellular cholesterol derivatives and long-chain fatty acids, and if ADAM9 induces the ligand-dependent signal transduction of lipid rafts on cell membranes in lung cancer cells.
Our study provides new insight into the underlying molecular mechanism by which ADAM9 stimulates immune suppression in the tumor microenvironment by releasing IL6. Furthermore, ADAM9 activated IL6-STAT3 signaling and promoted cholesterol biosynthesis, leading to immune suppression. Hence, ADAM9 displayed a novel biological activity to induce the immunosuppressive tumor microenvironment. Additionally, targeting ADAM9 potentially offers a new therapeutic strategy to treat lung cancer.
Acknowledgements
This study was supported by the grants from the Ministry of Science and Technology, Taiwan (108-2314-B-039-054-MY3, 111-2320-B-039-010, and 111-2314-B-039-044-MY3), the National Science and Technology Council, Taiwan (NSTC 112-2622-B-039-006 and NSTC 112-2320-B-039-006), the National Health Research Institutes (EX109-10706BI and EX112-11219BI), and China Medical University (CMU110-MF-32, CMU111-MF-32, and CMU112-MF-30) to Y.P.S. This work was supported by the “Cancer Biology and Precision Therapeutics Center, China Medical University” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, Li Y. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6:263. doi: 10.1038/s41392-021-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021;33:127–148. doi: 10.1093/intimm/dxaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19:145. doi: 10.1186/s12943-020-01258-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, Lee SJ, Yamashita M, Hughes SG, Fayad L, Piha-Paul S, Nadella MV, Mohseni M, Lawson D, Reimer C, Blakey DC, Xiao X, Hsu J, Revenko A, Monia BP, MacLeod AR. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 6.Hartmann P, Trufa DI, Hohenberger K, Tausche P, Trump S, Mittler S, Geppert CI, Rieker RJ, Schieweck O, Sirbu H, Hartmann A, Finotto S. Contribution of serum lipids and cholesterol cellular metabolism in lung cancer development and progression. Sci Rep. 2023;13:5662. doi: 10.1038/s41598-023-31575-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mao W, Cai Y, Chen D, Jiang G, Xu Y, Chen R, Wang F, Wang X, Zheng M, Zhao X, Mei J. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non-small cell lung cancer. JCI Insight. 2022;7:e161940. doi: 10.1172/jci.insight.161940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omoigui S. The interleukin-6 inflammation pathway from cholesterol to aging--role of statins, bisphosphonates and plant polyphenols in aging and age-related diseases. Immun Ageing. 2007;4:1. doi: 10.1186/1742-4933-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nawawi H, Osman NS, Annuar R, Khalid BA, Yusoff K. Soluble intercellular adhesion molecule-1 and interleukin-6 levels reflect endothelial dysfunction in patients with primary hypercholesterolaemia treated with atorvastatin. Atherosclerosis. 2003;169:283–291. doi: 10.1016/s0021-9150(03)00193-x. [DOI] [PubMed] [Google Scholar]
- 10.Shintani Y, Higashiyama S, Ohta M, Hirabayashi H, Yamamoto S, Yoshimasu T, Matsuda H, Matsuura N. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res. 2004;64:4190–4196. doi: 10.1158/0008-5472.CAN-03-3235. [DOI] [PubMed] [Google Scholar]
- 11.Lin CY, Chen HJ, Huang CC, Lai LC, Lu TP, Tseng GC, Kuo TT, Kuok QY, Hsu JL, Sung SY, Hung MC, Sher YP. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74:5229–5243. doi: 10.1158/0008-5472.CAN-13-2995. [DOI] [PubMed] [Google Scholar]
- 12.Lin CY, Cho CF, Bai ST, Liu JP, Kuo TT, Wang LJ, Lin YS, Lin CC, Lai LC, Lu TP, Hsieh CY, Chu CN, Cheng DC, Sher YP. ADAM9 promotes lung cancer progression through vascular remodeling by VEGFA, ANGPT2, and PLAT. Sci Rep. 2017;7:15108. doi: 10.1038/s41598-017-15159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh S, Park Y, Lee HJ, Lee J, Lee SH, Baek YS, Chun SK, Lee SM, Kim M, Chon YE, Ha Y, Cho Y, Kim GJ, Hwang SG, Kwack K. A disintegrin and metalloproteinase 9 (ADAM9) in advanced hepatocellular carcinoma and their role as a biomarker during hepatocellular carcinoma immunotherapy. Cancers (Basel) 2020;12:745. doi: 10.3390/cancers12030745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji H, Chang EY, Lin KY, Kurman RJ, Pardoll DM, Wu TC. Antigen-specific immunotherapy for murine lung metastatic tumors expressing human papillomavirus type 16 E7 oncoprotein. Int J Cancer. 1998;78:41–45. doi: 10.1002/(sici)1097-0215(19980925)78:1<41::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Lu TL, Sher YP, Chen HC, Cheng WC, Hsu LH, Lee CC. Articulatin B chain induced dendritic cells maturation and driven type I T helper cells and cytotoxic T cells activation. Life Sci. 2022;302:120635. doi: 10.1016/j.lfs.2022.120635. [DOI] [PubMed] [Google Scholar]
- 16.Cheng WC, Chang CY, Lo CC, Hsieh CY, Kuo TT, Tseng GC, Wong SC, Chiang SF, Huang KC, Lai LC, Lu TP, Chao KSC, Sher YP. Identification of theranostic factors for patients developing metastasis after surgery for early-stage lung adenocarcinoma. Theranostics. 2021;11:3661–3675. doi: 10.7150/thno.53176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fainaru O, Shay T, Hantisteanu S, Goldenberg D, Domany E, Groner Y. TGFbeta-dependent gene expression profile during maturation of dendritic cells. Genes Immun. 2007;8:239–244. doi: 10.1038/sj.gene.6364380. [DOI] [PubMed] [Google Scholar]
- 18.Pasquin S, Laplante V, Kouadri S, Milasan A, Mayer G, Tormo AJ, Savin V, Sharma M, Martel C, Gauchat JF. Cardiotrophin-like cytokine increases macrophage-foam cell transition. J Immunol. 2018;201:2462–2471. doi: 10.4049/jimmunol.1800733. [DOI] [PubMed] [Google Scholar]
- 19.Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. 2015;53:676–688. doi: 10.1165/rcmb.2015-0012OC. [DOI] [PubMed] [Google Scholar]
- 20.Bauer M, Redecke V, Ellwart JW, Scherer B, Kremer JP, Wagner H, Lipford GB. Bacterial CpG-DNA triggers activation and maturation of human CD11c-, CD123+ dendritic cells. J Immunol. 2001;166:5000–5007. doi: 10.4049/jimmunol.166.8.5000. [DOI] [PubMed] [Google Scholar]
- 21.Lin CC, Huang YK, Cho CF, Lin YS, Lo CC, Kuo TT, Tseng GC, Cheng WC, Chang WC, Hsiao TH, Lai LC, Shih JY, Liu YH, Chao KSC, Hsu JL, Lee PC, Sun X, Hung MC, Sher YP. Targeting positive feedback between BASP1 and EGFR as a therapeutic strategy for lung cancer progression. Theranostics. 2020;10:10925–10939. doi: 10.7150/thno.49425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509–W514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-γ production by T helper 1 cells. Eur J Immunol. 1996;26:659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 24.Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kämpgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 27.Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy. 2010;6:1125–1138. doi: 10.4161/auto.6.8.13547. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. 2017;8:93. doi: 10.3389/fimmu.2017.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein I, Paakinaho V, Baek S, Sung MH, Hager GL. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat Commun. 2017;8:1849. doi: 10.1038/s41467-017-02055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou CW, Huang YK, Kuo TT, Liu JP, Sher YP. An overview of ADAM9: structure, activation, and regulation in human diseases. Int J Mol Sci. 2020;21:7790. doi: 10.3390/ijms21207790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyakoshi M, Yamamoto M, Tanaka H, Ogawa K. Serine 727 phosphorylation of STAT3: an early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol Carcinog. 2014;53:67–76. doi: 10.1002/mc.21949. [DOI] [PubMed] [Google Scholar]
- 32.Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) J Biol Chem. 2013;288:18707–18715. doi: 10.1074/jbc.R113.479808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madison BB. Srebp2: a master regulator of sterol and fatty acid synthesis. J Lipid Res. 2016;57:333–335. doi: 10.1194/jlr.C066712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21:225–245. doi: 10.1038/s41580-019-0190-7. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, Chen L, Sun B, Liu D, He Y, Qi L, Li G, Han Z, Zhan L, Zhang S, Zhu K, Luo Y, Chen L, Zhang N, Guo H. LDLR inhibition promotes hepatocellular carcinoma proliferation and metastasis by elevating intracellular cholesterol synthesis through the MEK/ERK signaling pathway. Mol Metab. 2021;51:101230. doi: 10.1016/j.molmet.2021.101230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol. 2014;25:387–93. doi: 10.1097/MOL.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, Takahashi N, Taketomi A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017;108:1947–1952. doi: 10.1111/cas.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu KL, Lin YS, Kuo TT, Lo CC, Huang YK, Chang HF, Chuang EY, Lin CC, Cheng WC, Liu YN, Lai LC, Sher YP. ADAM9 enhances CDCP1 by inhibiting miR-1 through EGFR signaling activation in lung cancer metastasis. Oncotarget. 2017;8:47365–47378. doi: 10.18632/oncotarget.17648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dreymueller D, Uhlig S, Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol. 2015;308:L325–L343. doi: 10.1152/ajplung.00294.2014. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Wang Y, Chen Z, Zhu Y, Wang J, Guo J. Unfavorable immunotherapy plus tyrosine kinase inhibition outcome of metastatic renal cell carcinoma after radical nephrectomy with increased ADAM9 expression. Immunogenetics. 2023;75:133–143. doi: 10.1007/s00251-022-01292-3. [DOI] [PubMed] [Google Scholar]
- 41.Terrell M, Morel L. The intersection of cellular and systemic metabolism: metabolic syndrome in systemic lupus erythematosus. Endocrinology. 2022;163:bqac067. doi: 10.1210/endocr/bqac067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Zhang P, Xu J, Lv G, Li Y. Lipid metabolism in tumor microenvironment: novel therapeutic targets. Cancer Cell Int. 2022;22:224. doi: 10.1186/s12935-022-02645-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni W, Mo H, Liu Y, Xu Y, Qin C, Zhou Y, Li Y, Li Y, Zhou A, Yao S, Zhou R, Huo J, Che L, Li J. Targeting cholesterol biosynthesis promotes anti-tumor immunity by inhibiting long noncoding RNA SNHG29-mediated YAP activation. Mol Ther. 2021;29:2995–3010. doi: 10.1016/j.ymthe.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi Y, Kawazoe Y, Murakami K, Misawa H, Uesugi M. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J Biol Chem. 2003;278:7320–7324. doi: 10.1074/jbc.M210283200. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther. 2014;13:855–866. doi: 10.1158/1535-7163.MCT-13-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, Zanotti I, Lupo MG, Adorni MP, Cicero AFG, Fogacci F, Corsini A, Ferri N. PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep. 2018;8:2267. doi: 10.1038/s41598-018-20425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu NQ, Shi HW, Li JJ. Proprotein convertase subtilisin/kexin type 9 and inflammation: an updated review. Front Cardiovasc Med. 2022;9:763516. doi: 10.3389/fcvm.2022.763516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.