

Abstract
Background
Temple syndrome (TS14) is a rare imprinting disorder caused by maternal UPD14, imprinting defects or paternal microdeletions which lead to an increase in the maternal expressed genes and a silencing the paternally expressed genes in the 14q32 imprinted domain. Classical TS14 phenotypic features include pre- and postnatal short stature, small hands and feet, muscular hypotonia, motor delay, feeding difficulties, weight gain, premature puberty along and precocious puberty.
Methods
An exon array comparative genomic hybridization was performed on a patient affected by psychomotor and language delay, muscular hypotonia, relative macrocephaly, and small hand and feet at two years old. At 6 years of age, the proband presented with precocious thelarche. Genes dosage and methylation within the 14q32 region were analyzed by MS-MLPA. Bisulfite PCR and pyrosequencing were employed to quantification methylation at the four known imprinted differentially methylated regions (DMR) within the 14q32 domain: DLK1 DMR, IG-DMR, MEG3 DMR and MEG8 DMR.
Results
The patient had inherited a 69 Kb deletion, encompassing the entire DLK1 gene, on the paternal allele. Relative hypermethylation of the two maternally methylated intervals, DLK1 and MEG8 DMRs, was observed along with normal methylation level at IG-DMR and MEG3 DMR, resulting in a phenotype consistent with TS14. Additional family members with the deletion showed modest methylation changes at both the DLK1 and MEG8 DMRs consistent with parental transmission.
Conclusion
We describe a girl with clinical presentation suggestive of Temple syndrome resulting from a small paternal 14q32 deletion that led to DLK1 whole-gene deletion, as well as hypermethylation of the maternally methylated DLK1-DMR.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13148-024-01652-8.
Keywords: Temple syndrome (TS14), Methylation, DLK1, Deletion, DMR
Background
Temple syndrome (TS14, OMIM #616222) was described more than 30 years ago in a patient with a maternal uniparental disomy 14 (UPD(14)mat) due to a Robertsonian translocation (13;14) [1]. Classical TS14 phenotypic features include pre- and postnatal short stature, small hands and feet, muscular hypotonia, motor delay, feeding difficulties, weight gain, and precocious puberty [2, 3]. Clinical review of published cases showed that most patients, especially in infancy, were initially suspected to have Prader-Willi syndrome (PWS) or Silver-Russell syndrome (SRS), which is explained by the phenotypic overlap between TS14 and PWS (growth failure, hypotonia, and small hands and feet) and between TS14 and SRS (growth failure and feeding difficulties) [4–7].
TS14 is a rare imprinting disorder that is caused by genetic and epigenetic alterations including UPD(14)mat, imprinting defects and paternal microdeletions which lead to maternalization (i.e. the paternally derived chr14 resembles the maternal inherited copy) at the imprinted domain on chromosome 14q32 [3]. The 14q32 region harbors the paternally expressed genes DLK1 and RTL1 as well as the maternally expressed genes MEG3 (also known as GTL2), RTL1as, MEG8 and large sno- and microRNA gene clusters [8–10].
The parent-of-origin-specific expression is regulated by two differentially methylated regions (DMRs) acting as imprinting control centers, the germline primary IG-DMR (MEG3/DLK1:IG-DMR; following the nomenclature recommended in Monk et al.[11]) located between the DLK1 and MEG3 gene and the somatic secondary MEG3-DMR (MEG3:TSS-DMR) overlapping the MEG3 promotor region [2]. The germline-derived IG-DMR acts upstream of the secondary-derived somatic MEG3 DMR and governs it in a hierarchical fashion [12, 13]. Two somatically acquired DMRs methylated on the maternal allele have recently been described, the MEG8 DMR (MEG8: Int2-DMR) located in the intron 2 of the MEG8 gene [14, 15] and the DLK1 DMR (DLK1:Int1-DMR) overlapping the second exon of the DLK1 gene [16]. The function of these two DMRs is currently unknown.
Previously, patients with TS14 have shown hypomethylation at the IG-DMR and MEG3 DMR and hypermethylation at the MEG8 DMR with an increased expression of the maternally expressed transcripts and silencing of paternally expressed genes. Currently, 128 patients with TS14 have been described in the literature allowing the frequency of the underlying molecular causes to be defined. The majority present with UDP(14)mat (57%), epigenetic defects (31%) and microdeletions (12%) [3, 7, 17–22]. The size of microdeletions vary in length with the majority extending for ~ 1 Mb encompassing the EML1, EVL, YY1, SCL25A29, SLC325A47, BEGAIN, DLK1, MEG3, RTL1 genes, as well as the sno- and microRNA clusters, among others [18, 23].
Here, we report a family with a ~ 69 Kb microdeletion that led to a whole-gene deletion of DLK1. Paternal transmission resulted in clinical features consistent with Temple syndrome. The proband exhibited relative hypermethylation at the DLK1 and MEG8 DMR and normal methylation at the IG-DMR and MEG3-DMR.
Results
The proband is the first daughter of non‐consanguineous parents and was born at 42 weeks following an uncomplicated pregnancy with a weight of 2900 g (p7,− 1.6SD), length of 50 cm (0 SDS) and head circumference at birth of 34 cm (+ 1SD), which increased when the patient was one month old (+ 2SD), consistent with relative macrocephaly at birth. She presented stereotypies at 6 months old, and an electroencephalogram was performed with a normal result. She was followed up at the Childhood Development and Early Intervention Centre (CDIAP) due to muscular hypotonia. Autonomous sitting was done at 18 months old and she was able to walk independently at 21 months.
The proband was referred to the Clinical Genetic Unit at 2 years old due to psychomotor and language delay. The patient also presented with growth failure (height 82.7 cm, − 2.0 SDS) and muscular hypotonia. At 4 years old, a clinical reassessment noticed several other clinical features resembling Temple syndrome (Table 1), such as prominent forehead, almond-shaped eyes, a broad nasal tip, ears with attached lobes, hyperextensible joints, small hands and mild intellectual disability (Fig. 1). She manifested shyness and motor clumsiness and attended a school with educational support. At 5 years, an umbilical hernia was repaired. Follow-up at the endocrinologist unit evidenced thelarche at 7 years old with an increase in growth velocity (9 cm/year last 6 months) followed by premature reactivation of the hypothalamic-pituitary–gonadal axis, compatible with central precocious puberty (CPP). The LHRH stimulation test showed an LH peak of 10.5 U/L and FSH peak of 15.4 U/L with a LH/FSH relationship > 0.6. Her cousin was prenatally diagnosed with the deletion, who presented IUGR in the third trimester. At birth (40 weeks of gestational age), his weight was 2770 g (− 1.6SDS), length 46 cm (− 2.2SD) and showed muscular hypotonia. At 20 months old, he presented clinical traits of Temple as muscular hypotonia, hyperextensible joints, and facial traits (broad and prominent forehead, short nose with flat nasal root and wide tip, downturned corners of mouth, ears large and posteriorly rotated and micrognathia) and small hands.
Table 1.
Clinical traits present in patients with Temple syndrome and in patients with small deletions reported
| TS14 features | Present case | Dauber A [23] n = 4 (family) | Kagami M [2] n = 2 (family) n = 1 | Sabria-Back [24] n = 1 | TS14 features revised in Ioannides [3] and Kagami [18] series n = 83 patients | ||
|---|---|---|---|---|---|---|---|
| Size of deletion | 69 kb | 14 kb | 108 kb | 411 kb | 108 KB | 1 Mb to 6 Mb | |
| Genetic etiology | DLK1 whole-gene deletion | exon 1 DLK1 deletion | DLK1, MEG3/DLK1:IG-DMR and MEG3:TSS-DMR deletion | DLK1, RTL1, MEG3/DLK1: IG-DMR and MEG3:TSS-DMR deletion |
DLK1, MEG3/DLK1:IG-DMR and MEG3:TSS-DMR deletion |
Deletion = 8 | UPD = 63 Epimutation = 12 |
| Sex Age | Female 4 years old | Females 6/8/6/7 years old | Female/Male 36/62 years old | Female 28 years old | Female NR (adulthood) | 2 males:6 females | 39 males:36 females |
| Pregnancy and delivery | |||||||
| Premature birth | 42 w | 39–40 w | 39w/NP | 40w | NR | 0% (0/6) | 28% (18/65) |
| Growth | |||||||
| Intrauterine growth retardation | - | NR | +/NR | + | NR | 80% (4/5) | 79% (49/62) |
| Low birth weight SDS ≤ -2.0 | -1.6 | -0.52/1.37/-0.07/0.98 | -2.5/NR | − 2.2 | NR | 100% (4/4) | 92% (58/63) |
| Short stature SD ≤ -2.0 | -2.0* | 0.6/-0.6/-0.6/0.7** | -2.2/-2.9 | − 4.4 | + | 100% (8/8) | 86% (62/72) |
| Obesity (BMI SDS) |
Too Young (-0.8–0.6) |
2/4 (2, > 2)** | -/ + | − | NR | 20% (1/5) | 41% (21/51) |
| Head and neck | |||||||
| Relative macrocephaly at birth | + | - | NR/NR | + | NR | NR | 52% (14/27) |
| Frontal bossing/Prominent forehead | + | - | + / + | + | NR | 50% (1/2) | 64% (18/28) |
| High palate | − | - | NR/NR | − | NR | 0% (0/1) | 54% (14/26) |
| Skeletal | |||||||
| Hyperextensible joints | + | NR | -/NR | − | NR | 0% (0/3) | 48% (22/46) |
| Small hands (percentile) | + (p < 3) | − | + / + | + | NR | 100% (3/3) | 86% (31/36) |
| Small feet | + | − | + / + | + | NR | 100% (1/1) | 96% (25/26) |
| Clinodactyly | − | NR | -/NR | − | NR | 0% (0/2) | 42% (11/26) |
| Central Nervous System | |||||||
| Hypotonia | + | - | -/NR | + | NR | 80% (4/5) | 82% (55/67) |
| Motor development delay | + | - | NR/NR | NR | NR | 100% (3/3) | 82% (31/38) |
| Speech delay | + | - | -/- | − | NR | 100% (2/2) | 56% (14/25) |
| Feeding problems | − | - | -/NR | − | NR | 50% (1/2) | 64% (18/28) |
| Mild Intellectual disability | + | - | -/- | − | NR | 25% (1/4) | 34% (14/41) |
| Endocrine features | |||||||
| Early onset puberty |
Precocious thelarche (7 years old) |
4/4 | + /NR | - | + | 60% (3/5) | 70,6% (29/34) |
W: weeks of gestation, NR: not reported. *at 2 years old;** adult
Fig. 1.
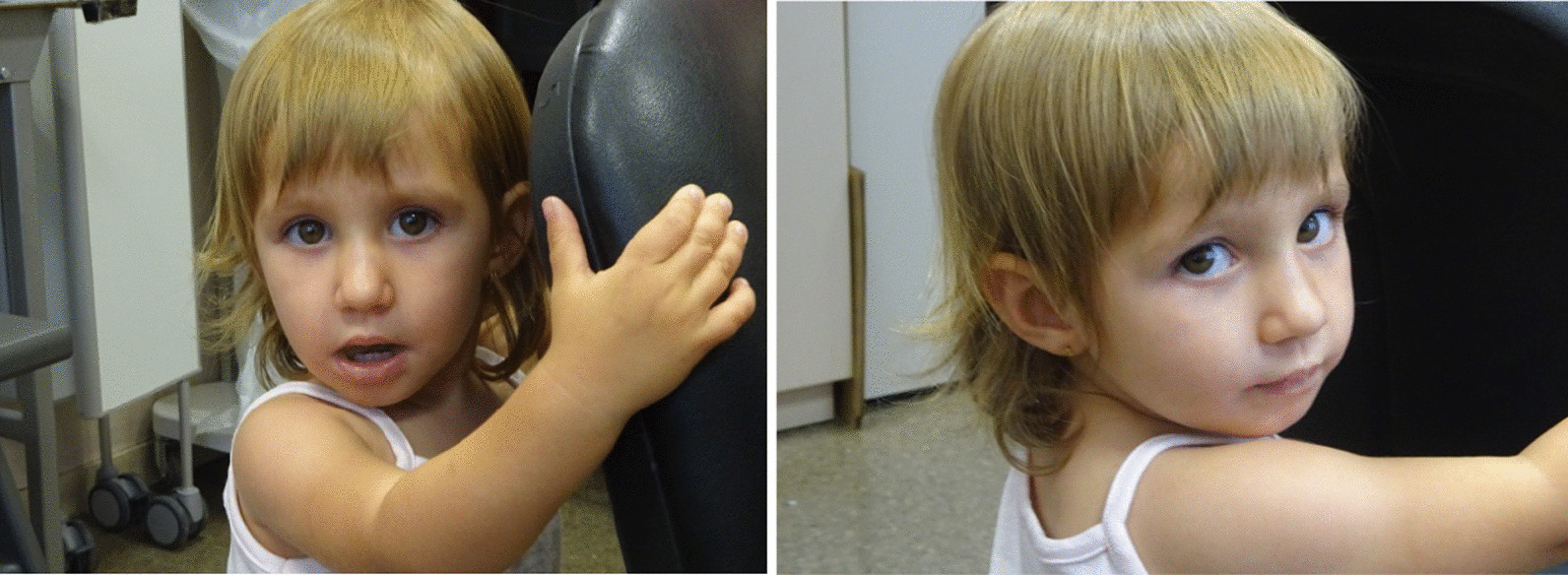
Clinical traits present in our case. The photographs show prominent forehead, almond-shaped eyes, broad nasal tip small hand and attached lobe in ear
Copy-number variation was assessed using aCGH. This revealed a normal female profile with an interstitial microdeletion at chromosome chr14q32.2 (arr[GRCh37] 14q32.2(101184595_101253432) × 1) with an approximate length of 69 Kb (Fig. 2). This deletion included the DLK1 gene, consistent with the suspicion of TS14. The deletion was detected in her father (II.3), uncle (II.2) and grandmother (I.2), all of them with normal phenotype and her cousin recently born was prenatally diagnosed with the deletion (Fig. 3) and born with a low birthweight consistent with early features of TS14. Subsequently, MS-MLPA was used to test the methylation status at the MEG3 DMR which resulted in a normal methylation profile in the proband and all deletion carriers. It must be noted that the MLPA kit used (ME021) does not include probes within other imprinted DMR on chromosome 14.
Fig. 2.

Deletions involving the DLK1 gene identified in this study and those reported in the literature
Fig. 3.

Family tree. III.2 is the TS14 index case. The remaining family members have normal phenotypes, with I.2, II.2, II.3 carrying the DLK1 microdeletion on the maternal allele
Extending characterization of allelic methylation throughout chr14q32 was performed in the index case (III.2), cousin (III.1), carriers (I.2, II.2, II.3) and non-deleted (II.4, I.1) family members using bisulfite PCR followed by pyrosequencing. This revealed IG-DMR and MEG3 DMR profiles comparable with 19 healthy control samples, in agreement to MS-MLPA methylation observations, indicating allelic methylation was maintained correctly at these regions in the presence of the deletion (Fig. 4a; Additional file 1: Supplementary Table 1). The index patient showed relative hypermethylation at the DLK1 DMR consistent with DLK1 deletion on the paternal allele. Carrier members (II.2 and II.3) were relatively hypomethylated at this interval consistent with maternal transmission. Interestingly, the MEG8 DMR was relatively hypomethylated (II.2 and II.3) in a similar way as the DLK1 DMR, despite not being within the deleted interval (Fig. 4a). These methylation profiles were confirmed in the cloned PCR products (Fig. 4b). The grandmother (I.2) presented with discordant methylation profiles at these two maternally methylated intervals, with hypomethylation at the MEG8 DMR but methylation within the normal range at the DLK1 DMR. Pyrosequencing in the grandmother was independently replicated with similar profiles. It remains to be determined if methylation at these two somatically acquired maternally methylated intervals are mechanistically linked. Profiles obtained from the healthy control samples were subject to large inter-individual variation which may be attributed to the fact that allelic methylation at these two DMRs is not absolute and that the maternal allele is more methylated than the paternal allele [8, 9], plots and the range of methylation as defined by the UPD(14)pat and UPD(14)mat control samples (Fig. 4a). Furthermore, the normal unmethylated paternal allele at the DLK1 DMR is associated with residual methylation, as indicated by the ~ 72% average methylation in the control cohort. Therefore, the normal methylation at the DLK1 DMR in the grandmother, despite being at the lower end of the control range, may reflect variability which may become more evident with age.
Fig. 4.

Methylation profiling of the TS14 index case and familial members. A Pyrosequencing was used to quantify methylation of CpG dinucleotides in the DLK DMR (maternally methylated), IG-DMR (paternally methylated), MEG DMR (paternally methylated) and the MEG8 DMR (maternally methylated). Violin plots represent the average methylation profiles of 19 control individuals, with data points for family members located alongside. The black and white boxes above and below the violin plots represent methylation values for UPD(14)pat and UPD(14)mat control samples respectively. B Bisulfite PCR followed sub-cloning for amplicons from proband III.1 and a control leukocyte DNA sample. Each circle represent a single dinucleotide on the DNA stand. (•) methylated cytosine, (o) unmethylated cytosine. Each row corresponds to an individual cloned sequence. CpG dinucleotides in red box indicated those quantified by pyrosequencing
On the basis of the presence of the microdeletion on the paternal allele and concomitant hypermethylation at the DLK1 DMR, we confirmed the diagnosis of TS14 in the index case, despite normal methylation level at the MEG3 and IG-DMR which are used for routine molecular diagnosis of this rare imprinting disorder.
Discussion
Temple syndrome can result from different molecular anomalies. The majority of cases described have a UPD(14)mat, while imprinting defects and deletions are much rarer [3, 18]. Clinical findings are grossly similar among patients with UPD(14)mat, epimutations and deletions and are present in more than 60% of patients with TS14 [3, 18]. Phenotypic comparison among the few cases with deletion showed two phenotypes. Common features of TS14 are observed in DLK1 whole deletions including short stature, small hands and feet, relative macrocephaly at birth, prominent forehead in infancy, hypotonia and CPP with motor development and intellectual disability. However, deletions restricted to the first exon of DLK1 result in CCP only [23].
We have identified a child of healthy non-consanguineous parents with a phenotype of TS14 carrying a small deletion on 14q32 encompassing the DLK1 gene with hypermethylation at the DLK1 and MEG8 DMR intervals and normal methylation at IG-DMR and MEG3 DMR (Fig. 5). So far only eight patients with small deletions encompassing DLK1 have been reported in the scientific literature (Fig. 2). Following Dauber et al. [23] description, rare small intragenic deletion in DLK1 with deficiency of DLK1 measured in serum has been recognized as a genetic cause of familial CPP when paternally inherited.
Fig. 5.

Structure of the imprinted 14q32 region, paternal and maternal DLK1 gene deletion. Maternally expressed genes are indicated in red boxes and paternally expressed genes are indicated by blue boxes. The methylation is indicated by black lollipops. The DLK1-DMR is maternally methylated (Mat), MEG3/DLK1:IG-DMR and MEG3:TSS-DMR are paternally methylated (Pat) and MEG8: Int2-DMR is maternally methylated (Mat)
The application of MS-MLPA in the affected individuals did not detect any additional copy number variation or abnormal methylation. Kagami et al. [2] described two families with three adult patients that were ascertained through familial studies following the birth of children with Kagami-Ogata syndrome (KOS14) in whom paternally transmitted DLK1 deletions were identified. The first family (one male and one female) had a deletion of 108 Kb from DLK1 to exon three of MEG3, including IG-DMR and MEG3 DMR (https://www.ncbi.nlm.nih.gov/clinvar/variation/162007/). The affected patients did not demonstrate additional clinical TS14 features, except for a high prevalence of metabolic abnormalities, such as overweight/obesity and insulin resistance. In the second family, a female had a longer 411 Kb deletion containing WD25, BEGAIN, DLK1, MEG3, RTL1, MEG8 including IG-DMR and MEG3 DMR (https://www.ncbi.nlm.nih.gov/ clinvar/variation/ 162,009/). This case had pre-natal growth failure, severe short stature, frontal bossing, relative macrocephaly and small hands and feet. Neither of them exhibits motor development delay nor speech delay, clinical findings that are present in our patient and are present in more than 60% of TS14 cases. The final TS14 case was associated with a different 108 Kb deletion that removed the DLK1 gene and both the paternally methylated DMRs. This individual was also found following intensive molecular analysis following the birth of a child with KOS14. This patient was initially undiagnosed, but had a constellation of TS14 phenotypic features including short stature, CPP and diabetes [24].
The index case described in this report had CPP as well as short stature, two main characteristics of TS14. Furthermore, she presented with many other clinical features of TS14 present in ≥ 50% of cases, such as relative macrocephaly, prominent forehead, small hands and feet, hypotonia, motor development delay and speech delay but contrary to the clinical review of Ioannides et al. [3] and Kagami et al. [18] she did not show feeding difficulties, IUGR, low birth weight, even though the birth weight was in p7 (− 1.5SD). Moreover, she showed hyperextensible joints, almond-shaped eyes, broad nasal tip and ears with attached lobe described in a lower frequency in TS14 [3, 18]. At present, a male cousin prenatally diagnosed with the 14q32 deletion who presented IUGR, at 20 months showed TS14 phenotype with prominent forehead, hyperextensible joints, small hands, and muscular hypotonia already present at birth. Together this suggests that lack of other paternally expressed genes, such as RTL1 (or its epigenetic silencing) may be primarily responsible for the perinatal growth phenotypes as observed in the knock-out mouse model [25].
Rare heterozygous mutations in DLK1 have also recently been recognized as a genetic cause of familiar CPP when paternally inherited. Indeed, three frameshift mutations in exon 5 of DLK1 (p.Gly199Alafs*11, p.Val271Cysfs*14, and p.Pro160Leufs*50) were described in four women from three families with CPP. The segregation analysis was consistent with the maternal silencing of DLK1 [26]. The affected patients did not demonstrate additional clinical TS14 features. Montenegro et al. [27] described a de novo heterozygous indel (c.401_404 + 8del) affecting exon 4 of DLK1 gene with CPP. Importantly, the 3 mentioned studies that identified DLK1 loss-of-function mutations as a monogenic cause of CPP (23,26,27) found undetectable serum levels of DLK1 in all affected individuals compared to controls. At present, patients with TS14 phenotype have not been described to carry DLK1 point mutations. Nevertheless, five very rare cases with loss-of-function variants in DLK1 are described in the GnomAD database and it is unknown whether the carriers of these variants who are reported in the database have any phenotypic abnormality.
The smallest overlapping region in patients with recognizable TS14 phenotype, based on the present case and those described above [18, 24], contains DLK1 as the solely disrupted gene. The loss of function of the paternal copy of DLK1 caused by whole-gene deletion, intragenic deletions, or frameshift mutation could lead to variable clinical features such as early pubertal signs (4.6–7 years) and additional TS14 associated features. DLK1 encodes a transmembrane protein that is important for adipose tissue homeostasis and neurogenesis [28]. The exact mechanism by which DLK1 regulates pubertal timing is not yet understood; however, it has been demonstrated in mouse that Dlk1 is essential for neuroendocrine control of GnRh and the timing of puberty onset, in part through kisspeptin neurons [29]. DLK1 could play a role in the regulation of neurogenesis within the hypothalamus, indirectly interfering with kisspeptin neuron formation, maturation, and/or secretion of kisspeptin through the activation or inhibition of Notch target genes. The disruption of the Notch signalling pathway, involved in neurogenesis and development of the hypothalamus, particularly in the reproductive axis, could be associated with central precocious puberty [28]. Moreover, DLK1 has a wide fetal expression but in postnatal life, the expression decreases, except in endocrine glands, mainly in adrenals, ovaries, adipose tissue, pituitary, and hypothalamic nuclei. These data seemed to be consistent with the pubertal and metabolic phenotypes presented by patients described with DLK1 deficiency. Recently, despite Dlk1-null mice displayed fetal growth restriction, in humans, DLK1 variants were not associated with being born small for gestational age in a large cohort of patients with pre- and postnatal growth failure (Silver-Russell phenotype) [30].
Psychomotor delay, mild intellectual disability and language delay present in our patient and reported in all the molecular etiologies of TS14 could be indirectly regulated by DLK1. Using the STRING tool (Search tool for Retrieval of Interacting Genes / Proteins) (https://string-db.org/) to obtain information about molecular networks and removing text mining with a high confidence score (> 0.7) revealed that DLK1 is connected with NOTCH1 and FIBP genes reporting confidence scores of 0.93 and 0.75 respectively. These genes are considered as intellectual disability candidate genes in the SysID database with mild or limit intellectual functioning. Pathogenic variants in NOTCH1 are associated with Adams-Oliver syndrome, a third of whom have intellectual disability [31] and loss-of-function mutation in the FIBP gene underlies an autosomal recessive syndromic overgrowth (mainly in height) associated with macrosomia, learning disabilities/developmental delay, and other congenital defects [32]. In addition, DLK1 is necessary for hippocampal neurogenesis in the regulation of neural stem cells maintenance in the long term modulation and for normal cognitive functions such as spatial memory consolidation [33].
Detailed methylation analysis by pyrosequencing confirmed the results obtained by MS-MLPA with normal methylation levels at the IG-DMR and MEG3 DMR. Normal methylation at these paternally methylated DMRs has not been reported in TS14 patients previously [14, 34], including patients with large and small deletions of the 14q32 region. Patients with paternal deletions of DLK1-MEG3, UPD(14)mat and epimutations all possess hypomethylation at the IG-DMR and MEG3 DMR. The relative hypermethylation at DLK1 DMR we observed is consistent with the paternally inherited deletion eliminating the unmethylated paternal allele of DLK1. Furthermore, the relative hypomethylation at the DLK1 DMR in the proband’s father and uncle is consistent with maternal transmission.
Interestingly, the MEG8 DMR also showed a reciprocal methylation pattern depending on transmission similar to that observed at the DLK1 DMR, despite not being deleted. The relative hypomethylation of these two somatically acquired DMRs suggests that methylation of MEG8 is dependent on, or secondary to the appropriate methylation at the DLK1 DMR. Such hierarchical dynamics have been described previously for the DMRs within the Igf2-H19 domain [28]. Recent targeted manipulation of allelic methylation within the orthologous domain in mouse using the dCas9-fusion system has revealed complex interactions between DMRs [35]. Although DNA methylation at the Dlk1 DMR was not directly targeted, dCas9GCN4-scFv-TETCD mediated IG-DMR hypomethylated resulted in down-regulation of both Dlk1 and Rtl1 and Meg8 DMR hypermethylation on the paternal allele. Together this suggests altered methylation at the IG-DMR causes domain-wide epigenotype switching, endorsing the existence of hierarchical allelic methylation similar to what we report in this family.
The function of the DLK1 DMR is unknown, but it is unlikely to directly influence the monoallelic expression since it is methylated on the expressed allele; however, this may reflect gene body methylation associated with silencing of cryptic transcript initiation. Recently, the Meg8 DMR was deleted in a mouse cell line model [36] and embryos [37]. This revealed that removal of the DMR, or only a CTCF insulator site, in MLTC-1 cells resulted in significant reduction in the paternally expressed Dlk1 and Rtl1 genes, and upregulation of the Meg3 and Rian. Similar results were observed in knock-out mice, with the exception that Dlk1 was not altered. The resulting knock-out did not grossly affect embryonic development [37]. Analysis of allelic methylation revealed that Meg8 DMR deletion did not result in changes within the cluster. Therefore, methylation at the IG-DMR, Meg3 DMR or Dlk1 DMR may influence Meg8 DMR establishment, but the reciprocal is not true, implying that Meg8 is the last DMR to be established, which is supported by the methylation profile in our proband. Interestingly, in humans, the MEG8 DMR is located only 4 Kb away of the RTL1 leader exon (NM_001134888), suggesting the MEG8 DMR may function as a cis-regulatory element. Interrogation of ENCODE ChIP-seq data revealed conserved CTCF binding within the MEG8 DMR, consistent with this maternally methylated interval contributing to allele-specific chromatin confirmation throughout the domain.
Irrespective of the molecular mechanism, individuals with TS14 generally lack DLK1 and RTL1 expression and it is highly likely that both of these paternally expressed genes are related to the growth, muscular and behavioral phenotypes reported. Despite the fact paternal RTL1 is intact in our proband, expression is likely reduced due to hypermethylation of the MEG8 DMR. Unfortunately, due to the tissue restricted expression of RTL1, we could not test this hypothesis in the proband.
Conclusion
Here, we described a small 14q32 paternal microdeletion encompassing the whole DLK1 gene in a patient with phenotypes resembling TS14. This patient has normal methylation at the paternally methylated imprinting control regions, IG-DMR and MEG3 DMR; therefore, normal DNA methylation testing of MEG3 DMR was inconclusive for TS14 in this case. Our results suggested that the maternally methylated DMRs located within the DLK1 and MEG8 genes could be also tested in patients with suspected TS14 to determine relative hypermethylation that could result from paternal microdeletions of DLK1.
Methods
Microarray
An exon array comparative genomic hybridization (aCGH) 4 × 180 K ISCA (Oxford Gene Technology, OGT) was carried out for genomic-wide copy number analysis following the manufacturer’s protocol. Data analysis was performed with Cytosure software.
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)
Gene dosage and methylation analyses of the 14q32 chromosomal region, including the MEG3 DMR, were carried out using the SALSA MLPA Kit ME032-A1 (MRC Holland, Amsterdam, The Netherlands) according to the manufacturer’s manual. Amplification products were run on an ABI3130 Genetic Analyzer (Applied Biosystems, California, USA) and analyzed using the GeneMapper software (Applied Biosystems).
Bisulfite PCR and pyrosequencing
Five hundred nanograms of genomic DNA were subjected to sodium bisulfite treatment and purified using the EZ GOLD methylation kit (ZYMO) following the manufacturer’s recommendations. DNAs from 19 normal controls were utilized to define the normal range of methylation while patients carrying UPD(14)pat and UPD(14)mat were used to detect extreme methylation values at the imprinted DMRs assessed. Approximately 50 ng of bisulfite converted DNA was used as template for PCR amplification of the MEG8 DMR, DLK1 DMR, IG-DMR and MEG3 DMR using Immolase Taq polymerase (Bioline, London, UK) at 45 cycles in which one primer was biotinylated (details of primer sequences available in Sabria-Back et al. [24]). One ul of each PCR productions from proband III.1 was cloned into pGEMT-easy vector (Promoga) for strand-specific methylation analysis, and the remainder was used in the pyrosequencing reaction. The entire biotinylated PCR product (diluted to 40 μl) was mixed with 38 μl of binding buffer and 2 μl (10 mg/ml) streptavidin-coated polystyrene beads. After incubation at 65◦C, DNA was denaturated with 50 μl 0.5MNaOH. The single-stranded DNA was hybridized to 40-pmol sequencing primers dissolved in 11 μl annealing buffer at 90◦C. For sequencing, a primer was designed on the opposite strand to the biotinylated primer used in the PCR reaction. The pyrosequencing reaction was carried out on a PyroMark Q24 instrument (Qiagen, Hilden, Germany). The peak heights were determined using Pyro Q-CpG1.0.9 software (Qiagen).
Supplementary Information
Additional file1: Supplementary table 1. Pyrosequencing results for the affected family and controls.
Acknowledgements
The authors would like to thank the family for participating in this study. The authors would also like to thank Professor Masayo Kagami for getting information about small deletion of cases with TS14.
Abbreviations
- TS14
Temple syndrome
- DMR
Differentially methylated regions
- UPD(14)mat
Maternal uniparental disomy 14
- PWS
Prader-Willi syndrome
- SRS
Silver-Russell syndrome
- SD
Standard deviation
- aCGH
Array comparative genomic hybridization
- MS-MLPA
Methylation-specific multiplex ligation-dependent probe amplification
- KOS14
Kagami-Ogata syndrome
- CpG
Cytosine-guanine dinucleotide
Author contributions
NB and DM wrote the original draft of the manuscript under the supervision of MG. NB, DM, CA, MFF, AGFF, and AR performed experiments and analyzed data. EG, RC, JPT, NC obtained clinical data. NB and DM were major contributors. All authors read and contributed to the preparation of the final manuscript. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Data are available from the corresponding author on request.
Declarations
Ethics approval and consent to participate
The protocol for the study has been approved by the institutional Ethics Committee of Institut d’InvestigacIó i Innovació Parc Taulí I3PT and written informed consent was obtained from the parents.
Consent for publication
The consent form for publication was obtained from the parents.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Temple IK, Cockwell A, Hassold T, Pettay D, Jacobs P. Maternal uniparental disomy for chromosome 14. J Med Genet. 1991;28(8):511–514. doi: 10.1136/jmg.28.8.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, Yamamori S, Kishimoto H, Nakayama M, Tanaka Y, Matsuoka K, Takahashi T, Noguchi M, Tanaka Y, Masumoto K, Utsunomiya T, Kouzan H, Komatsu Y, Ohashi H, Kurosawa K, Kosaki K, Ferguson-Smith AC, Ishino F, Ogata T. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40(2):237–242. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- 3.Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51(8):495–501. doi: 10.1136/jmedgenet-2014-102396. [DOI] [PubMed] [Google Scholar]
- 4.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi Syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 5.Eggermann T. Russell-Silver syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):355–364. doi: 10.1002/ajmg.c.30274. [DOI] [PubMed] [Google Scholar]
- 6.Kagami M, Mizuno S, Matsubara K, Nakabayashi K, Sano S, Fuke T, Fukami M, Ogata T. Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver-Russell Syndrome-compatible phenotype. Eur J Hum Genet. 2015;23(8):1062–1067. doi: 10.1038/ejhg.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geoffron S, Abi Habib W, Chantot-Bastaraud S, Dubern B, Steunou V, Azzi S, Afenjar A, Busa T, Pinheiro Canton A, Chalouhi C, Dufourg MN, Esteva B, Fradin M, Geneviève D, Heide S, Isidor B, Linglart A, Morice Picard F, Naud-Saudreau C, Oliver Petit I, Philip N, Pienkowski C, Rio M, Rossignol S, Tauber M, Thevenon J, Vu-Hong TA, Harbison MD, Salem J, Brioude F, Netchine I, Giabicani E. Chromosome 14q32.2 imprinted region disruption as an alternative molecular diagnosis of silver-russell syndrome. J Clin Endocrinol Metab. 2018;103(7):2436–2446. doi: 10.1210/jc.2017-02152. [DOI] [PubMed] [Google Scholar]
- 8.Charlier C, Segers K, Wagenaar D, Karim L, Berghmans S, Jaillon O, Shay T, Weissenbach J, Cockett N, Gyapay G, Georges M. Human-ovine comparative sequencing of a 250-kb imprinted domain encompassing the callipyge (clpg) locus and identification of six imprinted transcripts: DLK1, DAT, GTL2, PEG11, antiPEG11, and MEG8. Genome Res. 2001;11(5):850–862. doi: 10.1101/gr.172701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavaillé J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP. Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet. 2002;11(13):1527–1538. doi: 10.1093/hmg/11.13.1527. [DOI] [PubMed] [Google Scholar]
- 10.Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14(9):1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monk D, Morales J, den Dunnen JT, Russo S, Court F, Prawitt D, Eggermann T, Beygo J, Buiting K, Tümer Z. Nomenclature group of the European network for human congenital imprinting disorders Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. 2018;13(2):117–121. doi: 10.1080/15592294.2016.1264561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beygo J, Elbracht M, de Groot K, Begemann M, Kanber D, Platzer K, Gillessen-Kaesbach G, Vierzig A, Green A, Heller R, Buiting K, Eggermann T. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur J Hum Genet. 2015;23(2):180–188. doi: 10.1038/ejhg.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kagami M, O'Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, Matsuoka K, Fukami M, Matsubara K, Kato F, Ferguson-Smith AC, Ogata T. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. Plos Genet. 2010;6(6):e1000992. doi: 10.1371/journal.pgen.1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beygo J, Küchler A, Gillessen-Kaesbach G, Albrecht B, Eckle J, Eggermann T, Gellhaus A, Kanber D, Kordaß U, Lüdecke HJ, Purmann S, Rossier E, van de Nes J, van der Werf IM, Wenzel M, Wieczorek D, Horsthemke B, Buiting K. New insights into the imprinted MEG8-DMR in 14q32 and clinical and molecular description of novel patients with Temple syndrome. Eur J Hum Genet. 2017;25(8):935–945. doi: 10.1038/ejhg.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV, Keirstead H, Sanchez-Mut JV, Kaneki E, Lapunzina P, Soejima H, Wake N, Esteller M, Ogata T, Hata K, Nakabayashi K, Monk D. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24(4):554–569. doi: 10.1101/gr.164913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez Mora JR, Tayama C, Sánchez-Delgado M, Monteagudo-Sánchez A, Hata K, Ogata T, Medrano J, Poo-Llanillo ME, Simón C, Moran S, Esteller M, Tenorio J, Lapunzina P, Kagami M, Monk D, Nakabayashi K. Characterization of parent-of-origin methylation using the Illumina Infinium MethylationEPIC array platform. Epigenomics. 2018;10(7):941–954. doi: 10.2217/epi-2017-0172. [DOI] [PubMed] [Google Scholar]
- 17.Severi G, Bernardini L, Briuglia S, Bigoni S, Buldrini B, Magini P, Dentici ML, Cordelli DM, Arrigo T, Franzoni E, Fini S, Italyankina E, Loddo I, Novelli A, Graziano C. New patients with Temple syndrome caused by 14q32 deletion: Genotype-phenotype correlations and risk of thyroid cancer. Am J Med Genet A. 2016;170A(1):162–169. doi: 10.1002/ajmg.a.37346. [DOI] [PubMed] [Google Scholar]
- 18.Kagami M, Nagasaki K, Kosaki R, Horikawa R, Naiki Y, Saitoh S, Tajima T, Yorifuji T, Numakura C, Mizuno S, Nakamura A, Matsubara K, Fukami M, Ogata T. Temple syndrome: comprehensive molecular and clinical findings in 32 Japanese patients. Genet Med. 2017;19(12):1356–1366. doi: 10.1038/gim.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillessen-Kaesbach G, Albrecht B, Eggermann T, Elbracht M, Mitter D, Morlot S, van Ravenswaaij-Arts CMA, Schulz S, Strobl-Wildemann G, Buiting K, Beygo J. Molecular and clinical studies in 8 patients with Temple syndrome. Clin Genet. 2018;93(6):1179–1188. doi: 10.1111/cge.13244. [DOI] [PubMed] [Google Scholar]
- 20.Kimura T, Kagami M, Matsubara K, Yatsuga S, Mukasa R, Yatsuga C, Matsumoto T, Koga Y. Temple syndrome diagnosed in an adult patient with clinical autism spectrum disorder. Clin Case Rep. 2018;7(1):15–18. doi: 10.1002/ccr3.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagami M, Yanagisawa A, Ota M, Matsuoka K, Nakamura A, Matsubara K, Nakabayashi K, Takada S, Fukami M, Ogata T. Temple syndrome in a patient with variably methylated CpGs at the primary MEG3/DLK1:IG-DMR and severely hypomethylated CpGs at the secondary MEG3:TSS-DMR. Clin Epigenetics. 2019;11(1):42. doi: 10.1186/s13148-019-0640-2.PMID:30846001;PMCID:PMC6407230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brück J, Begemann M, Dey D, Elbracht M, Eggermann T. Molecular characterization of temple syndrome families with 14q32 epimutations. Eur J Med Genet. 2020;63(12):104077. doi: 10.1016/j.ejmg.2020.104077. [DOI] [PubMed] [Google Scholar]
- 23.Dauber A, Cunha-Silva M, Macedo DB, Brito VN, Abreu AP, Roberts SA, Montenegro LR, Andrew M, Kirby A, Weirauch MT, Labilloy G, Bessa DS, Carroll RS, Jacobs DC, Chappell PE, Mendonca BB, Haig D, Kaiser UB, Latronico AC. Paternally Inherited DLK1 Deletion Associated With Familial Central Precocious Puberty. J Clin Endocrinol Metab. 2017;102(5):1557–1567. doi: 10.1210/jc.2016-3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabria-Back J, Monteagudo-Sánchez A, Sánchez-Delgado M, Ferguson-Smith AC, Gómez O, Pertierra Cartada A, Tenorio J, Nevado J, Lapunzina P, Pereda Aguirre A, Giménez Sevilla C, Toro Toro E, Perez de Nanclares G, Monk D. Preimplantation genetic testing for a chr14q32 microdeletion in a family with Kagami-Ogata syndrome and Temple syndrome. J Med Genet. 2021;59(3):253–261. doi: 10.1136/jmedgenet-2020-107433. [DOI] [PubMed] [Google Scholar]
- 25.Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, Hino T, Suzuki-Migishima R, Kohda T, Ogura A, Ogata T, Yokoyama M, Kaneko-Ishino T, Ishino F. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet. 2008;40(2):243–248. doi: 10.1038/ng.2007.51. [DOI] [PubMed] [Google Scholar]
- 26.Gomes LG, Cunha-Silva M, Crespo RP, Ramos CO, Montenegro LR, Canton A, Lees M, Spoudeas H, Dauber A, Macedo DM, Bessa DS, Maciel GA, Baracat EC, Jorge AAL, Mendonca BB, Brito VN, Latronico AC. DLK1 Is a novel link between reproduction and metabolism. J Clin Endocrinol Metab. 2019;104(6):2112–2120. doi: 10.1210/jc.2018-02010. [DOI] [PubMed] [Google Scholar]
- 27.Montenegro L, Labarta JI, Piovesan M, Canton APM, Corripio R, Guillén L, Travieso-Suárez L, Martín-Rivada A, Barrios V, Seraphim C, Brito VN, Latronico AC, Argente J. Novel genetic and biochemical findings of DLK1 in children with central precocious puberty: a Brazilian-Spanish study. J Clin Endocrinol Metab. 2020;104(10):3165–3172. doi: 10.1210/clinem/dgaa461. [DOI] [PubMed] [Google Scholar]
- 28.Macedo DB, Kaiser UB. DLK1, notch signaling and the timing of puberty. Semin Reprod Med. 2019;37(4):174–181. doi: 10.1055/s-0039-3400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villanueva C, Jacquier S, de Roux N. DLK1 is a somato-dendritic protein expressed in hypothalamic arginine-vasopressin and oxytocin neurons. PLoS ONE. 2012;7(4):e36134. doi: 10.1371/journal.pone.0036134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pham A, Sobrier ML, Giabicani E, Le Jules FM, Mitanchez D, Brioude F, Netchine I. Screening of patients born small for gestational age with the Silver-Russell syndrome phenotype for DLK1 variants. Eur J Hum Genet. 2021;29(12):1756–1761. doi: 10.1038/s41431-021-00927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudoignon B, Huber C, Michot C, Di Rocco F, Girard M, Lyonnet S, Rio M, Rabia SH, Daire VC, Baujat G. Expanding the phenotype in Adams-Oliver syndrome correlating with the genotype. Am J Med Genet A. 2020;182(1):29–37. doi: 10.1002/ajmg.a.61364. [DOI] [PubMed] [Google Scholar]
- 32.Akawi N, Ben-Salem S, Lahti L, Partanen J, Ali BR, Al-Gazali L. A recessive syndrome of intellectual disability, moderate overgrowth, and renal dysplasia predisposing to Wilms tumor is caused by a mutation in FIBP gene. Am J Med Genet A. 2016;170(8):2111–2118. doi: 10.1002/ajmg.a.37741. [DOI] [PubMed] [Google Scholar]
- 33.Montalbán-Loro R, Lassi G, Lozano-Ureña A, Perez-Villalba A, Jiménez-Villalba E, Charalambous M, Vallortigara G, Horner AE, Saksida LM, Bussey TJ, Trejo JL, Tucci V, Ferguson-Smith AC, Ferrón SR. Dlk1 dosage regulates hippocampal neurogenesis and cognition. Proc Natl Acad Sci U S A. 2021;118(11):e2015505118. doi: 10.1073/pnas.2015505118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes S, Lewis A, Hajkova P, Dean W, Oswald J, Forné T, Murrell A, Constância M, Bartolomei M, Walter J, Reik W. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum Mol Genet. 2003;12(3):295–305. doi: 10.1093/hmg/ddg022. [DOI] [PubMed] [Google Scholar]
- 35.Kojima S, Shiochi N, Sato K, Yamaura M, Ito T, Yamamura N, Goto N, Odamoto M, Kobayashi S, Kimura T, Sekita Y. Epigenome editing reveals core DNA methylation for imprinting control in the Dlk1-Dio3 imprinted domain. Nucleic Acids Res. 2022;50(9):5080–5094. doi: 10.1093/nar/gkac344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X, He H, Shao L, Cui S, Yu H, Zhang X, Wu Q. Deletion of Meg8-DMR enhances migration and invasion of MLTC-1 depending on the CTCF binding sites. Int J Mol Sci. 2022;23(15):8828. doi: 10.3390/ijms23158828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Han Z, He H, Zhang X, Zhang M, Li B, Wu Q. Meg8-DMR as the secondary regulatory region regulates the expression of MicroRNAs while it does not affect embryonic development in mice. Genes (Basel) 2023;14(6):1264. doi: 10.3390/genes14061264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file1: Supplementary table 1. Pyrosequencing results for the affected family and controls.
Data Availability Statement
Data are available from the corresponding author on request.