

Abstract
RNA viruses have 5′ and 3′ untranslated regions (UTRs) that contain specific signals for RNA synthesis. The coronavirus genome is capped at the 5′ end and has a 3′ UTR that consists of 300 to 500 nucleotides (nt) plus a poly(A) tail. To further our understanding of coronavirus replication, we have begun to examine the involvement of host factors in this process for two group II viruses, bovine coronavirus (BCV) and mouse hepatitis coronavirus (MHV). Specific host protein interactions with the BCV 3′ UTR [287 nt plus poly(A) tail] were identified using gel mobility shift assays. Competition with the MHV 3′ UTR [301 nt plus poly(A) tail] suggests that the interactions are conserved for the two viruses. Proteins with molecular masses of 99, 95, and 73 kDa were detected in UV cross-linking experiments. Less heavily labeled proteins were also detected in the ranges of 40 to 50 and 30 kDa. The poly(A) tail was required for binding of the 73-kDa protein. Immunoprecipitation of UV-cross-linked proteins identified the 73-kDa protein as the cytoplasmic poly(A)-binding protein (PABP). Replication of the defective genomes BCV Drep and MHV MIDI-C, along with several mutants, was used to determine the importance of the poly(A) tail. Defective genomes with shortened poly(A) tails consisting of 5 or 10 A residues were replicated after transfection into helper virus-infected cells. BCV Drep RNA that lacked a poly(A) tail did not replicate, whereas replication of MHV MIDI-C RNA with a deleted tail was detected after several virus passages. All mutants exhibited delayed kinetics of replication. Detectable extension or addition of the poly(A) tail to the mutants correlated with the appearance of these RNAs in the replication assay. RNAs with shortened poly(A) tails exhibited less in vitro PABP binding, suggesting that decreased interactions with the protein may affect RNA replication. The data strongly indicate that the poly(A) tail is an important cis-acting signal for coronavirus replication.
Coronaviruses are members of the newly described order Nidovirales, a group of positive-sense, single-stranded RNA viruses that synthesize a nested set of subgenomic mRNAs during infection (reviewed in reference 9). Coronaviruses possess the largest known RNA virus genome, which is 26 to 30 kb long and contains 9 or 10 open reading frames (ORFs) as well as short untranslated regions (UTRs) at the 5′ and 3′ ends (reviewed in reference 27). The genome is capped at the 5′ end and polyadenylated at the 3′ end. Virus replication occurs entirely in the cytoplasm, with RNA synthesis being carried out by viral ribonucleoprotein (RNP) complexes that presumably also include cellular proteins.
Following coronavirus entry into cells, the plus-strand RNA genome serves as the initial template for both translation of the viral replicase proteins, including the RNA-dependent RNA polymerase, and synthesis of full-length minus-strand RNA. A nested set of subgenomic mRNAs that contain 5′ and 3′ ends identical to those on the genomic RNA are also synthesized during infection (reviewed in reference 27). A minus-strand RNA complement to each subgenomic RNA is present in infected cells (17, 18, 48). The mechanism for the synthesis of the subgenomic RNAs is not fully understood, but several models have been proposed to explain mRNA transcription (2, 24, 47, 58).
Previous reports have implicated a role for the coronavirus UTRs in genome replication. Mouse hepatitis coronavirus (MHV) defective genomes lacking the 3′-terminal 55 nucleotides (nt) of the 3′ UTR and the poly(A) tail were unable to serve as templates for minus-strand synthesis (33). In addition, UTRs from both the 5′ and 3′ ends of the genome were necessary for defective genome plus-strand synthesis (23, 32). These UTRs must therefore serve as cis-acting signals for defective genome replication and, in this capacity, recruit viral factors and possibly cellular proteins for formation of the replication complex.
Several studies have investigated whether host proteins specifically bind to the UTRs of MHV (10, 64). Proteins with molecular masses of 142, 120, 100, 103, 81, 70, 55, and 33 kDa bound the MHV 3′ UTR (64). To date, none of these proteins have been identified. However, two cellular proteins that bind to the 5′ end of the MHV genome have been identified. Heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), a cellular protein involved in alternative splicing of cellular mRNAs, binds the MHV minus-strand complement of the leader sequence (30). Polypyrimidine tract binding protein (PTB), also known as heterogeneous nuclear RNP I (hnRNP I), interacts with positive strand MHV leader RNA (29). Recently it was shown that PTB also binds the minus strand of the MHV 3′ UTR (20). It was suggested that hnRNP A1 and PTB may play a role in coronavirus transcription by helping mediate 5′- and 3′-end interactions (25, 26).
We began our studies on the role of host proteins in coronavirus genome transcription and replication by examining whether host proteins specifically bind the 3′ UTR of bovine coronavirus (BCV). Our results show that poly(A) binding protein (PABP) and other host proteins specifically interact with the 3′ UTR. As expected, the poly(A) tail was required for PABP interaction with the 3′ UTR. These results led us to extend our study and investigate the importance of the poly(A) tail in coronavirus defective genome replication. We demonstrate that the poly(A) tail is an important cis-acting signal for RNA replication. A BCV defective RNA replicon that lacked a poly(A) tail was not competent for replication, whereas replicon RNAs that contained 5-, 10-, and 68-nt poly(A) tails were replicated during BCV infection. MHV MIDI-C defective interfering (DI) RNAs containing poly(A) tails of 0, 5, and 10 nucleotides were also examined and found to undergo replication and amplification over virus passage at delayed rates compared to wild-type MIDI-C DI RNA. Replication of MIDI-C RNA with shortened tails or lacking a tail altogether appeared to be mediated by poly(A) repair. Detectable tail extension of poly(A) mutants correlated with the appearance of these RNAs in the DI replication assay. Binding of PABP to the 3′ UTR of the replicons correlated with their ability to be replicated. These findings are consistent with current thinking that plus-strand RNA virus replication has evolved to depend on elements of the cellular translation machinery.
MATERIALS AND METHODS
Viruses and cell lines.
Human ileocecal adenocarcinoma (HCT-8) cells (American Type Culture Collection) were used for BCV infections. A clonal line of mouse L cells (17Cl1) was used for MHV-A59 infections. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS). Stocks of plaque purified Mebus strain BCV and MHV-A59 were grown and subjected to titer determination in HCT-8 and 17Cl1 cells, respectively.
Preparation of S10 cytoplasmic extracts.
Subconfluent cell monolayers were infected in serum-free DMEM at a multiplicity of infection of 5. After infection, the cells were cultured in DMEM containing 5% FCS. Mock-infected cells were maintained in parallel. Mock- and BCV-infected cytoplasmic lysates were prepared 10 h after infection. The cells were washed three times with ice-cold phosphate-buffered saline and twice with ice-cold hypotonic buffer (10 mM HEPES [pH 7.6], 10 mM KCl, 1.5 mM MgCl2). The cells were resuspended in hypotonic lysis buffer that contained 1 mM phenylmethylsulfonyl fluoride, EDTA-free 1× protease inhibitor cocktail (Boehringer Mannheim), 0.5% Nonidet P-40 (NP-40), and 0.5% sodium deoxycholate. After being incubated on ice for 10 min, the cell lysates were centrifuged at 10,000 × g for 15 min at 4°C to remove nuclei and cell debris. Following centrifugation, the supernatant was dialyzed overnight at 4°C against hypotonic buffer supplemented with 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail, and 5% glycerol. Protein concentrations were determined using a bicinchonic acid protein assay (Pierce). Aliquots of the dialyzed supernatants were stored at −80°C. All lysates were monitored for the viral nucleocapsid protein by Western blotting. The lysates were stable for 1 month when stored at −80°C.
Plasmid construction.
All oligonucleotides used for plasmid constructions in this study are listed in Table 1. Plasmid pBCV3′UTR was constructed in two steps. First, the 426-nt PstI fragment from pDrep (5) was ligated into PstI-cut pGEM3Zf(+) to create pDrep3′. PCR primers SY-Eco and SY-Sty were used to amplify 217 nt from the 3′ UTR of pDrep. This fragment was digested with EcoRI and StyI before being ligated into EcoRI-StyI-cut pDrep3′ plasmid to create pBCV3′UTR (Fig. 1).
TABLE 1.
Oligonucleotides used in this study
| Name | Sequencea | Use | Polarity |
|---|---|---|---|
| SY-Eco | 5′ GGA GAA TTC ACC TTA TGT CG 3′ | BCV 3′ UTR | + |
| SY-Sty | 5′ CTT CCC TTG GGC ACT TG 3′ | BCV 3′ UTR | − |
| JS2 | 5′ GGC GAT CTA GAT GGG TAA CG 3′ | MHV 3′ UTR | − |
| JS3 | 5′ GAG AAT TCA TCC TAT GTC GGC G 3′ | MHV 3′ UTR | + |
| JS8 | 5′ AAT TGG AAG AAT CAC A 3′ | BCV 3′ A1, Drep A1 | + |
| JS9 | 5′ CGC GTG TGA TTC TTC C 3′ | BCV 3′ A1, Drep A1 | − |
| JS10 | 5′ AAT TGG AAG AAT CAC AAA AA 3′ | BCV 3′ A5, Drep A5 | + |
| JS11 | 5′ CGC GTT TTT GTG ATT CTT CC 3′ | BCV 3′ A5, Drep A5 | − |
| JS12 | 5′ AAT TGG AAG AAT CAC AAA AAA AAA A 3′ | BCV 3′ A10, Drep A10 | + |
| JS13 | 5′ CGC GTT TTT TTT TTG TGA TTC TTC C 3′ | BCV 3′ A10, Drep A10 | − |
| JS16 | 5′ AAT TGG AAG AAT CAC G 3′ | MIDI-C A0, MHV3′ A0 | + |
| JS17 | 5′CTA GCG TGA TTC TTC C 3′ | MIDI-C A0, MHV3′ A0 | − |
| JS18 | 5′ AAT TGG AAT AAT CAC AAA AAG 3′ | MIDI-C A5, MHV3′ A5 | + |
| JS19 | 5′ CTA GCT TTT TGT GAT TCT TCC 3′ | MIDI-C A5, MHV3′ A5 | − |
| JS20 | 5′ AAT TGG AAT AAT CAC AAA AAA AAA AG 3′ | MIDI-C A10, MHV3′ A10 | + |
| JS21 | 5′ CTA GCT TTT TTT TTT GTG ATT CTT CC 3′ | MIDI-C A10, MHV3′ A10 | − |
| JS24 | 5′ CTG AAT CTA AAG TGT GTG TTT GG 3′ | MHV poly(A) PCR | + |
| Oligo(dT)12–18 | MHV poly(A) PCR | − | |
| M144-163 | 5′ TAG CAT GTT TAT TTA TGT TG 3′ | MHV M PCR | + |
| M648-633 | 5′ GTT TGA GGG CAG TCG G 3′ | MHV M PCR | − |
| TGEV | 5′ AGA TCC ATG GCA CCA TCC TTG GCA ACC CAG 3′ | Drep Northern blotting | − |
Restriction sites added during cloning are underlined.
FIG. 1.

Schematic of BCV and MHV-A59 wild-type and defective genomes. ORFs are labeled and depicted as white boxes. The UTRs are shaded, with the 5′ UTR shown in black and the 3′ UTR shown in grey. Portions of the genome which are present in Drep, a cloned BCV defective genome (5), or MIDI-C, a cloned MHV-A59 DI (8), are indicated by dotted lines. Drep contains a 30-nt TGEV (T) reporter sequence. P indicates the MHV packaging signal. An expanded view of the BCV 3′ UTR and restriction sites used in this study is also shown.
Plasmid pMHV3′UTR was created by PCR amplifying a 394-nt region from pMIDI-C (8) using primers JS3 and JS2. EcoRI and XbaI sites were introduced at the 5′ and 3′ ends, respectively, during amplification. The amplified region was digested with EcoRI and XbaI and ligated into EcoRI-XbaI-cut pGEM3Zf(+) plasmid.
To make plasmid pBCV3′A1, oligonucleotides JS8 and JS9 were annealed to create a linker containing the authentic sequence between the unique MunI site and the 3′ end of the BCV 3′ UTR minus 67 nt of the poly(A) tail. Annealing was performed with equivalent amounts of each oligonucleotide at 37°C for 30 min. The linker was subsequently phosphorylated with T4 polynucleotide kinase (Promega) for 1 h at 37°C. Ligation of the linker into MunI-MluI-cut pBCV3′UTR resulted in deletion of 67 of the 68 A residues from the poly(A) tail.
Plasmid pBCV3′A5 was created in the same manner as described for pBCV3′A1, using oligonucleotides JS10 and JS11. Oligonucleotides JS12 and JS13 were used to make pBCV3′A10.
Plasmids pDrepA1, pDrepA5, and pDrepA10 were also constructed using JS8-JS9, JS10-JS11, and JS12-JS13 oligonucleotide pairs, respectively. The oligonucleotides were annealed, and the phosphorylated linkers were ligated into MunI-MluI-digested pDrep DNA.
pMHV3′UTR plasmid DNA was also manipulated by linker mutagenesis to contain poly(A) tails of 0 (pMHV3′A0), 5 (pMHV3′A5), and 10 (pMHV3′A10) A residues. Complementary oligonucleotides JS16-JS17, JS18-JS19, and JS20-JS21 were annealed to create linkers containing the designated poly(A) sequence flanked by MunI and NheI restriction site overhangs and were cloned into MunI-NheI-digested pMIDI-C DNA.
Plasmids pMIDI-C A0, pMIDI-C A5, and pMIDI-C A10 were constructed by replacing the NruI-NheI fragment from MIDI-C with the NruI-NheI fragments from pMHV3′A0, pMHV3′A5, and pMHV3′A10, respectively.
All plasmid sequences involving oligonucleotide synthesis and PCR amplification were verified by dideoxynucleotide sequencing using Sequenase (U.S. Biochemicals).
Preparation of RNA transcripts.
32P-labeled RNAs were prepared by in vitro runoff transcription of linearized plasmid DNAs with T7 RNA polymerase as specified by the manufacturer (Promega). pBCV3′UTR, pBCV3′A1, pBCV3′A5, pBCV3′A10, pDrepAwt, pDrepA1, pDrepA5, and pDrepA10 were linearized with MluI. pMHV3′UTR, pMHV3′A0, pMHV3′A5, pMHV3′A10, pMIDI-C Awt, pMIDI-C A0, pMIDI-C A5, and pMIDI-C A10 were linearized with NheI. Vector pGEM3Zf(+) was linearized with ScaI. Plasmid pGEM3HA that contains the influenza virus hemagglutinin gene (16) was digested with StyI. DNA templates used to transcribe RNAs for replication, stability, and streptavidin pulldown experiments were mung bean nuclease treated after endonuclease digestion to remove extraneous nucleotides on the noncoding strand of the DNA. Transcription reactions were carried out for 1 h at 37°C. Template DNA was destroyed by incubation for an additional 15 min with 20 U of RNase-free DNase. Unincorporated nucleotides were removed from labeled transcripts using micro Bio-Spin columns (Bio-Rad) which had been washed extensively with diethylpyrocarbonate-treated water. Transcripts were monitored on 5% polyacrylamide gels containing 8 M urea. The specific activity of each probe was determined using standard methods. Unlabeled RNAs were transcribed using the T7 MEGAscript kit (Ambion). RNAs generated for transfection were transcribed and capped using the MEGAscript kit, DNase treated, LiCl precipitated, quantitated spectrophotometrically at an optical density of 260 nm (OD260), and monitored on 1% agarose–0.25× Tris-borate-EDTA (TBE) gels.
To generate biotinylated RNAs, biotin-21-UTP (Clontech) was included in MEGAscript (Ambion) transcription reactions at a biotin-21-UTP/UTP ratio of 1:5. Biotinylated RNAs were processed as described for unlabeled RNA transcripts.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed as follows. RNA-protein binding reactions were performed by preincubating 1 to 10 μg of S10 cytoplasmic extracts with 20 μg of tRNA and a 50-fold molar excess of influenza virus HA RNA for 10 min at room temperature in reaction buffer (25 mM KCl, 5 mM HEPES [pH 7.6], 2 mM MgCl2, 0.1 mM EDTA, 3.8% glycerol, 2 mM dithiothreitol, 1 U of RNase inhibitor). Influenza virus HA RNA and tRNA were included to reduce nonspecific binding. After preincubation with the nonspecific RNAs, 1 ng of 32P-labeled BCV 3′ UTR RNA probe was added and the reaction mixtures were incubated an additional 10 min. Heparin (100 μg) was added, and the reaction mixtures were incubated further for 10 min. The final reaction volume was 10 μl. For competition assays, unlabeled competitor RNA was included in the preincubation step. RNA-protein complexes were resolved by electrophoresis through a 5% nondenaturing polyacrylamide gel in 0.5× TBE buffer (50 mM Tris, 45 mM boric acid, 0.5 mM EDTA) at constant voltage (350 to 450 V) at room temperature. The gels were preelectrophoresed for 45 min before the samples were loaded. The gels were fixed in a solution of 10% methanol and 5% acetic acid, dried, and autoradiographed.
UV-induced cross-linking of RNA-protein complexes.
RNA-protein binding reaction mixtures were assembled in microtiter plate wells. The binding reactions were set up as described for EMSAs, except that reaction volumes were tripled to give a final volume of 30 μl. Following binding, the reaction mixtures were placed on ice and UV irradiated at 254 nm (UV Stratalinker; Stratagene) at a distance of 10.5 cm for 30 min. After cross-linking, 25 μg of RNase A and 250 U of RNase T1 were added to each reaction mixture, and the mixtures were incubated at 37°C for 30 min. Laemmli sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer containing β-mercaptoethanol was added to the reaction mixtures. Samples were heated at 95°C before being resolved by SDS-PAGE. Gels were electrophoresed at 35 mA each, fixed in a solution of 10% methanol and 10% acetic acid, dried, and autoradiographed. For immunoprecipitation experiments, UV-cross-linked reaction mixtures from infected lysates were diluted in immunoprecipitation buffer (50 mM Tris [pH 7.6], 150 mM NaCl, 20 mM EDTA, 1% Triton X-100). The reaction mixtures were then incubated at 4°C for 2 to 4 h with antibody 61925, a polyclonal antibody against a human PABP (3), and then incubated with protein G-agarose (Pierce) for 1 h. Immunoprecipitates were washed three times with immunoprecipitation buffer and once with immunoprecipitation buffer lacking detergent. Immune complexes were eluted in Laemmli sample buffer, heated at 95°C for 3 min, and analyzed by SDS-PAGE.
Defective genome replication.
HCT cells at ∼70% confluency (∼106 cells) in 60-mm-diameter plates were infected with BCV at a multiplicity of infection of 5. Following infection, the inoculum was replaced with 1 ml of Opti-MEM (Life Technologies), and the cells were transfected immediately with 1 μg of capped RNA transcripts using Lipofectin (Life Technologies) as recommended by the manufacturer. At 4 h posttransfection, the medium was replaced with 2 ml of DMEM that contained 5% FCS. For virus passage experiments, cell culture supernatants were collected at 24 h after transfection and centrifuged for 15 min at 13,000 rpm in a microcentrifuge before being used to infect new cells.
MHV experiments were carried out in a similar manner using 17Cl1 cells. For virus passage experiments, cell culture supernatants were collected at 12 h after transfection and centrifuged for 15 min at 13,000 rpm in a microcentrifuge before being used to infect new cells.
Northern blot analysis.
To recover total intracellular RNA, cells were washed twice with phosphate-buffered saline and lysed with TRIzol reagent (Life Technologies) as specified by the manufacturer for isolation of RNA. RNA pellets were resuspended in diethylpyrocarbonate-treated water. ODs were determined and RNA concentrations were calculated for each sample. Equivalent amounts of total cytoplasmic RNA were denatured prior to electrophoresis at 80 V for 5 h (MHV) or 6 h (BCV) in 1% agarose gels containing 2.2 M formaldehyde. After electrophoresis, gels were vacuum blotted onto nylon membranes in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Following a brief soak in 6× SSC, the membranes were cross-linked using 0.12 J (UV Stratalinker). Blots were prehybridized in buffer containing 6× SSC, 5× Denhardt's solution, 50 μg of sheared salmon sperm DNA per ml, and 0.1% SDS for 2 h at 65°C for the transmissible gastroenteritis virus (TGEV) reporter probe or 68°C for the BCV N-specific or MHV N-specific probe prior to the addition of 32P-labeled probe. After an overnight incubation with probe, the membranes were washed and autoradiographed at −80°C for 1 to 24 h. A 5′-end-labeled oligonucleotide probe (Table 1) was used to specifically detect the TGEV reporter sequence in Drep and the Drep poly(A) tail mutants. To measure BCV or MHV replication 32P-labeled N-gene-specific riboprobes were used. The specific activities of the 32P-labeled probes ranged from 4.3 × 107 to 2.6 × 108 cpm/μg.
RNA stability.
HCT or 17Cl1 cells were seeded and transfected as described above. At the indicated times posttransfection, RNAs were extracted with TRIzol and analyzed by Northern blotting using the TGEV reporter oligonucleotide probe for Drep or an MHV N-gene-specific riboprobe for MIDI-C. Quantitation was performed by PhosphorImager (Molecular Dynamics) analysis.
Streptavidin pulldown assay.
Binding-reaction mixtures (50 μl) were assembled as described for EMSAs with the following modifications: 150,000 cpm of in vitro-translated luciferase or human PABP (hPABP) was used as a protein source, 1 μg of biotinylated BCV3′UTR or MHV3′UTR RNA was used as a probe, and the binding buffer contained 250 mM KCl, 5 mM HEPES [pH 7.6], 2 mM MgCl2, 0.1 mM EDTA, 3.8% glycerol, 2 mM dithiothreitol, 1 U of RNase inhibitor, and 0.2% NP-40. After the final 10-min incubation with heparin, 50 μl of a 20% Ultralink streptavidin (Pierce) suspension (packed volume/volume) was added to each sample and incubated for 15 min at room temperature with occasional mixing. Samples were washed five times with 1× binding buffer, eluted in 2× Laemmli sample buffer, and resolved by SDS-PAGE. To examine the recovery of biotinylated RNA, samples were assembled in double volume and, following the fifth wash with 1× binding buffer, divided in two. One half was processed as described for protein detection, and the other half was incubated with streptavidin conjugated to alkaline phosphatase (Zymed) for 15 min at room temperature in 200 μl of 1× binding buffer. The reaction mixtures were again washed five times with 1× binding buffer and resuspended in 100 μl of diethanolamine buffer (10% diethanolamine, 3 mM NaN3, 0.5 mM MgCl2) containing 1 mg of p-nitrophenyl phosphate (Sigma) per ml. The reaction mixtures were incubated for 30 min at 37°C, and the reactions were terminated by adding 50 μl of 3 M NaOH to each sample. The mixtures were briefly centrifuged, the supernatants were transferred to a microtiter plate, and the absorbance was read at OD410.
RT-PCR assay for poly(A) tail extension.
To determine whether poly(A) tail repair had occurred during the course of our replication experiments, a reverse transcription-PCR (RT-PCR) protocol was developed using oligo(dT)12–18 (Amersham Pharmacia) and a 5′ oligonucleotide primer, JS24 (Table 1). The 5′ PCR primer was designed to span the polymerase 1b-N junction found specifically in MIDI-C RNA (Fig. 1) to avoid amplification of viral genomic and subgenomic RNAs present in the total intracellular RNA samples. An 847-bp product was expected from successful RT-PCR. In vitro-transcribed MIDI-C RNA (200 ng) was reverse transcribed using 20 μM oligo(dT)12–18 with 8 to 10 U of avian myeloblastosis virus reverse transcriptase (Promega) at 37°C for 50 min. One-quarter of each reaction mixture was then subjected to 40 cycles of PCR amplification using Deep Vent polymerase (New England Biolabs) with 50 μM JS24 primer and oligo(dT)12–18 and the following cycling conditions: 94°C for 30 s, 40°C for 2 min, and 72°C for 1 min. A 5-min extension at 72°C was included following 40 cycles of PCR amplification. For replication samples, 500 ng of total cytoplasmic RNA from virus passages 0 to 4 was reverse transcribed in the presence of 20 μM oligo(dT)12–18 and 20 μM M648-633 primer (Table 1). One-quarter of every reaction mixture was then used in each of two parallel PCRs: poly(A) PCR [using JS24 primer and oligo(dT)12–18] and M PCR (using primers M144-163 and M648-633). The PCR products were monitored on 1% agarose–1× TBE gels.
RESULTS
Binding of host proteins to BCV 3′ UTR RNA.
To determine whether cellular proteins interact with the BCV 3′ UTR, a gel mobility shift assay was developed. We initially used an in vitro-generated transcript representing the entire 3′ UTR of BCV genomic RNA, including a 68-nt poly(A) tail, as a probe (Fig. 1). Uniformly labeled probes were incubated with cytoplasmic extracts from mock- and BCV-infected HCT cells. Binding-reaction mixtures were incubated at room temperature in the presence of excess noncoronavirus RNA to minimize nonspecific interactions. Protein-RNA complexes were analyzed by nondenaturing PAGE. Two distinct RNA-protein complexes were detected in both mock- and coronavirus-infected cytoplasmic extracts (Fig. 2A). Proteinase K sensitivity experiments demonstrated that both complexes contained protein components (data not shown).
FIG. 2.

Gel mobility shift and specificity of protein interactions with BCV3′UTR RNA. Cytoplasmic lysates were mock- or BCV-infected HCT cells. (A) Cytoplasmic lysates were preincubated with nonspecific competitor RNAs (influenza C virus HA RNA and yeast tRNA) for 10 min at room temperature prior to the addition of 32P-labeled BCV3′UTR RNA and further incubation for 10 min. Heparin was added, and incubation was continued for 10 min. RNA-protein complexes were resolved by nondenaturing PAGE (5% polyacrylamide). Lanes: 1, free probe; 2 to 5, addition of 5 μg (even-numbered lanes) and 10 μg (odd-numbered lanes) of cytoplasmic lysate from mock- and coronavirus-infected cells. The positions of free probe and protein complexes I and II are indicated. (B to D) Competition experiments were performed as described for the mobility shift assay, except that unlabeled competitor RNAs were also preincubated with cytoplasmic lysates prior to addition of 32P-labeled probe. Competitions were performed with 25- and 50-fold molar excesses of unlabeled BCV3′UTR RNA (B), 100- and 500-fold molar excesses of unlabeled pGEM RNA (C), or 25- and 50-fold molar excesses of unlabeled MHV3′UTR RNA (D).
The specificity of the RNA-protein complexes was determined by competition experiments with unlabeled competitor RNAs. Prior to the addition of labeled probe, unlabeled competitor RNAs were preincubated with cytoplasmic lysates. A 25-fold molar excess of unlabeled BCV 3′ UTR RNA efficiently competed for both complexes in mock-infected and infected cell lysates (Fig. 2B, lanes 3 and 4 and lanes 6 and 7). However, no competition was observed when a 100-fold molar excess of the pGEM RNA was included as the competitor (Fig. 2C, lanes 3 and 6). Addition of a 500-fold molar excess resulted in only very minor competition (lanes 4 and 7). The results clearly demonstrate that the BCV 3′ UTR-protein complexes are specific.
To further assess specificity and determine if the cellular proteins bound a closely related coronavirus 3′ UTR genomic RNA, the MHV A59 3′ UTR was used as the unlabeled competitor. A 25-fold molar excess of unlabeled MHV 3′ UTR RNA competed as efficiently as the BCV 3′ UTR RNA for complex formation (Fig. 2D, lanes 3 and 4 and lanes 6 and 7). Identical results were obtained when mock- and MHV A59-infected 17Cl1 cell lysates were used. Taken together, these results indicate that the 3′ UTR-protein complexes are coronavirus specific, at least for the group II viruses.
Size estimation of bound proteins by UV cross-linking.
To begin to identify the proteins that bound the BCV 3′ UTR, UV-cross-linking experiments were performed to estimate the sizes of the bound proteins. Gel shift reactions with the BCV 3′ UTR probe were set up in triple the normal volume. Competitions were performed with a 100- to 500-fold molar excess of unlabeled competitor RNAs. Protein-RNA complexes were UV cross-linked and extensively digested with a mix of RNase A and RNase T1 prior to analysis by SDS-PAGE. Several prominently labeled species were detected (Fig. 3, lane 2). The slowest-migrating species had estimated molecular masses of 99 and 95 kDa. The most heavily labeled protein was estimated to have a molecular mass of approximately 73 kDa. At least five or six additional faster-migrating species in the estimated molecular mass range of 30 to 60 kDa were also observed.
FIG. 3.

UV cross-linking of cellular proteins that bind to the BCV 3′ UTR RNA. 32P-labeled BCV3′UTR RNA probe was incubated in the absence of cell extract (lane 1) or in the presence of 30 μg of BCV-infected HCT cell extract (lanes 2 to 8). Samples were UV cross-linked for 30 min, RNase treated, heated at 95°C for 3 min, and resolved by SDS-PAGE (12% polyacrylamide). The positions of protein standards are indicated on the left. The arrow indicates the position of the p73 protein. To map the p73 protein binding site, UV cross-linking was performed in the presence of a 100-fold molar excess of coronavirus-specific unlabeled RNAs (BCV3′UTR, MHV3′UTR, BCV3′A1, BCV3′BalI, and BCV3′StyI) or 500-fold molar excess of pGEM RNA. Competitor RNAs were preincubated with lysate prior to the addition of 32P-labeled BCV3′UTR probe.
Mapping of the region bound by p73.
Since the 73-kDa species exhibited the strongest UV cross-linking signal, we chose to focus our initial efforts on further characterization of this protein. We first sought to determine the location of the p73 binding site(s) on the 3′ UTR. The unique restriction sites StyI and BalI within the 3′ UTR (Fig. 1) were used to create constructs from which truncated transcripts were transcribed. Transcription of BCV3′StyI yielded an RNA which lacked the 3′-most 172 nt [104 nt of UTR plus 68 nt of the poly(A) tail] of the 3′ UTR. BCV3′BalI transcripts lacked the 3′-most 85 nt [17 nt of UTR plus 68 nt of poly(A) tail] of the 3′ UTR. Results from UV-cross-linking experiments with the truncated RNA transcripts indicated that p73 bound the region encompassing the 3′ 17 nt and poly(A) tail (data not shown).
To further map the binding site(s) and determine the specificity of the UV-cross-linking experiments, a series of RNAs were used as competitors. BCV3′UTR, MHV3′UTR, BCV3′BalI, BCV3′StyI, pGEM, and BCV3′A1 RNA transcripts were used as competitors against labeled BCV3′UTR (Fig. 3). The BCV3′A1 transcript lacked all of the poly(A) tail, except for one A residue that was retained during construction. Preincubation of cell lysates with a 100-fold molar excess of unlabeled BCV3′UTR and MHV3′UTR prior to the addition of 32P labeled BCV3′UTR abolished detectable cross-linking of p73 to the labeled probe (Fig. 3, lanes 3 and 4). However, the same molar excess of BCV3′A1, BCV3′BalI, and BCV3′StyI, as well as a 500-fold molar excess of pGEM RNA, all failed to compete for p73 binding (lanes 5 to 8). This strongly suggested that p73 was binding to the poly(A) tail.
We were surprised to detect a protein that bound the poly(A) tail, since our assay was not designed to detect interactions with this region of our probe. Our full-length 3′ UTR probe was labeled with [α-32P]UTP, and we expected the unlabeled poly(A) tail and any cross-linked poly(A)-binding proteins to be cleaved away with the combination of RNase A and RNase T1. Therefore, we concluded that p73 must be recruited by the poly(A) tail sequence but must also be sufficiently close to or base paired with upstream labeled U residues to be detected by cross-linking. Alternatively, the tail may fold back such that p73 would be in the vicinity of upstream, labeled U residues. To address this issue, we labeled probes with [α-32P]ATP. However, the results of UV cross-linking experiments were never conclusive, since we were unable to completely digest the labeled probe, even when increasing amounts of multiple RNases were used (data not shown).
The BCV3′UTR and MHV3′UTR probes, both of which included the poly(A) tail, appeared to compete for some of the lower-molecular-weight cross-linked proteins (Fig. 3, lanes 3 and 4). However, binding of most of these proteins did not map to the poly(A) tail (Fig. 4, lane 5). It is possible, as was seen for the 3′ UTR of human rhinovirus, that protein binding is enhanced in the presence of a poly(A) tail (54). It also appeared that the BCV3′StyI probe partially competed for binding of the p99 and p95 species (Fig. 3, lane 7). The significance of these competitions will require more detailed mapping and analysis of the binding of these proteins to the 3′ UTR.
FIG. 4.

Identification of the UV cross-linked p73 protein. UV cross-linking was performed with 32P labeled BCV3′UTR RNA that contained (+) or lacked (−) the 68-nt poly(A) tail. RNA probes were incubated in the absence (lanes 1 and 4) or presence (lanes 2 and 5) of BCV-infected HCT cytoplasmic lysates. Double reactions were assembled for samples 2 and 5. Following RNase digestion, one-half of each reaction was immunoprecipitated (IP) with antibody 61925, a polyclonal antipeptide antibody against PABP (3) (lanes 3 and 6). The positions of protein standards are indicated on the left. The arrow indicates the position of PABP.
Identification of p73 as PABP.
After concluding that the poly(A) tail was necessary for binding of p73 to the 3′ UTR, it was logical to assume that the protein might be the cytoplasmic PABP. PABP is an abundant cytoplasmic protein of ∼70 kDa which interacts with the poly(A) tail that is present on most eukaryotic mRNAs (13). Nucleotide residues cross-linked to the protein probably account for the difference in the estimated sizes of p73 and PABP. To address whether p73 was indeed PABP, a polyclonal anti-peptide antibody against human PABP (3) was used to immunoprecipitate the protein from BCV-infected HCT lysates after UV cross-linking. Cell lysates from BCV-infected HCT cells were used in cross-linking experiments with both full-length BCV3′UTR RNA and BCV3′A1 RNA, which lacked the poly(A) tail. After the cross-linking step, equal samples from the infected-cell lysates were analyzed directly (Fig. 4, lanes 2 and 5) or after immunoprecipitation with the anti-PABP antibody (lanes 3 and 6). When analyzed directly, the p73 protein cross-linked to full length BCV3′UTR RNA (lane 2). However, a similar RNA-protein complex was not detected when the poly(A)-minus probe was used, further confirming that the tail is required for this complex to form (lane 5). When the lysates were immunoprecipitated after cross-linking, the antibody to PABP precipitated p73 that was cross-linked to the poly(A)-containing probe (lane 3). Proteins cross-linked to the BCV3′A1 RNA probe were not immunoprecipitated with the PABP antibody (lane 6). No proteins were immunoprecipitated after cross-linking to either probe using rabbit preimmune or anti-BCV nucleocapsid control sera (data not shown). The data indicated that p73 is PABP and confirmed that the poly(A) tail is necessary for PABP to interact with the BCV 3′ UTR.
The amount of PABP that was immunoprecipitated was greatly reduced compared to that of the p73 signal detected by direct UV cross-linking analysis (Fig. 4, compare lanes 2 and 3). Several possibilities may account for this. First, it is possible that more than one protein species may be cross-linked to the BCV 3′ UTR and that PABP is only one of the proteins that constitute the 73-kDa cross-linking signal. However, since PABP is found in vast excess within cells (13), it is more likely that free PABP protein that was not bound to the BCV 3′ UTR probe competed for immunoprecipitation of the cross-linked protein. Finally, the anti-PABP antibody has a low titer. Since we had a limited amount of the antibody for these experiments, our immunoprecipitations were probably not quantitative. Nevertheless, immunoprecipitation of UV-cross-linked p73 protein with PABP-specific antibody demonstrated that PABP interacts with the BCV 3′ UTR.
Requirement of the poly(A) tail for coronavirus defective genome replication.
After identifying PABP as one of the proteins that cross-linked to the 3′ UTR, this raised questions about the role of the poly(A) tail in coronavirus genome replication and the possibility that PABP might also play a role in RNA replication in addition to its role in translation. Previously published data indicated that the poly(A) tail is important for coronavirus RNA replication (33). We thus sought to more precisely define the requirement of the poly(A) tail for replication. Since an infectious clone is not available for coronaviruses, we used the BCV defective genome Drep (5) and MHV DI MIDI-C RNA (8) to address this question. Drep contains 498 nt from the 5′ end of BCV genomic RNA, the entire coding region of the N gene, 30 nt from TGEV that was engineered into the defective genome to function as a reporter sequence, and all of the 3′ UTR plus a 68-nt poly(A) tail (Fig. 1). The defective genome is replicated in BCV-infected cells (5). MIDI-C RNA is 5.5 kb long and contains 3.9 kb from the 5′ end of the MHV-A59 genome, including the entire 5′ UTR and a portion of ORF 1a sequence, 0.8 kb of ORF 1b sequence, and 0.8 kb from the 3′ end of the MHV genome including the carboxy terminus of the N ORF, the entire 3′ UTR, and a poly(A) tail (Fig. 1) (8).
To determine if the poly(A) tail is necessary for coronavirus RNA replication, we generated pDrep constructs containing poly(A) tails with 1 A residue (pDrepA1), 5 A residues (pDrepA5), or 10 A residues (pDrepA10), as well as pMIDI-C constructs containing poly(A) tails of 0 A residues (pMIDI-C A0), 5 A residues (pMIDI-C A5), or 10 A residues (pMIDI-C A10). Our mutants were designed based on the fact that the minimal binding site for PABP is 5 A residues in the context of a larger oligonucleotide (13). In vitro-generated transcripts from these constructs and wild-type Drep (DrepAwt) or MIDI-C (MIDI-C Awt) were transfected into mock- and BCV-infected HCT cells or MHV-infected 17Cl1 cells. Intracellular RNAs were extracted at 2 and 24 h (BCV P0) or 12 h (MHV P0) after transfection. Supernatants from virus-infected or -transfected cells were collected at P0, centrifuged to remove cellular debris, and used to infect new cells. Intracellular RNAs were harvested for four subsequent passages at 24 h (BCV) or 12 h (MHV) after infection. Total intracellular RNAs were quantified, and equal amounts of RNA were analyzed by Northern blotting. For the BCV experiments a 5′-end-labeled oligonucleotide probe that recognizes the TGEV reporter sequence in Drep and distinguishes between the defective genome and the viral subgenomic mRNAs was used (Fig. 5). An MHV N gene-specific riboprobe that recognizes MIDI-C and all viral genomic and subgenomic RNAs was used for the MHV experiments (Fig. 6).
FIG. 5.
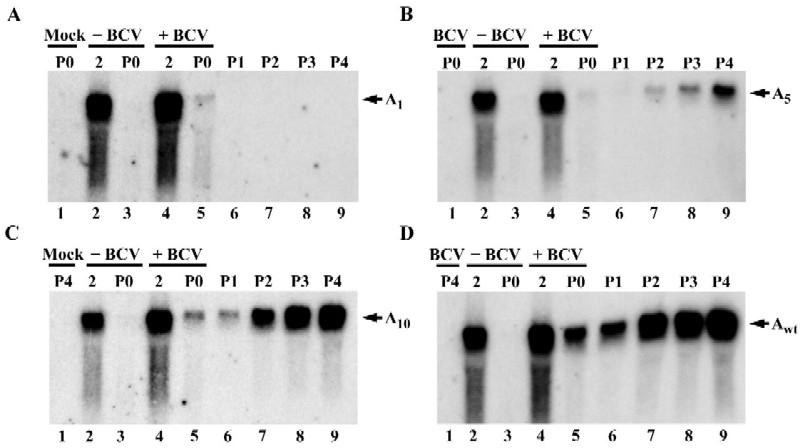
Replication of BCV defective genomes containing different lengths of poly(A) tail. Mock- or BCV-infected HCT cells were transfected with 1 μg of Drep RNAs. DrepA1 (A), DrepA5 (B), DrepA10 (C), and DrepA68 (D) contained poly(A) tails consisting of 1, 5, 10, and 68 A residues, respectively. Total intracellular RNA was extracted, resolved on denaturing agarose gels, and vacuum blotted onto nylon membranes as described in Materials and Methods. The blots were probed with a 5′-end-labeled oligonucleotide complementary to the TGEV reporter sequence in Drep. Lanes: 1, RNA from uninfected untransfected cells at 24 h after mock infection (A), RNA from BCV-infected untransfected cells at 24 h after infection (B), RNA from uninfected cells at 24 h after infection during P4 (C), and RNA from BCV-infected untransfected cells at 24 h after infection during P4 (D); 2 and 3, RNAs from uninfected cells transfected with the indicated RNAs; 4 and 5, RNAs from BCV-infected cells transfected with the indicated RNAs; RNAs were harvested at 2 or 24 h (P0) posttransfection as indicated across the top of each panel; 6 to 9, RNAs from cells at 24 h after infection with progeny virus. P1 to P4 indicates virus passage numbers.
FIG. 6.

Replication of MHV defective genomes containing different lengths of poly(A) tail. Mock- or MHV-infected 17Cl1 cells were transfected with 1 μg of MIDI-C RNAs. MIDI-C A0 (A), MIDI-C A5 (B), MIDI-C A10 (C), and MIDI-C Awt (D) contained poly(A) tails consisting of 0, 5, 10 and >50 A residues, respectively. Total intracellular RNA was extracted, resolved on denaturing agarose gels, and vacuum blotted onto nylon membranes. The blots were probed with a MHV N-gene specific riboprobe. Lanes: 1, RNA from uninfected untransfected cells at 12 h (P0) after mock infection (A), RNA from MHV infected untransfected cells at 12 h after infection (B), RNA from uninfected untransfected cells at 12 h after infection during P4 (C), and RNA from MHV-infected untransfected cells at 12 h after infection during P4 (D); 2, 20 ng of in vitro-transcribed MIDI-C Ax RNA to serve as a marker for the DI RNA; 3 and 4, RNAs from uninfected cells transfected with the indicated RNAs; 5 and 6, RNAs from MHV-infected cells transfected with the indicated RNAs; RNAs were harvested at 2 or 12 h (P0) posttransfection as indicated across the top of each panel; 7 to 10, RNAs from cells at 12 h after infection with progeny virus. M denotes marker RNA. P1 to P4 indicate virus passage numbers.
The results showed that little or none of the transfected Drep RNAs persisted in mock-infected cells at 24 h after transfection, indicating that signals detected in the BCV-infected cells represented newly replicated Drep (Fig. 5, lanes 2 and 3). In BCV-infected cells, all Drep RNAs were detected at 24 h; however only DrepA5, DrepA10, and DrepA68 were detected upon serial passage (Fig. 5B to D, lanes 6 to 9). Only a minor amount of what appeared to be residual transfected DrepA1 RNA was detected at the P0, 24-h time point (Fig. 5A, lane 5). No DrepA1 was detected upon passage (Fig. 5A, lanes 6 to 9). DrepA1 appeared not to be replicated or was so severely impaired for replication that it was undetectable upon passage. However, DrepAwt, DrepA10, and DrepA5 were clearly replication competent and were detected in passages 1 through 4 (Fig. 5B to D, lanes 6 to 9). In all experiments, comparable levels of virus replication were confirmed by reprobing blots with an N-gene-specific probe that recognizes genomic and all subgenomic viral RNAs (data not shown).
All MIDI-C RNAs were present at 2 h posttransfection in mock- and MHV-infected cells (Fig. 6, lanes 3 and 5). In mock-infected controls, none of the MIDI-C RNAs were detected at the P0 time point (lanes 4). In MHV-infected cells, replication of MIDI-C A10 (Fig. 6C, lanes 6 to 10) and MIDI-C Awt (Fig. 6D, lanes 6 to 10) was detected at all virus passages. Replication of MIDI-C A5 (Fig. 6B, lanes 6 to 10) was readily detectable from P1 through P4. However, MIDI-C A0 RNA was undetectable at early passages but replication was consistently observed at P3 or P4 (Fig. 6A, lanes 6 to 10).
The stability of the transfected Drep RNAs in the absence of viral infection was determined by harvesting total cytoplasmic RNA between 2 and 24 h following transfection and analyzing it by Northern blotting. All Drep RNAs exhibited similar decay rates (Fig. 7A), with a half-life of approximately 10 h. All MIDI-C RNAs exhibited similar decay rates (Fig. 7B) and possessed half-lives of approximately 5.3 h. It therefore appeared that there was no striking difference in the stabilities of the Drep and MIDI-C RNAs that could account for the observed differences in replication.
FIG. 7.

Stability of coronavirus defective genomes containing different lengths of poly(A) tail. (A) HCT cells transfected for 4 h with 1 μg of Drep RNAs. (B) 17Cl1 cells transfected for 4 h with 1 μg of MIDI-C RNAs. At various times posttransfection, total intracellular RNA was extracted and analyzed for the presence of Drep RNA by Northern blotting. The times when RNA was harvested are denoted above each panel. (A) Upper panel, lanes 1 to 5, DrepA1 RNA; upper panel, lanes 6 to 10, DrepA5 RNA; lower panel, lanes 1 to 5, DrepA10 RNA; lower panel, lanes 6 to 10, DrepAwt RNA. (B) Upper panel, lanes 1 to 7, MIDI-C A0 RNA; upper panel, lanes 8 to 14, MIDI-C A5 RNA; lower panel, lanes 1 to 7, MIDI-C A10 RNA; lower panel, lanes 8 to 14, MIDI-C Awt RNA.
PABP binding correlates with coronavirus defective genome replication.
To determine whether binding of the host factor PABP correlated with the observed replication phenotypes of our replicons, a streptavidin capture assay was developed. In vitro-translated, 35S-labeled PABP or luciferase was incubated with biotinylated 3′ UTR RNAs from the wild type and shortened-poly(A)-tail mutants used in the replication experiments described above. Immobilized streptavidin (Pierce) was added, and following further incubation, complexes were centrifuged and washed. Samples were divided in half. One aliquot was assayed for RNA recovery using an alkaline phosphatase-based colorimetric assay; RNA was recovered in all cases, as determined by the OD410 reading (data not shown). The second aliquot was assayed for protein binding by SDS-PAGE (Fig. 8). The levels of background binding in the absence of biotinylated RNA probe were negligible for the negative control luciferase and for PABP (lanes 2 and 7, respectively). No binding above background was detected between luciferase and any of the 3′ UTR RNAs (lanes 3 to 6).
FIG. 8.

In vitro binding of PABP to coronavirus 3′ UTR RNAs. In vitro-translated luciferase or PABP was incubated with 1 μg of biotinylated BCV3′UTR RNAs containing poly(A) tails of 1, 5, 10, or 68 A residues (A) or MHV3′UTR RNAs containing poly(A) tails of 0, 5, 10, or >50 A residues (B). Immobilized streptavidin was added to recover biotinylated RNA complexes, and samples were washed to remove any unbound RNA or protein. Samples were analyzed by SDS-PAGE (8% polyacrylamide). M (lanes 1 and 12) denotes marker and corresponds to the input amount of radiolabeled luciferase (lane 1) or PABP (lane 12) in each reaction. Lanes 2 and 7 represent the level of background protein binding in the absence of biotinylated RNA for luciferase (lane 2) and PABP (lane 7). (A) Lanes 3 to 6, luciferase recovered from interaction with BCV3′UTR RNAs; lanes 8 to 11, PABP recovered from interaction with BCV3′UTR RNAs. (B) Lanes 3 to 6, luciferase recovered from interaction with MHV3′UTR RNAs; lanes 8 to 11, PABP recovered from interaction with MHV3′UTR RNAs.
Binding of PABP to BCV3′A5, BCV3′A10, and BCV3′Awt was detected (Fig. 8A, lanes 9 to 11) and was calculated to be in the range of 14- to 100-fold greater than background. The interaction between PABP and BCV3′A1 was essentially that of background, at only 1.5- to 2-fold greater than the binding observed in the absence of RNA (compare lanes 7 and 8). PABP also readily interacted with MHV3′A10 and MHV3′Awt (Fig. 8B, lanes 10 and 11); however, interactions with MIDI-C A0 and MIDI-C A5 were comparably weak, being only slightly above background binding between PABP and immobilized streptavidin in the absence of biotinylated RNA (compare lanes 8 and 9 with lane 7). It was expected that MHV3′A0 would not interact with PABP, since this RNA lacks the minimal PABP binding site of 5 contiguous A residues (13). However, MHV3′A5 was expected to bind PABP, since we observed an interaction between BCV3′A5 RNA and PABP. It is possible that the structure assumed by the 3′ UTR RNA accounts for these results. Nonetheless MHV3′A10 and MHV3′Awt RNAs, which replicated the most efficiently in our DI replication assay, clearly interacted with PABP.
Poly(A) tail extension of truncated defective genomes.
We thought it likely that the eventual accumulation of RNAs containing severely truncated poly(A) tails was due to reestablishment of replication competency through poly(A) tail repair. To determine whether poly(A) tail repair had occurred during the course of our MIDI-C replication experiments, an RT-PCR method was developed using oligo(dT)12–18 and a 5′ oligonucleotide primer, JS24 (Table 1), spanning the junction between MIDI-C polymerase 1b and nucleocapsid ORF sequences. The 5′ PCR primer was designed to span the polymerase 1b-N junction found specifically in MIDI-C RNA to avoid amplification of viral genomic and subgenomic RNAs present in the total intracellular RNA samples. An 847-bp product was expected from successful RT-PCR.
Initially MIDI-C A0, MIDI-C A5, MIDI-C A10, and MIDI-C Awt in vitro-transcribed, capped RNAs that had been DNase treated were used as templates for RT-PCR to ascertain whether they contained poly(A) tails of sufficient length to be amplified by our method. A 200-ng portion of each RNA was reverse transcribed with avian myeloblastosis virus reverse transcriptase (Promega) at 37°C for 50 min. One-quarter of each reaction mixture was then subjected to 40 cycles of PCR amplification using Deep Vent polymerase with JS24 primer and oligo(dT)12–18. As shown in Fig. 9A, only MIDI-C Awt in vitro-transcribed RNA was amplified by RT-PCR (lane 4). MIDI-C A0, MIDI-C A5, and MIDI-C A10 (lanes 1 to 3, respectively) therefore did not contain poly(A) tails of sufficient length for priming by oligo(dT)12–18 during RT. This allowed us to track poly(A) addition to these RNAs over subsequent virus passages during DI replication experiments.
FIG. 9.

Poly(A) tail repair of MIDI-C mutant RNAs during DI replication over four virus passages. (A) In vitro-transcribed MIDI-C RNAs were used as templates to establish the RT-PCR assay for poly(A) tail repair. A poly(A) tail of more than 10 A residues was necessary to amplify MIDI-C using oligo (dT)12–18 and primer JS24 (Table 1) (lanes 1 to 4). (B to E) RT-PCR analysis of RNAs from the DI replication experiment in Fig. 6. An 847-bp RT-PCR product was expected using primers oligo(dT)12–18 and JS24 (lanes 1 to 5). Primers M648-633 and M144-163 (Table 1) were used as a control to amplify a 500-bp portion of the M gene (lanes 6 to 10). PCR products were analyzed by electrophoresis through 1% agarose gels containing ethidium bromide. M denotes DNA markers corresponding to the following sizes (in kilobases) from top to bottom: (A) 1.2, 0.8, and 0.4; (B to E) 2.0, 1.2, 0.8, and 0.4. C denotes control for the size of the expected poly(A) PCR product and corresponds to 10 μl of PCR product from RT-PCR of in vitro-transcribed MIDI-C Awt RNA.
RNA samples from DI replication experiments were then examined by RT-PCR. A 500-ng portion of total cytoplasmic RNA from virus passages 0 to 4 of the experiment in Fig. 6 was reverse transcribed in the presence of oligo(dT)12–18 as well as M648-633 primer (Table 1). M648-633 primer was included for further amplification of part of the MHV M gene. Amplification of MHV M sequence was expected from all RNA samples, whereas amplification of the poly(A) mutants was not. One-quarter of every reaction mixture was then used in each of two parallel PCR amplifications: poly(A) PCR [using JS24 primer and oligo(dT)12–18] and M PCR (using primers M144-163 and M648-633).
A 500-nt MHV M fragment was amplified from all samples (Fig. 9B to E, lanes 6 to 10), indicating that none of the samples contained RT-PCR inhibitors. The appearance of the extended tails, at least as detected by this assay, paralleled the results for MIDI-C Awt and the mutants from the DI replication assay. As expected, MIDI-C Awt samples yielded the predicted 847-bp amplicon from all virus passages (Fig. 9E, lanes 1 to 5) following RT-PCR, consistent with replication of the DI RNA at each virus passage. RT-PCR products for all of the mutants were also observed concurrent with the appearance of definitively detectable replication (compare Fig. 9B to D with Fig. 6A to C). The results indicated that upon transfection into MHV-infected cells, MIDI-C A0, MIDI-C A5, and MIDI-C A10 underwent poly(A) extension and were replicated.
Many attempts were made to determine whether poly(A) tail extension occurred on Drep-A5 and Drep-A10 from the replication experiments. However, for technical reasons that we do not understand, we were unable to successfully amplify a specific product using oligo(dT)12–18 and several Drep-specific upstream primers. All attempts to optimize the RT-PCR method for Drep did not alleviate this problem.
Taken together, the overall results with both systems support the same general conclusion. The coronavirus poly(A) tail is an important cis-acting signal for efficient DI and, by inference genomic, RNA replication. The ability of the defective genomes to be more efficiently propagated appears to correlate with binding of PABP to coronavirus 3′ UTR RNAs.
DISCUSSION
Results from this study provide further evidence that the poly(A) tail is an important cis-acting signal for coronavirus RNA replication. The data are the first to demonstrate that the poly(A) tail is required for BCV RNA replication. In addition, the results extend the results of a previous study in which it was shown that the poly(A) tail is required for MHV minus-strand synthesis (33). The results are also the first to demonstrate that host proteins specifically interact with the 3′ UTR of BCV genomic RNA.
Defective genomes containing shortened poly(A) tails were used to determine the importance of the poly(A) tail in viral RNA replication. Deletion of all but one A residue from the poly(A) tail resulted in failure of BCV Drep to be replicated, since little if any of the replicon RNA was detected at the P0 time point and none was detected from four subsequent virus passages. We have not determined whether the block is at the level of minus- or plus-strand synthesis, but we assume, based on previous results from Lin et al. (33), that the initial block to replication was at the level of minus-strand synthesis.
In contrast to BCV DrepA1, MHV MIDI-C A0 replication was detected at late virus passages. Cursory comparison of our results with those of Lin et al. (33) would indicate that the two are in conflict. However, differences in the protocols used in the two studies most probably account for this. The earlier study was unable to detect minus strands derived from a different poly(A) tail-lacking DI RNA at 6 h after transfection (33). We assayed for RNA replication and performed subsequent viral passages at 12-h time points. The additional 6 h following transfection (P0) probably allowed for either very inefficient replication that was not measurable by Northern blotting or PCR or, more likely, repair of the mutant poly(A) tails. Nonetheless, our data confirm that MHV DI replication is dependent on the presence of a poly(A) tail and extend the results of the previous work by showing that when DI RNAs with a shortened or deleted poly(A) tail are transfected into MHV-infected cells, the RNAs are not lost but are replicated over time. Our data strongly indicate that there is selective pressure for repair or restoration of the missing or truncated poly(A) tails, since the replication efficiency dramatically increased for all DIs once their tails were elongated.
We clearly demonstrated that the poly(A) tail was restored on MIDI-C A0. Both MIDI-C A5 and MIDI-C A10 underwent poly(A) extension, which appears to have contributed to the ability of these RNAs to be replicated, since the kinetics of the extension and replication were directly correlated. Replication of DrepA5 and DrepA10 could also be attributable to poly(A) tail repair, even though we were unable to demonstrate this. Repair could potentially occur by recombination with helper virus. This is the most likely explanation for the rescue of MIDI-C A0, as well as possibly MIDI-C A5 and MIDI-C A10. However, we do not support this explanation for Drep, since it is difficult to imagine that DrepA1 would not also be equally capable of recombination, assuming that A residues at the 3′ end are not involved in the mechanism of recombination. An alternate mechanism by which repair might occur for the shortened tails is through poly(A) extension by cytoplasmic poly(A) polymerase, which recognizes preformed tails of a minimum length and extends them (61). It is also possible that the shortened poly(A) tails of both MIDI-C and Drep were extended during the first round of plus-strand synthesis. Poly(A) tail extension of viral RNAs with shortened tails or lacking tails altogether has also been observed with other viruses (14, 15).
Even though Drep and MIDI-C RNAs with shortened tails were replicated, the overall amount of replication for these RNAs was lower than for wild-type Drep and MIDI-C. The amount of RNA at P4 mirrored the lengths of the starting RNAs, with A5 < A10 < Awt. The efficiency of any of the above-mentioned mechanisms during P0 and P1, which may have allowed the truncated RNAs to be replicated, could account for the results.
The poly(A) tail is important for replication of other plus-strand viruses. The presence of a poly(A) tail increases the infectivity of poliovirus RNA (46, 51). In vitro studies with encephalomyocarditis virus suggest that 3′-poly(A) may play a role in viral RNA template selection by the viral polymerase (7). The presence of a poly(A) tail on Sindbis virus RNA appears to be important, but not absolutely required, for replication (15).
What roles might the poly(A) tail play in coronavirus replication? Following entry into cells, the viral replicase proteins, including the polymerase, are translated from the polyadenylated genome. Therefore, the poly(A) tail must play its first role at the point of translation. Following translation, the genomic RNA is used as a template for minus-strand synthesis. During this step, the poly(A) tail may function as part of the promoter that is recognized by the polymerase. The requirement for 55 nt at the 3′ end of the genome plus the poly(A) tail for minus-strand synthesis is consistent with the notion that the tail might be part of a promoter (33). Also, the presence of a short poly(U) tract at the 5′ end of BCV minus-strand RNAs suggests that initiation of minus-strand synthesis may occur within the poly(A) tail (17). The poly(A) tail is generally predicted to be single stranded. Data suggest that there may be a general requirement for a single-stranded region for initiation of minus-strand RNA synthesis (43). Since neither the BCV 3′ UTR nor the MHV 3′ UTR contains the polyadenylation motif (AAUAAA) that is highly conserved in eukaryotic mRNAs (44), a short stretch of A residues may have to be copied during minus-strand synthesis to ensure that a poly(A) tail is added during the subsequent rounds of plus-strand synthesis.
Another possibility is that part of the poly(A) tail might interact with upstream nucleotides to form a functional promoter for polymerase recognition. Our cross-linking data suggest that, at least in the context of the 3′ UTR probe, the poly(A) tail can interact with upstream nucleotides. It was recently reported that part of the bamboo mosaic potexvirus RNA poly(A) tail is involved in the formation of a potential pseudoknot that is required for efficient replication (55). A pseudoknot is present within the BCV 3′ UTR that appears to be involved in Drep RNA replication; however, the poly(A) tail is not predicted to be part of this structure (62).
The first study to report an analysis of host protein interactions with the 3′ UTR of MHV failed to detect proteins that bound a 90-nt probe representing the very 3′ end of the genome (10). More recently, host protein binding to the MHV 3′ UTR was detected (64). Proteins with molecular masses of 142, 120, 100, 103, 81, 55, and 33 kDa were reported to bind the MHV JHM 3′ end. Two protein binding elements that bound the majority of these proteins were later mapped within the MHV 3′ UTR (34, 63). These elements are completely conserved among MHV strains (40). The 3′-most element (5′ UGAAUGAAGUU 3′) is also completely conserved between MHV and BCV, whereas the 5′-most element (BCV, 5′ UUGGAGAAAGU 3′; MHV, 5′ UGAGAGAAGUU 3′) of the two viruses has only 64% homology.
It remains to be determined if any of the proteins that bound the 3′ UTR in our study are the same as those observed in the study by Yu and colleagues. With the exception of the >100-kDa species that bound the MHV 3′ end, the proteins that cross-linked to the BCV 3′ UTR and those that bound the MHV 3′ UTR are in the same general molecular mass range. We have also examined the MHV 3′ UTR by UV cross-linking using 17Cl1 cell lysates and obtained profiles that were similar to our results with the BCV 3′ UTR (data not shown). The apparent differences in protein molecular masses previously reported for MHV (64) and in our results are most probably due to differences in the methods used to detect the 3′ UTR binding proteins. The probes used by Yu et al. (64) did not contain a poly(A) sequence and in some cases were significantly shorter than the full-length 3′ UTR used in our study. The MHV study required the use of RNase TI treatment to sufficiently resolve RNA-protein complexes for mobility shift assays (63, 64), whereas this procedure was not required in our assays. Our results strongly suggest that the same or similar host protein-RNA interactions may take place in BCV- and MHV-infected cells, since the 3′ UTR of MHV was able to compete as efficiently as the homologous 3′ end for host protein interactions with the BCV 3′ UTR.
We assumed that PABP would interact with our full-length 3′ UTR probe, since it included a 68-nucleotide poly(A) tail. However, as mentioned above, our protocol was designed to avoid detection of this interaction. Our results indicate that PABP can cross-link to non-A residues in vitro, consistent with previously demonstrated in vivo results (1, 13), but the poly(A) tail was clearly necessary for this interaction in our study. Further analysis is required to determine the structure of the 3′ UTR in order to explain this.
PABP is a highly abundant cytoplasmic protein (13) that binds the 3′ poly(A) tail on eukaryotic mRNAs and helps promote both efficient translation initiation and mRNA stability (reviewed in references 11, 22, and 45). It interacts with the translation factor eukaryotic initiation factor 4G eIF-4G in yeast (52, 53) and with eIF-iso4G and eIF-4B in plants (28). Mammalian PABP interacts with PABP-interacting protein (PAIP-1), a protein with homology to eIF-4G (6). Recently, mammalian PABP was also found to directly interact with eIF-4G (21). eIF-4G is part of a three-subunit complex, eukaryotic initiation factor 4F (eIF-4F), that binds mRNA cap structures during translation (49, 50). PABP binding to eIF-4G, and possibly other initiation factors, mediates interactions between the 5′ and 3′ ends of mRNAs. This interaction is known as the closed-loop model of translation initiation (reviewed in references 11, 22, and 45). Interactions between the 5′ and 3′ ends of yeast mRNAs were recently visualized by atomic force microscopy (59).
There is no question that PABP must be involved in translation of the coronavirus genome upon entry into the cell, but it is also possible that through this role it is either directly or indirectly involved in RNA replication. What supports this idea? All naturally occurring coronavirus defective genome RNAs that have been isolated contain ORFs (5, 36–38, 41, 42, 56). Translation is required for efficient replication of several of the defective genomes including Drep (4) and MIDI-C (57). The encoded sequence is not important for MHV DI RNA replication (31, 57), whereas it appears that either the encoded protein or an RNA element is required for BCV DI RNA replication (4; R. Cologna and B. G. Hogue, unpublished data). Infectious bronchitis coronavirus DI RNAs do not require a long ORF for efficient replication; however, the importance of a small ORF in the DI RNA has not been determined (42).
Since translation is required for efficient replication of coronavirus defective genomes, a lack of or decrease in interactions between PABP and the poly(A) tail may compromise translation. As a result, replication efficiency could be affected. Our analyses demonstrated that PABP binding is decreased when the poly(A) tail is shortened. Binding to the poly(A) tails of different lengths appeared to correlate with the replication phenotypes of the RNAs. Thus, it is reasonable to speculate that the PABP-poly(A) interaction may be important for replication. The coronavirus genomic RNA essentially resembles a large mRNA. Presumably, interactions between the ends of the genome are established during translation of the viral replicase proteins after the virus enters a cell. The juxtaposition of the 5′ and 3′ ends of the genome may be important for assembly of the viral replicase complexes, with the viral transcription and replication machinery having evolved to take advantage of the interaction that is initially established for translation. This could also explain, at least in part, the apparent requirement for both the 5′ and 3′ ends of the coronavirus genome for DI RNA replication (23). The data for coronaviruses and other viral systems supporting the closed-loop model were recently reviewed by Lai (26). Our data suggest that PABP may be an additional cellular factor that plays a role in the proposed interaction of the ends and should be included in the model.
Other plus-strand viruses also require translation in cis for RNA replication (35, 39, 60). Coupling between translation and replication has been demonstrated for poliovirus (39). Insight into how poliovirus controls the switch from translation to replication was recently gained when it was shown that the cellular protein poly(C)-binding protein (PCBP) upregulates viral translation whereas the viral protein 3CD represses viral translation and promotes minus-strand synthesis (12). Identification of cellular and viral proteins that interact with both the 5′ and 3′ ends of the genome and direct proof of their function(s) should provide insight into how the processes are controlled by coronaviruses.
The work reported here represents an important step in identifying the host proteins that specifically bind and play roles in group II coronavirus RNA replication. The precise role(s) of the poly(A) tail and the relevance of the PABP interaction(s) with the coronavirus 3′ UTR in the regulation of translation and replication warrant further study. Ongoing studies are directed toward demonstrating whether PABP plays a functional role in replication and, if so, the nature of this role. The other host factors that interact with the 3′ UTR and the sequences or motifs within the 3′ UTR with which the proteins interact are also being determined. Recently, bulged stem-loop (19) and pseudoknot (62) structures within the coronavirus 3′ UTR were described. Motifs such as these are potential binding sites for the proteins shown to interact with the 3′ UTR in this study.
ACKNOWLEDGMENTS
We thank Ray Cologna and Vinh-Phuc Nguyen for many helpful discussions and suggestions. We also thank Rick Lloyd, Frank Ramig, and members of their laboratories for insightful comments and suggestions. We are grateful to David Brian and Willy Spaan for providing us with the Drep and MIDI-C clones, respectively. We thank Shyan-Yuan Kao for constructing pBCV3′UTR during a rotation in the laboratory and Ian Hogue for helping prepare the figures.
This work was supported by Public Health Service NIH grant AI33500 to B.G.H. from the National Institute of Allergy and Infectious Diseases. J.F.S. was supported by training grant AI07471 from the National Institutes of Health.
REFERENCES
- 1.Adam S A, Choi Y D, Dreyfuss G. Interaction of mRNA with proteins in vesicular stomatitis virus-infected cells. J Virol. 1986;57:614–622. doi: 10.1128/jvi.57.2.614-622.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brian D A, Chang R Y, Hofmann M A, Sethna P B. Role of subgenomic minus-strand RNA in coronavirus replication. Arch Virol Suppl. 1994;9:173–180. doi: 10.1007/978-3-7091-9326-6_17. [DOI] [PubMed] [Google Scholar]
- 3.Campbell L H, Borg K T, Haines J K, Moon R T, Schoenberg D R, Arrigo S J. Human immunodeficiency virus type 1 Rev is required in vivo for binding of poly(A)-binding protein to Rev-dependent RNAs. J Virol. 1994;68:5433–5438. doi: 10.1128/jvi.68.9.5433-5438.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang R Y, Brian D A. cis requirement for N-specific protein sequence in bovine coronavirus defective interfering RNA replication. J Virol. 1996;70:2201–2207. doi: 10.1128/jvi.70.4.2201-2207.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang R Y, Hofmann M A, Sethna P B, Brian D A. A cis-acting function for the coronavirus leader in defective interfering RNA replication. J Virol. 1994;68:8223–8231. doi: 10.1128/jvi.68.12.8223-8231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig A W, Haghighat A, Yu A T, Sonenberg N. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature. 1998;392:520–523. doi: 10.1038/33198. [DOI] [PubMed] [Google Scholar]
- 7.Cui T, Sankar S, Porter A G. Binding of encephalomyocarditis virus RNA polymerase to the 3′-noncoding region of the viral RNA is specific and requires the 3′-poly(A) tail. J Biol Chem. 1993;268:26093–26098. [PubMed] [Google Scholar]
- 8.de Groot R J, van der Most R G, Spaan W J. The fitness of defective interfering murine coronavirus DI-a and its derivatives is decreased by nonsense and frameshift mutations. J Virol. 1992;66:5898–5905. doi: 10.1128/jvi.66.10.5898-5905.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries A A, Horzinek M C, Rottier P J M, de Groot R J. The genome organization of the Nidovirales: similarities and differences between arteri-, toro-, and coronaviruses. Semin Virol. 1997;8:33–47. doi: 10.1006/smvy.1997.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furuya T, Lai M M. Three different cellular proteins bind to complementary sites on the 5′-end-positive and 3′-end-negative strands of mouse hepatitis virus RNA. J Virol. 1993;67:7215–7222. doi: 10.1128/jvi.67.12.7215-7222.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallie D R. A tale of two termini: a functional interaction between the termini of an mRNA is a prerequisite for efficient translation initiation. Gene. 1998;216:1–11. doi: 10.1016/s0378-1119(98)00318-7. [DOI] [PubMed] [Google Scholar]
- 12.Gamarnik A V, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorlach M, Burd C G, Dreyfuss G. The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp Cell Res. 1994;211:400–407. doi: 10.1006/excr.1994.1104. [DOI] [PubMed] [Google Scholar]
- 14.Guilford P J, Beck D L, Forster R L. Influence of the poly(A) tail and putative polyadenylation signal on the infectivity of white clover mosaic potexvirus. Virology. 1991;182:61–67. doi: 10.1016/0042-6822(91)90648-u. [DOI] [PubMed] [Google Scholar]
- 15.Hill K R, Hajjou M, Hu J Y, Raju R. RNA-RNA recombination in Sindbis virus: roles of the 3′ conserved motif, poly(A) tail, and nonviral sequences of template RNAs in polymerase recognition and template switching. J Virol. 1997;71:2693–2704. doi: 10.1128/jvi.71.4.2693-2704.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiti A L, Davis A R, Nayak D P. Complete sequence analysis shows that the hemagglutinins of the H0 and H2 subtypes of human influenza virus are closely related. Virology. 1981;111:113–124. doi: 10.1016/0042-6822(81)90658-9. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann M A, Brian D A. The 5′ end of coronavirus minus-strand RNAs contains a short poly(U) tract. J Virol. 1991;65:6331–6333. doi: 10.1128/jvi.65.11.6331-6333.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann M A, Sethna P B, Brian D A. Bovine coronavirus mRNA replication continues throughout persistent infection in cell culture. J Virol. 1990;64:4108–4114. doi: 10.1128/jvi.64.9.4108-4114.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsue B, Masters P S. A bulged stem-loop structure in the 3′ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J Virol. 1997;71:7567–7578. doi: 10.1128/jvi.71.10.7567-7578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang P, Lai M M. Polypyrimidine tract-binding protein binds to the complementary strand of the mouse hepatitis virus 3′ untranslated region, thereby altering RNA conformation. J Virol. 1999;73:9110–9116. doi: 10.1128/jvi.73.11.9110-9116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imataka H, Gradi A, Sonenberg N. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 1998;17:7480–7489. doi: 10.1093/emboj/17.24.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson A. Poly(A) metabolism and translation: the closed-loop model. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 451–480. [Google Scholar]
- 23.Kim Y N, Jeong Y S, Makino S. Analysis of cis-acting sequences essential for coronavirus defective interfering RNA replication. Virology. 1993;197:53–63. doi: 10.1006/viro.1993.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai M M. Coronavirus: organization, replication, and expression of genome. Annu Rev Microbiol. 1990;44:303–333. doi: 10.1146/annurev.mi.44.100190.001511. [DOI] [PubMed] [Google Scholar]
- 25.Lai M M. RNA-protein interactions in the regulation of coronavirus RNA replication and transcription. Biol Chem. 1997;378:477–481. doi: 10.1515/bchm.1997.378.6.477. [DOI] [PubMed] [Google Scholar]
- 26.Lai M M. Cellular factors in the transcription and replication of viral RNA genomes: a parallel to DNA-dependent RNA transcription. Virology. 1998;244:1–12. doi: 10.1006/viro.1998.9098. [DOI] [PubMed] [Google Scholar]
- 27.Lai M M, Cavanagh D. The molecular biology of coronaviruses. Adv Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le H, Tanguay R L, Balasta M L, Wei C C, Browning K S, Metz A M, Goss D J, Gallie D R. Translation initiation factors eIF-iso4G and eIF-4B interact with the poly(A)-binding protein and increase its RNA binding activity. J Biol Chem. 1997;272:16247–16255. doi: 10.1074/jbc.272.26.16247. [DOI] [PubMed] [Google Scholar]
- 29.Li H P, Huang P, Park S, Lai M M. Polypyrimidine tract-binding protein binds to the leader RNA of mouse hepatitis virus and serves as a regulator of viral transcription. J Virol. 1999;73:772–777. doi: 10.1128/jvi.73.1.772-777.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H P, Zhang X, Duncan R, Comai L, Lai M M. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription-regulatory region of mouse hepatitis virus RNA. Proc Natl Acad Sci USA. 1997;94:9544–9549. doi: 10.1073/pnas.94.18.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao C L, Lai M M. A cis-acting viral protein is not required for the replication of a coronavirus defective-interfering RNA. Virology. 1995;209:428–436. doi: 10.1006/viro.1995.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin Y J, Lai M M. Deletion mapping of a mouse hepatitis virus defective interfering RNA reveals the requirement of an internal and discontiguous sequence for replication. J Virol. 1993;67:6110–6118. doi: 10.1128/jvi.67.10.6110-6118.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin Y J, Liao C L, Lai M M. Identification of the cis-acting signal for minus-strand RNA synthesis of a murine coronavirus: implications for the role of minus-strand RNA in RNA replication and transcription. J Virol. 1994;68:8131–8140. doi: 10.1128/jvi.68.12.8131-8140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Yu W, Leibowitz J L. A specific host cellular protein binding element near the 3′ end of mouse hepatitis virus genomic RNA. Virology. 1997;232:74–85. doi: 10.1006/viro.1997.8553. [DOI] [PubMed] [Google Scholar]
- 35.Mahajan S, Dolja V V, Carrington J C. Roles of the sequence encoding tobacco etch virus capsid protein in genome amplification: requirements for the translation process and a cis-active element. J Virol. 1996;70:4370–4379. doi: 10.1128/jvi.70.7.4370-4379.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makino S, Shieh C K, Soe L H, Baker S C, Lai M M. Primary structure and translation of a defective interfering RNA of murine coronavirus. Virology. 1988;166:550–560. doi: 10.1016/0042-6822(88)90526-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makino S, Yokomori K, Lai M M. Analysis of efficiently packaged defective interfering RNAs of murine coronavirus: localization of a possible RNA-packaging signal. J Virol. 1990;64:6045–6053. doi: 10.1128/jvi.64.12.6045-6053.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendez A, Smerdou C, Izeta A, Gebauer F, Enjuanes L. Molecular characterization of transmissible gastroenteritis coronavirus defective interfering genomes: packaging and heterogeneity. Virology. 1996;217:495–507. doi: 10.1006/viro.1996.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novak J E, Kirkegaard K. Coupling between genome translation and replication in an RNA virus. Genes Dev. 1994;8:1726–1737. doi: 10.1101/gad.8.14.1726. [DOI] [PubMed] [Google Scholar]
- 40.Parker M M, Masters P S. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology. 1990;179:463–468. doi: 10.1016/0042-6822(90)90316-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Penzes Z, Tibbles K, Shaw K, Britton P, Brown T D, Cavanagh D. Characterization of a replicating and packaged defective RNA of avian coronavirus infectious bronchitis virus. Virology. 1994;203:286–293. doi: 10.1006/viro.1994.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penzes Z, Wroe C, Brown T D, Britton P, Cavanagh D. Replication and packaging of coronavirus infectious bronchitis virus defective RNAs lacking a long open reading frame. J Virol. 1996;70:8660–8668. doi: 10.1128/jvi.70.12.8660-8668.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pogue G P, Huntley C C, Hall T C. Common replication strategies emerging from the study of diverse groups of positive-strand RNA viruses. Arch Virol Suppl. 1994;9:181–194. doi: 10.1007/978-3-7091-9326-6_18. [DOI] [PubMed] [Google Scholar]
- 44.Proudfoot N. Poly(A) signals. Cell. 1991;64:671–674. doi: 10.1016/0092-8674(91)90495-k. [DOI] [PubMed] [Google Scholar]
- 45.Sachs A B, Sarnow P, Hentze M W. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 46.Sarnow P. Role of 3′-end sequences in infectivity of poliovirus transcripts made in vitro. J Virol. 1989;63:467–470. doi: 10.1128/jvi.63.1.467-470.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawicki S G, Sawicki D L. Coronavirus transcription: subgenomic mouse hepatitis virus replicative intermediates function in RNA synthesis. J Virol. 1990;64:1050–1056. doi: 10.1128/jvi.64.3.1050-1056.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sethna P B, Hung S L, Brian D A. Coronavirus subgenomic minus-strand RNAs and the potential for mRNA replicons. Proc Natl Acad Sci USA. 1989;86:5626–5630. doi: 10.1073/pnas.86.14.5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sonenberg N. mRNA 5′ cap binding protein eIF4E and control of cell growth. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 245–270. [Google Scholar]
- 50.Sonenberg N, Morgan M A, Merrick W C, Shatkin A J. A polypeptide in eukaryotic initiation factors that crosslinks specifically to the 5′-terminal cap in mRNA. Proc Natl Acad Sci USA. 1978;75:4843–4847. doi: 10.1073/pnas.75.10.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spector D H, Baltimore D. Requirement of 3′-terminal poly(adenylic acid) for the infectivity of poliovirus RNA. Proc Natl Acad Sci USA. 1974;71:2983–2987. doi: 10.1073/pnas.71.8.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarun S Z J, Sachs A B. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 1996;15:7168–7177. [PMC free article] [PubMed] [Google Scholar]
- 53.Tarun S Z J, Wells S E, Deardorff J A, Sachs A B. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc Natl Acad Sci USA. 1997;94:9046–9051. doi: 10.1073/pnas.94.17.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Todd S, Nguyen J H, Semler B L. RNA-protein interactions directed by the 3′ end of human rhinovirus genomic RNA. J Virol. 1995;69:3605–3614. doi: 10.1128/jvi.69.6.3605-3614.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsai C H, Cheng C P, Peng C W, Lin B Y, Lin N S, Hsu Y H. Sufficient length of a poly(A) tail for the formation of a potential pseudoknot is required for efficient replication of bamboo mosaic potexvirus RNA. J Virol. 1999;73:2703–2709. doi: 10.1128/jvi.73.4.2703-2709.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Most R G, Bredenbeek P J, Spaan W J. A domain at the 3′ end of the polymerase gene is essential for encapsidation of coronavirus defective interfering RNAs. J Virol. 1991;65:3219–3226. doi: 10.1128/jvi.65.6.3219-3226.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Most R G, Luytjes W, Rutjes S, Spaan W J. Translation but not the encoded sequence is essential for the efficient propagation of the defective interfering RNAs of the coronavirus mouse hepatitis virus. J Virol. 1995;69:3744–3751. doi: 10.1128/jvi.69.6.3744-3751.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Most R G, Spaan W J. Coronavirus replication, transcription, and RNA recombination. In: Siddell S J, editor. The Coronaviridae. New York, N.Y: Plenum; 1995. pp. 11–31. [Google Scholar]
- 59.Wells S E, Hillner P E, Vale R D, Sachs A B. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 60.White K A, Bancroft J B, Mackie G A. Coding capacity determines in vivo accumulation of a defective RNA of clover yellow mosaic virus. J Virol. 1992;66:3069–3076. doi: 10.1128/jvi.66.5.3069-3076.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickens M. In the beginning is the end: regulation of poly(A) addition and removal during early development. Trends Biochem Sci. 1990;15:320–324. doi: 10.1016/0968-0004(90)90022-4. [DOI] [PubMed] [Google Scholar]
- 62.Williams G D, Chang R Y, Brian D A. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J Virol. 1999;73:8349–8355. doi: 10.1128/jvi.73.10.8349-8355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu W, Leibowitz J L. A conserved motif at the 3′ end of mouse hepatitis virus genomic RNA required for host protein binding and viral RNA replication. Virology. 1995;214:128–138. doi: 10.1006/viro.1995.9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu W, Leibowitz J L. Specific binding of host cellular proteins to multiple sites within the 3′ end of mouse hepatitis virus genomic RNA. J Virol. 1995;69:2016–2023. doi: 10.1128/jvi.69.4.2016-2023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]