

Significance
The neural crest cells (NCC), a highly multipotent vertebrate-specific population induced in the gastrula ectoderm, emerges by an epithelial–mesenchymal transition (EMT) in late neurulas. NCC induction and diversification prior to EMT remains only partially explored. Using single-cell transcriptomes with fine temporal resolution, machine learning, and experimental validation in Xenopus, we here provide a global scaffold of the gene programs driving anterior NCC induction during gastrulation and its diversification during neurulation. Importantly, we select two key transcription factors and use high-throughput strategies (RNAseq and ChIPseq on dissected ectoderm tissue) to validate their connectomes and actions in gene program switches predicted in silico. This allowed us to propose an architecture for transcriptome regulations driving premigratory NCC formation.
Keywords: single-cell transcriptomes, gene regulatory networks, Xenopus, neural border, neural crest
Abstract
Neural crest cells exemplify cellular diversification from a multipotent progenitor population. However, the full sequence of early molecular choices orchestrating the emergence of neural crest heterogeneity from the embryonic ectoderm remains elusive. Gene-regulatory-networks (GRN) govern early development and cell specification toward definitive neural crest. Here, we combine ultradense single-cell transcriptomes with machine-learning and large-scale transcriptomic and epigenomic experimental validation of selected trajectories, to provide the general principles and highlight specific features of the GRN underlying neural crest fate diversification from induction to early migration stages using Xenopus frog embryos as a model. During gastrulation, a transient neural border zone state precedes the choice between neural crest and placodes which includes multiple converging gene programs. During neurulation, transcription factor connectome, and bifurcation analyses demonstrate the early emergence of neural crest fates at the neural plate stage, alongside an unbiased multipotent-like lineage persisting until epithelial–mesenchymal transition stage. We also decipher circuits driving cranial and vagal neural crest formation and provide a broadly applicable high-throughput validation strategy for investigating single-cell transcriptomes in vertebrate GRNs in development, evolution, and disease.
Neural crest cells form a population of multipotent and migratory progenitors found in vertebrate embryos, essential for the peripheral and enteric nervous system, craniofacial structures, endocrine, and pigment cells among others. Together with ectodermal placodes, neural crest (NC) cells are evolutionary inventions that support many cell and tissue innovations promoting the vertebrate predatory lifestyle. Shortly after gastrulation, NC cells are induced from the dorsal-lateral “neural border zone” (NB), an ectoderm domain located between the nonneural ectoderm and the neural plate ectoderm (1, 2). In addition to the NC, the NB territory also gives rise to posterior placodes, nonneural ectoderm, and the dorsal part of the neural tube (3, 4). Whether these four cell types arise from a common and multipotent early progenitor state, and how the sequence of fate decisions is orchestrated at the NB during gastrulation remain open questions. During neurulation, NC specification and induction progresses as an anterior-to-posterior wave along the edges of the neural plate, with gene programs that define early and immature neural crest cells (e.g. expression of snail2, foxd3, and sox8 genes) followed by later premigratory programs presaging emigration of NC cells from the NB epithelium as the neural folds elevate and close (e.g. expression of sox10, twist1, and cdh2 (N-cadherin) genes) (5, 6). In addition to this pan-NC program, several regional molecular modules are activated along the anterior–posterior body axis and define subpopulations with specific potential (7, 8). How these programs are interconnected with the pan-NC module and how and when they are activated in premigratory NC cells are poorly described. Later, at the end of neurulation, NC cells leave the dorsal ectoderm by a stereotypical epithelium-to-mesenchymal transition (EMT) followed by extensive migration toward a variety of target tissues.
NC biology has been scrutinized during development and evolution, leading to the elucidation of elaborate gene regulatory networks (GRNs) during the last decade (9–12). These networks, however, remain incomplete and do not account for most of the defects observed in human neurocristopathies (13, 14). This problem is ripe for single-cell (SC) transcriptomics, which would enable a full description of NC development over sequential developmental stages, and which would define the developmental genetic trajectories of the complete NC lineage tree, compared to progenitor cell states at the neural border. Many recent SC studies on NC cells have explored NC after emigration (SI Appendix, Table S1). In contrast, premigratory NC single cells and the links with earlier ectoderm patterning have received very limited exploration and validation (7, 15, 16). Previously, formation of the NB territory has been defined by the relatively specific expression of a few genes during gastrulation (e.g. pax3 and pax7) and by the overlapping expression of ventral (tfap2a, msx1) and dorsal (zic1) genes (2, 9, 17). However, whether NB represents a distinct cell state, the timing of its specification from the rest of the dorsal ectoderm and the circuits driving fate decisions between the four NB-derived cell fates (NC, placodes, nonneural ectoderm, and dorsal neural tube) remain to be established (3, 18, 19). Furthermore, the subsequent timing of lineage decisions in the premigratory NC along the anterior–posterior axis, the potential long-term maintenance of a multipotent NC subpopulation, and the molecular mechanisms driving each state of the premigratory NC lineage remain mostly unexplored. Here, we used single-cell transcriptomes from eight consecutive developmental stages of Xenopus tropicalis, featuring 6,135 NC cells and 17,138 early ectoderm cells, to provide a comprehensive developmental profiling of the NB and the premigratory NC. During neurulation, we define 16 NC subpopulations and highlight the transcriptomic trajectories between “early” premigratory NC states and “late” NC subpopulations emigrating from cranial to vagal levels of the body axis. Interestingly, we find that distinct signatures for prospective NC fates emerge much earlier than previously anticipated, that NC state diversity is maintained upon EMT, contrasting with reports in mouse cranial NC (15), and that further diversification occurs at the onset of migration. We then connect ectoderm patterning during gastrulation to NC and placode specification. Interestingly, rather than individual clusters, we find a series of transitional clusters supported by distinct molecular trajectory predictions. At each stage, we propose a temporal sequence of molecular events underlying these successive transcriptomic states and identify key candidate transcription factors for the branching between states. Importantly, we validate selected regulatory predictions using transcriptomes of dissected NB or NC explants, transcription factor binding to chromatin (TF-ChIP-seq) and in vivo approaches, supporting the pertinence of in silico predictions. Last, we propose a model of “dual convergence” for parallel transcriptomic routes driving neural border specification and a “diversification tree” for premigratory NC. Altogether, we provide an extensive gene regulatory network describing the emergence of the neural crest from the ectoderm of vertebrate embryos.
Results
Defining the Diversity of Premigratory Neural Crest States.
We used the single-cell (SC) series from Briggs et al. (20) and performed deeper resequencing of the libraries from whole X. tropicalis embryos taken at eight consecutive developmental stages, followed by updated genome annotation and improved sequence alignment. The quality of the dataset is compared to previous NC datasets in other species (data processing is described in Materials and Methods and SI Appendix, Part B and Table S1). Through unsupervised Leiden clustering (21) of the whole embryo dataset at each stage (a total of 177,250 cells), we identified NC clusters by coexpression of well-established NC genes, such as snai2, TFAP2B, and SOXE genes, from induction to migration (gene-supervised approach, Fig. 1 A and B and SI Appendix, Fig. S1A and Table S2). This allowed us to scrutinize 6,135 neural crest (NC) cells from early gastrulation to early migration stages. The large cell number of our dataset allowed greater assessment of the cellular diversity in the NC population during early induction (at late gastrulation stages 12 to 13 and neural plate stages 13 to 14), during neural fold elevation (stages 16 to 18), during EMT (neural tube stages 18 to 20) and at the earliest stages of NC cells emigration (tailbud stage 22). To validate the gene-supervised approach used to define NC cells, signatures of NC/non-NC cells from the whole annotated frog dataset were then used to train a classifier which could faithfully detect NC cells when applied to an independent and poorly related zebrafish whole embryo dataset (22). This test indicated that the initial gene-supervised selection had retrieved bona fide NC cells (SI Appendix, Part B and Fig. S1 B–E). We then used the NC dataset (and not the whole embryo dataset as above) to define which genes would best characterize the NC cell population compared to other embryonic cells at each developmental stage: an ideal pan-NC marker can be defined based on its highly NC-specific expression compared to non-NC cells, and low variation in expression across the NC population (Fig. 1C). This highlighted that the early NC is best labeled by snai2 at stages 13 to 16, followed by tfap2b from stage 14 to 20, and c3, c9, sox8 between stages 14 to 18. Comparatively among these genes, tfap2b was the most specific NC marker, albeit not strictly exclusive as pronephros and other cells express tfap2b at tailbud stages (SI Appendix, Part A). However, collectively, these genes thus define an early and generic “pan-NC” signature.
Fig. 1.
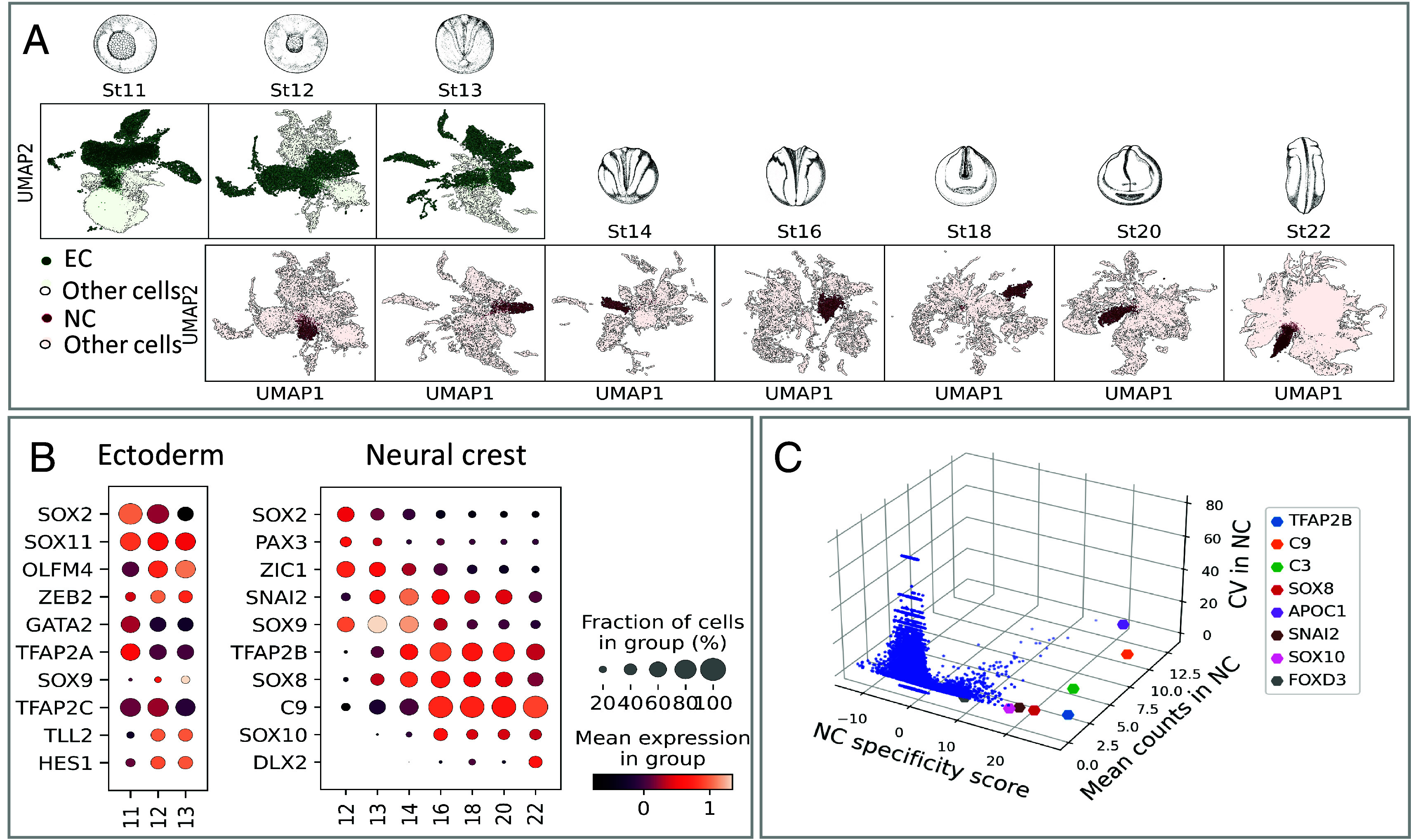
Cell selection for neural crest, ectoderm, and neural border. (A) Ectoderm (EC, stages 11 to 13, green) and NC cells (stages 12 to 22, brown) were selected from a whole embryo SC transcriptome dataset. (B) Expression dynamics of well-referenced transcripts (for example, (1, 2, 7, 9–11, 16)) identify EC and NC. Dot size: number of cells expressing the gene, color shade: average expression level. Neural border was defined as stages 11 to 13 tfap2a+, zic1+ cells. (C) 3D scatter plot of NC z-scores (specificity), mean gene expression (counts) and coefficient of variation (CV), defining a few highly expressed pan-NC genes, compared to the whole embryo dataset.
We then explored transcriptome diversity of the premigratory NC cell population and whether transitional states could be identified during neurulation (Fig. 2 and SI Appendix, Fig. S2). From late gastrulation to emigration stages, we defined 16 different NC states (c1 to c16), compared to the eight described previously (20) (Fig. 2A). All NC cells expressed the generic “pan-NC” signature (Fig. 2B), either alone (c5, c14, thereafter called “unbiased” clusters) or in combination with other markers (Fig. 2 B–D). Partition-based graph abstraction analysis (PAGA) (23) showed that most NC clusters were highly interconnected, with stronger connectivity within early (c1–c4), vagal (c8, c9, c12, and c13), and cranial (c10, c7, and c11) clusters (Fig. 2E). The clusters could be identified based on unique combinations of marker gene expression, although subsets of these markers were shared between closely related clusters, particularly those containing cell states transitioning from one to another (such as mafb+ vagal c9 and c12, Fig. 2 C and D). Further clustering lost significance as the resulting subclusters expressed similar markers. Each cluster’s gene signature and proposed biological characteristics are detailed in SI Appendix, Part A and Table S3. We thus only highlight here a few interesting features of this premigratory/early migrating NC dataset. First, we have identified 11 clusters (c6–c16), composed mainly of stage 18 to 22 cells, with an explicit mesenchymal NC signature, i.e., various levels of itga4/integrin-a4, vim/vimentin or fn1/fibronectin (SI Appendix, Fig. S2B). This indicated that those cells were preparing or engaging EMT and early migration, and thus provided the opportunity to explore gene expression dynamics and other molecular characteristics of EMT in vivo. For example, we compared EMT in cranial and vagal NC, using hox gene signatures to position each cluster along the body axis: cranial-most clusters were hox-negative while vagal clusters expressed a range of anterior (rhombencephalon, hoxa-d 1-3) to posterior (spinal cord, hoxa-d 7-11) hox genes (SI Appendix, Fig. S2C). With this knowledge of distinct spatial positions spanning cranial to vagal NC, we queried whether all NC cells adopted a similar “stem-like” state upon EMT as proposed recently in the mouse (15). Instead, we observed clear transcriptomic diversity across the mesenchymal clusters: For example, c10, c13, and c15 all undergo a transition to a mesenchymal vim+ state but c10 and c15 expressed the ectomesodermal marker twi1, while c13 highly expressed tnc (Fig. 2D and SI Appendix, Fig. S3). Moreover, there has been a debate in the field, especially in trunk NC, about the appearance of lineage-specific markers prior to EMT (24–26). As one key expectation from SC transcriptomics is to identify and characterize rare or new cell states, we specifically searched for NC lineage determination markers in premigratory cranial and vagal NC. We found no sign of cell determination toward the melanoblast, neural, or glial lineage. Instead, we clearly identified clusters enriched in markers essential for NC cell migration (semaphorin coreceptors nrp1 in c12 and c15, and nrp2 in c10 and c11). Shortly after EMT, we also found a few cells with enriched myogenic marker expression (actc1, myl, mylpf, des) in c16, along with tfap2b, sox10, twi1, mmp14 expression (Fig. 2D and SI Appendix, Fig. S2). The identity of this small population remains unclear, as discussed in SI Appendix, Part A. This finding aligns with recent cranial NC lineage tracing which indicates that the majority of NC cell fates are established after EMT (26–29).
Fig. 2.

The premigratory neural crest displays high transcriptomic heterogeneity. (A) Leiden clustering revealed 16 distinct states (clusters) before and during EMT (developmental stages 12 to 22) (SI Appendix, Supplementary Text) (B) Dotplot depicting the expression of the top NC-specific genes within the different clusters. (C) Top three enriched genes for each cluster (Top and Bottom lines respectively), with their expression in the other clusters and hierarchical clustering between clusters. (D) Expression of key cluster-specific genes, including rpe65 (cluster 10), cyp26c1 (clusters 4/6/15), itga4, dlx2 (cluster 11), egr2, mafb (cluster 12), tnc (cluster 13), early olig4, hnf1b (cluster 3) and muscle-like NC specific myl1 (cluster 16). Gjb1 is expressed by all clusters but is highest in cluster 2. Genes expressed broadly in NC cells define a “canonical NC” signature: early tfap2b, tfap2c, sox9, snai2, and c9. Multipotency-related genes are present mostly until mid-neurula stage (pou5f1-1). (E) PAGA estimates cluster connectivity where line thickness increases with stronger connections.
This dataset’s main asset is that it follows NC cell states over a large developmental time series spanning gastrulation to EMT. We looked how the emigrating cell states were related to induction-stage states, in order to define the potential trajectories underlying NC early diversification. Through PAGA, we observed the relationships between clusters and were able to define multiple progenitor states linked to the later clusters defined above. Consequently, we delineated transcriptional dynamics of cranial, vagal, and enteric nervous system progenitors between late gastrulation and EMT. These clusters mainly include cells from late gastrula (st12) to mid-neurula (st16) stages. Late cranial c10 (nrp2+rpe65+hox−) and c11 (nrp2+dlx2+hox−) were related to c7 (nrp2+ hox−) while late vagal c12 (mafb+epha4+) and c13 (tnc+wnt11+ltbp1+) were linked to c9 (nrp1+). C13 was also linked to the early c3 (hnf1b+) indicating a possible converging trajectory. In turn, c7 and c9 were related to the unbiased c5 (tfap2c+sox9+). The nrp1+ late cranial c15 was linked to c6 and c4, all of which specifically expressed cyp26c1, epha2, and efnb2. C16 (myl1+, SI Appendix, Fig. S4) was related to unbiased c14, which was in turn linked to unbiased c5, c1 (c9+sox9+) and c2 (gjb1+) (SI Appendix, Figs. S5–S7). Several genes found enriched in each progenitor cluster were consistent with previous experimental analyses, validating our SC-transcriptome-based predictions (see detailed description in SI Appendix, Part A). This analysis allowed defining potential progenitor state larger signatures compared to previous in vivo experiments and selecting the clusters pertinent for further trajectory analysis (see below).
Interestingly, most clusters gathered cells from multiple stages, indicating that a given transcriptome state can be reached by different cells over long developmental periods (SI Appendix, Fig. S3). For example, cranialmost cells of c10 are generated over the entire duration of neurulation from neural plate stage 13 to migration stage 22. This reflects how NC cells of a similar state can be continuously generated over the course of development at a given portion of the neuraxis. In contrast, cranial c11 and c15, two migratory clusters highly expressing vimentin (vim+), are suddenly generated at post-EMT stage 22. This could reflect an abrupt step of mesenchymalization in the epithelial-to-mesenchymal transition process of cranial NC, as reflected in their transcriptomes.
Last, we also detected cells with an immature pan-NC signature throughout neurulation and EMT, representing a long-lasting, potentially “multipotent” and “stem-like” population (c1, c5, c14; Fig. 3). These “unbiased” progenitors formed 72% of all NC cells during induction (gastrula and neural plate stages 12 to 14), 15% at premigratory and EMT stages 16 to 18, and 9% among tailbud stage 22 NC cells (SI Appendix, Fig. S2 D and E). Compared to c1–c5, we find that c14 cells express medium levels of pluripotency markers sox2, cmyc, and pou5f3.1, along with pan-NC markers. As c14 is formed of cells from stages 13 to 22 (SI Appendix, Fig. S3), we detail that this expression is found not only in the early (stages 13 to 16) cells but also the later ones (stages 18 to 22, Fig. 3 A and B). This is compared to higher levels in early “unbiased” clusters c1–c5 and lower levels in c11 and c15 taken as examples of biased ones. We confirmed pou5f3.1 expression by in situ hybridization in NC explants until the end of neurulation (stage 18). Last, c14 cells express mmp14 suggesting engagement in the EMT process (SI Appendix, Fig. S2). This suggests that a small population of naïve NC cells may be maintained as highly plastic throughout neurulation and NC EMT. Further functional studies, including lineage tracing, are needed to define the potential of these cells in vivo.
Fig. 3.

Characteristics of the “unbiased” NC clusters c1-c5-c14. (A) Expression of pluripotency (cmyc, sox2, pou5f3.1) and early unbiased NC (tfap2b, c3, c9, sox9, pax3, zic1, tfacp2c) genes in clusters c1, c5, c14, c11, c15. Each bar is a bin of 40 cells. The “stage” line indicates cells’ developmental stage, the “dpt-pseudotime” cell advancement along their transcriptional path. (B) Highly significant differential expression of Pou5f1-1 and sox2 in clusters c1-c5 (early stage, high expression), c14 (mid-neurula stage, moderate expression) and c11-c15 (late stage, low expression). (C) Sustained expression of unbiased NC markers tfap2b and cmyc and pluripotency marker pou5f3.1 in microdissected NB (stage 14), NC (stages 16 to 18) and whole embryos (stage 18). AS—antisense probe, S—sense probe control.
In summary, with its large cell number and temporal series, our data details the continuum of NC states along a large segment of the body axis, from the anteriormost cranial to the vagal levels. We highlight three important characteristics of NC cells around the time of induction to initiation of migration. First, there is a large diversity of cell states along the cranio-caudal axis throughout this developmental period, including spatially defined cell groups (regional modules), transitional states, and multipotent-like cells. Second, state diversity is maintained across stages including during EMT, with cells of different developmental time presenting similar transcriptome states. Last, a small “stem-like” unbiased NC subpopulation persists as a distinct cluster until EMT.
Connectome Analysis by Intersecting SC-Transcriptomics Modeling with In Vivo Morpholino RNA-Seq and Transcription-Factor-Based ChIP-Seq.
To expand the network of genes involved in the NC-GRN, we used an input list of 1,417 transcription factors (TFs) (30) and applied GRNboost2 on the NC dataset to link transcription factors to their potential targets in the NC transcriptome. The principle of GRNBoost2 relies on analysis of gene synexpression dynamics (31). We retrieved a vast network of 16,978 potential TF-targets with a median of 22 connections per TF. Among the most connected genes for each stage (SI Appendix, Table S4), we retrieved the previously known neural border specifiers pax3 and zic1 at NC induction stages, as well as zic3, olig4, and sox9 and the anterior NC marker dmbx1. At premigratory stages, the prominent nodes included the NC specifiers tfap2b, sox10, and snai2, the anterior NC markers rpe65 and alx1, and the hindbrain hox gene hoxb3. At migration stages, tfap2e, mycn, dlx2, and egr2 (krox20) displayed both high expression and multiple connections.
To confront the predicted network with experimental data obtained in vivo, we chose two well-known regulators of NC induction, Pax3 (9, 32) and TFAP2e (33) also identified as major nodes by GRNBoost2. We sequenced microdissected NB/NC explants after in vivo knockdown of either pax3 or tfap2e, using previously validated antisense morpholino oligonucleotides (MO) (Fig. 4 A and E). Expression was decreased for 1,179 transcripts in Pax3 morphant explants, confirming that Pax3 is essential for NB/NC development (zic1, msx1, msx2, and sox9) and identifying a large series of new targets (olig4, hnf1b, vim, olfm4, and dhrs3) (Fig. 4A and SI Appendix, Table S6). We also identified genes putatively directly bound by Pax3 in vivo using ChIP-seq (Fig. 4 B and C) and identified 657 potential targets in the whole stage 14 embryo, of which we selected the 475 were expressed in the SC NC dataset (SI Appendix, Table S8), including known direct targets, e.g. cxcr4 and prtg (2, 34) and potential novel ones (e.g. efna3 and psen2) (Fig. 4C and SI Appendix, Fig. S8). In contrast to the previously known role of TFAP2e in NC induction (33), GRNBoost2 identified TFAP2e as a major node later on, at neural fold stage. We thus used a similar approach on NC explants at a later stage (stages 17 too 19) and validated TFAP2e as an important regulator of NC EMT markers, decreased expression of 586 genes including twist1, sox10, sncaip, and fn1 (Fig. 4E and SI Appendix, Table S7). Using ChIP-seq for TFAP2e, we identified 642 potential targets expressed in the NC dataset, among 805 potential targets for the whole stage 19 embryo, including top-scored rbm20, pim1, adam33, and tfap2a (Fig. 4F and SI Appendix, Fig. S4 and Table S9). Controls for these ChIP experiments are presented in SI Appendix, Fig. S4. By themselves, these validation datasets based on microdissected morphant samples and transcription-factor-based ChIPseq provide two genome-wide NC-GRNs centered on Pax3 and TFAP2e respectively, ready for later functional exploration.
Fig. 4.
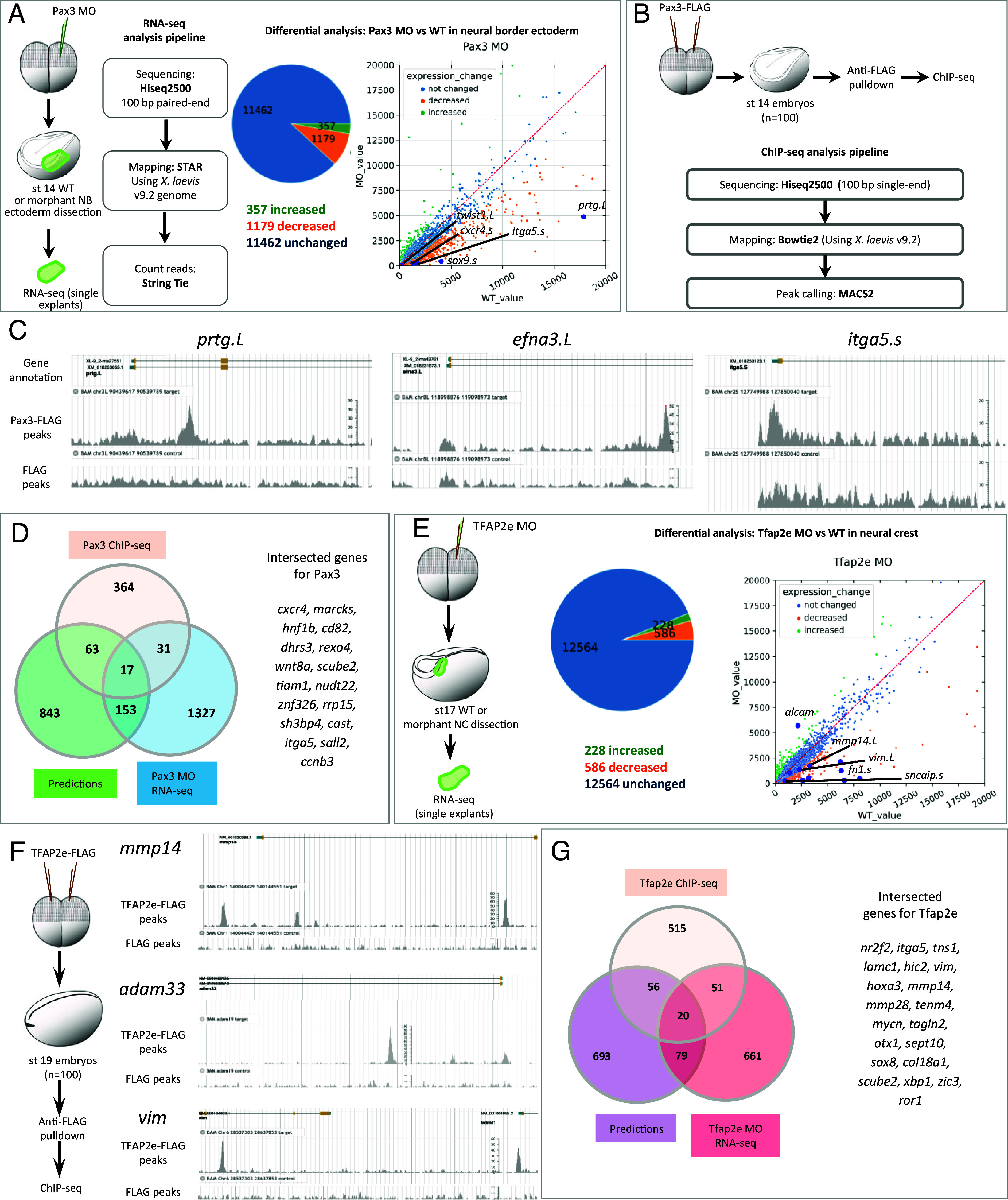
Pax3 and TFAP2e GRNs. (A) RNA-sequencing on microdissected NB ectoderm either wild-type (wt) or following Pax3 depletion in vivo (Pax3 MO) identified 1,179 downregulated genes (SI Appendix, Table S6). (B) Pax3 ChIP-seq analysis identified 657 candidate direct targets, among which 475 expressed in NC. (C) Chromatin peaks enriched in Pax3-FLAG in prtg.L, efna3.L, and itga5.s genes. (D) Venn diagram compares Pax3 target genes validated by ChIP-seq, MO-RNA-seq, and GRN-boost2 modeling. List of the 17 genes found by all three methods. (E) Similarly, 586 genes were downregulated after TFAP2e depletion. (F) TFAP2e ChIP-seq found 805 candidate direct target genes, among which 642 were expressed in NC, such as mmp14, vim, and adam33 (adam13). (G) Venn diagram compares TFAP2e putative target genes validated by ChIP-seq, MO-RNA-seq, and GRN-boost2 modeling, list of the 20 genes found by all three methods.
We then compared these in vivo data to GRNBoost2 predictions. Due to their use of different parameters (e.g. putative target gene binding by ChIP, direct and indirect regulation by knock-down, stage-wise gene expression levels for synexpression, etc.), we expected to find only a partial overlap between the potential target genes identified by each method. However, we also wondered whether intersecting those approaches would highlight common, potential targets to prioritize in follow-up studies. We thus explored gene list intersections in each case. For example, among 848 putative targets of TFAP2e predicted from scRNA-seq, 99 showed changed expression after TFAP2e knockdown in NC explants in vivo (e.g., tfap2b, sox10, sncaip, zic3; SI Appendix, Fig. S4), while 80 of the Pax3 ChIPseq-validated potential targets were predicted by GRN-Boost2 modeling including psmd4, psen2, sp7, notch1, and hnf1b. In both Pax3 and TFAP2e cases, we find that the three approaches partially overlap, identifying 17 and 20 candidate gene targets for Pax3 and Tfap2e respectively, validated through all three techniques. To compare these results to a random chance we utilized bootstrapping. To do this, we randomly selected the same number of genes for different methods (ChIP-seq, MO, GRNboost2) 1,000 times from the total number of genes (Xenopus laevis and X. tropicalis). We obtained a median intersection value of 3, confirming that our analyses were not obtained at random and that we obtained a statistical enrichment (Fig. 4 D and G and SI Appendix, Fig. S4H), which encourages special future focus on those genes.
Branching Toward Biased Premigratory Neural Crest Subpopulations Is Controlled by Key Transcription Factors.
To explore the temporal dynamics of TF expression that may specify decision points in the development of premigratory NC, we used tree inference (ElPiGraph) and advanced pseudotime downstream analysis focused on fate biasing using scFates (35, 36). We thus explored potential branch-specific transcriptional regulation from the calculated pseudotime, in order to determine not only the temporal order in which their functions may be accomplished but also genes which may bias the NC toward a fate. ElPiGraph approximates datasets with complex topologies to build the graph structure, with the limitation that it cannot be applied to large datasets with many potential branches. Thus, using the principal graph constructed with PAGA (Fig. 2B), we subselected cells around three main bifurcation points in the NC lineage tree and then applied scFates branching analysis (Fig. 5).
Fig. 5.

Gene programs driving NC fate trajectories. Transcriptomes of cells subselected around a chosen bifurcation point were analyzed using tree inference and pseudotime downstream analysis, yielding gene programs accompanying each trajectory. Gene programs for bifurcation (A and B) of premigratory unbiased cluster 5 into cluster 9 (A) and cluster 7 (B); (C and D) of migratory bipotent vagal cluster 9 into clusters 12 (C) and 13 (D); (E and F) of migratory bipotent cranial cluster 7 into clusters 10 (E) and 11 (F).
Cranial vs. vagal bifurcation at the end of neural plate stage.
Cranial NC cells emerge from the neural tube anterior to the otic vesicle while vagal NC cells form from the hindbrain region adjacent to somites 1 to 7 (37). Our data indicated that the first cells biased toward vagal or cranial populations arose from the unbiased cluster c5 around early neural fold stage 14. Although we did not see a separation of c5 into two populations at the chosen level of clustering, we still observe an early internal bias marked by the increased expression of early cranial (nrp2) and vagal (mafb) markers in distinct subregions of c5: c5 cells that highly express nrp2 were seven times closer to the cranial state, while cells that highly express mafb were 1.5 times closer to the vagal state (SI Appendix, Fig. S5). Through branching analysis, we uncovered gene programs potentially governing the bifurcation, consisting of “early” genes activated before bifurcation and “late” genes with continued expression in each branch: nrp2, alk, rnd1, adam19 (early) and shisa3, frdm6 (late) for the cranial branch, and mafb, klb, mdk (early) and prdm1, cf1, hoxd3 (late) for the vagal branch (Fig. 5 A and B and SI Appendix, Fig. S5). By their high specificity and early expression, nrp2 and mafb seemed the best early predictors of branching between the cranial and vagal populations (Fig. 2E). These results match in vivo analyses on expression of key NC regulators, such as Mafb during cardiac NC specification or Alk in cranial NC migration (38, 39) and support the validity of this modeling.
Vagal to enteric split and cranial subdivisions at neural fold stages.
From the apparently homogeneous clusters c7 and c9, branching analysis predicted candidates regulating subsequent bifurcation. The vagal c9 seems to split into cardiac c12 and enteric nervous system progenitors (ENSp) c13. For c12, early-enriched genes were mafb, mycn, prdm1, nolc1, and eef1d, late ones being egr2, hoxd3, epha4, and abtb2 (Fig. 5C). For c13, early genes were olig4 and fbn2. Due to its significantly increased expression in later c13 cells, pax3 was identified as a late actor, but the expression-pseudotime heatmap showed that pax3 was already expressed prior to branching in ENSp progenitors and increased afterward (Figs. 2D and 5D). Therefore, we also assigned pax3 as an early c13 gene. Later c13 genes consist of tnc, ltbp1, wnt11, and hoxb6. Interestingly, through ChIP-seq and depletion analyses of the early TF Pax3, we found that some late genes were directly bound by Pax3 as well as affected in morphants (cfb, bmp5), while others showed increased (krt8) or decreased (cldn1, hoxb6, hoxa7) expression after Pax3 depletion (Fig. 6A and SI Appendix, Fig. S6). Thus, using RNA-seq after a TF’s depletion and ChIP-seq data produced in vivo for a given TF allows a first level of validation of the branch-specific candidate signatures modeled by scFates.
Fig. 6.

Branching analyses uncover undescribed later-stage roles of Pax3 and TFAP2e in NC at mid-neurula stage. (A) NC-specific Pax3-MO-mediated RNA-seq and Pax3-ChIP-seq identify vagal (c13) or cranial (c10) branch-specific genes affected by Pax3 depletion (plain) or bound by Pax3 (bold) in vivo. Similarly TFAP2e affects cranial branch (c11) migratory signature in vivo. (B) Experimental paradigm to test TFAP2e function in cranial NC migration. (C) While Tfap2e depletion abrogates grafted NC migration (gfp staining), reactivating TFAP2e after EMT partially rescues it. (D) Quantification of NC migration as the farthest NC migration distance from the dorsal boundary [red bracket in (C)] relative to head width (dotted line). (E) To test Pax3 putative role in driving branching of cranial NC toward rpe65+ branch 10 and reducing dlx2+ branch 11, Pax3 was activated at mid-neurula stage 14. Branch-specific genes’ expression was tested at EMT/migration stage in microdissected NC: Pax3 activated branch-10 gene dmbx1, while reducing branch-11 genes mmp14, fn1, twist1. (F) Model of Pax3 participation to cranial NC branching. SEM, *P < 0.05, **P < 0.01, ****P < 0.0001.
Similar analysis on cranial NC clusters (c7, c10, and c11) showed that c10-specific early genes were rpe65, dmbx1, rgr, lmx1a, and zfhx4, while late genes included alx1 and bmper (Fig. 5E). Interestingly, pax3 was also expressed early in the cranial bifurcation from c7 toward c10 and c11 and later and was specifically enriched in rpe65+ c10. The Pax3 ChIP-seq and MO datasets revealed that rpe65+branch-specific transcripts comt, slc23a2, and rpe65 were decreased after pax3 depletion while others, bmper, f10, sema3a, dmbx1, slc16a12, and kif26a, were both depleted in Pax3 morphants and bound by Pax3 in vivo (Fig. 6A). On the other hand, tfap2e expression initiated before bifurcation and was enriched in hox−dlx2+ c11 relative to c10. Using a similar strategy, we validated that TFAP2e depletion reduced the expression of nine of the hox−dlx2+ branch-specific genes (e.g., early gene mef2c, and late genes dlx2, mmp14, vim), and that Tfap2e regulated and directly bound four other genes in the c11 signature (c9, vim, mmp14, and mycn, Fig. 6A and SI Appendix, Fig. S6). These data suggested by an independent strategy than GRNBoost2 that TFAP2e, important for NC induction, also directly regulates cranial NC EMT and migration by controlling vim and mmp14. We tested this late-stage function in vivo using low-level depletion of TFAP2e which still allowed partial NC induction and tested for migration ability of the induced cells (Fig. 6 and SI Appendix, Fig. S7). Specifically, a pre-EMT NC explant coinjected with low levels of TFAP2e MO, mRNA encoding a dexamethasone-inducible TFAP2e and a lineage tracer, was grafted into a wild-type control embryo prior to EMT (stage 17, Fig. 6B and SI Appendix, Fig. S7). While grafted control NC cells efficiently populated the craniofacial areas, morphant NC remained at the grafted site (Fig. 6C). Importantly, when TFAP2e was reactivated in morphant NC at EMT stage, cell migration was partly restored, and lineage-traced cells were found along NC cranial migration routes (Fig. 6 C and D). As predicted, we thus validate a later role for TFAP2e in NC cell migration. Last, we tested the branching predictions, looking if the early expression of Pax3 affected the choice between rpe65+ c10 and dlx2+ c11. For this, we activated pax3 expression using an inducible form at the time of predicted branching (mid-neurula stage 14) and dissected NC explants to analyze them by qRT-PCR at migration stage. We found that pax3 activated expression of c10 marker dmbx1 while it repressed expression of c11 migratory markers mmp14 and fn1 (Fig. 6E). We did not observe significant regulation for the other markers tested such as c10 zfhx4 and c11 twist. This indicates that the scFates predictions have pointed at Pax3, which plays a role in some of the reciprocal regulations occurring during branching between two cranial cluster states (Fig. 6F).
In conclusion, using computational approaches we define and analyze three main bifurcation points in the premigratory NC dataset: from unbiased c5 to vagal and cranial NC, from vagal NC (c9) to cardiac (c12) and ENSp (c13) fates, and from early cranial (c7) to rpe65+ (c10) and dlx2+ (c11) cranial NC. For each branch, we define specific gene programs, including early actors predicted to trigger specific states. Last, we provide several validation elements which link numerous branch-specific genes and two of the early regulators, Pax3 and TFAP2e. Importantly, these analyses reveal novel late-stage functions of Pax3 and TFAP2e in regulating NC development, at stages later than what was previously studied. Pax3 was found to regulate branch-specific genes in both ENSp and cranial NC cells, including genes in cranial Hox-negative clusters while TFAP2e was shown to regulate genes involved in EMT and migration in the cranial NC cells. Altogether, these results indicate that scFates branching analysis predictions identify important candidate regulators for late functions in NC development, independently from their roles during earlier stages (induction) and allow testing their functions in driving state choices during later NC diversification in vivo.
Coexisting Neural Border, Ventral, and Dorsal Ectoderm Gene Programs Specify Neural Crest and Placodes.
Neural border cell signature displays enriched ventral and dorsal gene expressions which are regulated by Pax3.
Next, we decided to look at earlier stages, specifically during gastrulation, when most ectodermal tissues start to be specified. To unveil the molecular mechanisms that induce and distinguish NC and the neighboring cranial placodes at the ectoderm during gastrulation, we gathered data for all ectoderm cells from stages 11 to 13 and identified 10 distinct cell clusters (c’0 to c’9; Figs. 1A and 6A). The “neural border” model suggests that the NB zone is an ectodermal area located in-between sox2+ neuroepithelium (NE) and epidermal keratin+ nonneural ectoderm (NNE), coexpresses tfap2a, zic1, and pax3 and gives rise to both NC and placodes (Fig. 7 A–C and SI Appendix, Figs. S8A and S10) (40–43). The early developmental dynamics of this ectodermal area has not yet been described at the single-cell level in frogs (later stages described in ref. 44) and by only two studies in chick embryos (16, 45). Moreover, it remains unclear whether NB cells resemble adjacent progenitors or if they exhibit a specific gene signature. From the force-directed graph plot, we selected cells coexpressing high levels of tfap2a and zic1 as the presumptive neural border (46–48). These 503 cells, depicted as zone 3, did not appear as an individual cluster, rather as cells spread out among clusters c’1, c’2, c’5, and c’6 (Fig. 7A and SI Appendix, Figs. S8A and S10). While we observed low cell density specifically for the NB zone between stages 11 and 12, we excluded a stage-related sampling issue since NE/NNE areas presented normal density. Rather this could relate to a faster transcriptional transition of NB cells compared to NNE cells: if early ectoderm cells transit through the NB state quickly before switching toward NC or placode states, fewer cells would be captured. Alternatively, such a plot could be obtained if the NB zone was highly heterogeneous and contained a mosaic of states (NB, NNE, or NE).
Fig. 7.

Gastrula-stage ectoderm branching indicates undescribed Pax3-Sox9 epistasis during NC induction. (A) Ectoderm force-directed graph: clusters (Top), stages (Bottom). c’0—early ectoderm, c’1—neural ectoderm, c’2—st11 nonneural ectoderm (NNE), c’3—neural border, c’4—stage 12/13 NNE, c’5—placodes (PC), c’6—NC, c’7—neurons, c’8—eye primordium, c’9—neural plate. (B) Top-3 genes for each cluster. (C) Neural border-enriched gene signature (c’3). (D) Forced-directed graph-suggested paths for NC and PC. (E) NNE1 → NP program, (F) NE → NC program, (G) NB → PC program, (H) NB → NC program, (I) Connectome and branching analyses suggest pax3/sox9 epistasis upstream of NC specifiers snail2, c3, cmyc. (J) Validation of Pax3-Sox9-NC specifiers epistasis using iNC animal cap (AC) assay at stage 12.5. (K) Sox9 activates snail2 precociously in iNC. (J and K) A: wt whole embryo, B: wt AC, C: pax3-GR; zic1-GR-injected AC, D: pax3 -GR; zic1-GR; sox9 MO-injected AC, E: pax3-GR; zic1-GR; sox9-injected AC. All conditions were normalized to C at stage 12.5. SEM, *P < 0.05, **P < 0.01, ***P < 0.01, ****P < 0.0001.
We then identified a detailed signature for the tfap2a+zic1+ cells. Several transcripts were enriched from stage 11: tfap2c, pax3, sox9, hes1, gmnn, and myc (Fig. 7C). Most of these transcripts also formed main nodes in the whole Ectoderm connectome generated with GRNBoost2 (10,085 gene connections, SI Appendix, Table S5). We examined whether Pax3, previously shown to appear as early as stage 10.5 in vivo (46), triggers expression of NB zone signature genes in vivo; in Pax3 morphant NB transcriptome, we found decreased expression of the other NB genes sox9, axin2, zic3, and zic1, while early NE and NNE marker lhx5.l was increased (SI Appendix, Fig. S8 B and C). These results identify a gene signature enriched in tfap2a+zic1+ NB cells, consolidate Pax3 as a major regulator of NB formation, and importantly, as an activator of the NB signature.
The NB zone contributes to NC and PC in parallel to convergent contributions from neural plate and nonneural ectoderm progenitors.
Based on the force-directed graph, we hypothesized three possible developmental routes leading to NC and placodes from stage 11 ectoderm cells: a) NE → NC; b) NB zone → PC and NC; c) NNE → PC (Fig. 7D); these possibilities are consistent with current models of NC and PC formation, the “neural border zone model” (route b) and the “neural vs. nonneural” model (routes a & c) (12, 19, 40, 41). Interestingly using branching analysis, no direct route was found between early NNE (nne1) and NC, or between NE and PC, confirming biological features, such as the partial neuralization needed for NC induction or the close relationship between placodes and NNE (20, 42, 49). We applied branching analysis to each potential route and compared the resulting gene programs underlying NC vs. PC fate decisions. For route (a), branching toward NP from the NE state involved early genes elav3 and sall2 and late genes nkx6 and tubb2b, consistent with current knowledge on neural plate induction (sall2, nkx6) and primary neurogenesis (elav3, tubb2b) (Fig. 7E) (50). Route (a) branching toward NC involved early genes zic1 and foxd3 and late genes c3, myc, sox8, sox9, pcdh8, and snai2 (Fig. 7F). In comparison, NC cells emerging from the NB zone by route (b) expressed c3 and sox9 before bifurcation, while NC markers foxd3, sox8, pcdh8, snail2 were enriched after splitting (Fig. 7H). Similarly, we explored both proposed routes of placodal development: route (b) from the NB zone showed early enrichment of tcf7l1, hesx1, and late for stmn1, pax6, pitx1/2 (Fig. 7G) while route (c) from NNE exhibited early enrichment of placode specifiers six1, otx2, and late expression of placode markers egflam, pax6, pitx1/2, SI Appendix, Fig. S8D). Last, the route (c) branching for NNE formation confirmed known developmental dynamics with early enrichment for gata2 and late for ectoderm stem cell marker tp63 and epithelial cells cldn1 (SI Appendix, Fig. S8E) (51). This hierarchy of gene expression along the different branches thus opens avenues to further elaborate each of the NC- and PC-GRNs, and the signaling that acts on them.
Different gene programs can lead distinct progenitors toward a similar state.
Interestingly, according to the route studied, we found that i) distinct genes were activated to obtain the same state, and that ii) some genes were activated with different expression dynamics relative to different bifurcations. For example, during the NB → NC transition (route b, Fig. 7H), sox9 and c3 were activated early (before bifurcation) suggesting that they could play a part in the fate decision network from NB progenitors. In contrast, during the NE → NC gene program (Fig. 7F) sox9 and c3 were late genes while foxd3 and zic1 were expressed early. This observation suggested a new model of fate decisions in the developing ectoderm, where parallel and distinct genetic programs activated in distinct ectoderm progenitors may lead to a similar state.
Last, by branching analysis, NB zone-specific gene pax3 was expressed prior to bifurcation in the route (b), NB → NC gene program, and proposed to activate expression of late NC branch markers sox9, sox8, zic1, pcdh8, and c3 (Fig. 7H). Interestingly, by GRNBoost2 analysis, the Ectoderm connectome described NC genes (snai2, sox8, cmyc, c3) connected to the rest of the network through Pax3 and Sox9 (Fig. 7I), therefore suggesting that Sox9 might play a yet undescribed function immediately downstream of Pax3 in NC induction and upstream of the other late NC-branch markers. So far, Sox9 was known to be essential for NC induction, but its epistasis with Pax3 remained unknown. We tested the epistasis relationships between Pax3 and Sox9 in NC induction (snai2, cmyc, c3 expression) by combining Sox9 depletion or gain-of-function in the induced-neural crest assay (iNC is Pax3/Zic1-based NC induction from pluripotent ectoderm cells, SI Appendix, Fig. S8G). Early NC marker snail2 expression starts from gastrula stage 12.5 both in vivo and in iNC and increases at neurula stage 14 (SI Appendix, Fig. S8H). In this experiment, Sox9 depletion reduced snail2 activation as early as at stage 12.5 while coactivation of Sox9 strongly increased snail2 expression in iNC (Fig. 7 J and K and SI Appendix, Fig. S8 H and I), indicating that Sox9 is required for efficient NC induction by Pax3 and Zic1. Similar results were observed on cmyc and c3 activation when Sox9 was depleted (SI Appendix, Figs. S7J and S9). Interestingly, when iNC explants were analyzed prior to the normal onset of snail2 expression, at mid-gastrula stage 11/11.5, Sox9 activation led to premature snail2 expression (Fig. 7K), suggesting that Sox9 synergizes with Pax3 and Zic1 at the onset of NC induction, and may act earlier than previously observed (52–54).
In conclusion, we have defined the transcriptional signature for the incompletely described Neural Border zone, established a global Ectoderm connectome and experimentally validated NC-related nodes, in particular highlighting how Sox9 enhances NC induction immediately downstream of Pax3 and Zic1. We characterized three different transcriptional programs branching from neural, neural border, and nonneural ectoderm progenitors toward NC and PC and propose a model in which multiple coexisting paths lead to the early NC or placode states in gastrula-stage ectoderm.
Discussion
In this work, we exploit the resolution of high-density single-cell transcriptomes collected from 8 frog developmental stages to unravel the emergence of the neural crest lineage from the ectoderm during gastrulation, followed by diversification of neural crest progenitors during neurulation, and upon EMT. Modeling gene transcription dynamics around each cell state allows the inference of the underlying molecular networks. We selected several important nodes for large-scale and in vivo experimental validation (Fig. 8 A and B). This study highlights the temporal sequence of states in premigratory neural crest development. First, we characterize neural crest activation either from a transient neural border state, or from a neural plate state during mid-gastrulation, and suggest a model that reconciles current debates upon multiple possible routes leading from immature ectoderm to neural crest and placodes (Fig. 8C). Second, we delineate the early and later neural crest transcriptome trajectories during neurulation and define key regulators of branching, leading to eight transitional states and eight early migration states (Fig. 8D).
Fig. 8.

Neural crest GRN and developmental transcriptome trajectories. (A) Connectomes for ectoderm and neural crest (https://github.com/Qotov/neucrest_grn). (B) Large GRNs of the major nodes Pax3 and TFAP2e. (C) In the “dual convergence model,” neural border cells present trajectories toward both placodes and NC, which converge with a trajectory from neural plate toward NC and another one from nonneural ectoderm toward placodes. Branching analyses suggest the top genes underlying transcriptome transitions. (D) Transcriptomic tree of NC cell states, across late gastrulation, neurulation, and epithelial–mesenchymal transition, ending at early migratory stage with gene signatures supporting each trajectory. Pax3 or Tfap2e expression prior to branching (in blue) prompted in vivo validation of their role on the branch-specific signature: branch-specific genes modulated by Pax3 or TFAP2e are shown in green.
A Reconciliatory “Model of Dual Convergence” Describes the Converging Trajectories Initiating Neural Crest and Placode States.
The molecular signature of the neural border ectoderm has been simple, with only Pax3 (in frogs and fish) or Pax7 (in chick) as relatively specific markers for this domain during gastrulation and early neurulation stages (9, 17). Here, we have characterized the neural border ectoderm state by two features: lower expression of genes expressed by adjacent neural (dorsal) and nonneural (ventral) cells (sox2, lhx2, sall2, gata2, ker19) and increased expression of a large gene list including zic1, tfap2a/c, pax3, sox9, hes1, cmyc. We find that this state seems more transient than other ectoderm states, suggesting that these fate decisions occur quickly. In frog embryos, the end of gastrulation is clearly defined by blastopore closure and allows more precise exploration of timing, compared to organisms with simultaneous gastrulation and neurulation such as chick embryos. In frogs, fate choices in the dorsal ectoderm happen during the second half of gastrulation (between stages 11 and 12.5). As the neural plate forms and the blastopore closes (stage 13), fate decisions are clearly established between neural, nonneural, placode, and neural crest with robust molecular signatures (Fig. 7C). Modeling neural crest emergence from the neural border cell state confirmed the central role for Pax3 and suggested a key epistatic relationships and functional synergy between Pax3 and Sox9 upstream of the definitive neural crest state, defined by snail2 expression. Importantly, we propose a novel model for the transcriptional pattern of decisions between the four main ectoderm fates, neural, nonneural, placodal, neural crest. Instead of the contrasting “neural border” and “nonneural vs. neural” hypotheses, we find that these routes may not be exclusive and find potential trajectories supporting the emergence of neural crest from either the neural border or the nascent neural ectoderm on one hand, as well as two putative trajectories leading to placodes from either the neural border or the nonneural ectoderm. In each case, the candidate gene programs underlying those alternative trajectories involve a subset of common genes and a few specific factors (Fig. 7F). For example, specific expression of tcf7l1 and stmn1 is found in placodes arising from the NB zone, compared to the NNE route (Fig. 7 J and K). For neural crest, early sox9 expression in the NB route contrasts with postbifurcation expression in the NP route (Fig. 7 H and I). Thus, our SC transcriptome modeling reconciles and combines previous alternatives in a “Dual convergence Model” of neural crest and placode patterning, together with specific gene signatures ripe for future lineage tracing studies, functional exploration (Fig. 8C) and higher resolution analyses which may or not uncover so far hidden differences between NC cells of different origins. Our model also aligns with a recent study in the chick embryo neural border zone, showing that a continuous gradient of early and transitional states converges on each of the four main ectoderm derivatives (NE, NNE, NC, PC) [“gradient border model” by Thiery et al. (45)]. Further, our branching analysis also provides candidate genes driving the possible transcriptional programs. From an evolutionary perspective, the dual convergence concept suggests that distinct developmental GRNs, in parallel or collaboratively, can achieve a same developmental outcome, serving as a redundancy mechanism or through convergent evolution for enhanced robustness in neural crest evolution.
A Combination of Omics and In Vivo Strategies Validates Large Sets of Gene Regulations Driving the Dynamics of Neural Crest Diversification.
The second outcome of our study is to define the temporal dynamics of trajectories that result in eight neural crest states present upon early migration stage along the cranial and vagal axial positions (Fig. 8D). The first key observation is the presence of a main population of NC unbiased toward any particular state, expressing markers of the immature neural crest cells, from which all the other trajectories emerge. This unbiased cell trajectory is maintained during and after EMT suggesting that a very plastic, stem-like NC cell population emigrates and is subjected to the signals from the microenvironment prior to fate choices. The second critical observation is that, for the anterior part of the body axis considered here, trajectories do not emerge in a spatially linear sequence from anterior to posterior as previously anticipated in a model where NC would follow an anterior–posterior wave of maturation. Two early trajectories arise from both the anterior (progenitors of posterior cranial NC—c15) and the posterior-most positions (minor vagal trajectory progenitors—c3) at neural plate stage (stages 13 and 14) prior to neural fold elevation. This is followed at mid-neurula stage (stages 15 and 16) by the emergence of the three other main cranial and vagal trajectories leading to cranial c10 and c11, and to vagal NC c12 and c13. Together with the maintenance of an immature stem-like cell population from induction to emigration, this sequence of trajectory determination suggests that the main cue controlling the temporal dynamics of states hierarchy in the cranial and vagal NC-GRN is not a function of the time elapsed since NC cell induction, or correlated to Hox gene positional information, but rather may involve response to external signals.
Our temporal analysis highlights three important points deepening our understanding of NC biology. First, there have been long-standing debates about the timing of NC fate decisions, prior or after EMT from the neural tube, in a variety of animal models (55). Importantly, we did not detect distinctive expression of predictive differentiation/determination fate markers before EMT. This suggested that, even if some NC progenitors were biased toward a given fate prior to EMT, they did not exhibit a detectable signature in our dataset. However, our observations are in agreement with several lineage tracing studies showing the high multipotency of most NC cells prior to EMT (28) and the determination of NC cells after EMT (26, 27). Second, our data support the early diversification into several distinct cell states prior, during, and after EMT, contrasting with the recent suggestion that upon EMT the NC progenitors would regroup into a single common multipotent state (15). The high cell content of our dataset proves otherwise, suggesting that this previous observation made on a smaller subset of cranial NC did not fully capture the diversity of premigratory NC states. Last, temporal trajectory analysis unravels the branch-specific dynamics of gene expression underlying bifurcations and state diversification. For each bifurcation, we provide a list of key genes likely to control branching choices (Figs. 5 and 6). We further validate these predictions in several instances, by experimental modulation of pivotal transcription factors function in the premigratory neural crest (Pax3, TFAP2e), followed by in vivo or deep sequencing analysis. In sum, our study provides a comprehensive view of the hierarchy of molecular decisions driving the cranial and vagal neural crest gene regulatory network from induction at the neural border to early migration, with unprecedented resolution and deep learning-aided experimental validation. We propose a new “dual convergence model” for neural crest and placode lineage emergence and provide a detailed roadmap of the main molecular events in the premigratory and early migrating NC-GRN. Using a dedicated interactive network visualization interface, any gene of interest can be queried (https://github.com/Qotov/neucrest_grn). Moreover, the detailed sequence of cell states provided here will prove an essential reference for monitoring induction of neural crest derivatives, for example from patient-derived induced pluripotent stem cells, when reliable specification protocols preferably recapitulate the steps of embryonic development.
Materials and Methods
Extended version in SI Appendix, Part B.
Single-Cell Sequencing.
Instead of collecting new materials, we resequenced the whole-embryo SC-RNA libraries for developmental stages NF11 to NF22 (56) used in ref. 20 generating a total of 12 billion reads. SC analysis used the X.tropicalis v.10 genome assembly, gene models v. 10.7, STAR aligner, and DropEst pipeline (57, 58). After filtration by counts and gene numbers (>200 genes; >300 counts), we gathered a dataset of 177,250 cells (NCBI Gene Expression Omnibus, GSE198494). In the cells of interest (Ectoderm and NC cells), mean counts number was 1,778, and mean gene number 1035. scRNA-seq postprocessing was done using Scanpy (59).
Clustering.
For each stage, we obtained independent standard dimensionality reduction with PCA, computing a neighborhood graph and UMAP (60) followed by clustering, [Leiden algorithm, (21)]. Cluster-specific genes were defined with differential expression analysis (scanpy t-test_overestim_var) Clusters the most similar to NC cells using NC signatures from ref. 20 were selected from the whole embryo dataset. The optimal level of NC subclustering was defined by increasing cluster number and checking biological meaning using specific gene expression.
GRN Generation.
With time-resolved gene expression dynamics and a list of TFs, we inferred genetic coregulation and targets with GRNBoost2 (30, 31).
Principal Graph Generation and Branching Analysis.
We generated the main tree with PAGA (23), revealing cluster–cluster relationships: Early stages showed the strongest connectivity. ElPiGraph indicated specific branches and bifurcation points. scFates (35) defined features significantly changing along the tree. Pseudotime values and differential expression analysis determined early and late branch-specific features. Insufficient sequencing depth prevented use of RNA velocity.
X. Laevis Injections, Microdissections, Grafting, and RNA-Seq on Explants.
Animal use followed recommendations of the European Community (2010/63/UE) and international guidelines (Authorization APAFiS#36928-2022042212033387-v1). In vivo injections, NB/NC dissections and grafting used X. laevis embryos (2, 61). Knockdown experiments used previously validated antisense morpholino oligonucleotides (MO) for pax3 or TFAP2e (9, 33). Individual anterior NB explant (wild-type or Pax3 morphant) or anterior premigratory NC (wild-type or TFAP2e-morphant) were dissected and then sequenced in triplicate. (RNAseq parameters in SI Appendix, Part B).
Chromatin Immunoprecipitation and Sequencing.
ChIPseq (62, SI Appendix, Part B) was done on mid-neurula stage 14 or early NC migration stage 19 embryos, injected with trace mRNA amounts (75 pg). Pax3-FLAG-HA: 100 X. laevis embryos/condition, three biological replicates; TFAP2e-FLAG, 100 X. tropicalis embryos/condition, one biological replicate, validation controls in SI Appendix, Fig. S4C.
iNC Assay.
The induced neural crest assay (iNC) uses coactivation of dexamethasone-inducible Pax3-GR and Zic1-GR at gastrula stage 10.5 (47, 63). iNC is here combined with Sox9 depletion or gain-of-function (53) and processed by RT-qPCR (49, SI Appendix, Table S10).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Drs. Zinoviev, Schlosser, Adameyko, and Walter for scientific discussions; C. Lantoine, Q. Thuillier, and Monsoro-Burq lab members for technical assistance and support; J. Briggs for single-cell sequencing, J.L. Plouhinec for preliminary RNAseq analysis. RNAseq and ChIPseq used ICGex NGS platform of Institut Curie (ANR-10-EQPX-03; ANR-10-INBS-09-08; Canceropole Ile-de-France; SiRIC-Curie INCa-DGOS-4654). This project received funding from European Union Horizon 2020 Marie Skłodowska-Curie grant no. 860635, NEUcrest ITN; Agence Nationale pour la Recherche ANR-15-CE13-0012-01, ANR-21-CE13-0028; Institut Universitaire de France (AHMB); NIH R01HD073104, R24OD031956 (L.P.); NIH NIH-R01-GM42341; NIH-R35GM127069 (R.H.).
Author contributions
A.K., S.S., M.A., S.M.R., L.P., and A.H.M.-B. designed research; A.K., S.S., M.A., V.K., S.M.R., and A.H.M.-B. performed research; A.K., S.S., M.A., V.K., S.M.R., H.A., R.M.H., L.P., and A.H.M.-B. analyzed data; and A.K., S.S., M.A., R.M.H., L.P., and A.H.M.-B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
MultiOmics sequencing data have been deposited in Gene Expression Omnibus (GEO) (GSE198491 (64); GSE198492 (65); GSE198493 (66); and GSE198494 (67)).
Supporting Information
References
- 1.Eames B. F., Medeiros D. M., Adameyko I., Evolving Neural Crest Cells (CRC Press, 2020). [Google Scholar]
- 2.Plouhinec J.-L., et al. , A molecular atlas of the developing ectoderm defines neural, neural crest, placode, and nonneural progenitor identity in vertebrates. PLoS Biol. 15, e2004045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steventon B., Mayor R., Early neural crest induction requires an initial inhibition of Wnt signals. Dev. Biol. 365, 196–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Streit A., Stern C. D., Establishment and maintenance of the border of the neural plate in the chick: Involvement of FGF and BMP activity. Mech. Dev. 82, 51–66 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharya D., Azambuja A. P., Simoes-Costa M., Metabolic reprogramming promotes neural crest migration via yap/tead signaling. Dev. Cell 53, 199–211.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueiredo A. L., et al. , PFKFB4 control of Akt signaling is essential for premigratory and migratory neural crest formation. Development 144, 4183–4194 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Ling I. T. C., Sauka-Spengler T., Early chromatin shaping predetermines multipotent vagal neural crest into neural, neuronal and mesenchymal lineages. Nat. Cell Biol. 21, 1504–1517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang W., Li Y., Li A., Bronner M. E., Clonal analysis and dynamic imaging identify multipotency of individual Gallus gallus caudal hindbrain neural crest cells toward cardiac and enteric fates. Nat. Commun. 12, 1894 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monsoro-Burq A.-H., Wang E., Harland R., Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell 8, 167–178 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Khudyakov J., Bronner-Fraser M., Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev. Dyn. 238, 716–723 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simões-Costa M., Tan-Cabugao J., Antoshechkin I., Sauka-Spengler T., Bronner M. E., Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res. 24, 281–290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roellig D., Tan-Cabugao J., Esaian S., Bronner M. E., Dynamic transcriptional signature and cell fate analysis reveals plasticity of individual neural plate border cells. Elife 6, e21620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watt K. E. N., Trainor P. A., “Neurocristopathies” in Neural Crest Cells, Trainor P. A., Ed. (Elsevier, 2014), pp. 361–394. [Google Scholar]
- 14.Medina-Cuadra L., Monsoro-Burq A. H., Xenopus, an emerging model for studying pathologies of the neural crest. Curr. Top. Dev. Biol. 145, 313–348 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Zalc A., et al. , Reactivation of the pluripotency program precedes formation of the cranial neural crest. Science 371, eabb4776 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams R. M., Lukoseviciute M., Sauka-Spengler T., Bronner M. E., Single-cell atlas of early chick development reveals gradual segregation of neural crest lineage from the neural plate border during neurulation. eLife 11, e74464 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basch M. L., Bronner-Fraser M., García-Castro M. I., Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 441, 218–222 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Groves A. K., LaBonne C., Setting appropriate boundaries: Fate, patterning and competence at the neural plate border. Dev. Biol. 389, 2–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maharana S. K., Schlosser G., A gene regulatory network underlying the formation of pre-placodal ectoderm in Xenopus laevis. BMC Biol. 16, 79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briggs J. A., et al. , The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360, eaar5780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traag V. A., Waltman L., van Eck N. J., From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner D. E., et al. , Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolf F. A., Angerer P., Theis F. J., SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krispin S., Nitzan E., Kalcheim C., The dorsal neural tube: A dynamic setting for cell fate decisions. Dev. Neurobiol. 70, 796–812 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Nitzan E., et al. , A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development 140, 2269–2279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison J. A., et al. , Single-cell transcriptome analysis of avian neural crest migration reveals signatures of invasion and molecular transitions. Elife 6, e28415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrison J. A., et al. , Single-cell reconstruction with spatial context of migrating neural crest cells and their microenvironments during vertebrate head and neck formation. Development 148, dev199468 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Baggiolini A., et al. , Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem. Cell 16, 314–322 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Tatarakis D., et al. , Single-cell transcriptomic analysis of zebrafish cranial neural crest reveals spatiotemporal regulation of lineage decisions during development. Cell Rep. 37, 110140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blitz I. L., et al. , A catalog of Xenopus tropicalis transcription factors and their regional expression in the early gastrula stage embryo. Dev. Biol. 426, 409–417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moerman T., et al. , GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Sato T., Sasai N., Sasai Y., Neural crest determination by co-activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development 132, 2355–2363 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Hong C.-S., Devotta A., Lee Y.-H., Park B.-Y., Saint-Jeannet J.-P., Transcription factor AP2 epsilon (Tfap2e) regulates neural crest specification in Xenopus. Dev. Neurobiol. 74, 894–906 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu M., et al. , PAX3 promotes cell migration and CXCR4 gene expression in neural crest cells. J. Mol. Neurosci. 64, 1–8 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Albergante L., et al. , Robust and scalable learning of complex intrinsic dataset geometry via ElPiGraph. Entropy 22, 296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faure L., Soldatov R., Kharchenko P. V., Adameyko I., scFates: A scalable python package for advanced pseudotime and bifurcation analysis from single-cell data. Bioinformatics 39, btac746 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Douarin N., Kalcheim C., The Neural Crest (Cambridge University Press, 2022), 10.1017/CBO9780511897948. [DOI] [Google Scholar]
- 38.Gonzalez Malagon S. G., Liu K. J., ALK and GSK3: Shared features of neuroblastoma and neural crest cells. J. Exp. Neurosci. 12, 1179069518792499 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tani-Matsuhana S., Inoue K., Identification of regulatory elements for MafB expression in the cardiac neural crest. Cells Dev. 167, 203725 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Seal S., Monsoro-Burq A. H., Insights into the early gene regulatory network controlling neural crest and placode fate choices at the neural border. Front. Physiol. 11, 608812 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thawani A., Groves A. K., Building the border: Development of the chordate neural plate border region and its derivatives. Front. Physiol. 11, 608880 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pieper M., Ahrens K., Rink E., Peter A., Schlosser G., Differential distribution of competence for panplacodal and neural crest induction to non-neural and neural ectoderm. Development 139, 1175–1187 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Schlosser G., Early embryonic specification of vertebrate cranial placodes. WIREs Dev. Biol. 3, 349–363 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Lee J., et al. , A single-cell, time-resolved profiling of Xenopus mucociliary epithelium reveals nonhierarchical model of development. Sci. Adv. 9, eadd5745 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery A., et al. , scRNA-sequencing in chick suggests a probabilistic model for cell fate allocation at the neural plate border. eLife 12, e82717 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Crozé N., Maczkowiak F., Monsoro-Burq A. H., Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc. Natl. Acad. Sci. U.S.A. 108, 155–160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milet C., Maczkowiak F., Roche D. D., Monsoro-Burq A. H., Pax3 and Zic1 drive induction and differentiation of multipotent, migratory, and functional neural crest in Xenopus embryos. Proc. Natl. Acad. Sci. U.S.A. 110, 5528–5533 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garnett A. T., Square T. A., Medeiros D. M., BMP, Wnt and FGF signals are integrated through evolutionarily conserved enhancers to achieve robust expression of Pax3 and Zic genes at the zebrafish neural plate border. Development 139, 4220–4231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alkobtawi M., Pla P., Monsoro-Burq A. H., BMP signaling is enhanced intracellularly by FHL3 controlling WNT-dependent spatiotemporal emergence of the neural crest. Cell Rep. 35, 109289 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Exner C. R. T., Kim A. Y., Mardjuki S. M., Harland R. M., sall1 and sall4 repress pou5f3 family expression to allow neural patterning, differentiation, and morphogenesis in Xenopus laevis. Dev. Biol. 425, 33–43 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Haas M., et al. , ΔN-Tp63 mediates Wnt/β-catenin-induced inhibition of differentiation in basal stem cells of mucociliary epithelia. Cell Rep. 28, 3338–3352.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Donnell M., Hong C.-S., Huang X., Delnicki R. J., Saint-Jeannet J.-P., Functional analysis of Sox8 during neural crest development in Xenopus. Development 133, 3817–3826 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Spokony R. F., Aoki Y., Saint-Germain N., Magner-Fink E., Saint-Jeannet J.-P., The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 129, 421–432 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Lee Y.-H., et al. , Early requirement of the transcriptional activator Sox9 for neural crest specification in Xenopus. Dev. Biol. 275, 93–103 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Kalcheim C., Kumar D., Cell fate decisions during neural crest ontogeny. Int. J. Dev. Biol. 61, 195–203 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Nieuwkoop P. D., Faber J., Eds., Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis (Garland Pub, 1994). [Google Scholar]
- 57.Baruzzo G., et al. , Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 14, 135–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petukhov V., et al. , dropEst: Pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stuart T., et al. , Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacomy M., Venturini T., Heymann S., Bastian M., ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the gephi software. PLoS One 9, e98679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milet C., Monsoro-Burq A. H., Dissection of Xenopus laevis neural crest for in vitro explant culture or in vivo transplantation. J. Vis. Exp. 85, 51118 (2014), 10.3791/51118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wills A. E., Gupta R., Chuong E., Baker J. C., Chromatin immunoprecipitation and deep sequencing in Xenopus tropicalis and Xenopus laevis. Methods 66, 410–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong C.-S., Saint-Jeannet J.-P., The activity of Pax3 and Zic1 regulates three distinct cell fates at the neural plate border. MBoC 18, 2192–2202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kotov A., et al. , From neural border epithelium to neural crest emigration: A comprehensive single cell roadmap of the timing and regulatory logic underlying cranial and vagal neural crest emergence [scRNAseq]. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198491. Deposited 12 March 2022.
- 65.Kotov A., et al. , From neural border epithelium to neural crest emigration: A comprehensive single cell roadmap of the timing and regulatory logic underlying cranial and vagal neural crest emergence [ChIPseq]. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198492. Deposited 12 March 2022.
- 66.Kotov A., et al. , From neural border epithelium to neural crest emigration: A comprehensive single cell roadmap of the timing and regulatory logic underlying cranial and vagal neural crest emergence [morpholinoRNAseq]. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198493. Deposited 12 March 2022.
- 67.Kotov A., et al. , From neural border epithelium to neural crest emigration: A comprehensive single cell roadmap of the timing and regulatory logic underlying cranial and vagal neural crest emergence. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198494. Deposited 12 March 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
MultiOmics sequencing data have been deposited in Gene Expression Omnibus (GEO) (GSE198491 (64); GSE198492 (65); GSE198493 (66); and GSE198494 (67)).