

Abstract
Portal hypertension (PH) is the main driver of cirrhosis decompensation, the main determinant of death in patients with cirrhosis. PH results initially from increased intrahepatic vascular resistance. Subsequently, increased inflow from splanchnic vasodilation and increased cardiac output lead to a further increase in portal pressure (PP). Reducing PP in cirrhosis results in better outcomes. Removing the cause of cirrhosis might improve PP. However, this is a slow process and patients may continue to be at risk of decompensation. Additionally, for some chronic liver diseases, such as nonalcoholic fatty liver disease (NAFLD), etiological treatments are not yet available. Therefore, there is a need to develop better therapies specifically aimed at reducing PP. For over 35 years, the mainstay of such therapy has been the use of nonselective beta-blockers (NSBBs) that act by reducing portal venous inflow. Recently, many drugs (mainly targeting intrahepatic mechanisms) have shown promise in preclinical and early clinical studies and may act alone or synergistically with NSBBs in reducing PP in cirrhosis. The objective of this position paper is to propose a novel framework for the design of clinical trials (phase 1, 2, and 3) in patients with cirrhosis and PH and to prioritize targets and pharmacological therapies in this setting. We have focused the discussion on patients with compensated cirrhosis. The paper summarizes discussions held at The American Association for the Study of Liver Diseases (AASLD) Industry Colloquium in January 2018, with the participation of clinical and translational investigators, regulatory professionals, and industry partners. (Hepatology 2019;69:1287–1299).
Portal hypertension (PH) is the main consequence of cirrhosis and is the main driver of cirrhosis decompensation.(1) Cirrhosis decompensation is defined by the presence of clinical complications (ascites, variceal hemorrhage, and encephalopathy) and constitutes the main determinant of death in patients with cirrhosis.(2)
PH results initially from increased intrahepatic resistance attributed to architectural distorsion and intrahepatic vasoconstriction.(3) At this stage, PH is mild but leads to increase in vasodilators in the splanchnic vasculature that result in increased portal venous inflow. This increased inflow leads to a further increase in portosystemic gradient, and, once it surpasses a certain level (clinically significant PH; CSPH), the patient becomes more susceptible to decompensation.(4)
Elimination of the underlying cause of cirrhosis may lead to a reduction in fibrosis and to an improvement in PH. For example, elimination of the hepatitis C virus (HCV) has been associated with a reduction in portal pressure (PP) in patients with cirrhosis.(5,6) However, once PH becomes clinically significant, it is unlikely to regress, at least in the short term, and the patient continues to be at risk of decompensation.(6,7) Additionally, there are etiologies (e.g., nonalcoholic fatty liver disease [NAFLD]) for which etiological treatment is not yet available.
Reducing PP in cirrhosis, independent of elimination of the cause of liver damage, results in better outcomes.(8,9) Therefore, there is still a need to develop therapies that reduce PP by targeting its main pathogenic vascular mechanisms. For over 35 years, the mainstay of such therapy has been the use of nonselective beta-blockers (NSBBs) that act by reducing portal venous inflow.
Recently, many drugs (mainly targeting intrahepatic mechanisms) have shown promise in preclinical and early clinical studies. The American Association for the Study of Liver Diseases (AASLD), at its yearly Industry Colloquium ( January 19–20,2018), invited members of the AASLD Portal Hypertension Special Interest Group (SIG) to participate in a symposium that had the objective of discussing novel therapies in PH from the perspective of clinical and translational investigators, with participation from regulatory professionals and industry partners.
The objective of this position paper, as discussed in the symposium, is to propose a framework for the design of clinical trials (phase 1, 2, and 3) in patients with cirrhosis and PH and to prioritize novel targets and pharmacological therapies for PH.
Preclinical Studies in Portal Hypertension (PH)
The initial mechanism in the pathogenesis of PH, an increase in intrahepatic vascular resistance, is attributed to a combination of structural changes (i.e., fibrosis, disruption of sinusoidal structure, and microthrombi) and increased sinusoidal resistance (i.e., vasoconstriction) that results from increased contraction of activated hepatic stellate cells (HSCs) in the setting of increased vasoconstrictors and to dysfunctional endothelial cells with decreased production of vasodilators such as nitric oxide (NO).(3) In response to intrahepatic changes and/or mild increases in PP, splanchnic and systemic vasodilation and angiogenesis follow, leading to increased portal venous inflow and worsening of PH.(10,11)
The complexity of PH is evident by the divergent actions of vasoconstrictive and vasodilating systems in the intrahepatic versus extrahepatic circulations. Vasoconstrictors, such as angiotensin II and norepinephrine, contribute to intrahepatic vasoconstriction.(10) Rho-kinase, a key pathway in the maintenance of basal vascular tone, is dysregulated in both the intrahepatic and extrahepatic circulations; whereas in the liver it contributes to vasoconstriction, in the extrahepatic vessels contractility is almost completely lost, leading to vasodilatation.(10) Conversely, availability of the vasodilator, NO, is reduced in the liver and increased in the extrahepatic tissues.(3) Because of these and other changes, even the strongest vasoconstrictors like angiotensin II or epinephrine fail to constrict extrahepatic vessels, whereas the intrahepatic vascular bed shows exaggerated contraction. Therefore, it becomes a major challenge to select systemic therapies for PH in cirrhosis, because vasoconstrictors (that would target splanchnic vasodilatation) will worsen intrahepatic resistance and vasodilators (that would target intrahepatic vasoconstriction) will lead to systemic hypotension and its downstream deleterious consequences.
Systemic and splanchnic vasodilatation is a key component of the hyperdynamic syndrome in cirrhosis and is characterized clinically by low blood pressure and increased cardiac output. It not only leads to the development of CSPH and portosystemic collaterals, but is also responsible for sodium and water retention, all of which eventually lead to the development of clinical decompensation.(12) In a stage of further progression of liver disease, systemic hypotension develops, mainly attributed to further vasodilatation and a relative decrease in cardiac output, leading to refractory ascites and hepatorenal syndrome.(13) Systemic hypotension on the one side and clinical immunological events (e.g., systemic inflammation, infections) on the other side lead to impaired organ perfusion and dysfunction, and the development of multiorgan failure in cirrhosis (so-called acute-on-chronic liver failure).(14) Therefore, the selection of targets could be different in compensated and decompensated cirrhosis depending on the objective of treatment (Fig. 1).
FIG. 1.
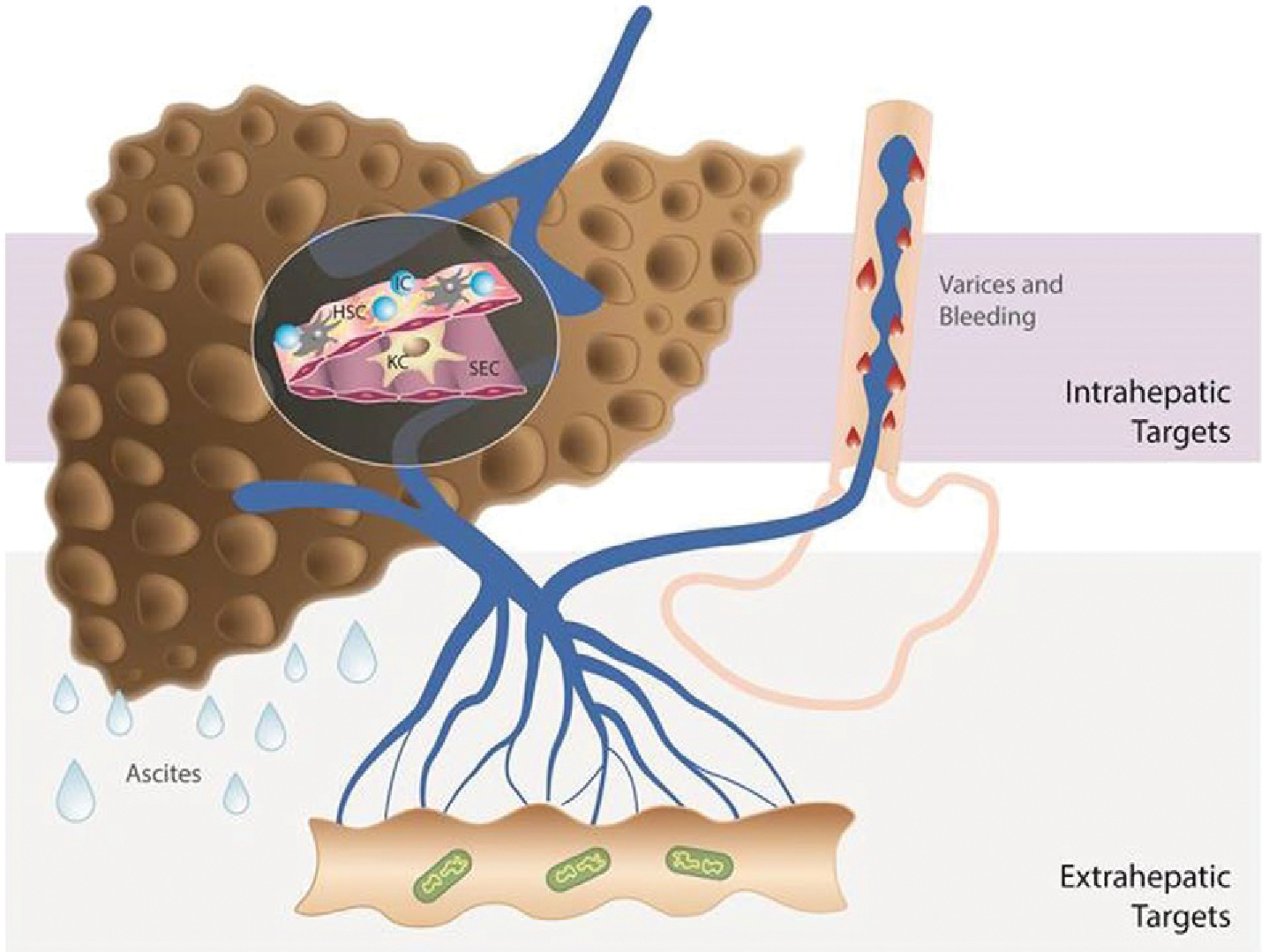
Different approaches to treat PH in cirrhosis. Strategies to decrease PP include drugs that act on intrahepatic targets: fibrosis, endothelial dysfunction, and inflammation and drugs that act on decreasing portal flow: splanchnic vasculature, microbiome, and others such as the nervous system, fat, etc., as recently reviewed.(109,110) A single drug may be able to affect more than one target (multitargeted approach): either two or more intrahepatic targets (e.g., statins) or one intrahepatic and one extrahepatic target (e.g., carvedilol). If a multitargeted drug is associated with unfavorable effects, the selective delivery either to the intrahepatic or to the extrahepatic circulation may be preferable. Drug therapy should also be tailored to the stage of cirrhosis. In compensated cirrhosis, the main treatment should target the underlying cause of cirrhosis in order to prevent progression of disease (or even to induce regression). Additionally, antifibrotic strategies together with drugs to decrease PP (if significant PH is present) are beneficial in compensated cirrhosis, given that fibrosis and PH are also self-maintaining processes. In decompensated cirrhosis (ascites, variceal hemorrhage), the main and short-/mid-term goal should be reduction in PP and prevention of further complications, given that antifibrotic and etiological therapies, although also important, might not have the necessary short-term impact. Abbreviations: IC, inflammatory cells; KC, Kupffer cells; SEC, sinusoidal endothelial cells. Additional references (#109 and #110) are provided as Supporting Information.
Table 1 provides a list of drugs that, in preclinical studies, have been shown to reduce PP, including their purported site of action and other potential benefits as well as potential deleterious effects. Of them, only statins have gone on to proof-of-concept (POC) clinical trials leading to a randomized controlled trial (RCT) with clinical outcomes.(15–17) Statins are approved drugs for the treatment of hypercholesterolemia, but they are pleiotropic drugs that are also Rho-kinase inhibitors and have antifibrotic effects. The use of statins is an example of how drugs that are in clinical use for other indications can be repurposed for the treatment of PH, and this constitutes a promising model.
TABLE 1.
Selected New Candidate Drugs With a Proven Effect in Reducing PP in Preclinical Studies and in Some Clinical Studies
| Preclinical Studies | POC Studies | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Drugs | Purported Site of Action (Intrahepatic, Extrahepatic, or both) | Effect Other Than ↓ in PP | Potential Deleterious Signal | Reduced HVPG | Positive Effect Other Than on HVPG | AEs Observed | Phase 3 Trials |
|
| |||||||
| Statins | lntrahepatic(10,24,46) | Antifibrotic(47) | Rhabdomyolysis(24) | Yes(16,48,49) | Increased in ICG clearance(16) | Rhabdomyolysls, liver toxicity(17) | Yes (decrease mortality(17) |
| PDE5 inhibitors | lntrahepatic(50–52) | Antifibrotic(52) | — | ± (discordant)(53–57) | Erectile dysfunction | Hypotension | No |
| Sorafenib (anti-VEGF) | Both(58–60) | Antifibrotic, antiangiogenic(69) | Hepatic Injury(61) | ± (discordant)(62–64) | Improved MAP | Multiple | No |
| Poorly absorbed antibiotics (rifaximin, norfloxacin) | Extrahepatic(65) | Decreased bacterial translocation(65) | — | ± (discordant)(66–70) | Less HE, less decompensation(66,67) | — | No? |
| Caspase inhibitors | Both(71,72) | Liver injury, flbrosis(71) | — | Yes(73) | Decrease AST(74) | — | No |
| FXR agonists | Both(76–77) | Antifibrotic, decreased bacterial translocation(78–81) | — | Yes(82) | — | No | |
| Serelaxin | Intrahepatic(83) | Improved renal perfusion(84) | — | No data(84) | Improved renal perfusion(84) | — | No |
| Enoxaparin | Both(86,86) | Antifibrotic(85) | — | Ongoing trial (NCT02643212) | Decreased decompensation, less portal vein thrombosis(87) | — | |
| Endothelin-A receptor antagonists | lntrahepatic(88,89) | Antifibrotic(89) Antiangiogenic(90) |
— | ± (discordant)(91,92) | Decreased pulmonary arterial pressure (treatment of portopulmonary hypertension(91) | Decreased MAP(91) | No |
| Thalidomide | Intrahepatic(93,94) | Antifibrotic, antiangiogenic, reduced systemic inflammation(93,94) | — | Yes(95) | Reduced systemic inflammation(95) | — | — |
| Angiotensin II type 1 receptor blocker | lntrahepatic(96,98) | Antifibrotic,(98) improved renal perfusion as low dose(99) | Arterial hypotension(96) | ± (discordant)(100–106) | Antifibrotic(100) | Arterial hypotension, renal dysfunction(105,106) | No |
| Taurine | Intrahepatic and extrahepatic(107) | Antifibrotic(107) | — | Yes(108) | — | — | — |
Additional references (from #46 on) are provided as Supporting Information.
Abbreviations: AST, aspartate aminotransferase; FXR, farnesoid X receptor; MAP, mean arterial pressure; PDE5, phosphodiesterase-5; PP, portal pressure; VEGF, vascular endothelial growth factor.
The complexity of the processes involved in PH and in the development of complications of cirrhosis would favor the pursuit of multitargeted therapies, such as statins or of combination therapies, addressing different targets. An attractive possibility is cell-specific targeting of important pathways. Further research and development is still needed, but there are currently several avenues to pursue (Table 1).
Pharmacokinetic and Pharmacodynamic Evaluation in PH Clinical Trials (Phase 1 and Phase 2)
Pharmacokinetic (PK) studies are generally undertaken during phase 1b and/or phase 2a clinical trials. In addition to investigating PK in healthy volunteers, there should be close attention to understanding the compound’s PK characteristics in hepatically impaired populations. Several pathophysiological changes, which regularly occur in individuals with cirrhosis and PH, can significantly impact drug disposition in this population (Table 2).(18–23) The 2003 U.S. Food and Drug Administration (FDA) Guidance for Industry(24) recommends a formal PK study in hepatically impaired individuals if (1) hepatic metabolism and/or excretion accounts for >20% of the absorbed drug’s or its active metabolite’s disposition, or (2) the test compound is either known or suspected to have a narrow therapeutic window, or (3) there is insufficient information about the compound’s metabolism and excretion and its predicted therapeutic window. However, a formal PK study in hepatically impaired individuals may not be necessary if a compound is entirely renally excreted with no involvement of the liver, or if a drug is gaseous or volatile and its active metabolite(s) are primarily eliminated by the lungs.(24)
TABLE 2.
Mechanisms Behind Altered PKs in Patients With Cirrhosis and PH
| Mechanism | Comments | |
|---|---|---|
|
| ||
| 1 | Reduced intrinsic capacity - Decreased enzyme levels - Decreased activity |
Liver disease etiology–specific variability in different CYPs may exist—for example, alcohol and infection can affect some CYPs selectively |
| 2 | Reduction in blood flow | May affect drug disposition through a number of mechanisms |
| 3 | Shunts | Intrahepatic shunts—may account for up to 65% of hepatic blood flow Spontaneous portosystemic shunts and TIPS can significantly modify |
| 4 | Changes in protein binding | Impacts delivery of drugs to hepatocytes and can also have an impact on PD response |
| 5 | Reduced delivery of oxygen | Oxygen is important for CYP activity |
| 6 | Altered transporter expression and function | Hepatocyte uptake as well as efflux into biliary canaliculi can be affected |
| 7 | Small bowel CYP activity | TIPS significantly reduces small bowel CYP3A activity |
| 8 | Drug-drug interactions | Altered PKs of one compound may have secondary effects on the PKs of coadminstered drugs |
| 9 | Impaired renal function | PKs of renally excreted drugs may be profoundly affected in individuals with cirrhosis |
| 10 | Altered intestinal absorption | There may be diminished absorption of orally administered drugs and may also influence the PKs of drugs which are bile excreted because of impaired enterohepatic circulation |
Abbreviations: CYP, cytochrome P450; TIPS, transjugular intrahepatic portosystemic shunts.
PK studies can have a basic full study design, a reduced study design, or a population PK approach. The basic full study design involves the entire Child-Pugh spectrum (at least 6 patients in each Child-Pugh category) and controls, whereas a reduced study design allows for the study to be limited to 8 patients with Child-Pugh B cirrhosis and 8 controls matched by age, sex, and comorbidities. The findings from a reduced study design conducted on Child-Pugh B patients, though considered applicable to individuals with Child-Pugh A cirrhosis, would be generally contraindicated in Child-Pugh C patients. Although widely utilized, limiting PK studies to a reduced study design is not recommendable given that individuals with Child-Pugh A (mostly compensated) and Child B (mostly decompensated) cirrhosis may have markedly different pathophysiological abnormalities (e.g., plasma protein binding, hepatic blood flow, or intrinsic clearance). One might consider a staged approach to a basic full study design where individuals with Child-Pugh A and B are investigated initially, assuming that initial efficacy and safety studies are limited to patients with mild-to-moderate Child-Pugh cirrhosis. Conducting PK studies in individuals with severe liver dysfunction is challenging because of their tenuous health status with frequent complications and hospitalizations and multiple concomitant medications. Wherever possible, controls in these studies should not be healthy volunteers, but should be matched for age, sex, comorbidities, and concomitant medications. A single-dose PK study might be sufficient if a compound and its metabolite exhibit linear and time-independent PK at concentrations anticipated in the patients to be studied and are not expected to change in patients with cirrhosis and PH. Although their validity is not fully established in patients with cirrhosis and PH, population PK analyses should be built into phase 3 and phase 4 studies wherever possible given that such data can be quite instructive in fully understanding the PK characteristics.
Patients with cirrhosis may respond differently than the general population to medications because of accompanying pathophysiological abnormalities. These include plasma protein binding, decreased skeletal muscle mass, alterations in blood–brain barrier, reduced renal blood flow, and increased QT interval. Some examples of altered pharmacodynamics (PD) in cirrhosis include: unresponsiveness to loop diuretics in patients with refractory ascites,(25) risk of acute kidney injury from nonsteroidal anti-inflammatory drugs in patients with cirrhosis, especially ascites,(26) and more recent description of worsened insulin sensitivity from nadolol in patients with cirrhosis.(27) PD assessments should be considered on a compound-by-compound basis, especially if concentration-response relationship is unknown, or if altered hepatic function and/or PH are suspected to alter PD response. It is our understanding that the FDA is in the midst of revising its guidance for PK studies in the hepatic impaired population. It is possible that Model for End-Stage Liver Disease score (rather than Child-Pugh classes) forms the basis for future PK studies in patients with cirrhosis.
SAFETY MONITORING IN CIRRHOSIS AND PH CLINICAL TRIALS
Monitoring for hepatic and extrahepatic toxicity is very important in drug development for cirrhosis and PH. Two important considerations in monitoring for hepatotoxicity are (1) challenges in biochemical monitoring as baseline aminotransferases and bilirubin may be abnormal, and (2) because of lower functional hepatic reserve, patients may be at risk for rapid decompensation and death, even with injuries that are otherwise not as serious (e.g., cholestatic liver injury).(28) Patients with cirrhosis may be at risk for extrahepatic (off-target) adverse events (AEs) attributed to systemic pathological changes such as decreased plasma protein, altered volume of distribution, decreased skeletal muscle mass, decreased renal blood flow, and increased blood-brain permeability, and altered gut permeability. Furthermore, these patients may already be taking other medications and thus may be exposed to drug-drug interactions. Every study should have a well-thought-out plan for safety monitoring and stopping rules. and the data safety and monitoring boards should include hepatologists with familiarity in drug safety and causality adjudication.
Proof of Concept Studies (Phase 2) Trials in PH
SELECTION OF RELEVANT POPULATION
Cirrhosis is subclassified in two main clinical stages, compensated and decompensated, each of them with different prognosis and different predominant pathogenic mechanisms and therefore different therapeutic targets.(29) Patients with compensated and decompensated cirrhosis should be investigated or analyzed separately in clinical trials.
Compensated cirrhosis is the asymptomatic and the longest stage of cirrhosis. In this stage, the patient with chronic liver disease and/or chronic risk factors for liver disease (e.g., alcohol ingestion, HCV infection) has histological, imaging, or elastographic evidence of cirrhosis, but has not developed any decompensating events (ascites, variceal hemorrhage, or encephalopathy). Median survival is around 15 years.(30)
Compensated cirrhosis is subclassified by severity of PH into those with mild portal hypertension (MPH) and those with CSPH. These two stages are different not only in the degree of PH, but also regarding their risk of progression to decompensation, the amount of hepatic fibrosis, and the predominant pathogenic mechanisms. Whereas patients with MPH have a very low risk of decompensation, have less liver fibrosis (and thinner fibrous septa), and the main pathogenic mechanism is increased intrahepatic resistance, those with CSPH have a 4-times higher risk of decompensation than patients with MPH,(1) have more liver fibrous tissue,(31) and, in addition to intrahepatic resistance, a main contributor to PH is increased splanchnic blood flow.(4) Because NSBBs act by decreasing flow, the PP response to NSBBs is much lower in patients with MPH compared to those with CSPH.(4)
Treatment of underlying disease, if possible, is the most effective and appropriate treatment at the compensated stage. In patients with MPH, it is conceivable that this treatment (or antifibrotic treatment, when available) could lead to regression to a noncirrhotic stage or at least to halting the progression to CSPH. However, although treatment of the primary liver disease may slow progression to decompensation in patients with CSPH, the risk is not eliminated and a substantial number of patients remain with CSPH and at risk for decompensation. It is in these patients and in those for whom specific etiologic therapies are unavailable where measures to reduce PP will be more important.
Decompensated cirrhosis is the symptomatic and shorter stage of cirrhosis. This stage is characterized by the presence of clinical complications, specifically ascites, variceal hemorrhage, encephalopathy, and jaundice (although the latter is very rare as a first decompensating event).(29) Median survival is 1.5 years.(30) Type and number of decompensating events is of prognostic importance as is the presence of further complications of decompensating events such as hepatorenal syndrome (“further” decompensation”). Decompensated patients have, by definition, CSPH. Although worsening vasodilatation and inflammation contribute to outcomes in these patients, PH still plays a major role as outcomes improve with reduction in PP.(8,32)
MEASUREMENT OF PP AND DEFINITION OF MILD VERSUS CLINICALLY SIGNIFICANT PORTAL HYPERTENSION
The hepatic venous pressure gradient (HVPG) is the main method used to measure PP. It consists of the percutaneous catheterization of a hepatic vein (mostly by the transjugular route) with measurements of the wedged hepatic vein pressure (WHVP) and the free hepatic vein pressure (FHVP).(33) The WHVP is obtained by either advancing the catheter until it is wedged in the most distal portion of the hepatic vein or, preferably, by inflating a balloon at the end of a catheter in a more proximal portion of the vein. It is a measure of hepatic sinusoidal pressure and correlates closely with PP in viral, alcoholic, and nonalcoholic steatohepatitis cirrhosis where there is sinusoidal and/or postsinusoidal fibrosis. The FHVP is a measure of systemic pressure and is used as an internal zero to “correct” the WHVP. The HVPG is therefore obtained by subtracting the FHVP from the WHVP. Normal HVPG is 3–5 mm Hg. An HVPG >5 mm Hg indicates the presence of PH, and an HVPG ≥10 mm Hg indicates the presence of CSPH.
Although all patients with cirrhosis should theoretically have an abnormal portal pressure (i.e., HVPG >5 mm Hg), the HVPG is normal in ~10% of patients with histological/clinical cirrhosis. This could be attributed to sampling error in the biopsy, a predominantly presinusoidal fibrotic component, or to an inadequate occlusion of the hepatic vein attributed to the presence of intrahepatic shunts.
Subclassification of patients into MPH versus CSPH is based on the HVPG. MPH is defined as an HVPG >5 mm Hg, but less than 10 mm Hg. CSPH is defined as an HVPG ≥10 mm Hg. Because patients with varices and/or decompensation have an HVPG of at least 10–12 mm Hg, patients with varices (or collaterals on imaging) and/or clinical decompensation by definition have CSPH.(12)
Noninvasive approaches to assess the presence of CSPH include measurements of liver stiffness with or without additional parameters such as platelet count and/or spleen size.(34) The presence of varices that have not bled or a history of variceal hemorrhage can be used to select candidates for studies of patients with CSPH. However, standard of care would require that some of these patients (those with moderate/large varices) receive therapies to prevent variceal hemorrhage (either NSBB and/or ligation) that could confound the interpretation of results. If included in a single study, patients with compensated and decompensated cirrhosis would require separate analysis.
DEFINITION OF HVPG RESPONSE
Drugs identified in preclinical studies with a PP-reducing effect (candidate drugs) should be tested on their ability to reduce HVPG in patients with CSPH. Unfortunately, until now, there is not a single noninvasive test (e.g., Doppler, liver stiffness) whose changes correlate with changes in HVPG and that would be acceptable alternatives to paired HVPG measurements.
The magnitude of change in the HVPG required to predict that a drug has potential clinical utility in patients with CSPH is still controversial. In patients with a history of variceal hemorrhage, a decrease in HVPG to <12 mm Hg or a decrease >20% from baseline is associated with a significant reduction in the risk of recurrent hemorrhage, ascites, encephalopathy, and death.(8) Reductions in HVPG >10% from baseline have been associated with a reduction in variceal development,(35) first variceal hemorrhage, and death.(9)
It is important to note that these threshold HVPG reductions apply to NSBB-based therapies, which act by decreasing portal blood inflow and may not apply to drugs that decrease intrahepatic resistance, which, though decreasing PP only modestly may have a mild effect in preventing bleeding, but by improving liver perfusion may have a more important effect on survival.(16,17)
Importantly, using the most common cutoffs (reduction of HVPG to <12 mm Hg or >20% from baseline) to classify patients in a dichotomous fashion into HVPG “responders” and “nonresponders” may underestimate the value of a candidate drug. Although responders have in fact been shown to have better clinical outcomes than nonresponders, the definition lacks sensitivity given that many patients not meeting these criteria may still have a good clinical response to therapy.(36) It would be unwise to rule out a potential therapeutic benefit for a drug that achieves a consistent decrease in PP, even if this decrease in PP does not reach the threshold used to define a good response.
On the other hand, preliminary analyses of a dynamic HVPG model obtained from patients with compensated cirrhosis without varices included in the timolol study demonstrates that, at any time during follow-up, a 1 mm Hg increase (or decrease) in HVPG is associated with a 1.19-times higher (or lower) risk of decompensation/death (95% confidence interval, 1.11–1.27).(37) This model provides much more granularity to changes in HVPG and, if confirmed, a POC study could be powered on a predefined specific reduction in HVPG associated with a certain percentage reduction in decompensation in the patient population to be analyzed. Furthermore, if the POC study includes clinical outcomes, results could inform a potential effect size for a phase 3 trial.
COMPARATOR TO BE USED
In POC studies that investigate the effect of a candidate drug on HVPG, the use of a placebo of the candidate drug(s) is mandatory.
If the investigational drug has a different mechanism of action from the standard of care (NSBBs in patients with medium/large varices), patients on these drugs should be stratified separately to investigate the presence of a synergistic or additive effect of the candidate drug.
READOUTS OTHER THAN HVPG TO BE INVESTIGATED IN POC STUDIES
Candidate drugs with a purported intrahepatic mechanism of action (i.e., reduction of intrahepatic resistance) may have a more important effect by improving liver perfusion while demonstrating an only modest effect on PP (Table 3). Therefore, analysis of these drugs should include quantitative tests of liver perfusion, such as the indocyanine green (ICG) clearance test, or breath tests that analyze clearance of ingested or injected markers.
TABLE 3.
Readouts (Positive or Negative) to Be Evaluated in POC Studies of Candidate Drugs for PH
| Positive Readouts | Negative Readouts |
|---|---|
|
| |
| • Decrease in HVPG | • Decrease in mean arterial pressure/renal perfusion |
| • Improvement in liver perfusion/liver synthetic function (breath tests, ICG, HepQuant) | • Decrease in liver perfusion/liver synthetic function (breath tests, ICG) |
| • Tendency for improvement in clinical outcomes (including mortality) | • Evidence of hepatotoxicity |
| • Results support a mechanism of action. | • Drug interactions |
| • Definition of determinants of response | • Other adverse effects (low tolerance) |
| • Improvement in patient-reported outcomes | • Applicability |
Other readouts that should be prospectively collected are the development of clinical outcomes during the course of the study (clinically overt decompensating events) as well as potentially negative readouts, such as reduction in mean arterial pressure (MAP) or cardiac output or development of renal dysfunction, and other AEs.
In addition, analyses that would support the purported mechanism of action of the candidate drug would be desirable given that a correlation between the hemodynamic effect and mechanistic markers would support advancing to a phase 3 trial. These analyses may disclose useful biomarkers of the response to this specific drug.
As in any clinical trial, evidence of hepatotoxicity or other AEs should be collected in the knowledge that the study population (compensated cirrhosis) may have low tolerance to even mild AEs.
OTHER CONSIDERATIONS
Duration of therapy: Depends on the proposed mechanism of action of the candidate drug. If it has direct vasoactive action, the HVPG response could be assessed acutely after a short period as has been demonstrated after the intravenous administration of propranolol.(9) However, it would be necessary to continue the drug and have a follow-up HVPG after longer administration to demonstrate that the effect is maintained and is not offset by compensatory mechanisms (as occurs with long-acting nitrates). Drugs targeting the structural component of the increased hepatic resistance (e.g., antifibrotic drugs) may take much longer (e.g., 12 months) to demonstrate HVPG reductions.
Confounders, such as concomitant treatment of underlying cause of liver disease (e.g., weight loss, antiviral therapy), comorbidities (e.g., alcohol use, cardiovascular disease), or treatment of comorbidities that may affect HVPG (e.g., statins, vasoactive medications), should be considered.
In summary, POC studies in PH should include patients with cirrhosis and CSPH, with the main readout being a decrease in HVPG. Other readouts (positive and negative) and findings should inform the decision to move on to phase 3 trials.
Phase 3 Trials in PH
Most clinical trials in PH have been directed toward treating or preventing variceal hemorrhage. However, varices and variceal hemorrhage are not isolated events, and they must be considered in the context of the presence (or absence) of other decompensating events.(2,12) Therefore, the aim of clinical trials in PH should be to prevent not only variceal hemorrhage, but also any form of decompensation. Unfortunately, a paucity of data currently exist in this area, and RCTs are needed. Trial design in patients with decompensated cirrhosis has been extensively discussed elsewhere,(2) and we therefore focus the discussion on patients with compensated cirrhosis.
PRIMARY ENDPOINT
In agreement with the consensus achieved at the 2015 Baveno workshop on PH,(2) the development of a decompensating event (ascites, variceal hemorrhage, and/or encephalopathy) should be the main primary endpoint in phase 3 trials. Such a composite endpoint provides certain advantages. Most important, the combination of clinical variables provides the most clinically relevant measure of clinical well-being. Additionally, in RCTs, a composite endpoint will enhance the ability to detect differences in outcomes. It should be noted that ascites is the most common first decompensating event (followed by variceal hemorrhage and encephalopathy(38)). An objective and standardized definition of all these events is needed to adequately assess the main endpoint. Ascites, or free fluid in the peritoneal cavity, should be documented by imaging techniques and confirmed by diagnostic paracentesis (which is otherwise indicated in patients first presenting with ascites), given that clinical signs of ascites are not accurate.(39) Variceal hemorrhage should follow the definitions of the Baveno VI consensus conference.(2) “Covert” hepatic encephalopathy (HE) is difficult to detect outside of specialized dedicated units, and reproducibility of this diagnosis is low.(40) Therefore, encephalopathy should be “overt” (at a minimum confusion and asterixis, or West Haven Grade ≥2) and documented by a physician to be considered an endpoint. Many patients with compensated cirrhosis are taking sedatives, anxiolytics, or antidepressant drugs that may interfere with the recognition of mild encephalopathy.
DEVELOPMENT OF VARICES AS A PART OF THE MAIN PRIMARY ENDPOINT
Although the development of varices without decompensation is a sign of disease progression, this clinical feature should not be included as part of the primary endpoint. Even though the 2-year risk of decompensation in patients with and without varices is different (~15% vs. ~8%, respectively),(1,30,38,41) there are several operational reasons for not considering varices as part of the primary endpoint. First, the trial would be limited to patients without varices, which have a much lower risk of decompensation than patients with varices. Second, the interobserver agreement for small varices is low.(42) Third, if development of varices is a component of a composite primary endpoint, development of varices would lead to the discontinuation of the study drug. This could result in missed opportunities when evaluating the efficacy of drugs that might prevent decompensation, but not the development of varices. In fact, in the study comparing tImolol or placebo for the prevention of varices,(35) the development of varices led to drug discontinuation, precluding assessment of a potential effect on the development of ascites or other decompensating events. Very recent data, suggest that NSBBs might have a strong effect preventing ascites in patients with cirrhosis and CSPH.(43)
The development of varices should be assessed as a secondary endpoint, but should not be part of the primary endpoint and should not lead to study drug discontinuation. This secondary endpoint could be analyzed as an exploratory ordinal outcome (considering death as the worst potential outcome, followed by decompensation and varices; Fig. 2). Patients with varices fulfilling current recommendations for treatment either at baseline or during the trial should receive standard therapy to prevent first variceal hemorrhage, which, in the context of a phase 3 drug-therapy–based trial, should preferably be endoscopic therapy. Ideally, this should be standardized across centres.
FIG. 2.
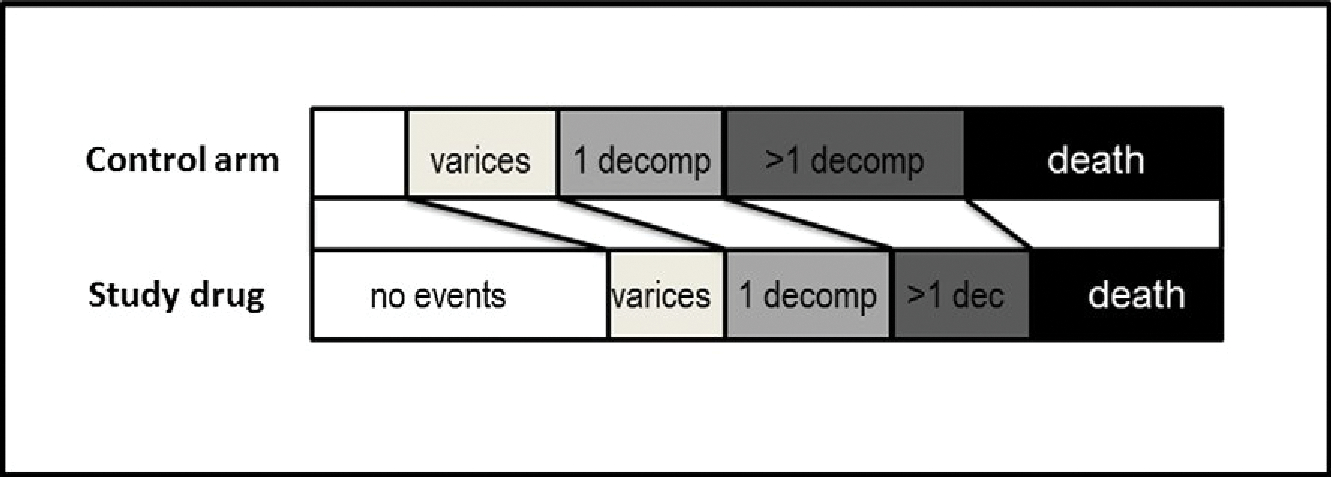
Example of a potential exploratory ordinal outcome including the development of varices. The development of varices would be hierarchically the less-severe endpoint, followed by a first decompensation with only one event, and a first decompensation with more than one simultaneous decompensating event (i.e., variceal hemorrhage plus ascites or plus encephalopathy), which is associated with a much higher risk of death than a single decompensating event.(41) Death (without decompensation) would be the most severe outcome. The analysis would test whether the study drug induces a significant “category shift” as compared to the control arm.
USE OF A SURROGATE ENDPOINT
Using an outcome surrogate is attractive for studies where a low event rate is expected within the study time frame. A surrogate would not only reduce sample size and study duration, but could also be considered appealing because it would be a substitute for a more-severe clinical outcome. Because patients with compensated cirrhosis have an overall low rate of decompensation,(30,38) it has been suggested that changes in HVPG could be used as a surrogate endpoint to support the approval of new treatments for PH (within the FDA accelerated approval framework(44)).
Although, as mentioned previously, demonstrating a decrease in HVPG is the main goal of phase II trials, several issues would need to be resolved before a drug could be approved based on HVPG response. First, the magnitude of HVPG response considered clinically relevant remains to be determined. Second, most of the data demonstrating an association between an HVPG decrease and a reduction in clinical outcomes were obtained in studies using NSBBs,(8) drugs that decrease PP by decreasing portal blood inflow, or in studies using treatments for etiology, such as alcohol abstinence and antivirals.(6,7,45) New drugs with different mechanisms of action (i.e., acting by decreasing hepatic resistance) would certainly decrease HVPG, but the association between the change in HVPG and clinical benefit might be different and require validation. This validation could be achieved by incorporating HVPG measurements in phase 3 trials (of drugs other than NSBBs) with clinical endpoints. Thus, if the beneficial effect of a reduction in HVPG was to be validated in a phase 3 trial (i.e., correlated with an improvement in clinical outcome), this would strongly favor the approval of new therapies for PH purely based on their ability to reduce PP, as is the case for therapies for arterial hypertension.
FEASIBILITY OF RCTs WITH CLINICAL ENDPOINTS IN PATIENTS WITH COMPENSATED CIRRHOSIS: THE RELEVANCE OF PATIENT SELECTION
The target population to evaluate drugs that would prevent decompensation is that of patients with cirrhosis and CSPH. However, among these patients, the risk of decompensation varies widely. Indeed, data from the cohort included in the timolol trial showed that for every mm Hg increase in baseline HVPG, there was a 13% increase in the risk of decompensation (Fig. 3).(1) This indicates that setting the threshold HVPG for inclusion in the study at a level above that defining CSPH (10 mm Hg), for example, at 14 mm Hg, would result in a higher rate of events in a shorter time period and a lower required sample size. Selecting patients based on HVPG would be feasible, as shown by recent studies,(43) but may prove difficult in large phase 3 trials. Examples of potential sample sizes according to the minimum expected effect size, length of follow-up, and predicted rates of decompensation in the control group are provided in Table 4.
FIG. 3.

Nomogram showing the 1-, 2-, and 3-year decompensation risk according to baseline HVPG (constructed with data published in Ripoll et al.(1); details are provided in Supporting Information File S1). Note the very different incidence of decompensation over time with increasing baseline HVPG values.
TABLE 4.
Examples of Approximate Sample Sizes That Would Be Needed in Trials for the Prevention of Decompensation, According to the Minimum Effect Size (Which Determines the Number of Events Needed), and the Rate of Decompensation and Length of Follow-up (Which Determines the Total Sample Size Needed to Achieve That Number of Events)
| Minimum Effect Size (HR) | No. of Events Needed | Median Follow-up (Years) | Rate Decompensation per Year in the Control Group | Total Sample Size |
|---|---|---|---|---|
|
| ||||
| 0.7 | 247 | 1 | 15% | ~1,500 |
| 0.7 | 247 | 2 | 15% | 1,000 |
| 0.7 | 247 | 2 | 20% | ~700 |
| 0.6 | 120 | 1 | 15% | ~1,000 |
| 0.6 | 120 | 2 | 15% | ~500 |
| 0.6 | 120 | 2 | 20% | ~380 |
Calculations were performed as proposed by Schoenfeld (Biometrics 1983;39:499–503).
Abbreviation: HR, hazard ratio.
There are several noninvasive tools that could substitute for HVPG in its prediction of clinical outcomes in patients with compensated cirrhosis, including laboratory values and imaging-based techniques such as transient elastography. Unfortunately, these have not yet been well validated and will likely need to be disease specific given that the association between these noninvasive tests and the risk of decompensation might be quantitatively different among patients with viral and alcoholic liver disease and NAFLD.
Supplementary Material
Abbreviations:
- AASLD
American Association for the Study of Liver Diseases
- AEs
adverse events
- CSPH
clinically significant portal hypertension
- FDA
U.S. Food and Drug Administration
- FHVP
free hepatic vein pressure
- HSCs
hepatic stellate cells
- HVPG
hepatic venous pressure gradient
- ICG
indocyanine green
- MAP
mean arterial pressure
- MPH
mild portal hypertension
- NAFLD
nonalcoholic fatty liver disease
- NO
nitric oxide
- NSBBs
nonselective beta blockers
- PD
pharmacodynamics
- PH
portal hypertension
- PK
pharmacokinetic
- POC
proof-of-concept
- PP
portal pressure
- RCT
randomized controlled trial; SIG, Special Interest Group
- WHVP
wedged hepatic vein pressure
Footnotes
Potential conflict of interest: Dr. Gonzalez-Abvraldes consults for Theravance and Pfizer. Dr. Rockey receives grants from Cumberland Pharm, Ironwood Pharm, Gilead, Galectin, and Conatus. Dr. Bosch consults for, advises for, and received grants from Conatus. He owns stocks in, consults for, advises for, receives grants for, and has property rights with Exalenz. He consults for and received grants from Gilead.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.30314/suppinfo.
REFERENCES
Author names in bold designate shared co-first authorship.
- 1).Ripoll C, Groszmann R, Garcia-Tsao G, Grace N, Burroughs A, Planas R, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology 2007;133:481–488. [DOI] [PubMed] [Google Scholar]
- 2).de Franchis R, Baveno VI Faculty. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol 2015;63:743–752. [DOI] [PubMed] [Google Scholar]
- 3).Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J Hepatol 2015;62(1 Suppl):S121–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Villanueva C, Albillos A, Genesca J, Abraldes JG, Calleja JL, Aracil C, et al. Development of hyperdynamic circulation and response to beta-blockers in compensated cirrhosis with portal hypertension. Hepatology 2016;63:197–206. [DOI] [PubMed] [Google Scholar]
- 5).Mandorfer M, Kozbial K, Schwabl P, Freissmuth C, Schwarzer R, Stern R, et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J Hepatol 2016;65:692–699. [DOI] [PubMed] [Google Scholar]
- 6).Lens S, Alvarado-Tapias E, Mariño Z, Londoño MC, LLop E, Martinez J, et al. Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus-associated cirrhosis. Gastroenterology 2017;153:1273–1283.e1. [DOI] [PubMed] [Google Scholar]
- 7).Lens S, Rincón D, García-Retortillo M, Albillos A, Calleja JL, Bañares R, et al. Association between severe portal hypertension and risk of liver decompensation in patients with hepatitis C, regardless of response to antiviral therapy. Clin Gastroenterol Hepatol 2015;13:1846–1853.e1. [DOI] [PubMed] [Google Scholar]
- 8).D‘Amico G, Garcia-Pagan JC, Luca A, Bosch J. HVPG reduction and prevention of variceal bleeding in cirrhosis. A systematic review. Gastroenterology 2006;131:1611–1624. [DOI] [PubMed] [Google Scholar]
- 9).Villanueva C, Aracil C, Colomo A, Hernandez-Gea V, Lopez-Balaguer JM, Alvarez-Urturi C, et al. Acute hemodynamic response to beta-blockers and prediction of long-term outcome in primary prophylaxis of variceal bleeding. Gastroenterology 2009;137:119–128. [DOI] [PubMed] [Google Scholar]
- 10).Hennenberg M, Trebicka J, Sauerbruch T, Heller J. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut 2008;57:1300–1314. [DOI] [PubMed] [Google Scholar]
- 11).Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol 2009;50:604–620. [DOI] [PubMed] [Google Scholar]
- 12).Garcia-Tsao G, Abraldes J, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis and management—2016 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2017;65:310–335. [DOI] [PubMed] [Google Scholar]
- 13).Turco L, Garcia-Tsao G, Magnani I, Bianchini M, Costetti M, Caporali C, et al. Cardiopulmonary hemodynamics and C-reactive protein as prognostic indicators in compensated and decompensated cirrhosis. J Hepatol 2018;68:949–958. [DOI] [PubMed] [Google Scholar]
- 14).Wong F. Recent advances in our understanding of hepatorenal syndrome. Nat Rev Gastroenterol Hepatol 2012;9:382–391. [DOI] [PubMed] [Google Scholar]
- 15).Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernandez M, Garca-Pagan JC, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 2004;126:749–755. [DOI] [PubMed] [Google Scholar]
- 16).Abraldes JG, Albillos A, Banares R, Turnes J, Gonzalez R, Garcia-Pagan JC, Bosch J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651–1658. [DOI] [PubMed] [Google Scholar]
- 17).Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez-Guerra M, Genesca J, et al. Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology 2016;150:1160–1170. [DOI] [PubMed] [Google Scholar]
- 18).Li R, Barton HA, Maurer TS. A mechanistic pharmacokinetic model for liver transporter substrates under liver cirrhosis conditions. CPT Pharmacometrics Syst Pharmacol 2015;4:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Liu H, Sahi J. Role of hepatic drug transporters in drug development. J Clin Pharmacol 2016;56(Suppl 7):S11–S22. [DOI] [PubMed] [Google Scholar]
- 20).Le Couteur DG, Fraser R, Hilmer S, Rivory LP, McLean AJ. The hepatic sinusoid in aging and cirrhosis: effects on hepatic substrate disposition and drug clearance. Clin Pharmacokinet 2005;44:187–200. [DOI] [PubMed] [Google Scholar]
- 21).Chalasani N, Gorski JC, Patel NH, Hall SD, Galinsky RE. Hepatic and intestinal cytochrome P450 3A activity in cirrhosis: effects of transjugular intrahepatic portosystemic shunts. Hepatology 2001;34:1103–1108. [DOI] [PubMed] [Google Scholar]
- 22).Thakkar N, Slizgi JR, Brouwer KL. Effect of liver disease on hepatic transporter expression and function. J Pharm Sci 2017;106:2282–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Edginton AN, Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet 2008;47:743–752. [DOI] [PubMed] [Google Scholar]
- 24).Rodriguez S, Raurell I, Torres-Arauz M, Garcia-Lezana T, Genesca J, Martell M. A nitric oxide-donating statin decreases portal pressure with a better toxicity profile than conventional statins in cirrhotic rats. Sci Rep 2017;7:40461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Gentilini P, La Villa G, Marra F, Carloni V, Melani L, Foschi M, et al. Pharmacokinetics and pharmacodynamics of torasemide and furosemide in patients with diuretic resistant ascites. J Hepatol 1996;25:481–490. [DOI] [PubMed] [Google Scholar]
- 26).Boyer TD, Zia P, Reynolds TB. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology 1979;77:215–222. [PubMed] [Google Scholar]
- 27).Lee WG, Murphy R, McCall JL, Gane EJ, Soop M, Tura A, Plank LD. Nadolol reduces insulin sensitivity in liver cirrhosis: a randomized double-blind crossover trial. Diabetes Metab Res Rev 2017;33. 10.1002/dmrr.2859. Epub 2016 Nov 10. [DOI] [PubMed] [Google Scholar]
- 28).Chalasani N, Regev A. Drug-induced liver injury in patients with preexisting chronic liver disease in drug development: how to identify and manage? Gastroenterology 2016;151:1046–1051. [DOI] [PubMed] [Google Scholar]
- 29).D‘Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis. A systematic review of 118 studies. J Hepatol 2006;44:217–231. [DOI] [PubMed] [Google Scholar]
- 30).D‘Amico G, Morabito A, D‘Amico M, Pasta L, Malizia G, Rebora P, Valsecchi MG. Clinical states of cirrhosis and competing risks. J Hepatol 2018;68:563–576. [DOI] [PubMed] [Google Scholar]
- 31).Nagula S, Jain D, Groszmann RJ, Garcia-Tsao G. Histological-hemodynamic correlation in cirrhosis-a histological classification of the severity of cirrhosis. J Hepatol 2006;44:111–117. [DOI] [PubMed] [Google Scholar]
- 32).Abraldes JG, Tarantino I, Turnes J, Garcia-Pagan JC, Rodes J, Bosch J. Hemodynamic response to pharmacological treatment of portal hypertension and long-term prognosis of cirrhosis. Hepatology 2003;37:902–908. [DOI] [PubMed] [Google Scholar]
- 33).Groszmann RJ, Wongcharatrawee S. The hepatic venous pressure gradient: Anything worth doing should be done right. Hepatology 2004;39:280–283. [DOI] [PubMed] [Google Scholar]
- 34).Abraldes JG, Bureau C, Stefanescu H, Augustin S, Ney M, Blasco H, et al. Anticipate Investigators. Noninvasive tools and risk of clinically significant portal hypertension and varices in compensated cirrhosis: the “Anticipate” study. Hepatology 2016;64:2173–2184. [DOI] [PubMed] [Google Scholar]
- 35).Groszmann RJ, Garcia-Tsao G, Bosch J, Grace ND, Burroughs AK, Planas R, et al. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med 2005;353:2254–2261. [DOI] [PubMed] [Google Scholar]
- 36).Thalheimer U, Bosch J, Burroughs AK. How to prevent varices from bleeding: shades of grey—the case for nonselective beta blockers. Gastroenterology 2007;133:2029–2036. [DOI] [PubMed] [Google Scholar]
- 37).Abraldes JG, Garcia-Tsao G, Ripoll C, Patch D, Grace N, Bosch J, Groszmann RJ. Dynamic prediction of the risk of decompensation/death in patients with compensated cirrhosis based on serial hepatic venous pressure gradient (HVPG) measurements. Hepatology 2018;68(Suppl 1):Abstract (in press). [Google Scholar]
- 38).D’Amico G, Morabito A, D’Amico M, Pasta L, Malizia G, Rebora P, Valsecchi MG. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol Int 2018;12:34–43. [DOI] [PubMed] [Google Scholar]
- 39).Zipprich A, Garcia-Tsao G, Rogowski S, Fleig WE, Seufferlein T, Dollinger MM. Prognostic indicators of survival in patients with compensated and decompensated cirrhosis. Liver Int 2012;32:1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Reuter B, Walter K, Bissonnette J, Leise MD, Lai J, Tandon P, et al. Assessment of the spectrum of hepatic encephalopathy: a multicenter study. Liver Transpl 2018;24:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).D‘Amico G, Pasta L, Morabito A, D‘Amico M, Caltagirone M, Malizia G, et al. Competing risks and prognostic stages of cirrhosis: a 25-year inception cohort study of 494 patients. Aliment Pharmacol Ther 2014;39:1180–1193. [DOI] [PubMed] [Google Scholar]
- 42).Bendtsen F, Skovgaard LT, Sorensen TI, Matzen P. Agreement among multiple observers on endoscopic diagnosis of esophageal varices before bleeding. Hepatology 1990;11:341–347. [DOI] [PubMed] [Google Scholar]
- 43).Villanueva C, Albillos A, Genesca J, Garcia-Pagan JC, Calleja JL, Aracil C, et al. Beta-blockers to prevent the decompensation of cirrhosis in patients with clinically significant portal hypertension: a randomized, double-blind, placebo-controlled, multi-centre trial. Lancet 2018. In press. [DOI] [PubMed] [Google Scholar]
- 44).Sanyal AJ, Friedman SL, McCullough AJ. Dimick-Santos L; American Association for the Study of Liver Diseases, United States Food and Drug Administration. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration Joint Workshop. Hepatology 2015;61:1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Vorobioff J, Groszmann RJ, Picabea E, Gamen M, Villavicencio R, Bordato J, et al. Prognostic value of hepatic venous pressure gradient measurements in alcoholic cirrhosis: a 10-year prospective study. Gastroenterology 1996;111:701–709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.