

Abstract
Background:
Clear cell renal cell carcinoma (ccRCC) tumors have low frequencies of genetic alterations compared with other malignancies, but very high levels of immune cell infiltration and favorable response rates to immunotherapy. Currently, the interplay between specific ccRCC somatic mutations and immune infiltration pattern is unclear.
Objective:
To analyze the associations between common ccRCC somatic mutations and immune cell infiltration patterns within the tumor immune microenvironment (TIME).
Design, setting, and participants:
The study included tumor samples (24 primary and 24 metastatic) from 48 patients with stage IV ccRCC. Targeted sequencing was performed for well-characterized recurrent somatic mutations in ccRCC, with the analysis focusing on the six most common ones: VHL, BAP1, PBRM1, SETD2, TP53, and KDM5C. For each sample, multiplex immunofluorescence (IF) was performed in lymphoid and myeloid panels, for seven regions of interest in three zones (tumor core, stroma, and tumor-stroma interface). IF-derived cellular densities were compared across patients, stratified by their somatic mutation status, using a linear mixed-model analysis. External validation was pursued using RNA-seq enrichment scoring from three large external data sources.
Results and limitations:
Tumors with SETD2 mutations demonstrated significantly decreased levels of FOXP3+ T cells in the tumor core, stroma, and tumor-stroma interface. PBRM1 mutations were associated with decreased FOXP3+ T cells in the tumor core. Primary KDM5C mutations were associated with significantly increased CD206+ macrophage tumor infiltration in the tumor core. A computational method estimating immune cell types in the TIME using bulk RNA-seq data, xCell scoring, failed to validate associations from the IF analysis in large external data sets. A major limitation of the study is the relatively small patient population studied.
Conclusions:
This study provides evidence that common somatic mutations in ccRCC, such as SETD2, PBRM1, and KDM5C, are associated with distinct immune infiltration patterns within the TIME.
Patient summary:
In this study, we analyzed tumor samples from patients with metastatic kidney cancer to determine whether common genetic mutations that arise from the cancer cells are associated with the density of immune cells found within those tumors. We found several distinct immune cell patterns that were associated with specific genetic mutations. These findings provide insight into the interaction between cancer genetics and the immune system in kidney cancer.
Keywords: Immune cell markers, Somatic mutations, Metastatic clear cell renal cell, carcinoma, Tumor immune, microenvironment, Tumor-associated macrophages
1. Introduction
Immune checkpoint inhibitors (ICIs), such as nivolumab, ipilimumab, avelumab, and pembrolizumab, have been shown to extend survival for a subset of patients with metastatic clear cell renal cell carcinoma (ccRCC) [1–3]. Across tumor types, response to ICIs correlates with higher frequencies of somatic mutations that are believed to give rise to tumor-specific neoantigens, stimulating a robust antitumor immune response [4]. In contrast, analysis of ccRCC tumors has demonstrated a relatively low frequency of somatic mutations, but high levels of immune cell infiltrate, suggesting that mutational burden is not solely responsible for inducing immune infiltration in ccRCC [5–7].
Inactivation of the VHL tumor suppressor gene was the first recognized genetic alteration in ccRCC and is found in the majority (57–80%) of ccRCC tumors [8,9]. Largely through efforts led by The Cancer Genome Atlas (TCGA) Research Network, additional common genetic alterations have been identified in kidney cancer, including PBRM1, BAP1, SETD2, TP53, and KDM5C mutations [10–12]. These somatic mutations have been investigated, with variable success, as genomic biomarkers for the prognostication of cancer-specific survival and response to targeted therapies (TTs) and ICI monotherapies [5,13,14].
As the management of metastatic ccRCC shifts toward ICI-based treatment strategies, investigations have begun to focus on better understanding immune cell interactions within the tumor immune microenvironment (TIME) [15]. Identification of associations between somatic mutations and immune infiltration patterns within the time TIME could provide clues about the underlying biological mechanisms that drive susceptibility and resistance to ICIs and systemic TT.
Our primary objective was to determine the associations between well-characterized somatic mutations and immune cell infiltration patterns within the TIME of patients with metastatic ccRCC.
2. Patients and methods
A full detailed description of the materials and methods can be found in the Supplementary material accompanying this manuscript. The following is an abbreviated description.
From the years 2000 to 2018, 48 tumor samples (24 primary and 24 metastatic samples) were obtained from 33 individual patients with stage IV ccRCC, by American Joint Committee on Cancer staging criteria, through protocols approved by the institutional review board (H. Lee Moffitt Cancer Center and Research Institute’s Total Cancer Care protocol [MCC# 14690]; Advarra IRB Pro00014441). Hereafter, this cohort will be referred to as the immunofluorescence (IF) cohort. Written informed consent was obtained from all tissue donors. Patients were included in this study if they developed stage IV ccRCC at any point during their disease course, provided written consent to the molecular characterization of their tissue, did not receive any systemic therapy prior to tissue collection, and received immunotherapy (IT; interleukin-2, nivolumab, pembrolizumab, or ipilimumab) after tissue collection.
Prior to multiplex IF, an experienced genitourinary pathologist (J.D.) reviewed each slide obtained from formalin-fixed paraffin-embedded tissue samples and annotated seven separate regions of interest (ROIs) from three predetermined tumor zones: two ROIs from the tumor core, three from the tumor-stroma interface, and two from the stroma. Tissue slides were sequentially stained in two panels using antibodies against CD3, CD8, CD68, CD163, CD206, forkhead box P3 (FOXP3), T-box transcription factor TBX21 (T-Bet; a T-box protein expressed in T-cells), and programmed death-ligand 1 (PD-L1). These markers were selected for their previously demonstrated frequency in ccRCC [16]. Digital image analysis of the IF images generated total cell count, positive cell counts, cell density (cells/mm2), fluorescence intensity, and percent of cells that were positive for each marker.
These same tissue blocks were used to extract tumor DNA for targeted sequencing, which determined the presence of specific somatic mutations in ccRCC tumors using the 275-gene Qiagen QIAseq Comprehensive Cancer Panel (Qiagen, Germantown, MD, USA) along with a booster panel enabling the interrogation of the TERT promoter region. Data analysis, including alignment and variant calling, was performed using the QiaSeq data analysis pipeline.
A linear mixed-model analysis was performed to test associations between somatic mutation status and immune cell density for each marker, stratified by zone (tumor core, stroma, and tumor-stroma interface). Multivariable Cox proportional hazard regression, adjusted for age, gender, and International Metastatic RCC Database Consortium (IMDC) risk score, was used to analyze the association of cell density with survival outcomes. The Kaplan-Meier method was used to estimate survival distribution and median overall survival (OS), by mutation status. For patients in the IF cohort, OS (days from diagnosis to death or censoring), OS since IT (days from first IT treatment to death or censoring), and OS since TT (days from first TT treatment to death or censoring) were determined.
Specimens of ccRCC and their associated RNA sequencing data were identified in three external data sources: TCGA, Clinical Proteomic Tumor Analysis Consortium (CPTAC), and the Moffitt Cancer Center Total Cancer Care (TCC) data set. The TCC patients are independent of the IF cohort. For each specimen in all three external cohorts, RNA sequencing data were analyzed using the xCell bioinformatic pipeline, generating an adjusted enrichment score for 64 cell types within the specimen, which is referred to as the xCell score [17]. The xCell scores were generated for each ccRCC tumor specimen and stratified by somatic mutation status. These scores were also generated for a subset of patients in the IF cohort (n = 12) who had tumor samples that had previously undergone bulk RNA sequencing.
The xCell scores from the TCC, CPTAC, and TCGA data sets were compared between mutant and wild-type groups for each somatic mutation using the Mann-Whitney test. Patients were then identified from the IF cohort whose tumor samples underwent RNA sequencing previously (n = 12). Correlations were identified between xCell enrichment scores and IF immune infiltrate patterns among these 12 patients using Spearman rank correlation.
Statistical significance for all analyses was defined as a two-tailed p < 0.05. Statistical analyses were performed using R program version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria).
3. Results
3.1. Patient and tumor characteristics (IF cohort)
Forty-eight tumor specimens obtained from ccRCC patients who presented with or developed metastatic disease were analyzed (24 primary and 24 metastatic tumors; Table 1). The most frequent somatic mutations were VHL (79.1%), BAP1 (41.7%), SETD2 (41.7%), PBRM1 (35.4%), TP53 (18.8%), and KDM5C (16.7%; Fig. 1). The distribution of somatic mutations in our cohort is similar to previously reported metastatic kidney cancer sequencing analyses [18]. When stratified by primary versus metastatic sample source, the mutation distributions demonstrated higher proportions of SETD2 mutations in metastatic samples than in primaries (54% vs 29%) and higher proportions of PBRM1 mutations in primaries than in metastatic samples (46% vs 25%).
Table 1 –
Baseline patient and specimen demographics
| Variable | N = 33 Patients a |
|---|---|
|
| |
| Median age at diagnosis (yr) | 55 (52–61) |
| Median follow-up after diagnosis (mo) | 55 (30–97) |
| Median maximal tumor dimension (cm) | 10 (7.5–11.5) |
| Gender | |
| Male | 19 (58%) |
| Female | 14 (42%) |
| Race | |
| White | 30 (91%) |
| Asian | 1 (3%) |
| Black | 0 (0%) |
| Other | 2 (6%) |
| ISUP grade of primary | |
| 2 | 3 (9%) |
| 3 | 12 (36%) |
| 4 | 18 (55%) |
| Laterality of primary | |
| Right | 21 (64%) |
| Left | 12 (36%) |
| pT b | |
| T1 | 3 (9%) |
| T2 | 5 (15%) |
| T3 | 19 (58%) |
| T4 | 6 (18%) |
| pN b | |
| N0 | 14 (42%) |
| N1 | 19 (58%) |
| pM b | |
| M0 | 11 (33%) |
| M1 | 22 (67%) |
| Sarcomatoid histology in primary | 2 (6%) |
| IMDC risk category c | |
| Favorable risk (0 criteria) | 0 (0%) |
| Intermediate risk (1–2 criteria) | 15 (45%) |
| Poor risk (≥3 criteria) | 18 (55%) |
| Tissue specimen collection site (N = 48) | |
| Kidney | 24 (50%) |
| Skin/soft tissue | 10 (21%) |
| Bone | 4 (8%) |
| Lung | 3 (6%) |
| Retroperitoneum | 2 (4%) |
| Brain | 2 (4%) |
| Liver | 1 (2%) |
| Colon | 1 (2%) |
| Adrenal | 1 (2%) |
| Lines of systemic therapy | 3 (1–5) |
| Types of systemic therapy | |
| Immunotherapy | 33 (100%) |
| Targeted therapy | 22 (67%) |
| Immune checkpoint inhibition | 12 (36%) |
| mTOR inhibitor | 11 (33%) |
IQR = interquartile range; ISUP = International Society of Urological Pathology; mRCC = metastatic renal cell carcinoma.
Results listed as median (IQR) or N (%).
Pathological staging is at the time of initial nephrectomy or metastasectomy. All patients in this study (n = 33) developed metastatic disease.
International Metastatic RCC Database Consortium risk score is relevant to mRCC patients undergoing systemic therapy, and several ongoing trials are using this model in prospective studies. The criteria include the following: <1 yr from the time of diagnosis to systemic therapy, Karnofsky performance status <80%, hemoglobin less than the lower limit of normal, calcium more than the upper limit of normal, neutrophil count more than the upper limit of normal, and platelet count more than upper limit of normal.
Fig. 1 –
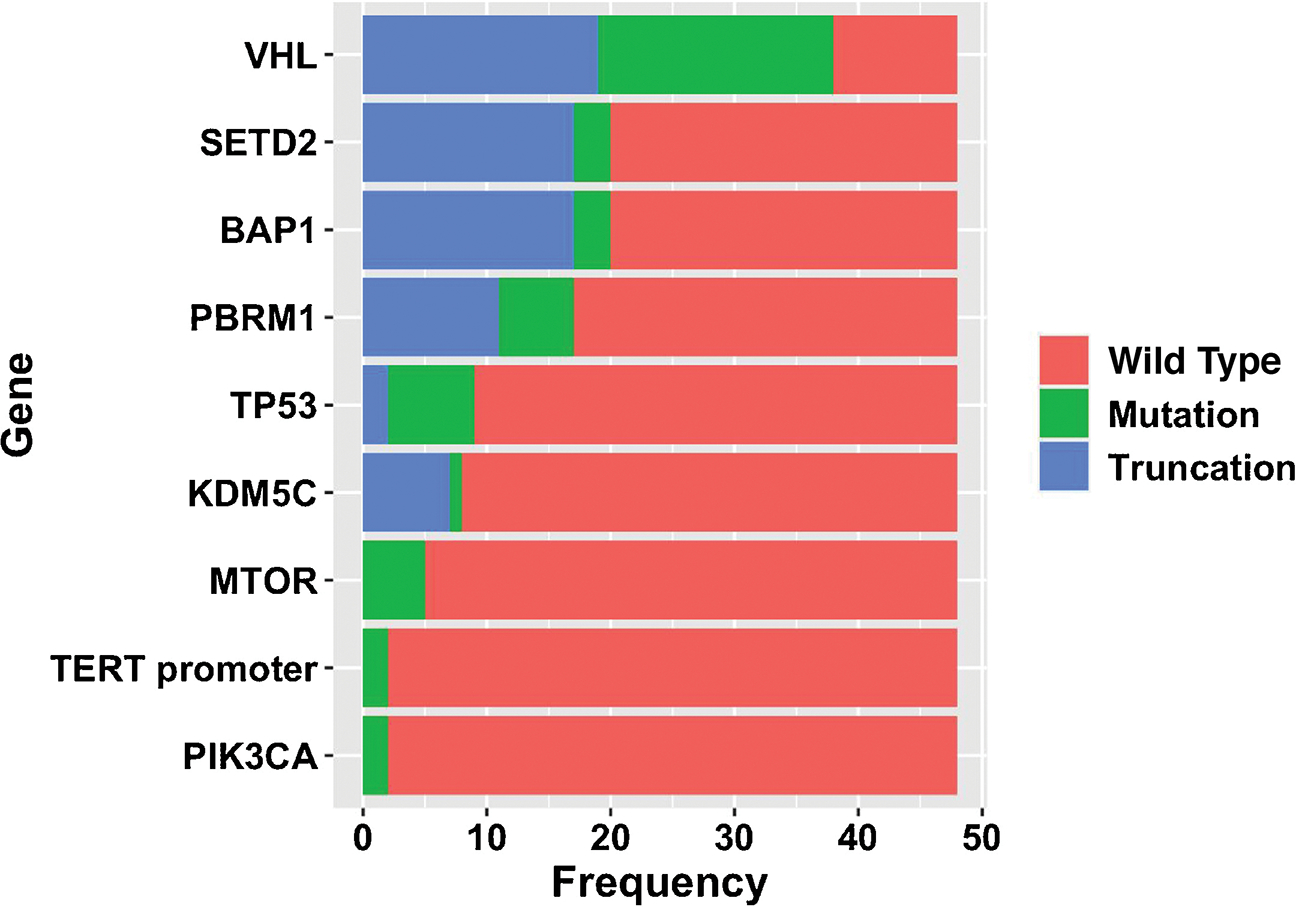
Bar diagram of identified somatic mutations among the entire cohort of tumor samples (N = 48).
3.2. Associations between somatic mutations and immune infiltration patterns (IF cohort)
Among both primary and metastatic tumors (n = 48), SETD2 mutations correlated with significantly decreased FOXP3+ T-cell density in the tumor core (p = 0.046), at the tumor-stroma interface (p = 0.037), and in the stroma (p = 0.046; Fig. 2). SETD2 mutations also correlated with increased CD206+/PD-L1+ macrophage density at the tumor-stroma interface (p = 0.036) and decreased stromal CD3+/CD8+ T-cell density (p = 0.022). KDM5C mutations correlated with increased CD206+ and CD68+/CD206+ macrophage densities within the tumor core (p = 0.013 and p = 0.047, respectively). KDM5C mutations also correlated with decreased CD68+/PD-L1+ macrophage density at the tumor-stroma interface (p = 0.020). PBRM1 mutations correlated with decreased FOXP3+ regulatory T-cell (Treg) density in the tumor core (p = 0.022). VHL mutations correlated with significantly decreased stromal CD3+/CD8+ and CD8+ cytotoxic T-cell density (p = 0.046 and p = 0.033, respectively; Fig. 3).
Fig. 2 –
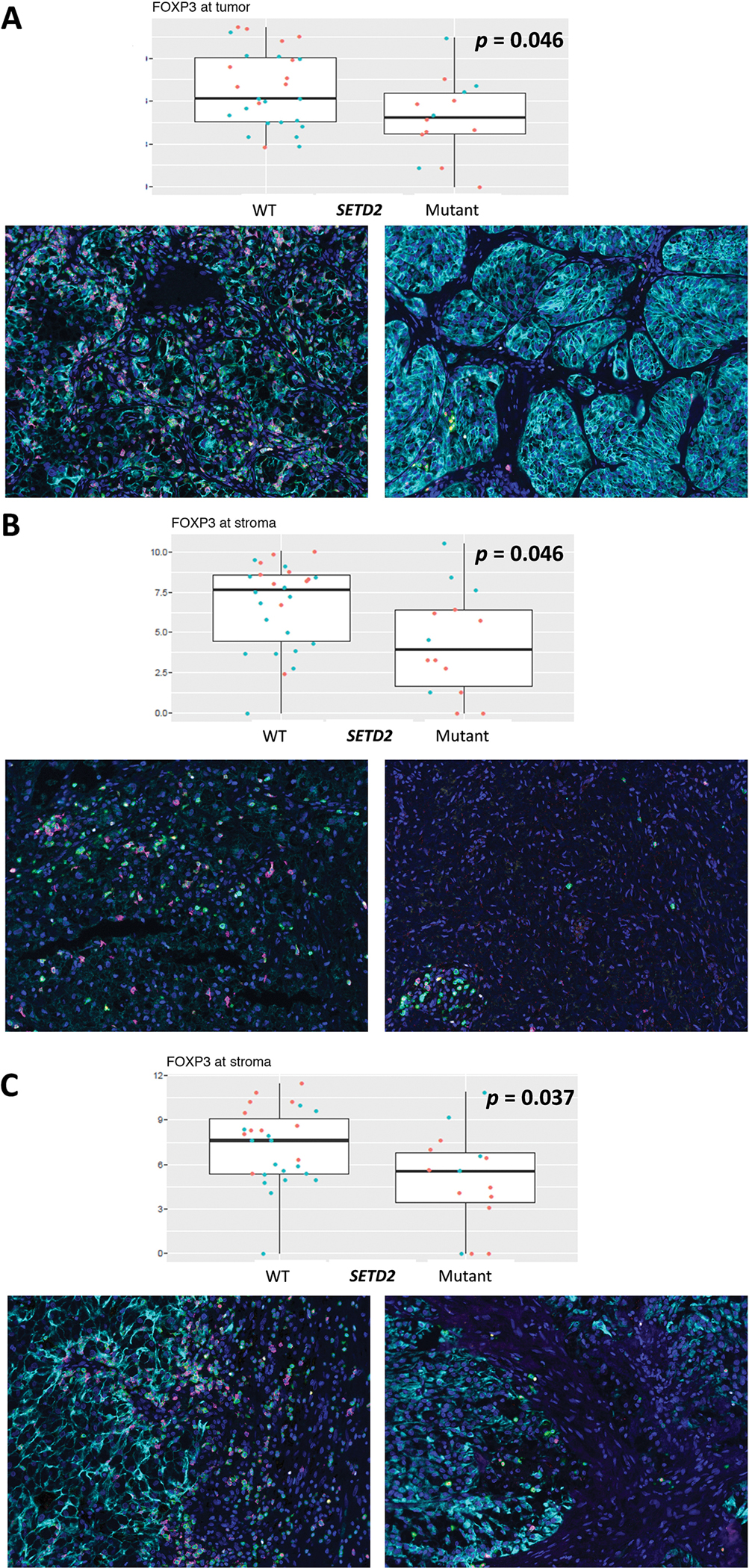
Box-plot diagrams for FOXP3+ cell density, stratified by SETD2 mutant status, with associated IF panels (FOXP3+ = pink, DAPI = dark blue, pan-cytokeratin = turquoise, CD3 = green, T-Bet = yellow). Red points in the box plot represent metastatic samples, and blue points represent primary samples. Wilcoxon rank-sum p values are listed in the plots. (A) Tumor core. (B) Stroma. (C) Tumor-stroma interface. DAPI = 40′, 6-diamidino-2-phenylindole; FOXP3 = forkhead box P3; IF = immunofluorescence; T-Bet = T-box transcription factor TBX21.
Fig. 3 –
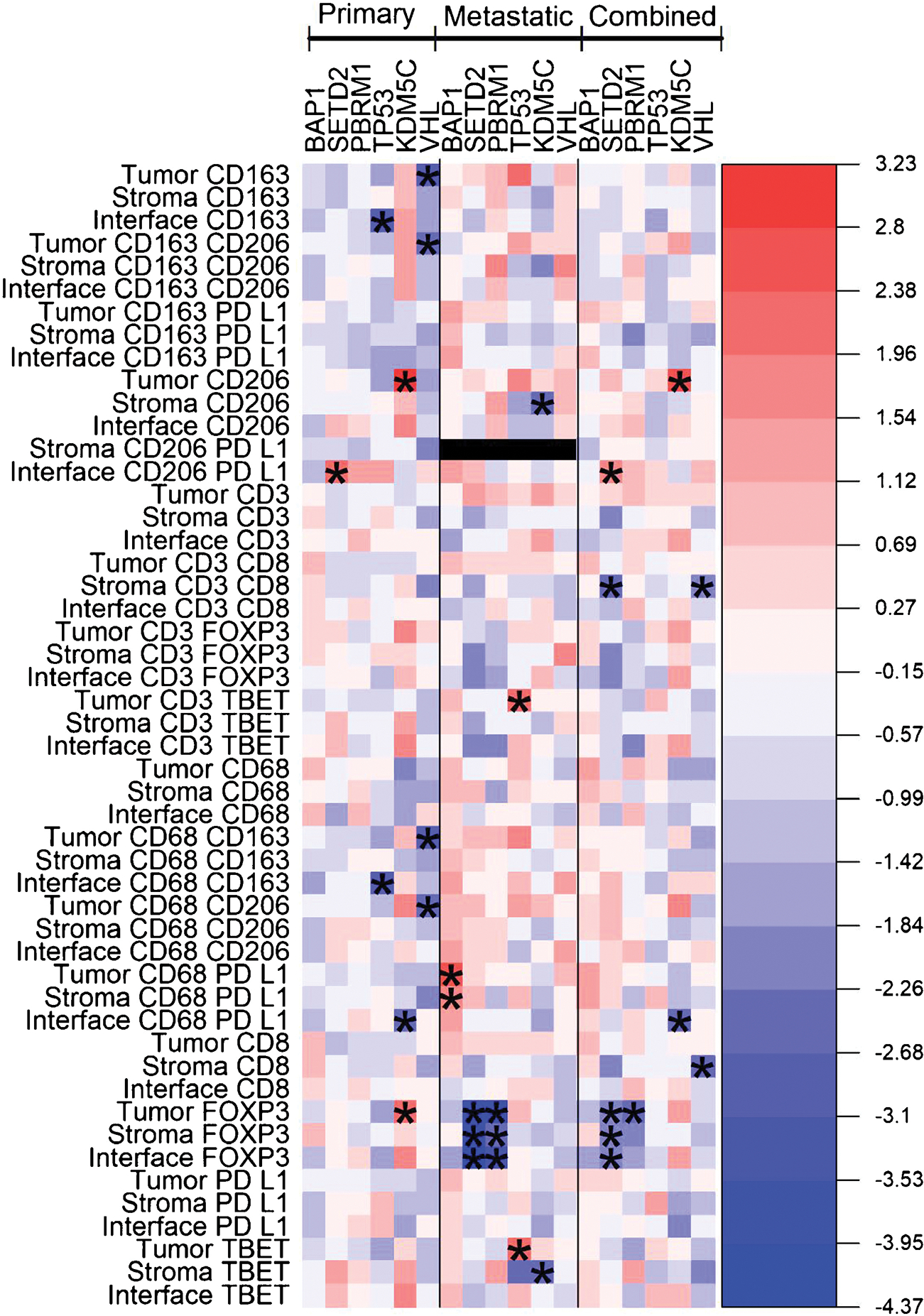
Heat-map diagram of immune cell density for all tumors, and primary and metastatic tumor subgroups. Color gradient represents linear mixed-model coefficients between the somatic mutation subgroups and immune infiltrate density for the cell type and region location listed in the row names (red = higher immune cell density, blue = lower immune cell density). The asterisks represent p < 0.05. Stromal CD206+/PD-L1+ was unable to be assessed for metastatic tumors; too few cells were identified. PD-L1 = programmed death-ligand 1.
3.2.1. Primary tumors only
In primary tumors (n = 24), SETD2 mutations correlated with increased CD206+/PD-L1+ macrophage density at the tumor-stroma interface (p = 0.017). KDM5C mutations correlated with decreased density of CD68+/PD-L1+ macrophages at the tumor-stroma interface (p = 0.029) and increased density of CD206+ macrophages and FOXP3+ T cells within the tumor core (p = 0.004 and p = 0.027, respectively). VHL mutations were associated with decreased CD163+/CD206+, CD163+, CD68+/CD163+, and CD68 +/CD206+ macrophage densities within the tumor core (p = 0.021, p = 0.014, p = 0.009, and p = 0.028, respectively). TP53 mutations correlated with decreased CD163+ and CD68+/CD163+ macrophage density at the tumor-stroma interface (p = 0.011 and p = 0.010, respectively; Fig. 3).
3.2.2. Metastatic tumors only
In metastatic tumors (n = 24), SETD2 mutations were associated with decreased FOXP3+ T-cell density in the tumor core, stroma, and tumor-stroma interface (p = 0.003, p = 0.004, and p < 0.001, respectively). KDM5C mutations correlated with decreased levels of stromal CD206+ macrophages (p = 0.033) and T-bet+ cells (p = 0.047). PBRM1 mutations correlated with lower levels of FOXP3+ Tregs within the tumor core and stroma (p = 0.003 and p = 0.046, respectively). TP53 mutations correlated with increased densities of CD3+/T-bet+ and T-bet+ Th1 cells within the tumor core (p = 0.027 and p = 0.037, respectively). BAP1 mutations correlated with increased densities of CD68+/PD-L1+ macrophages within the tumor core and stroma (p = 0.011 and p = 0.046, respectively; Fig. 3).
3.3. Comparison of xCell enrichment score and IF cell patterns
3.3.1. External ccRCC cohorts
The xCell enrichment score analysis for each of the three external data sources (TCGA, CPTAC, and TCC) is graphically represented in Figure 4, and the full data summary can be found in Supplementary Tables 1–3.
Fig. 4 –
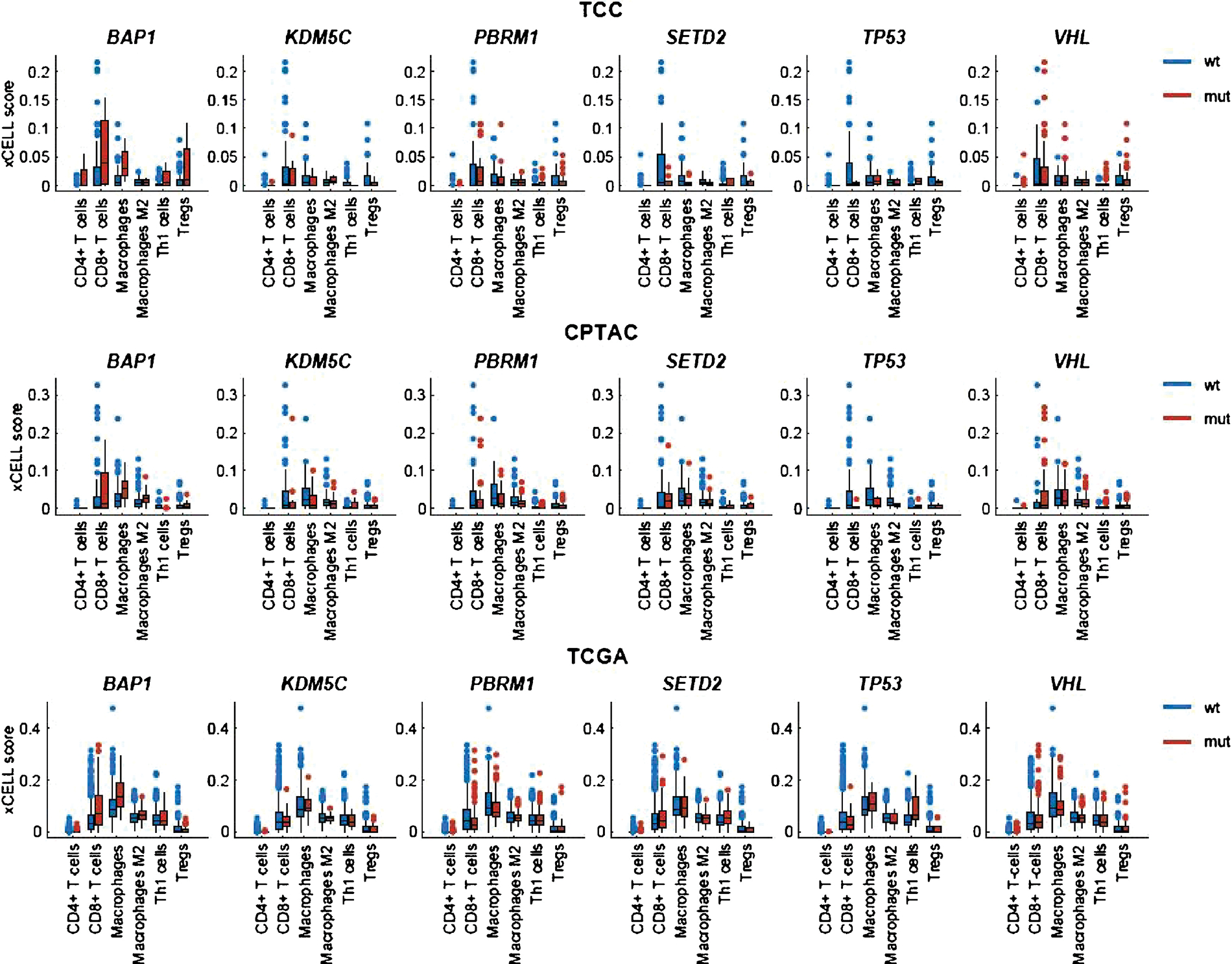
Box-plot diagrams for xCell enrichment scoring for selected cell types. RNA-seq data were obtained from TCC, CPTAC, and TCGA cohorts. CPTAC = Clinical Proteomic Tumor Analysis Consortium; mut = mutant; TCC = Moffitt Cancer Center Total Cancer Care; TCGA = The Cancer Genome Atlas; Treg = regulatory T cell; wt = wild type.
BAP1 mutations correlated with increased macrophage xCell scores in the TCGA and CPTAC data sets (p < 0.01 and p < 0.01, respectively), decreased Treg scores in TCGA (p = 0.02), and increased CD8 (p < 0.01) and M2 macrophage (p < 0.01) scores in TCGA. SETD2 mutations correlated with increased Th1 xCell scores in TCGA (p = 0.02). PBRM1 mutations correlated with decreased CD8 (p = 0.03) xCell scores in TCGA and decreased macrophage scores in TCGA and CPTAC (p < 0.01 and p = 0.01, respectively). KDM5C mutations correlated with decreased Th1 xCell scores in TCC (p = 0.02). TP53 mutations correlated with increased Th1 xCell scores in TCGA and TCC (p < 0.01 and p = 0.02, respectively) and decreased M2 macrophage scores in CPTAC (p = 0.045).
The xCell and IF cell types were matched to facilitate comparison (Supplementary Table 4). None of the significant cell-type–specific findings from the IF analysis were identified as significant in their matched xCell score counterparts for any of the three external data sources (Fig. 5).
Fig. 5 –
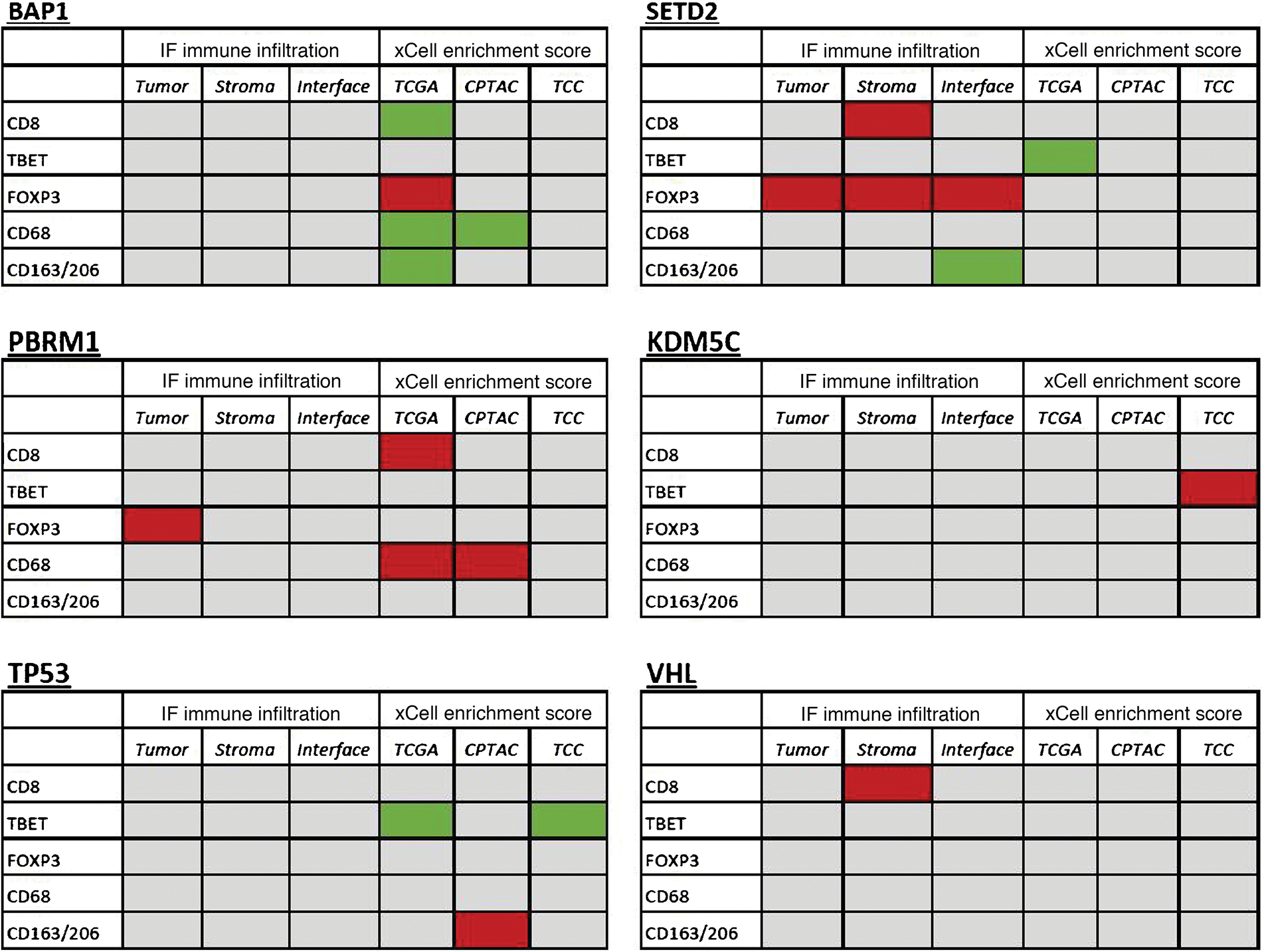
Tables comparing statistically significant findings from the IF-derived immune infiltrate analysis and xCell enrichment scoring, for all tumor samples. Green panels represent increased infiltration or xCell score for mutants, and red panels represent decreased infiltration or xCell score for mutants. CPTAC = Clinical Proteomic Tumor Analysis Consortium; FOXP3 = forkhead box P3; IF = immunofluorescence; TBET = T-box transcription factor TBX21; TCC = Moffitt Cancer Center Total Cancer Care; TCGA = The Cancer Genome Atlas.
3.3.2. IF cohort
In our IF cohort, 12 patients had tumor samples (nine primary and three metastatic tumor samples) that previously underwent RNA sequencing. We evaluated the ability of xCell to recapitulate the significant immune cell patterns associated with specific mutations in our IF cohort analysis. Overall, in this small matched-sample subset, the vast majority of correlations were weak between IF immune infiltration patterns and the relevant xCell score (R < 0.5), despite both values being measured with the same tumor samples, the only exception being CD8+ cells in the primary tumor subgroup (R = 0.58; Supplementary Fig. 2 and 3).
3.4. Survival analyses in IF cohort
On multivariable Cox regression, patients whose tumors had SETD2 mutations had better OS (hazard ratio [HR], 0.106 [95% confidence interval {CI}, 0.024–0.461], p < 0.01) and OS following IT (HR, 0.154 [95% CI, 0.038–0.63], p < 0.01; Supplementary Fig. 1A). Patients whose tumors had KDM5C mutations had significantly better OS (p = 0.027) and OS following IT (p = 0.048) on log-rank analyses, but the HR was not estimable using a Cox regression because there were no deaths in the mutant group (Supplementary Fig. 1B). The complete survival analysis summary is available in Supplementary Table 5. The median follow-up from the date of diagnosis until death or censoring at the last follow-up was 55 mo (interquartile range [IQR] 30–97) for the entire cohort, 81 mo (IQR 51–110) for patients diagnosed cM0, and 45 mo (IQR 28–65) for patients diagnosed cM1. Seventeen patients died during the follow-up period (52%).
4. Discussion
In this study, we report a granular analysis of the associations between common somatic mutations in ccRCC and immune infiltration patterns within the TIME of 48 tumor samples collected from patients with metastatic ccRCC. Using targeted sequencing, multiplex IF, and digital quantitative image analysis, we identified several novel associations.
One such association was that SETD2 mutations correlated with significantly lower levels of FOXP3+ T–cells in the tumor core, stroma, and tumor-stroma interface. PBRM1 mutations were also found to correlate with decreased FOXP3+ T-cell density, with this finding limited to the tumor core. FOXP3+ Tregs suppress host-versus-tumor immunity in the TIME through inhibition of antitumor cytotoxic T cells [19]. SETD2-mediated methylation pathways are critical for interferon-α–dependent immune processes, and dampened interferon-α/β receptor signaling may impair Treg development, providing a plausible explanation as to why SETD2-mutated tumors in our cohort consistently exhibited low densities of FOXP3+ Tregs [20,21].
We also identified several associations between somatic mutations and the distribution of tumor-associated macrophages (TAMs) within the TIME. TAMs, which typically express an M2 phenotype, are attracted to the TIME by chemokines and cytokines produced by tumor cells, and TAMs have been found to support tumor growth and metastasis through a variety of pathways [6,15,22,23]. Notably, we found that tumors with KDM5C mutations had higher levels of CD206+ and CD68+/CD206+ TAMs within the tumor core. The product of KDM5C is a lysine-specific histone demethylase, and mutations of this gene have been associated with a higher risk of cancer recurrence and death among patients with small renal masses [24,25]. Similar epigenetic mechanisms have been shown to play a role in modulating macrophage polarization to the M2 phenotype and their infiltration into tumor tissues, providing a biological rationale for our finding of increased tumor-core infiltration of CD206 + TAMs in tumors with KDM5C mutations [26,27].
We also sought to evaluate the ability of a commonly used immune cell deconvolution method, xCell, to recapitulate the significant associations we found in our IF cohort analysis using RNA sequencing data. None of the statistically significant IF-derived immune infiltrate findings were matched with a similar finding in their counterpart xCell enrichment score for any of the three external data sources (TCGA, CPTAC, and TCC). We were surprised that none of the cohorts demonstrated similar TIME associations with the five somatic mutations studied, even when assessing just the tumor-core TIME associations.
There are some possible reasons for this discordance. The 48 patients included in the IF cohort all had stage IV ccRCC, contrasting with the relatively lower-stage tumors in TCGA, CPTAC, and TCC (cM1 rates of 14.3%, 10.9%, and 21.4%, respectively). Additionally, xCell, similar to other deconvolution or enrichment pipelines, was tested and validated using flow cytometry cell counts as the reference standard [17]. Prior reports correlating sequencing-derived deconvolution or enrichment outputs to IF-derived cell counts have demonstrated decreased correlation compared with using flow cytometry–derived cell counts [28]. Braun et al [7] recently reported several misclassifications of hot versus cold CD8+ infiltration in ccRCC tumors when a sequencing-derived deconvolution pipeline was compared with IF, despite a statistically significant Spearman’s correlation. These examples of discordance may be attributable to the large difference in proportional sampling of the tumor between IF and flow cytometry, the reference standard upon which these pipelines have been tested and validated. Additionally, when studying the 12 patients who had data for both IF-derived immune infiltrate and xCell scores, without stratifying for mutation status, we found that xCell scores and IF-derived immune infiltrate correlated poorly, calling into question the use of either technique as a surrogate for the other. A similar phenomenon is noted in proteomics analyses, in which data demonstrates that protein abundance cannot be predicted reliably from DNA- or RNA-level measurements [29,30].
This study has several limitations. The sample size of the IF cohort, 48 total tumors, is relatively modest, particularly concerning the subgroup analyses of primary and metastatic tumors, with 24 tumors in each subgroup. As a pilot study, many comparisons were undertaken and correction for multiple comparisons was not performed as to limit the probability of a type-II error in this small discovery cohort. As such, the risk of a type-I statistical error is certainly >5%. Larger studies to validate these findings should be performed with correction for multiple comparisons, if applicable. Additionally, IF analysis samples a small portion of the overall tumor, limiting the immune cell infiltration analysis to the specific regions that were sampled. To mitigate this effect, multiple regions were examined per tumor (seven separate ROIs per specimen); however, this limitation will always persist in studies of this nature. As there is no standardized method of matching IF markers with xCell cell-type groups, our method may not have accurately classified the intended immune cell types, leading to discordant results. Finally, all patients included in the IF cohort had metastatic ccRCC and received IT, but the patients were heterogeneous regarding the exact type of IT received (interleukin-2, ipilimumab, nivolumab, or pembrolizumab), limiting the generalizability of the survival analysis.
5. Conclusions
Overall, this study provides evidence that common somatic mutations in ccRCC, such as SETD2, PBRM1, and KDM5C, may be driving alterations in immune cell infiltration patterns within the TIME. These novel findings have the potential to inform precision research and immunotherapeutic treatment strategies.
Supplementary Material
{kind=link}
Acknowledgments:
Editorial assistance was provided by the Moffitt Cancer Center’s Scientific Editing Department by Dr. Paul Fletcher & Daley Drucker. No compensation was given beyond their regular salaries.
Funding/Support and role of the sponsor:
This work was supported by the Urology Care Foundation Research Scholar Award Program and Society for Urologic Oncology (to Brandon J. Manley); the United States Army Medical Research Acquisition Activity Department of Defense (KC180139 to Brandon J. Manley); Total Cancer Care Protocol at Moffitt Cancer Center, which was enabled in part by the generous support of the DeBartolo Family; the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute designated Comprehensive Cancer Center (P30-CA076292); and the Tissue Core Facility at the H. Lee Moffitt Cancer Center & Research Institute (P30-CA076292). The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Urological Association or the Urology Care Foundation.
Footnotes
Financial disclosures: Nicholas H. Chakiryan certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Ali Hajiran, Ahmet M. Aydin, Logan Zemp, Jonathan Nguyen, James Mulé, Jad Chahoud, SZ, SF, Michelle Fournier, Jamie K. Teer, Jasreman Dhillon, SM, Carlos Moran-Segura, Esther Katende, WJS, and Youngchul Kim have no disclosures. Brandon J. Manley is an NCCN Kidney Cancer Panel Member. Philippe E. Spiess is an NCCN Bladder and Penile Cancer Panel Member and Vice-Chair. James Mulé is an Associate Center Director at Moffitt Cancer Center; has ownership interest in Fulgent Genetics, Inc., Aleta Biotherapeutics, Inc., Cold Genesys, Inc., Myst Pharma, Inc., and Tailored Therapeutics, Inc.; and is a consultant/advisory board member for ONCoPEP, Inc., Cold Genesys, Inc., Morphogenesis, Inc., Mersana Therapeutics, Inc., GammaDelta Therapeutics, Ltd., Myst Pharma, Inc., Tailored Therapeutics, Inc., Verseau Therapeutics, Inc., Iovance Biotherapeutics, Inc., Vault Pharma, Inc., Noble Life Sciences Partners, Fulgent Genetics, Inc., UbiVac, LLC, Vycellix, Inc., and Aleta Biotherapeutics, Inc.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.euf.2021.04.014.
Data Sharing:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Escudier B Combination therapy as first-line treatment in metastatic renal-cell carcinoma. N Engl J Med 2019;380:1176–8. [DOI] [PubMed] [Google Scholar]
- [4].Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miao D, Margolis CA, Gao W, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Braun DA, Hou Y, Bakouny Z, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 2020;26:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988;332:268–9. [DOI] [PubMed] [Google Scholar]
- [9].Gnarra JR, Tory K, Weng Y, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994;7:85–90. [DOI] [PubMed] [Google Scholar]
- [10].Creighton CJ, Morgan M, Gunaratne PH, et al. Comprehensive molecular characterizationof clear cell renal cell carcinoma.Nature 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 2016;374:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Manley BJ, Zabor EC, Casuscelli J, et al. Integration of recurrent somatic mutations with clinical outcomes: a pooled analysis of 1049 patients with clear cell renal cell carcinoma. Eur Urol Focus 2017;3:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hakimi AA, Ostrovnaya I, Reva B, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 2013;19:3259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Joseph RW, Kapur P, Serie DJ, et al. Clear cell renal cell carcinoma subtypes identified by BAP1 and PBRM1 expression. J Urol 2016;195:180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hakimi AA, Voss MH, Kuo F, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: data from a randomized phase III trial. Cancer Discov 2019;9:510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chevrier S, Levine JH, Zanotelli VRT, et al. An immune atlas of clear cell renal cell carcinoma. Cell 2017;169:736–49, e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Clark DJ, Dhanasekaran SM, Petralia F, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 2020;180:207. [DOI] [PubMed] [Google Scholar]
- [19].Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015;5:15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017;541:321–30. [DOI] [PubMed] [Google Scholar]
- [21].Metidji A, Rieder SA, Glass DD, Cremer I, Punkosdy GA, Shevach EM. IFN-alpha/beta receptor signaling promotes regulatory T cell development and function under stress conditions. J Immunol 2015;194:4265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kovaleva OV, Samoilova DV, Shitova MS, Gratchev A. Tumor associated macrophages in kidney cancer. Anal Cell Pathol (Amst) 2016;2016:9307549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee C, Jeong H, Bae Y, et al. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J Immunother Cancer 2019;7:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Niu X, Zhang T, Liao L, et al. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 2012;31:776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Manley BJ, Reznik E, Ghanaat M, et al. Characterizing recurrent and lethal small renal masses in clear cell renal cell carcinoma using recurrent somatic mutations. Urol Oncol 2019;37:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol 2013;34:216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Osawa T, Tsuchida R, Muramatsu M, et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res 2013;73:3019–28. [DOI] [PubMed] [Google Scholar]
- [28].Finotello F, Mayer C, Plattner C, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014;513:382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stewart PA, Welsh EA, Slebos RJC, et al. Proteogenomic landscape of squamous cell lung cancer. Nat Commun 2019;10:3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.