

Abstract
Nuclear receptor binding SET domain proteins (NSDs) catalyze the mono- or di-methylation of histone 3 lysine 36 (H3K36me1 and H3K36me2), using S-adenosyl-L-methionine (SAM) as a methyl donor. As a key member of NSD family proteins, NSD2 plays an important role in the pathogenesis and progression of various diseases such as cancers, inflammations, and infectious diseases, serving as a promising drug target. Developing potent and specific NSD2 inhibitors may provide potential novel therapeutics. Several NSD2 inhibitors and degraders have been discovered while remaining in the early stage of drug development. Excitingly, KTX-1001, a selective NSD2 inhibitor, has entered clinical trials. In this perspective, the structures and functions of NSD2, its roles in various human diseases, and the recent advances in drug discovery strategies targeting NSD2 have been summarized. The challenges, opportunities, and future directions for developing NSD2 inhibitors and degraders are also discussed.
Graphical Abstract

INTRODUCTION
As important epigenetic “writers”, histone lysine (K) methyltransferases (HKMTases) decorate the core histone proteins by catalyzing reversible addition of one, two, or three methyl groups to the ε-nitrogen of a specific lysine residue (H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20), using S-adenosyl-L-methionine (SAM) as a methyl donor, which results in the formation of mono-, di-, tri-methylated derivatives (Kme1, Kme2, and Kme3) and production S-adenosyl-L-homocysteine (SAH) (Figure 1a).1, 2 HKMTases play critical roles in gene transcription, DNA repair, DNA replication, and cell differentiation.1, 3 More importantly, HKMTases have been reported to be implicated in the causes of many diseases, including cancer, mental health disorders, and developmental disorders.2
Figure 1.
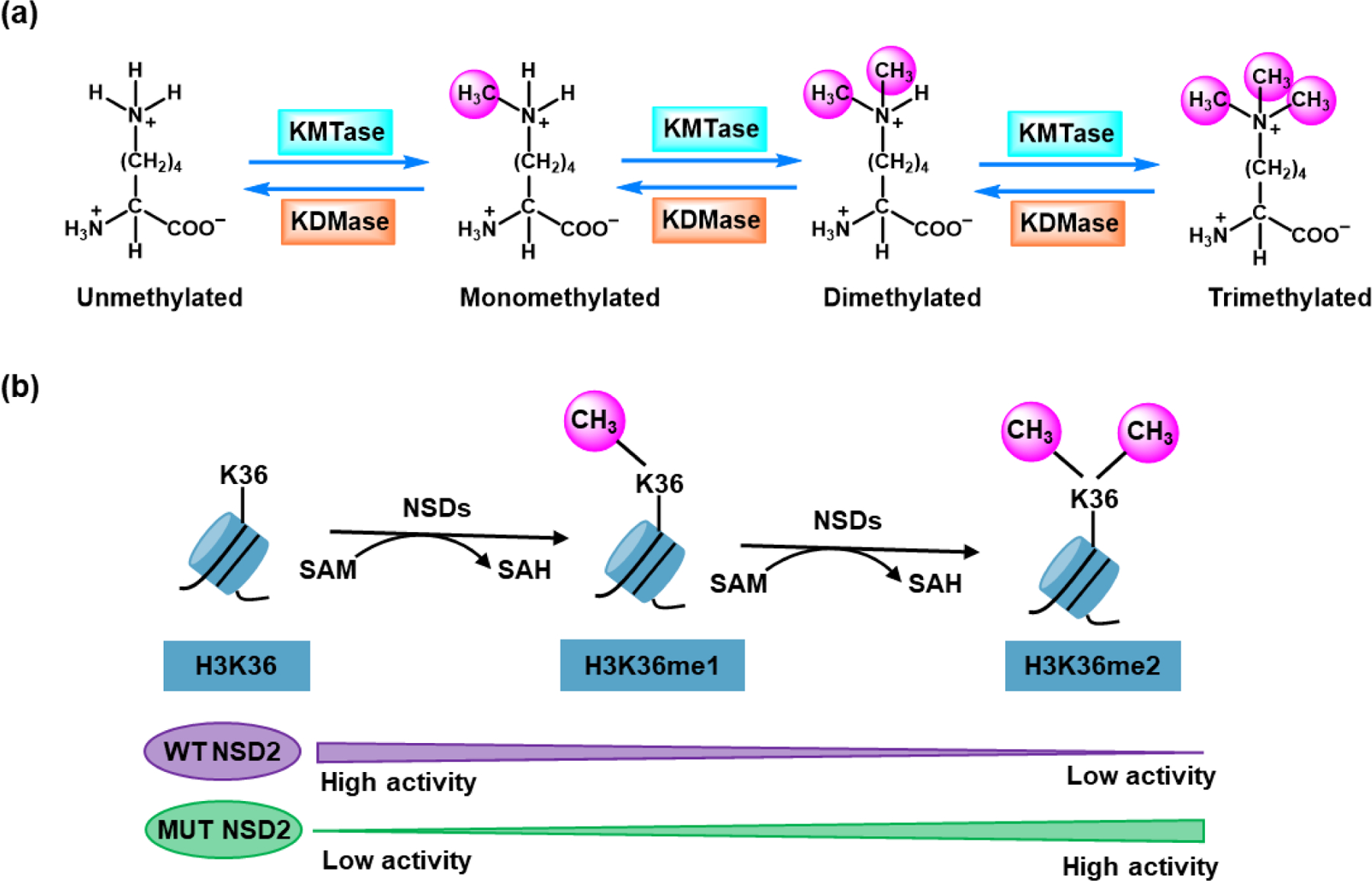
(a) The general process of the methylation (monomethylation, dimethylation, and trimethylation) and demethylation of histone lysine that is catalyzed by lysine methyltransferase (KMTase) and demethylase (KDMase), respectively. (b) The monomethylation and dimethylation process of histone lysine 36 (H3K36) catalyzed by lysine methyltransferase (KMTase) NSDs, with SAM as the methyl donor, producing H3K36me1, H3K36me2, and SAH.
Nuclear receptor binding SET domain protein 2 (NSD2), also known as Wolfe-Hirschhorn syndrome candidate 1 (WHSC1) or multiple myeloma SET domain (MMSET), is a protein lysine methyltransferase (PKMT) that predominantly catalyzes mono- and di-methylation of histone 3 lysine 36 (H3K36) with SAM as a methyl donor, and produces SAH (Figure 1b).4–6 NSD2 is an important member of the NSD family, which also includes NSD1 and NSD3 (also known as WHSC1L1, WHSC1 like 1).2, 7–9 NSD2 is associated with a broad spectrum of human diseases, especially various cancers.1 NSD2 is aberrantly overexpressed, amplified, or somatically mutated in multiple types of cancer and has been defined as an oncoprotein.10–13 Notably, NSD2 is overexpressed in multiple myeloma (MM) patients, predominantly harboring a t(4,14) translocation that leads to the aberrant upregulation of this gene.14–16 A recurrent gain-of-function mutation with the substitution of glutamic acid to lysine at residue 1099 (p.Glu1099Lys, p.E1099K) in the catalytic SET domain of NSD2 has been revealed in pediatric acute lymphoblastic leukemia (ALL) patients. NSD2 p.E1099K hyperactivates its methyltransferase activity, driving oncogenesis and progreesion.17–20 Accumulating evidence indicates that NSD2 is overexpressed in various cancers such as MM, skin, lung, bladder, brain, metaplastic breast, and prostate cancer (PCa).11, 21–23 NSD2 has been identified as a promising drug target, receiving more and more attention from both academia and pharmaceutical industry.24, 25 Therefore, developing potent and selective small molecule NSD2 inhibitors may provide potential novel therapies for patients carrying NSD2 overexpression, translocation, and/or mutation. Such compounds may also serve as useful pharmacological tools for exploring the critical roles of NSD2 in various human diseases.
As critical epigenetic regulators, the aberrant HKMTases functions are associated with many diseases. Developing HKMTases inhibitors as effective therapeutic agents have attracted a lot of attention.2, 7 Over the past decade, significant advances have been made in developing drugs targeting HKMTases for disease treatment by blocking the methylation of histone lysine.2 In 2020, a selective and potent EZH2 inhibitor 1 (tazemetostat, EPZ-6438), developed by Epizyme Inc., became the first HKMTase inhibitor approved by the United States Food and Drug Administration (U.S. FDA) for treating epithelioid sarcoma and follicular lymphoma.26 In addition to tazemetostat, several other EZH2 inhibitors have entered clinical trials at different stages. Moreover, a DOT1L histone lysine methyltransferase inhibitor 2 (pinometostat, EPZ-5676) has completed the phase I clinical trials for treating leukemia showing an acceptable safety and pharmacodynamics profile (ClinicalTrials.gov identifiers: NCT01684150 and NCT02141828).27, 28 In addition, several other HKMTases inhibitors have been developed (Figure 2), including 3 (UNC0642, a G9a/GLP inhibitor),29 4 (UNC0379, a SETD8 inhibitor),30, 31 5 ((R)-PFI-2, a SETD7 inhibitor),32 6 (BAY-598, a SMYD2 inhibitor),33 7 (EPZ028862, a SMYD3 inhibitor)34 8 (A-196, a SUV420H1 and SUV420H2 inhibitor),35 and 9 (MRK-740, a PRDM9 inhibitor).36
Figure 2.

Chemical structures of representative histone lysine methyltransferase (HKMTase) inhibitors 1–9 in clinical and pre-clinical trials.
Given the driving roles of NSD2 in various diseases and simultaneously inspired by the great success in devolving selective potent EZH2 and DOT1L inhibitors, as well as other HKMTases inhibitors, substantial efforts have been made to identify potent NSD2 inhibitors. To date, several small molecule NSD2 inhibitors and degraders have been reported, targeting either the catalytic SET domain or the proline-tryptophan-tryptophan-proline (PWWP) domain with representative chemical structures shown in Figure 3.24, 25 However, these inhibitors mainly target the catalytic SET domain, having little success likely owing to poor enzymatic and/or cellular potency, as well as the subtype selectivity, etc. Discovery and development of more potent and specific NSD2 inhibitors are urgently needed. Encouragingly, in August 2022, compound KTX-1001 as a novel NSD2 inhibitor targeting the catalytic SET domain, developed by K36 Therapeutics, Inc., has been advanced into the phase I clinical trial to treat patients with relapsed and refractory MM (ClinicalTrials.gov Identifier: NCT05651932). There is a lack of detailed information about KTX-1001, including its structure and preclinical data. Nevertheless, accumulating evidence supports that developing novel potent and selective NSD2 inhibitors may offer viable therapeutic approaches for treating human diseases, particularly various cancers. In this review, the structures and functions of NSD2, its critical role in various diseases, and the recent advances in developing NSD2 inhibitors have been comprehensively summarized. The relevant challenges, opportunities, and future directions for developing small molecule NSD2 inhibitors and degraders are also discussed.
Figure 3.

Chemical structures of representative NSD2 inhibitors and degraders 10–16.
STRUCTURES OF NSD2
The NSD family members (NSD1, NSD2, and NSD3) are large multidomain proteins containing a conserved catalytic SET domain that is subdivided into pre-SET (aka. AWS), SET, and post-SET domains,37, 38 two PWWP domains (PWWP1 and PWWP2) that are responsible for binding to methylated histone H3 and DNA,39 five PHD (plant homeodomain) zinc fingers (PHD1-PHD5) that are critical for exerting interactions with other methylated histones,40, 41 and a cysteine-histidine-rich (C5HCH) domain.42 NSD2 is the shortest member of the NSD family, displaying a complex expression pattern, which contains three isoforms, namely NSD2-long (MMSET-II) (containing 1365 amino acids), NSD2-short (MMSET-I) (containing 647 amino acids), and interleukin-5 response element II-binding protein (RE-IIBP) (containing 584 amino acids).23, 24, 43, 44 NSD2-long has multiple protein-protein interactions (PPI) domains, composed of a conserved catalytic SET domain with its AWS and post-SET domains, two PWWP domains, five PHD domains, and a putative DNA-binding high mobility group box (HMG-box) domain. NSD2-short isoform consists of a PWWP domain and an HMG-box domain, lacking histone methyltransferase activity due to the absence of a SET domain. Compared to the NSD2-short isoform, RE-IIBP initiates from the PHD domain of the NSD2-long isoform and contains two PHD finger domains, a PWWP domain, and a SET domain, but without an HMG-box domain (Figure 4).10 NSD family proteins (NSD1, NSD2, and NSD3) share a similar structural organization in the C-terminal block region (about 700 amino acids) and possess approximately 55%−68% identical amino acid sequences located among residues 703 and 1409.42 High sequence conservation is observed among the catalytic SET domains of all the NSD members. Sequence analysis revealed that the NSD2-SET domain shares 75.9% identity and 90.1% similarity with the NSD1-SET domain, while the NSD3-SET domain shares 72.9% identity and 85% similarity with the NSD1-SET domian.45
Figure 4.

(a) SAM crystal structure in complex with NSD2-SET domain (PDB ID: 5LSU). SAM is shown as cyan sticks; (b) Crystal structure of DNA in complex with NSD2-PWWP1 domain (PDB ID: 5VC8). The key residues LYS-304, LYS-309, and LYS-312 in NSD2-PWWP1 domain that form direct electrostatic interactions with the DNA phosphate backbone are shown as cyan sticks; and (c) The structures of three NSD2 isoforms (NSD2-long, NSD2-short, and RE-IIBP) that are composed of multiple domains, including PWWP domain, PHD domain, SET domain (AWS/pre-SET, SET, and post-SET), etc.
BIOLOGICAL FUNCTIONS OF NSD2
NSD proteins (NSD1, NSD2, and NSD3) share a highly homologous architecture composed of about 700 amino acid sequences; however, they display substantially different functions.5, 42 One study showed that embryonic lethality occurs in NSD1-knockout mice.46 Another study reported that although embryonic lethality was avoided, death occurs shortly after birth in NSD2-deficient mice.47 Functional diversity of three NSD members might attribute to the substrate specificities of their catalytic SET domains.48 Highly conserved catalytic SET domain of NSD2 plays an important role in transcriptional regulation through the catalytic methylation, predominantly mono- and di-methylation of histone 3 lysine 36 (H3K36).37, 38 In addition to the catalytic SET domain, other “reader” domains (five PHDs, two PWWPs, and an HMG-box) also play critical roles in the NSD2 function.39–42 However, their individual and/or cooperative roles have not yet been comprehensively elucidated.10 Five PHD fingers have been found to play a significant role in chromatin reorganization.40, 41 Studies reveal that PWWP domains can recognize and bind to di- and trimethylated H3K36 (H3K36me2 and H3K36me3) through a conserved aromatic cage and simultaneously interact with nucleosomal DNA adjacent to H3K36.49, 50 Moreover, PWWP domains often exert functions with other histone and DNA “reader” or “modifier” domains cooperatively to initiate the crosstalk between diverse epigenetic marks.49 The isolated N-terminal PWWP domain (PWWP1) of NSD2 binds H3K36me2/H3K36me3 presumably by a conserved aromatic cage and stabilizes NSD2 at chromatin. PHD fingers play a crucial role in NSD interactions with other methylated histones except for methylated histone H3.39 Among all structural domains of NSD2, the catalytic SET domain, an epigenetic writer, plays the most important role in post-translational gene modification and regulation via catalyzing the mono- and di-methylation of H3K36. As a cancer-driving protein, NSD2 is frequently overexpressed, translocated (SET domain), or mutated (SET domain) in many aggressive cancers, including malignant hematological and solid cancers. However, to date, the roles of NSD2 in oncogenesis and progression have not been fully elucidated. Notably, besides the critical roles in cancers, NSD2 also plays an essential role in the normal development of the human body. Loss-of-function of NSD2 is correlated with the developmental disorder Wolf-Hirschhorn syndrome (WHS) due to decreased histone methylation activity.51 Patients with this syndrome lack one copy of MMSET resulting from a partial deletion of the short arm of chromosome 4 (4p16.3). More importantly, these patients often suffer from some mental and developmental disorders, such as mental retardation, severe growth delays, craniofacial dysgenesis, etc.52–57 Consistently, NSD2 heterozygous mice display severe growth defects and WHS-like midline defects. Moreover, mice with homozygous NSD2−/− die shortly after birth owing to fetal growth retardation.47 It is critical to understand the biological functions of NSD2 and its corresponding mechanisms in various related diseases and the normal development of the human body, which will facilitate the development of effective therapies. The main biological functions of NSD2 and the underlying molecular mechanisms are depicted in Figure 5 and discussed below.
Figure 5.

The biological functions of NSD2 and underlying mechanisms.
NSD2 Facilitates DNA Damage Repair.
In addition to regulating gene expression, H3K36me2 plays a significant role in keeping genomic stability via engaging in DNA damage repair.58 Given that the hyperactivation of NSD2 caused by gene translocation or mutation increases the level of H3K36me2, it is reasonable to hypothesize that NSD2 is associated with DNA damage repair. Indeed, several groups have also revealed that NSD2 participates in DNA damage repair, as shown in Figure 5.59–61 The p53-binding protein 1 (53BP1) is a well-known important DNA damage response (DDR) mediator,62 which is recruited to the double-strand breaks (DSBs) with the critical assistance of di-methylation of histone H4 lysine 20 (H4K20me2).63, 64 In general, the mono-methylation of H4K20 (H4K20 me1) was catalyzed by SETD865, 66 and H4K20me1 can be transferred into di-methylated H4K20 (H4K20me2) and tri-methylated H4K20 (H4K20me3) mediated by SUV420H1 and SUV420H2 enzymes.67, 68 However, the absence of SUV420H1/2 and subsequent lack of most H4K20me2/3 did not abolish the accumulation of 53BP1 at DSBs but only slightly delayed, indicating that other HMTases may be responsible for methylating H4K20 specifically at DSBs.68
Interestingly, NSD2 was found to significantly upregulate H4K20 methylation at the site of DSBs in mammals.59 Moreover, the downregulation of NSD2 remarkably suppresses the methylation of H4K20 at DSBs, which subsequently leads to reduced accumulation of 53BP1. In addition, the phosphor-H2AX (γH2AX)-MDC1 (the mediator of DNA damage checkpoint protein 1) signaling pathway is engaged in the recruitment of NSD2 to DSBs via the specifical interaction between the BRCT domain of MDC1 and phosphorylated Ser 102 of NSD2. Thus, the γH2AX-MDC1-NSD2 signaling pathway regulates the H4K20 methylation at the site of DSBs, subsequently promoting the recruitment of 53BP1. NSD2 roles as a bridge that connects the γH2AX-MDC1 pathway and H4K20 methylation. Hyperactivation of NSD2 induced by translocation, mutation, or dysregulation contributes to the repairment of DNA damage, which implies that patients carrying t(4;14) translocation or E1099K mutation often show poor prognosis after treatment with DNA damage-inducing regimens, including chemotherapies and radiotherapies. DSBs may enhance the interaction between the C-terminal domain of NSD2 and phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which stimulates NSD2-mediated di-methylation of PTEN (PTENme2) at lysine 349 (K349).60 PTENme2 is recognized by the Tudor domain of 53BP1 and then recruited into DNA-damage sites to implement the timely repair of DSBs, partly through the dephosphorylation of γH2AX. More importantly, blocking NSD2-mediated methylation of PTEN by expressing methylation-deficient PTEN mutants or inhibiting NSD2 improves the sensitivity of cancer cells to the combination use of a PI3K inhibitor and DNA-damaging agents in both cell culture and in vivo xenograft models. Interestingly, NSD2-II (long) but not NSD2-I (short) display a high binding affinity with PTEN. These studies revealed a novel mechanism that PTEN regulates the DNA damage repair in a methylation- and protein-phosphatase-dependent manner. Therefore, the γH2AX-MDC1-NSD2 axis is critical in the DNA damage repair machinery.
NSD2 Regulates the Epithelial-Mesenchymal Transition (EMT) Process.
As the most common cause of death among cancer patients, tumor metastasis goes through multiple steps, during which cancer cells spread from the primary tumor site into distant organs and establish a secondary tumor site.69 EMT process has been reported to be critical in tumor development and progression.70–73 After activation of the EMT process, the adhesive epithelial cells convert into migratory and invasive mesenchymal cells, allowing them to migrate through the extracellular matrix. Thus, cancer cells disseminate to the whole body based on this mechanism. The highly conserved twist family bHLH transcription factor (TWIST), a well-known master regulator of embryonic morphogenesis, plays a crucial role in tumor metastasis.70 Inhibiting the expression of TWIST in highly metastatic breast cancer cells specifically suppresses the cell metastasis from the breast to the lung. In addition, the ectopic expression of TWIST decreases E-cadherin-mediated cell-cell adhesion, activates the mesenchymal markers, and promotes cell motility, indicating that TWIST facilitates tumor metastasis by enhancing the EMT process. TWIST is a crucial regulator in the development and progression of PCa, and its activation is closely associated with tumor metastasis.71 Additionally, inhibiting the activity of the TWIST gene is a potential therapeutic approach for suppressing cell growth, invasion, and metastasis of androgen-independent PCa cells.
Several studies have reported that hyperactivation of NSD2 promotes the metastasis and invasiveness of tumor cells by strongly activating TWIST and then activating the EMT process (Figure 5).15, 23, 74, 75 Moreover, down-regulation of NSD2 enhances the expression of E-cadherin protein, while decreases N-cadherin and vimentin proteins. The downregulation of NSD2 inhibits the metastasis of renal cell carcinoma (RCC) by blocking the EMT process.74 In addition, NSD2 was found overexpressed in PCa cells, and its knockdown in DU145 and PC-3 cells results in the suppression of cell proliferation and colony formation in soft agar and remarkably diminishes cell migration and invasion.75 In contrast, overexpression of NSD2 facilitates cell metastasis and invasion, accompanied by an EMT process. Mechanism studies showed that overexpressed NSD2 interacts with TWIST1 and strongly activates its activity, increasing H3K36me2 expression, which promotes the EMT process, migration, and invasion of PCa cells. NSD2 is a critical driver of an EMT process in PCa cells, partly depending on the activation of the TWIST1 gene. The t(4;14) chromosomal translocation, accounting for 15% in MM patients, could promote the overexpression of NSD2, which facilitates tumor dissemination and accelerates disease progression and relapse.15 Furthermore, TWIST, a downstream target of NSD2, was found to activate the expression of a more extensive EMT-like gene signature in t(4;14)-positive MM and promote tumor metastasis both in vitro and in vivo, which may partly be responsible for the aggressive features of MM patients harboring t(4;14) translocation. Notably, the expression level of mesenchymal markers N-cadherin and vimentin are also observed to be upregulated in t(4;14)-positive MM patients.76, 77 Interestingly, NSD2 is also detected to be aberrantly overexpressed in aggressive, metastatic solid tumors, such as neuroblastoma, hepatocellular carcinoma and ovarian carcinoma.13
NSD2 Regulates the EZH2-NSD2-Histone Methyltransferase (HMTase) Axis.
EZH2, an epigenetic regulator that mediates the tri-methylation of H3K27 (H3K27me3), plays a driving role in tumor initiation, progression, metastasis, and invasion and has been successfully validated as a druggable target.26, 78–80 Several studies have revealed that abnormal expression of EZH2 and NSD2 exert a synergetic effect on the occurrence and development of many malignant tumors, such as ovarian clear cell carcinoma (OCCC),81 myeloma,82 and breast cancer.83 Mechanism studies showed that there exists an HMTase axis linking EZH2 and NSD2 (EZH2-NSD2-HMTase axis), which is regulated by a network of microRNA (miR-203, miR-26a, and miR-31), and the oncogenic functions of EZH2 correlate with NSD2 activity (Figure 5). Activated EZH2 induces the expression of NSD2 through H3K27me3-mediated suppression of microRNAs, leading to the progression of tumors.84
NSD2 is overexpressed in OCCC cells and promotes cell proliferation by increasing the expression of H3K36me2.81 Moreover, there exists a significant correlation between NSD2 and EZH2 mRNA levels. Importantly, the NSD2 mRNA level and the levels of two important histone methylation markers, H3K36me2 and H3K27me3, that catalyzed by NSD2 and EZH2, respectively, are significantly decreased after EZH2 knockdown in OCCC cells. However, the knockdown of NSD2 showed almost no effect on the levels of EZH2 and H3K27me3. These results suggest that NSD2 is a downstream target of EZH2, whose expression is regulated by EZH2. The overexpression of NSD2 not only increases the expression level of H3K36me2 but also decreases H3K27me3, which subsequently stimulates the hyperactivation of EZH2, sensitizing NSD2-overexpressing cells to EZH2 inhibition.82 This study indicates the existence of an interplay between NSD2 and EZH2 in myeloma oncogenesis. NSD2 and EZH2 were found to be overexpressed in breast cancer, especially triple-negative breast cancer (TNBC), and tightly associated with pathological tumor grade and lymph node metastasis.83 Moreover, the knockdown of EZH2 significantly suppressed the proliferation, migration, and invasion of TNBC cells, reducing the expression levels of NSD2, H3K27me3, and H3K36me2. Furthermore, the down-regulation of NSD2 expression in EZH2-overexpressing cells alleviates the EZH2-caused oncogenic effects. More importantly, the EZH2-NSD2 axis participates in cell division, mitotic nuclear division, and the mitotic cell cycle transition in TNBC. Thus, these findings demonstrated that the EZH2-NSD2 axis plays key roles in tumor initiation, progression, migration, and invasion and may serve as a predictive marker for poor prognosis.
Other NSD2-Mediated Signaling Pathways.
In addition to the mechanisms mentioned above, some other NSD2-mediated signaling pathways have also been reported, such as the Wnt/β-catenin and NF-κB pathways, promoting tumorigenesis, proliferation, metastasis, and invasion (Figure 5).85, 86 It was reported that NSD2 could promote oncogenesis through regulation of the Wnt pathway in bladder and lung cancer cells. NSD2 interacts with β-catenin to initiate the Wnt pathway and then transcriptionally activates the β-catenin/Tcf-4 complex downstream gene CCND1.85 NSD2, a coactivator of NF-κB, directly interacts with NF-κB to activate the downstream genes, including interleukin-6 (IL-6), interleukin-8 (IL-8), vascular endothelial growth factor A (VEGFA), cyclin D, B-cell lymphoma 2 (Bcl-2), and survivin, in castration-resistant PCa cells.86 Interestingly, NSD2 is engaged in cytokine-mediated recruitment of NF-κB and acetyltransferase p300, contributing to the hyperacetylation of histone. Importantly, NSD2 overexpression in PCa cells is tightly associated with the activation of NF-κB. Notably, NSD2 overexpression plays an important role in cell proliferation, survival, and tumor growth, which is strongly induced by NF-κB target genes, including tumor necrosis factor-alpha (TNF-α) and IL-6. This study suggests that NSD2 contributes to cell proliferation, survival, and growth via activating the NF-κB signaling pathway. Several studies have reported that NSD2 also plays a critical role in normal DNA replication and cell-cycle progression, whose expression is dynamically regulated throughout the cell cycle.57, 87 NSD2 degradation was found to be induced during the S phase of cell mitosis in a cullin-ring ligase 4-Cdt2 (CRL4Cdt2)- and proteasome-dependent manner. Importantly, NSD2-depletion in cells resulted in DNA replication defects and a decreased association between pre-replication complex (pre-RC) factors and chromatin.57
THE ROLES OF NSD2 IN VARIOUS HUMAN CANCERS
Hyperactivation of NSDs, including NSD1, NSD2, and NSD3, caused by either overexpression or point mutations, have been reported to be tightly associated with the occurrence and progression of diverse cancer types,11, 21–23 including hematological malignancies, such as MM, ALL, etc.22 and solid tumors, such as colon cancer, breast cancer, lung carcinoma, etc.21, 23 Overexpression of NSD2 in MM patients contributes to tumor proliferation, dissemination, progression, and rapid relapses.14 NSD2 is one the most frequently mutated epigenetic regulators in some different cancer types, especially pediatric cancers, such as ALL.17 Mutation of NSD2 in tumor cells leads to reduced apoptosis and accelerated proliferation, clonogenicity, migration, and adhesion.18 Other than cancers, NSD2 is also associated with other diseases, such as inflammatory disorders, viral infections, and autoimmune diseases. Importantly, cancer is the most directly related and extensively studied disease associated with NSD2 abnormalities compared to other diseases. The functions and possible mechanisms of NSDs in the tumorigenesis of diverse cancers are summarized in Table 1. In this section, we mainly focus on the roles of NSD2 in various cancers.
Table 1.
Overview of various cancers associated with NSD lysine methyltransferases (KMTases) dysregulation.
| Cancer type | Enzyme | Function | Mechanism | |
|---|---|---|---|---|
| Acute lymphoblastic leukemia | NSD2 | Reduces apoptosis and enhances proliferation, clonogenicity, adhesion, and migration | NSD2 mutation activates enzyme activity and other transcriptional pathways17, 18 | |
| Multiple myeloma | NSD2 | Contributes to neoplastic transformation, and promotes cell growth and adhesion | Aberrant expression of genes involved in cell cycle, apoptosis, DNA repair, and adhesion43, 88 | |
| Mantle cell lymphoma | NSD2 | Promotes cell proliferation and regulates cell cycle89, 90 | Unknown | |
| Acute myeloid leukemia | NSD1 | Promotes tumorgenesis | Fusion of NUP98-NSD1 enforces the expression of proto-oncogenes HOXA7, HOXA9, HOXA10, and Meis191, 92 | |
| NSD2 | Promotes cell proliferation93, 94 | Unknown | ||
| NSD3 | Promotes tumorgenesis | Fusion of NUP98-NSD3 genes95 | ||
| Diffuse large B-cell lymphoma | NSD2 | Promotes cell proliferation17, 96 | Unknown | |
| Chronic myeloid leukemia | NSD2 | Promotes cell proliferation17, 96 | Unknown | |
| Hodgkin’s lymphoma | NSD2 | Promotes cell proliferation17, 96 | Unknown | |
| Breast cancer | NSD2 | Promotes TNBC cell survival and invasion; develops resistance to EGFR-targeted drugs | ADAM9-EGFR-AKT signaling pathway97 | |
| Promotes cell proliferation, migration, and metastasis | Wnt/β-catenin signaling pathway98 | |||
| Develops resistance to tamoxifen | Erα gene signaling pathway99; the whole glucose metabolism process100 | |||
| NSD3 | Promotes cell growth and survival | Wnt signaling pathway101 | ||
| Urinary cancers | Renal cancer | NSD2 | Promotes cell metastasis | EMT process74 |
| Prostate cancer | NSD1 | Promotes tumorigenesis102 | Unknown | |
| NSD2 | Cell proliferation, migration, and invasion | EMT process and TWIST175; PTEN and PI3K/AKT signaling pathway103 | ||
| Cell metastasis | AKT, RICTOR, and Rac1 signaling pathway103, 104 | |||
| Female reproductive cancers | Endometrial cancer | NSD2 | Promotes tumor progression105 | Unknown |
| Cervical cancer | NSD2 | Promotes cell proliferation, migration, and invasion | eNOS signaling pathway106; AKT/MMP-2 signaling pathway107; TGF-β1/TGF-βRI/SMADs signaling pathway108 | |
| Lung cancer | NSD1 | DNA hypomethylation | NSD1 inactivation109 | |
| NSD2 | Promotes cell proliferation | RAS-mediated transcriptional responses110 | ||
| NSD3 | Regulates MYC-driven tumors | BRD4-NSD3-MYC signaling pathway111 | ||
| Regulates tumorigenesis | Enhances the dimethylation of H3K36112 | |||
| Osteosarcoma | NSD2 | Promotes cell proliferation and metastasis | EMT process113 | |
| Reduces apoptosis and chemosensitivity | ERK-AKT signaling pathway114 | |||
| NSD3 | Promotes cell proliferation, migration, and invasion115 | Unknown | ||
| Hepatocellular carcinoma | NSD2 | Promotes DNA damage repair116 | Unknown | |
| Head and neck squamous cell carcinoma | NSD1 | Causes cell cycle abnormalities117 | Unknown | |
| NSD2 | Promotes oncogenesis | NEK7-driven signaling pathway118 | ||
| NSD3 | Drives cell cycle progression119, and enhances DNA synthesis and cell cycle progression120 | CDC6 and CDK2 regulation119; EGFR signaling pathway120 | ||
| Skin squamous cell carcinoma | NSD2 | Promotes cell proliferation | P53 signaling pathway121 | |
| Neuroblastoma and glioma | NSD1 | Promotes colony formation and cell growth | NSD1 inactivation caused by CpG island promotor hypermethylation122 | |
| NSD2 | Enhances aggressiveness123, 124 | Unknown | ||
| Colorectal cancer | NSD1 | Promotes cell growth | NF-κB signaling pathway125 | |
| NSD2 | Inhibits cell apoptosis | Targeting anti-apoptotic BCL2126, 127 | ||
| NSD3 | A putative cancer driver gene128 | Unknown | ||
| Stomach carcinoma and anal canal carcinoma | NSD2 | Promotes tumor progression13 | Unknown | |
| Pancreatic adenocarcinoma | NSD3 | Promotes cell proliferation, migration, and invasion129 | Unknown | |
| Bladder cancer | NSD3 | Promotes cell cycle progression at mitosis | CCNG1 and NEK7 signaling pathway130 | |
| Nuclear protein in testis midline carcinoma | NSD3 | Blocks differentiation and maintains tumor growth131 | Unknown | |
| Pelvic high-grade serous carcinoma | NSD3 | Promotes tumor progression | Activates the NSD3-BRD4-CHD8 signaling pathway132 | |
Hematological Malignancies.
Acute Lymphoblastic Leukemia (ALL).
ALL is a clonal malignant disease that affects the blood and bone marrow. ALL originates in a single cell and is characterized by the overproduction of immature B-cells (white blood cells), called lymphoblasts or leukemic blasts, that are phenotypically similar to the normal B-cell differentiation stages.133 Accumulated white blood cells finally suppress normal hematopoiesis and the infiltration of many vital organs.134 Relapsed ALL has been a leading cause of pediatric cancer mortality, accounting for 15–20%.20, 134–136 Cell lines harboring NSD2 translocations showed increased H3K36me2 level due to the enhanced methyltransferase activity.17, 88 NSD2 p.E1099K mutation was identified in non-translocated ALL cell lines, leading to accumulated H3K36me2.17 p.E1099K variant induced the hyperactivation of NSD2 and promoted cell proliferation. NSD2 knockdown selectively inhibited the proliferation of NSD2-mutant cell lines and hampered the tumor growth in an NSD2-mutant ALL xenograft mice model, suggesting NSD2 plays a vital role in ALL. The NSD2 p.E1099K alteration accounts for 14% of t(12;21) ETV6-RUNX1-containing ALLs. Point mutations taking place in NSD2 catalytic SET domain (E1099K, D1125N, and T1150A) enhanced its interaction with nucleosomes, causing the hyperactivation of its enzymatic activity, leading to higher proliferation rates.17, 18, 89, 137, 138 Recent studies conducted through high-throughput drug screens revealed that overactivation of NSD2 catalytic activity induced by E1099K mutation is closely associated with glucocorticoid resistance.138 Collectively, NSD2 may serve as a potential therapeutic target for pediatric ALLs.
Multiple Myeloma (MM).
MM is one type of hematological malignancy characterized by the uncontrolled growth of plasma cells in the bone marrow, accounting for approximately 10% of hematological malignancies and 1.6% of all U.S. deaths caused by cancer.139–141 As estimated in 2022, more than 34,470 new cases were diagnosed, and 12,640 deaths were caused in the United States.142 Both the incidence of new cases and deaths of MM are slightly higher in men than women and two-fold or more in African-Americans than Whites.143 The median diagnosis age of MM patients is about 65 years.144 Overexpression of NSD2 appeared in about 15–20% of MM patients, which was featured by a translocation in the chromosome 4 t(4;14). Overexpression of NSD2 leads to apparent H3K36me2 increase and significant H3K27me3 decrease, which enhances the recruitment of EZH2 and increases the sensitivity of cells to EZH2 inhibition.4, 43, 82 The dysregulation in H3K36me2 distribution has been considered as one of the first steps of MM oncogenic program, owing to the consequent alteration of gene expression participated in cell growth, adhesion, chromatin accessibility, and DNA damage response.4, 87, 88, 145, 146 Cancer cells with overexpressed NSD2 exert resistance to drug treatment, which the enhanced capacity of DNA damage repair may explain.147 The overexpression of NSD2 prompts the aberrant expression of the c-MYC gene through the aberrant repression of miR126.148 TP53, c-MYC, and other related genes have been supposed as the key targets of NSD2 in MM patients, whose highly aberrant expression levels are directly associated with the aggressiveness and high risk in different subtypes of myeloma patients (both NSD2-related and NSD2 non-related patients).149, 150
Mantle Cell Lymphoma (MCL).
MCL belongs to a subtype of B-cell non-Hodgkin lymphoma (NHL), which takes up 5%−10% of all lymphomas.151, 152 MCL is defined as a unique incurable, rare B-cell malignancy that possesses a generally aggressive and albeit heterogeneous clinical course and is subjected to produce resistance and relapse after the initial response to therapy.151, 152 Gene mutations occurring in MCL pose a big challenge for the diagnosis, pathogenesis, prognosis, and therapeutic management and response with MCL patients.90, 153, 154 As one of the frequently mutated genes in MCL patients, NSD2 is tightly related to the poor prognosis.154–156 Based on the analysis of patient-level data in 2,127 MCL patients, NSD2 mutations with a prevalence of 15% were observed.152 Intriguingly, accumulated evidence indicates that both E1099K and T1150A mutations take place in MCL patients, which accelerates the malignancy process of cancer cells through hyperactivating the catalytic potency of NSD2 enzyme. NSD2 can be hyperactivated by E1099K mutation alone, T1150A mutation alone, or a combination of E1099K and T1150A mutations, resulting in the overexpression of genes associated with cellular proliferation and cell cycle regulation.89, 90 Additional hydrogen bonds can be formed once the T1150A mutation occurs, which helps the histone to insert into the catalytic pocket of the NSD2 enzyme.157 Therefore, developing potent inhibitors that selectively target NSD2 proteins, including T1150A and E1099K mutations, will likely offer new therapeutic options for MCL patients.
Acute Myeloid Leukemia (AML).
AML is the most common type of leukemias among adults, accounting for about 80% of all leukemias, which is characterized by clonal expansion of immature “blast cells” in the peripheral blood and bone marrow, subsequently leading to ineffective erythropoiesis and bone marrow failure.158–160 AML, a highly heterogeneous disease with a variable prognosis, can result from genetic mutations, chromosomal translocations, or changes in molecular levels.161 Although many therapies have been developed for treating patients with AML, there still exists an unmet clinical need for new therapies. As mentioned above, NSD2 plays a significant role in developing AML patients.17 However, the role and function of NSD2 in other leukemias, such as AML, has not been fully explored. The loss of function of SETD2 was noticed to play a critical role in facilitating the initiation and progression of leukemias characterized by MLL fusion, MLL-PTD or AML1-ETO, through decreased H3K36me3.162 In addition, the reduction of H3K36me3 caused by SETD2 mutations may potentiate leukemic transformation and progression in cooperation with another genetic or epigenetic abnormality through an independent and distinct epigenetic mechanism.162 SETD2 mutations occurred in many hematopoietic malignancies and non-hematopoietic malignancies, indicating that the SETD2-H3K36me3 pathway may serve as a common tumor-suppressive mechanism for cancer, thereby providing a new chance for developing cancer diagnostics and therapeutics.162 Furthermore, AML patients carrying SETD2 mutations were reported to produce resistance to chemotherapy, partially attributed to cell cycle checkpoints’ alterations.163
While SETD2 and NSD2 share a similar catalytic domain, it is reasonable to believe that NSD2 mutations may have a close association with the transformation and progression of AML. Moreover, studies showed that NSD1 is essential for developing the human body and is easily mutated in AML patients. In 5% human AMLs, NSD1 fuses to nucleoporin-98 (NUP98) to form the NSD1-NUP98 fusion gene owing to the recurring t(5;11) (q35;p15.5) translocation.91 NUP98-NSD1 fusion was demonstrated to be able to induce AML in vivo, sustain self-renewal of myeloid stem cells in vitro, and enforce the expression of several proto-oncogenes, including HoxA7, HoxA9, HoxA10, and Meis1.92 Further mechanisms studies revealed that NUP98-NSD1 fusion binds genomic elements adjacent to HoxA7 and HoxA9 oncogenes, maintains histone H3K36 methylation and histone acetylation, and prevents EZH2-mediated transcriptional repression of the HoxA locus during the process of differentiation. As reported, the NUP98-NSD3 fusion gene in AML patients was associated with t(8;11)(p11.2;p15).95 Patients with radiation-associated myelodysplastic syndrome (r-MDS) were detected to carry chromosome abnormalities, including t(8;11)(p11;p15) and del(1)(p22p32).164 Therapy-related myelodysplastic syndrome (t-MDS), a heterogeneous disease of pluripotent hematopoietic stem cells, characterized by bone marrow (BM) failure, often evolves into AML and displays a poor prognosis. Simultaneously, the presence of NSD3-NUP98 fusion transcripts was observed and might be related to leukemogenesis. Collectively, NSD1 and NSD3 proteins play important roles in the development and progression of AML.
Since NSD family members share similar structural domains and functions, it is reasonable to believe that NSD2 also serves critical roles in AML. So far, the definitive relationship between NSD2 and AML remains unclear and needs further elucidation.165 Functional alterations of NSD1 and NSD2 were found to be associated with the development of erythroleukemia, a subtype of AML. Indeed, NSD1 has been reported to serve as a critical regulator in erythroid differentiation, and its knockout significantly suppressed the proper erythroblast maturation and promoted erythroleukemia in mice.93 Recently, it was reported that MOLM13 cells (a kind of AML cell line), harboring an FMS-like tyrosine kinase 3 (FLT3) mutation associated with a worse prognosis, show slower proliferation after the first a few days of treatment with NSD2 inhibitor IACS17596 (the structure was not disclosed), suggesting NSD2 is closely associated with the proliferation of AML cells.94 More detailed mechanism of action for NSD2 inhibitor slowing the proliferation of AML cells needs to be further investigated.
Other Hematological Malignancies.
NSD2 plays a key role in the development and evolution of many hematological malignancies. Overall, the main hematological tumors associated with the alterations of NSD2 function caused by overexpression, translocations, and/or mutations have been described above. With the MS profiling approach, global histone modifications were characterized in 115 cell lines from the Cancer Cell Line Encyclopedia (CCLE), a total of 1,000 human cancer lines that have received extensive genomic and pharmacologic annotation.17 Based on prior evidence of epigenetic dysregulation, they prioritized hematological malignancies, including ALL, MM, AML, and other hematological tumors, such as diffuse large B-cell lymphoma (DLBCL), chronic myeloid leukemia (CML), Hodgkin’s lymphoma, etc. Results showed that among 115 different cell lines, six ALL cell lines and one MM cell line harbor E1099K mutation, and six MM cell lines contain t(4;14) translocation. Besides E1099K, other NSD2 variants, including G945fs and K361Q, were found in AML (CMK115) and CML (MOLM6) cell lines. The AACR (American Association for Cancer Research) Project GENIE (Genomics, Evidence, Neoplasia, Information, Exchange), an international consortium, reveals that NSD2 mutations occasionally occur in a small percentage of other hematological tumors, such as non-Hodgkin lymphomas (2.86%), Hodgkin lymphoma (1.79%), diffuse large B-cell lymphoma (2.58%), and chronic lymphocytic leukemia/small lymphocytic lymphoma (0.53%),96 which is highly consistent with the data shown in COSMIC database.22, 166 These results collectively support that although NSD2 mutations are associated with the development and progression of many different types of hematological malignancies, the most relevant hematological cancers with the alteration of NSD2 function disclosed by current studies are still B-ALL, MM, MCL, and AML.
Solid Tumors.
Breast Cancer.
Breast cancer, one type of female malignancy, is characterized by high incidence and mortality.167, 168 Nowadays, the prevention, diagnosis, and treatment of breast cancers, especially triple-negative breast cancer (TNBC), remain a significant challenge worldwide,169 which deserves more efforts to develop more potent and efficient medications. Accumulated evidence suggests that NSD2 plays a significant role in the development of breast cancers and may act as a potential novel therapeutic target. Overexpression and hyperactivation of NSD2 have been found in both breast cancer tissues and cells. Aberrant alterations in the function of NSD2 were detected to be closely associated with earlier disease-related death, which is highly consistent with the published tumor datasets that NSD2 is overexpressed in TNBC and high levels of NSD2 mRNA correlates (P = 0.027) with the poor survival of TNBC patients.97
Mechanistically, NSD2 modulates the survival and invasion of TNBC cells by mediating the ADAM9-EGFR-AKT signaling pathway. The hyperactivation of NSD2 abnormally activates the EGFR-AKT signaling pathway, which contributes to the TNBC cell resistance to EGFR-targeted drugs.97 Additionally, based on the real-time PCR and western blotting analysis, NSD2 was observed to be significantly upregulated in breast cancer cells and tissues of clinical patients, and high level of NSD2 is tightly related to poor prognosis.98 Besides, downregulating the expression level of NSD2 via gene knockdown or silencing results in a remarkable decrease in cancer cell proliferation, migration, and metastasis through inhibiting the Wnt/β-catenin signaling pathway. Besides, the overexpression of NSD2 is highly associated with the therapy-resistance that occurred in breast cancer cells. Tamoxifen, one kind of endocrine therapy, is widely used to prevent and treat breast cancer patients with estrogen receptor alpha (ERα)-positive postmenopausal characteristics. Overexpressed NSD2 was noticed to upregulate the expression of ERα gene in tamoxifen-resistant breast cancer cells.99 NSD2 expression was observed to be highly elevated in tamoxifen-resistant breast cancer cell lines and clinical tumors, and correlates with disease recurrence and poor diagnosis,100 suggesting that the overexpression of NSD2 plays a critical role in drug-resistant breast cancers. A further study disclosed that NSD2-mediated tamoxifen-resistant in breast cancers is characterized by alterations of the whole glucose metabolism process, including the enhanced activity of pentose phosphate pathway (PPP), increased level of nicotinamide adenine dinucleotide phosphate (NADPH) and decreased level of ROS, which is realized by upregulating the genes expression of glucose-6-phosphate dehydrogenases (G6PD), hexokinase 2 (HK2), and TP53-mediated glycolysis regulatory phosphatase TIGAR.100 Recently, DNZep, an indirect small-molecule NSD2 inhibitor, was found to induce the degradation of NSD2 protein and inhibit the expression of NSD2 target genes, including G6PD, HK2, TIGAR, and GLUT1.170 In addition, another study revealed that NSD2 upregulates the expression of the ERα gene via bromodomain and extra-terminal (BET) proteins BRD3/4.99 Therefore, small molecule inhibitors targeting NSD2 may provide a novel therapeutic option for breast cancer patients, including those resistant to traditional therapies.
Urinary System Cancers.
Kidney and renal pelvis cancer has a high occurrence and lethality that seriously affects human health worldwide, with 431,288 new cases and 179,368 deaths estimated for 2020.167 Moreover, it was estimated to be the 6th and 8th most widespread cancer in men and women, respectively. Renal cell carcinoma (RCC) takes up about 85% of all primary kidney neoplasms, recognized as one of the top 10 prevalent cancers worldwide. Metastatic RCC (mRCC) frequently and easily resists traditional radiotherapy and chemotherapy.74 Developing new drugs with novel mechanisms of action is highly anticipated to improve the therapeutic outcomes of mRCC patients. Based on the bioinformatic analysis NSD2 expression was noticed to be considerably upregulated in many types of renal cancers, especially in metastatic clear cell RCC (ccRCC), illustrating that NSD2 plays a critical role in the process of RCC carcinogenesis.74 Furthermore, the silencing of NSD2 potently prevents cell metastasis and invasion by inhibiting the EMT process in RCC. The detailed mechanism that NSD2 participates in the progression of metastatic RCC via suppressing EMT remains largely unknown and will be further explored. Taken together, NSD2 displays a significant role in RCC metastasis, and inhibition of NSD2 may provide a promising therapeutic alternative for patients with mRCC. Highly potent and selective NSD2 inhibitors are urgently needed as pharmacological tools for the biological function studies of NSD2, underlying mechanism exploration, and the mRCC treatment.
In 2020, prostate cancer (PCa) was estimated to be the second most frequent cancer and the fifth leading cause of cancer deaths among men worldwide, with about 1.4 million new cases and 375,000 deaths.167, 171 Advanced metastatic PCa patients display high mortality once androgen-depletion therapy is ineffective.75 Comprehensive knowledge of the mechanisms of PCa progression and a better understanding of metastatic properties may contribute to identifying new therapeutic targets and drugs. NSD2 has been reported to be associated with the advancement of PCa.75, 86, 172 NSD2 was observed to be overexpressed in PCa tissue samples and cell lines, and its knockdown or inhibition by small molecules, such as MTCP-39, could significantly suppress the proliferation and invasion of DU145 cells.172 NSD2 was found to be overexpressed and hyperactivated in PCa cells, facilitating cell migration and invasion, accompanied by the EMT process.75 Excitingly, the depletion or silencing of NSD2 impairs the proliferation, migration, and invasion of cancer cells. Mechanism study manifested that the overexpression of NSD2 strongly activates twist family bHLH transcription factor 1 (TWIST1) to promote the invasion, metastasis, and EMT process of PCa cells. Moreover, when TWIST1 was depleted in NSD2-overexpressing PCa cells, it was observed that cell invasion and EMT were blocked, suggesting that TWIST1 is an important partner of NSD2.75 NSD2 is aberrantly overexpressed in human lethal PCa cells and silencing of NSD2 blocked the metastasis of mouse allografts in vivo, indicating that NSD2 is a driver of metastatic PCa progression.104 Likewise, a recent study disclosed that NSD2 serves as a critical modulator of PCa resident immune pathways, suggesting that NSD2 may be a potential novel therapeutic target for PCa treatment.173
PCa shares two significant hallmarks: loss of phosphatase and tensin homolog (PTEN) and activation of the PI3K/AKT signaling pathway.174, 175 Interestingly, it was found that these two alterations alone are insufficient for cells to acquire metastatic traits.103 Additionally, NSD2 is a critical driver for indolent PTEN-null tumors to become metastatic PCa. Molecular characterization unveiled that upregulated AKT due to PTEN loss directly phosphorylates NSD2 at the S172 site to inhibit its degradation by CRL4Cdt2 E3 ligase, which contributes to the effects of NSD2 on PCa metastasis. Overexpressed NSD2 transcriptionally enhances the expression of RICTOR, a key member of mTOR complex 2 (mTORC2), to further hyperactive AKT.103 Therefore, the AKT/WHSC1/mTORC2 signaling cascade is a vicious feedback loop that activates the AKT signaling continuously. Furthermore, NSD2 was found to increase the motility of PCa cells by positively regulating Ras-related C3 botulinum toxin substrate 1 (Rac1) transcription. The biological importance of an NSD2-mediated signaling cascade is substantiated by patient sample analysis in which WHSC1 signaling is tightly correlated with disease progression and recurrence. These findings manifest an unexpected but important correlation between an epigenetic regulator NSD2, and critical intracellular signaling molecules (AKT, RICTOR, and Rac1), to promote PCa metastasis.103 Therefore, NSD2 is a potential drug target, and its inhibition may serve as an efficient therapeutic strategy against PCa.
Female Reproductive System Cancers.
In 2020, invasive cervical cancer (CC) was estimated to be the fourth most commonly diagnosed cancer and the fourth leading cause of cancer deaths among women worldwide, with appropriately 604,000 new cases and 342,000 deaths.167 Radiotherapy and surgical therapy greatly benefit CC patients; however, numerous late-stage patients always suffer from metastasis and recurrence heavily, particularly in developing countries.176 Therefore, increasing attention has been focused on developing effective targeted therapies to treat CC. Several studies have unveiled that NSD2 is significantly overexpressed in the CC tissues and cells, is closely correlated with adverse prognosis, and promotes cervical cancer cell proliferation, migration, and invasion.106–108
NSD2 expression was found significantly upregulated in CC tissues and cells and tightly correlated with the International Federation of Gynecology and Obstetrics (FIGO) stage and differentiation.106 Moreover, they noticed that NSD2 knockdown prevents proliferation, migration, and invasion of endothelial C33A cells, characterized by overexpressed nitric oxide synthase (eNOS), suggesting that NSD2 may participate in regulating the progression of CC cells via the eNOS signaling pathway. Furthermore, the depletion of NSD2 inhibits angiogenesis in human umbilical vein endothelial cells (HUVECs). Therefore, NSD2 is a poor prognostic indicator of CC and is considered a novel potential therapeutic target for CC patients. Likewise, NSD2 mRNA levels were identified to be significantly overexpressed in CC cells, contributing to cell proliferation, migration, and invasion.107 NSD2 knockdown markedly suppressed CC cell proliferation, migration, invasion, and silencing of NSD2 inhibited tumor growth in a xenograft model. NSD2 overexpression may promote cervical carcinogenesis by activating the AKT/MMP-2 signaling pathway. NSD2 was found to exert its function in the development process of CC through a manner of gradual up-regulation from the normal cervix (NC) to cervical carcinoma in situ (CIS) and then to invasive cervical cancer (ICC).108 Moreover, NSD2 knockdown inhibited CC cell proliferation, and NSD2 deletion markedly suppressed CC cell migration and invasion. Inconsistent with in vitro results, in vivo experiments demonstrated that NSD2 knockdown inhibits tumor growth and suppresses the development of tumor metastasis. Furthermore, the overexpressed NSD2 regulates the progression of CC cells by activating the transforming growth factor-β1 (TGF-β1)/TGF-βRI/SMADs signaling pathway. Thus, targeting NSD2 inhibition may be a promising therapeutic strategy for overcoming metastasis in CC cells.
NSD2 was also found overexpressed in endometrial cancers compared to normal endometrium.105 Furthermore, the patients with high NSD2 expression showed poor overall survival and disease-free survival compared with patients with normal or low NSD2 expression, indicating that NSD2 overexpression may serve as a new prognostic biomarker and a potential therapeutic target for patients with endometrial cancer.
Lung Cancer.
Several studies have reported that all three NSD members are frequently overexpressed in lung cancer.109–112, 177, 178 However, the contribution of NSD2 to the development and progression of lung cancer is still poorly understood. To validate the expression level and the role of NSD2 in lung cancer, the data from The Cancer Genome Atlas (TCGA) were analyzed.110 Based on the analysis of mRNA levels, NSD2 is highly overexpressed in lung cancers, such as adenocarcinoma (AD) and squamous cell carcinoma (SCC), compared with normal lung tissue obtained from the same patients, which is in agreement with previous reports. Moreover, NSD2 promotes the proliferation of a series of lung cancer cell lines by enhancing oncogenic RAS-mediated transcriptional responses. Furthermore, the combinatorial therapy, composed of MEK or BRD4 inhibitors and NSD2 inhibition, effectively treats oncogenic RAS-driven lung cancers with NSD2 overexpression. Similarly, other NSD members, including NSD1 and NSD3, have also been reported to be overexpressed in a subset of lung cancers and play a critical role in the proliferation of tumor cells.109, 111, 112, 177, 178
Osteosarcoma (OS).
OS represents one of the most frequent primary malignant bone tumors, with an annual incidence of about one to three cases per million worldwide. OS mainly affects the health of children and adolescents and is characterized by rapid growth, a high tendency for invasion, metastasis, and poor prognosis.179, 180 The conventional therapies for OS patients are surgical resection combined with chemotherapy and/or radiotherapy, significantly improving the 5-year survival rate of approximately 60–70%. However, owing to the high frequency of recurrence and chemotherapy resistance, the survival time of OS patients is significantly shortened.181, 182 New promising target-based therapies are urgently needed for the survival improvement of OS patients. Several studies have shown that NSD2 is highly overexpressed in OS cells, and its overexpression is tightly associated with unfavorable prognosis and poor five-year overall survival.113, 114 It was reported that upregulated NSD2 could promote the proliferation and invasion of OS cells through a possible mechanism of suppressing E-cadherin and induction of the EMT.113 NSD2 expression was observed to be highly elevated in OS patients, especially in cisplatin-resistant patients, and patients with lower expression level of NSD2 display better prognosis.114 Furthermore, NSD2 knockdown can promote OS apoptosis both in vitro and in vivo. In addition, NSD2 knockdown significantly enhances the sensitivity of OS to cisplatin treatment and subsequently prevents properties closely correlated with cancer stem cells (CSCs). Mechanism analysis illustrated that NSD2 knockdown negatively regulates the expression of apoptosis regulatory proteins BCL2 and SOX2 via the ERK and AKT signaling pathways.114 Together, these results support that NSD2 may be a novel therapeutic target for overcoming OS resistance to chemotherapy.
Other Solid Tumors.
Except for the tumors discussed in the above sections, NSD2 also has been reported to be significantly overexpressed in other solid tumors, such as hepatocellular carcinoma (HCC),116 head and neck squamous cell carcinoma (HNSCC),118, 183 skin squamous cell carcinoma (SCC),121 colorectal cancer (CRC),126, 127 neuroblastoma and glioma,123, 124 stomach and anal canal carcinomas,13 etc. NSD2 was observed to be aberrantly overexpressed in HCC patients, and its overexpression is strongly related to the Edmondson stage and vascular invasion.116 Additionally, NSD2 upregulation in HCC patients is highly correlated with shorter overall survival and disease-free survival. Their study distinctly manifested that abnormal NSD2 upregulation is an independent prognostic factor associated with unfavorable survival in HCC patients. The expression levels of NSD2 and H3K36me2 were found to be significantly upregulated in HNSCC tissues compared to the normal epithelium and significantly associated with histologic grade.183 Moreover, NSD2 knockdown can remarkably suppress cell growth, induce cell apoptosis, and delay cell-cycle progression in multiple HNSCC cell lines, suggesting that NSD2 is crucial for cell proliferation. Furthermore, NSD2 affects cell growth, apoptosis, and cell-cycle progression in HNSCC cells with a mechanism of regulating its direct downstream target NIMA-related-kinase-7 (NEK7) via H3K36me2.118 NSD2 was also found significantly overexpressed in human skin SCC cells and that downregulation of both NSD2 and microRNA-154 resulted in proliferation suppression and apoptosis induction through blocking the P53 signaling pathway.121 NSD2 is also overexpressed in CRC tumoral tissue compared to normal colon tissues and inhibits CRC cell apoptosis by targeting anti-apoptotic BCL2126, 127. Therefore, the NSD2 gene may be a potential prognostic biomarker for CRC patients with poor prognosis. It was reported that NSD2 overexpression isfrequently occurres in neuroblastoma, accounting for 75% of all neuroblastomas (n = 164).123 Moreover, the overexpression of NSD2 in neuroblastomas is significantly associated with poor survival, unfavorable prognosis, and unexpected metastasis. Furthermore, the level of NSD2 in human neuroblastoma cells decreased strongly after retinoic acid-induced differentiation in vitro, which is highly consistent with the results obtained after chemotherapy. This study suggested that NSD2 may be a potential therapeutic target for neuroblastoma treatment. NSD2 was revealed to be overexpressed in stomach and anal canal carcinomas, etc.13 NSD2, along with 103 other proteins, was disclosed to exist only in the glioblastoma multiforme (GBM) tissue samples but not in normal brain cortex samples by mass spectrometric sequencing, which was also validated by western blot and immunohistochemistry experiments.124 Moreover, the expression of NSD2 is positively correlated with glioma cell proliferation activity. In addition, RNA interference (RNAi) can suppress glioma cell growth by inhibiting NSD2 expression. Collectively, these results support that NSD2 is involved in the progression of glioma, while a detailed mechanism remains to be elucidated.
THE ROLES OF NSD2 IN OTHER HUMAN DISEASES
In addition to cancers,184, 185 epigenetic targets also play significant roles in other human diseases,186 such as inflammation,187 viral infections,188 central nervous system (CNS) disorders.189 For example, bromodomain-containing protein 4 (BRD4), a key epigenetic regulator, has been implicated in cancers,190 viral infections,191–194 and inflammations195–197 and their function inhibition by small molecules resulted in anticancer, antiviral, and anti-inflammatory effects,198, 199 which has also been clarified in our previous work.200–203 NSD2 also plays pivotal roles in the pathogenesis of inflammations, viral infections, and autoimmune diseases beyond cancers.204, 205
Both NSD2 and its target gene HDAC2 were revealed to activate the NF-κB signaling pathway inducing the occurrence and progression of inflammation by promoting the release of proinflammatory cytokines.204 Meanwhile, NSD2 can modulate the envelope protein (protein E) of SARS-CoV2 via interactions with BRD4, suggesting that NSD2 may play an important role in the progression of SARS-CoV2.204 Proteolysis protein chimeras (PROTACs) targeting NSD2 degradation are being developed as valuable tools to explore the role of NSD2 in SARS-CoV2 and/or as potential therapeutic agents to treat COVID-19, a SARS-CoV2-related coronavirus disease (https://www.mitacs.ca/en/projects/development-targeted-degradation-nuclear-receptor-binding-set-domain-protein-2-nsd2). Verticillin A was found to be able to awake latent virus and its combined use with one or more antiretroviral drugs may significantly improve the antivirus potency, including human immunodeficiency virus (HIV).206 NSD2 was observed to play an essential role in pathogen infection by regulating the differentiation of follicular helper T (Tfh) cells essential for humoral immune responses, and the deficiency of NSD2 ultimately resulted in delayed viral clearance.205 In addition, both Crotty and Ueno groups found that Tfh cells are also implicated in autoimmune diseases, especially autoantibody-mediated autoimmune diseases.207, 208 Therefore, regulating the activity of NSD2 with small molecules or vaccines may serve as a potential therapeutic strategy for pathogen infection and auto-immune diseases. Notably, beyond an association with various cancers and other diseases mentioned above, NSD2 also plays important roles in the normal development of the human body. NSD2 defects caused by genetic depletion display Wolf-Hirschhorn syndrome (WHS), resulting in mental retardation, severe growth delays, craniofacial dysgenesis, congenital cardiovascular anomalies, and other developmental disorders.51, 57
TARGETING NSD2 AS A POTENTIAL THERAPEUTIC STRATEGY FOR CANCERS AND OTHER DISEASES
As discussed earlier, HKMTases are key epigenetic modulators that catalyze the mono-, di-, or tri-methylation of specific lysine on histone H3 and H4. HKMTases have received increasing attention for their significant role in gene regulation, DNA replication, and cell differentiation. HKMTases are associated with cancer and other diseases and have been demonstrated as potential therapeutic targets. NSD2, one member of HKMTases, serves as an important epigenetic regulator that catalyzes the formation of H3K26me1 and H3K26me2. Studies have shown that NSD2 participates in several cellular processes, including DNA damage repair, EMT and cell cycle regulation, etc. Its translocation, mutations, and/or overexpression strongly correlate with cell proliferation, migration, invasion, and apoptosis. Furthermore, NSD2 depletion can suppress cell proliferation, induce cell proptosis, and delay cell-cycle progression. Moreover, the abnormal expression of NSD2 is an oncogenic driver in diverse cancers, including hematological tumors (e.g., ALL, MM, MCL, AML) and solid tumors (e.g., PCa, breast cancer, lung cancer, and renal cancer). Therefore, selectively targeting NSD2 with small molecules may offer a potentially viable therapeutic approach for treating various cancers and other relevant diseases.
To date, several small molecule NSD2 inhibitors have been reported. However, most currently available NSD2 inhibitors have limited success due to either poor selectivity, limited potency, or less optimal drug-like properties. Therefore, the development of potent and selective NSD2 inhibitors remains an unmet need. Several challenges exist in the discovery of NSD2 inhibitors, significantly impacting the efficiency and outcomes of drug development. These challenges include: 1) The exact roles and mechanisms of action of NSD2 in various cancers and other related diseases are still not fully understood; 2) A unique autoinhibitory loop exists in the catalytic SET domain of all NSD family members that blocks the binding of small molecule inhibitors with the active site, thereby significantly impeding the development of potent and selective NSD2 inhibitors; 3) Since all NSD family proteins (NSD1, NSD2 and NSD3) and other HKMTases contain a conserved catalytic SET domain, most of currently available NSD2 inhibitors show poor selectivity, which may cause some side effects and hinder the pharmacological studies on the exact effects with specific NSD2 targeting and inhibition; 4) While several NSD2 inhibitors showed excellent enzymatic inhibitory activity, their cellular activity and/or in vivo efficacy were poorly translated likely owing to their unfavorable druglike properties such as low cellular permeability, poor solubility, poor oral bioavailability, or some other unknown reasons. With the rapid development of innovative technologies and more extensive studies of biological functions and mechanisms, these challenges may be overcome, eventually leading to exciting outcomes. Therefore, drug discovery and development of more potent and selective NSD2 inhibitors with ideal druggability properties appear to be a viable approach for novel therapies in the near future. For example, such efforts are inspired by the successful advancement of KTX-1001 into clinical trials. Herein, the recent advances in developing small molecule NSD2 inhibitors were summarized, and the relevant strategies targeting NSD2 are classified into three categories: NSD2 inhibitors, NSD2 protein degraders, and combinatorial therapies.
Small Molecule NSD2 Inhibitors.
Based on the targeted domain, small molecule NSD2 inhibitors are divided into three types: inhibitors targeting the catalytic SET domain of NSD2 (NSD2-SET), inhibitors targeting the PHD domain of NSD2 (NSD2-PHD), and inhibitors targeting the PWWP1 domain of NSD2 (NSD2-PWWP1). NSD2 inhibitors targeting the SET domain can be classified into SAM coenzyme-competitive inhibitors and or histone-tail substrate-competitive inhibitors.
Inhibitors Targeting the Catalytic SET Domain of NSD2.
HMTase inhibitors are generally classified into three categories based on their mechanism of action, including allosteric inhibitors, SAM coenzyme-competitive inhibitors, and histone-tail substrate-competitive inhibitors.209 The SET domains of HMTases comprise two binding pockets: the large histone-tail binding/active groove and the small coenzyme SAM binding cavity, which are linked by a narrow channel where the histone lysine substrate extends into.210 Due to a highly conserved SET domain shared by NSDs, it is challenging to design compounds specifically targeting NSDs to explore their biological functions.
Several compounds have been characterized as NSD2 inhibitors through screening and structural optimization, such as SAM-competitive inhibitors Sinefungin and its derivatives211–214, 3-Deazaneplanocin A (DZNep)170, and MCTP-39 and its derivatives172, and substrate-competitive inhibitors Suramin212, Chaetocin212, 215, Verticillin A206, UNC0638214, PF-03882845215, TC LPA5 4215, and ABT-199.215 Although these compounds may serve as useful tools for exploring the function of NSD2 and its relationship with diseases, their further drug development value is greatly limited due to the poor activity, target specificity, membrane permeability, bioavailability, and other drug-like properties. This section describes several substrate-competitive NSD2-SET inhibitors with potential further study value.
The catalytic SET domains of all HMTases are conserved, especially among the NSD subfamily members (NSD1, NSD2, NSD3). In the catalytic SET domain, NSD2 is found to share extremely high structural identity and similarity with G9a and 9a-like protein (GLP) (identity: 29.6% with G9a vs. 27.7% with GLP; similarity: 48.3% with G9a vs. 46.1% with GLP), indicating that NSD2 and G9a/GLP have similar structural features in their SET domains.209 BIX-01294 (17, Figure 6), a histone-tail mimetic inhibitor, was first reported as a potent G9a/GLP inhibitor (G9a: IC50 = 180 nM; GLP: IC50 = 34 nM), resulting in the reduction of K3H9me2/3 by inhibiting H3K9 methylation activity.216–219 Given that NSD2 and G9a/GLP share similar structural features in their SETs, compound 17 was further identified to display inhibition of NSD2.209 The in vitro study demonstrated that 17 can potentially suppress the level of H3K36me1 catalyzed by NSD-SETs (NSD1-SET: IC50 = 112 ± 57 μM; NSD2-SET: IC50 = 41 ± 2 μM; NSD3-SET: IC50 = 95 ± 53 μM), suggesting that 17 is a pan-NSD inhibitor with a poor selectivity towards NSD2. At that time, the co-crystal structures of HMTase inhibitors in complex with NSD2-SET or NSD3-SET were not available, an accurate binding mode and detailed interactions between 17 and NSD2-SET could not be determined. Among all three NSD members, 17 exhibits the strongest inhibition against NSD2, suggesting that it might be further optimized into a potent, selective NSD2 inhibitor.
Figure 6.

Discovery of compounds 10 and 11 based on lead compound 17.
The larger histone-tail binding pocket in the SET domain of NSD1 has greater sequence heterogeneity and is more easily accessible than the small conserved SAM binding pocket.220 It was hypothesized that selective inhibition against NSDs with small molecules could be realized through targeting the histone-tail binding pocket rather than SAM binding pocket. Based on virtual ligand screening against an NSD2 homology mode, compound LEM-06 (10, Figure 6) was further identified as a hit inhibitor of NSD2, which specifically binds to the histone-tail binding pocket in the SET domain of NSD2.220 Compound 10 suppressed the H3K36me1 in vitro with an IC50 value of 0.89 ± 0.25 mM, which is much weaker than MTCP-39 (IC50 = 3 μM).221 10 served as a valuable hit to be further optimized into a selective NSD2 inhibitor for exploring the role of NSD2 and treating malignancies. The continuous efforts210 discovered compound LEM-14 (11, Figure 6) as the first selective NSD2 inhibitor by virtual ligand screening. Compound 11 can inhibit the in vitro H3K36 methylation mediated by NSD2 (IC50 = 132 μM) while interestingly showing very weak potency against NSD1 (IC50 >1000 μM) and not detectable activity against NSD3. Therefore, more optimization efforts are imperative to further improve the potency and selectivity of 11.
Researchers from Epizyme Inc. validated compound PTD2 (18, Figure 7a), a norleucine-containing peptide that derived from histone H4 (H4K44) substrate sequence, as an NSD2 inhibitor (Kd = 3.0 ± 0.3 μM).222 Compound 18 showed an inhibitory activity against both NSD2 and NSD3 with IC50 values of 22 ± 2 μM and 3.2 ± 0.2 μM, respectively, while its effect on NSD1 was unclear. Moreover, a crystal structure (Figure 7b, PDB ID: 6CEN) of 18 in complex with SAM and the SET domain of NSD3 was determined, revealing that the autoinhibitory loop plays a critical role in protein substrate binding and catalytic cycle of NSD3. The discovery of 18 is an important step for sorting out the binding modes of NSDs in obtaining potent and selective NSD2 inhibitors for better understanding its complicated biological functions associated with various diseases, especially cancers.
Figure 7.

(a) Chemical structure of compound 18 (PTD2). (b) Crystal structure of compound 18 (PTD2) in complex with NSD3 SET domain and SAM (PDB ID: 6CEN). Red dashed lines highlight the hydrogen bonds between 18 and the key residues in the NSD3 SET domain. Compound 18 is shown as yellow sticks. Key residues THR-1203, THR-1232, ASN-1262, TYR-1261, LEU-1263, ASP-1264, and MET-1201 in the NSD3 SET domain are shown as green sticks. SAM is shown as cyan sticks.
Based on the structural analysis of available HMTase inhibitors and the effective application of HTS, compound 19 (Figure 8) with a urea moiety was successfully identified as a potent NSD2 inhibitor (IC50 = 8.3 μM).223 To obtain more potent NSD2 inhibitors, a systematical structural optimization was conducted around 19. As a result, a series of di-aryl urea derivatives (20, Figure 8) were synthesized and disclosed as potent NSD2 inhibitors for treating various cancers, especially PCa, lung, breast, and ovarian cancer.223 Excitingly, compounds 21–25 (Figure 8) showed superior or comparable NSD2 inhibitory activity compared to 19 with an inhibition rate of 96%, 92%, 95%, 92%, and 99%, respectively, at 50 μM. Their IC50 values were further determined as low micromolar potency (IC50 = 4.8 μM, 7.5 μM, 3.7 μM, 8.1 μM, and 7.3 μM, respectively). Compound 23, with the strongest NSD2 inhibitory activity, was chosen for further study. 23 robustly suppressed the proliferation of 22Rv-1 cells, one type of PCa cells, in a dose- and time-dependent manner, and the cell growth inhibition rate was determined as 66% after treatment with 23 at 10 μM for 10 days. Moreover, 23 reduced the methylation of H3K36, including H3K36me and H3K36me2 in 22Rv-1 cells in a dose-dependent manner and it showed an inhibitory effect on the expression of NSD2, suggesting that 23 also can inhibit the activity of NSD2 in cells. These results illustrated that 23 suppressed the proliferation of cells via inhibiting the activity of NSD2 to reduce H3K36me and H3K36me2. In addition, 23 showed potent NSD2 inhibition (97% @ 10 μM) and modest MLL-1 and SETDB1 (79% @ 10 μM and 85% @ 10 μM, respectively), while almost no effect on PRDM9, PRMT5, PRMT7, and SMYD3, suggesting 23 displayed excellent selectivity for NSD2 compared with other HMTases. Furthermore, 23 displayed robust and broad-spectrum anti-proliferation activity in a panel of cancer cells (IC50s < 10 μM), including PCa, lung, ovarian, and tri-negative breast cancer (TNBC). All these results indicate that 23 may be further developed as a potential therapeutic agent for treating or preventing various tumors.
Figure 8.

Chemical structures of compounds 19–25.
The benzothiazole derivatives (26, Figure 9) were reported as nonspecific NSD inhibitors for treating various diseases, especially cancers.224 Most of them showed modest NSD inhibitory activities with IC50 values of 20–200 μM. Some of them, such as compound BT3, displayed a moderate NSD2 inhibition with IC50 values of < 20 μM. Later, they reported the detailed discovery process of compounds BT1 (27), BT2 (28), BT3 (29), and BT5 (30) (Figure 9).225 Compound 27 was identified as an NSD1 inhibitor that binds to the catalytic SET domain by nuclear magnetic resonance (NMR)-based fragment screening of about 1,600 in-house compounds with fragment-like properties. Several analogs of 27 were subsequently synthesized and tested, of which compound 28 with 4-hydroxyl and 6-bromo substituents was demonstrated as the most potent compound in NMR experiments. 28 exhibited a modest binding affinity to NSD1 SET domain (Kd = 10.4 μM) and NSD1 enzyme inhibitory activity (IC50 = 66 μM). To obtain irreversible NSD1 ligands, further structural optimization was performed around 28, leading to the discovery of compound 29 with a thiocyanate at the 6-position instead of bromide. The crystal structure of NSD1 SET in complex with 29 (Figure 10, PDB ID: 6KQQ) indicates that 29 covalently binds to the NSD1 SET domain via a disulfide bond formed between 29 and the side chain of C2062. Moreover, compound 29 is almost entirely buried in the SET domain, and the exerted hydrophobic interactions with L2081 and F1996 and hydrogen bond network interactions formed with the carbonyl group of T1994, hydroxyl group of the SAM cofactor, and an internal water molecule. Furthermore, the crystal structure revealed covalent binding caused a conformational change in the autoinhibitory loop, forming a unique, channel-like pocket suitable for accommodating small molecules to access. Based on the analysis of the crystal structures, to covalently target amino acid residue C2062, more structural exploration was conducted, and several analogs of 29 with an aziridine group were synthesized. Compound 30 with a methyl-aziridine was validated to covalently bind to NSD1 via MS and NMR experiments. Compound 30 showed potent NSD1 inhibitory activity (IC50 = 5.8 μM at 4 h and improved to 1.4 μM at 16 h due to irreversible binding) as well as moderate NSD2 (IC50 = 26.7 μM at 4 h) and NSD3 (IC50 = 14.3 μM at 4 h). In addition, compound 30 was demonstrated to selectively engage the NSD1 SET domain in eukaryotic cells (HEK293T cells) transfected with Flag-NSD1 SET. Moreover, 30 selectively suppressed the growth of NUP98-NSD1 cells in a time-dependent manner (GI50 = 1.3 μM and 0.87 μM at days 3 and 7, respectively). The mechanistic study revealed that 30 selectively suppressed the global expression levels of H3K36me2 and reduced the expression levels of NUP98-NSD1 target genes (Hoxa9, Hoxa5, Hoxa7, and Meis1) in leukemia cells through covalent binding and followed by impairing the activity of NUP98-NSD1. Overall, compound 30 may serve as a valuable lead compound for further optimization to obtain the next generation of more potent and selective NSD inhibitors.
Figure 9.

Chemical structures of compounds 26–30.
Figure 10.

Crystal structure of compound 29 (BT3) in complex with NSD1 SET domain and SAM (PDB ID: 6KQQ). Hydrogen bonds formed between 29 and the key residues in the NSD1 SET domain are highlighted by red dashed lines. Compound 29 is shown as yellow sticks. Key residues THR-1994 and CYS-2062 in the NSD1 SET domain are shown as green sticks. SAM is shown as cyan sticks.
To develop potent NSD2 inhibitors with new scaffolds, HTS was carried out to screen the Chinese National Compound Library, a total of 296,080 compounds.226 As a result, 26 compounds at 20 μg/mL were successfully identified as hits, of which compound 31 (Figure 11) displayed the most potent inhibitory activity against NSD2 with an IC50 value of 19.4 ± 6.5 μM. To find out more potent NSD2 inhibitors, scaffold exploration around compound 31 was conducted. Compound 32 (Figure 11) with a sulfonamide to replace the amide in 31 showed a 7-fold improvement in inhibitory activity against NSD2 (IC50 = 2.7 ± 0.3 μM). 32 was chosen as the lead compound with a systematical structure-activity relationship (SAR) study on the 5-aminonaphthalene-1-sulfonamide scaffold. Results showed that most of the compounds displayed much weaker potency compared to 32, and only 33 (IC50 = 3.4 ± 0.4 μM) and 34 (IC50 = 3.2 ± 0.8 μM) (Figure 11) exhibited comparable activity with that of 32 (IC50 = 2.7 ± 0.3 μM), suggesting that 32 is the most potent NSD2 inhibitors among all these synthesized compounds. Interestingly, 32 exhibited distinct selectivity towards NSD2 inhibition over other human methyltransferases, including not only SET-domain-containing methyltransferases, such as NSD1, SETD2, EZH2, MLL1, and MLL4, but also non-SET domain-containing methyltransferases, such as DOT1L, PRMT4, and PRMT5. Moreover, 32 significantly prevented the methylation of H3K36me2 in a dose-dependent manner and did not affect the level of histone H3 and NSD2 in RS4:11, MV4;11, KMS11, and OPM-2 cell lines, indicating that the reduction of H3K36me2 is achieved by directly inhibiting NSD2 methyltransferase activity but not by inducing the degradation of histone H3 or NSD2. Furthermore, 32 suppressed the proliferation of ALL cell line RS4:11 (IC50 = 0.52 μM) and MM cell line KMS11 (IC50 = 1.88 μM) in a time- and dose-dependent manner. Additionally, 32 can induce the apoptosis of RS4:11 and KMS11 cells in a dose-dependent manner by arresting the cell cycle mainly at G0/G1 phase. 32 can also completely block the transcriptional activation of NSD2 target genes in KMS11 cells, including TGFA, MET, PAK1, and RRAS2. Importantly, in vivo studies demonstrated that 32, when administered intraperitoneally (i.p.) at 25 mg/kg QD for successive 21 days, can significantly suppress the tumor growth of RS4:11 xenografts mice (TGI = 43.6%, T/C = 56.3%). Moreover, no obvious side effect on the body weight change of SCID mice was observed, suggesting that 32 shows safety at the testing dose. Collectively, 32 may serve as a useful lead compound with the potential to be further developed as a novel therapy for hematological malignancies.
Figure 11.

Chemical structures of compounds 31–34.
Researchers from Novartis Inc. filed a patent on a series of purineamine derivatives containing a piperidine moiety (35, Figure 12) as NSD2 inhibitors to treat or prevent various NSD2-mediated diseases or disorders, especially for cancers, including MM and thyroid carcinoma.227 Most of the example compounds in this patent exhibit potent NSD2 (amino acids 1–1365) inhibitory activity (IC50s = 0.01–1.0 μM), of which compounds 36–42 (Figure 12) even display more potent activity (IC50s = 0.001–0.01 μM). Moreover, 36–42 significantly decrease the H3K36me2 levels in MM cell line KMS11-Par as determined by the FRET assay (36–40: IC50 = 0.214 μM, 0.16 μM , 0.248 μM, 0.225 μM and 0.27 μM, respectively) and in thyroid carcinoma cell line CGTH-W-1 as determined by ELISA (41–42: IC50 = 0.041 μM and 0.035 μM, respectively).
Figure 12.

Chemical structures of compounds 35–42.
In 2022, a potent and selective NSD2 inhibitor KTX-1001 held by K36 Therapeutics was approved by the FDA for conducting clinical trials. KTX-1001 selectively binds to NSD2-SET domain and inhibits its catalytic function as a methyltransferase. KTX-1001, was initially licensed from Novartis Inc., and its structure has not been disclosed. Now, KTX-1001 is in phase I clinical trials for treating relapsed and refractory MM (ClinicalTrials.gov Identifier: NCT05651932). Inspired by this milestone success of KTX-1001, more and more potent and selective NSD2 inhibitors are anticipated to be developed into clinical trials as promising therapeutic agents in the near future.
Compound DA3003–1 (12, Figure 13) was identified as a potent NSD2 inhibitor through the HTS assays.215 Compound 12 inhibited the NSD2 activity in vitro by directly binding to the catalytic SET domain. 12 inhibits both WT NSD2, and mutated NSD2 E1099K and T1150A with sub-micromolar potency (IC50 = 0.17 μM, 0.11 μM, and 0.17 μM, respectively). 12, a non-selective NSD inhibitor, also displays inhibitory activity against NSD1 (IC50 = 0.11 μM) and NSD3 (IC50 = 0.26 μM). Moreover, 12 shows obvious inhibitory activity against other HMTases, indicating that it is a non-specific HMTase inhibitor. Furthermore, 12 can decrease H3K36me2 level in a dose-dependent manner (IC50 = 545 nM) in human osteosarcoma cells (U2OS); however, the cytotoxicity occurs at a higher concentration (CC50 = 270 nM). Although 12 shows potent NSD2 inhibition and significantly reduces H3K36me2 level in U2OS cells, the exact mechanisms of action remain to be fully elucidated, which may be complicated and associated with multiple targets given its quinone scaffold. Further optimization to improve the target specificity of 12 is imperative.
Figure 13.

Chemical structures of compounds 12 and 13.
In 2023, a comprehensive SAR study around the lead compound 12 targeting NSD2 inhibition was conducted based on its quinoline-5,8-dione scaffold.228 Newly designed compounds displayed potent inhibitory activity against NSD2 (IC50s < 0.5 μM). Compound 13 (Figure 13) exhibited superior NSD2 inhibitory activity (IC50 = 0.23 μM) and significantly suppressed the proliferation of several hematological cell lines (IC50s < 0.5 μM), including MM cell KMS-11 with NSD2 t(4;14) translocation (IC50 = 0.27 ± 0.02 μM) and AML cell RS4;11 with E1099K variant (IC50 = 0.304 ± 0.00 μM). Moreover, 13 can induce cell apoptosis and suppress the mRNA expression of NSD2-mediated downstream target genes in the KMS-11 cells. Importantly, 13 showed good pharmacokinetic (PK) properties with high oral bioavailability (F = 275%). Furthermore, 13 (75 mg/kg and 150 mg/kg) displayed significant antitumor efficacy with (TGI = 56% and 58.4%, respectively), and showed desirable safety profiles (no obvious body weight loss and no adverse effects at the testing dosages) in the MM xenograft mice model. These findings suggest that 13 may be a potential NSD2 inhibitor as an advanced lead for further optimization. Interestingly, a good dose-dependent effect was not observed when testing at a higher dose (150 mg/kg), likely owing to the dynamic nature of epigenetic modulation and the difference in NSD2-mediated genes. In addition, the NSD2-associated histone modification effect has also been reported to regulate the tumor suppressor genes except for oncogenes. Similar results were also obtained to show that 13 at higher concentrations can simultaneously reduce the expression of tumor suppressor genes TRIB3 and DDIT4. However, the precise mechanistic understanding of the poor dose response of 13 remains to be unraveled. Other than NSD2 inhibition, 13 also displayed strong inhibitory activities against NSD1 (IC50 = 0.69 ± 0.04 μM), NSD3 (IC50 = 0.75 ± 0.01 μM), and EZH2 (IC50 = 0.25 ± 0.02 μM), indicating a limited selectivity profile of this series of compounds.
Targeting the PHD Domain of NSD2.
PHD fingers are non-catalytic Zn2+ binding domains found in more than 170 human chromatin-related proteins.229 They are epigenetic readers, typically regulating the transcriptional activity of its hosting protein through recognizing the specific histone marks, including H3K4me0, H3K4me3, H3K9me3.230–232 The mutations of PHD fingers have been reported to associate with many diseases, such as neurological and developmental diseases, cancers, and immunological disorders.41 Therefore, PHD fingers are evolving as attractive domains to be targeted for epigenetic drug development, providing a promising therapeutic alternative for classical inhibitors targeting inhibition of enzymatic activity.233–237 In 2020, based on NMR binding assays and ITC measurements, a virtual screening of a small Zinc Drug Database (ZDD) with a total of 2,924 compounds was performed against PHD domain of NSD1 protein (PHDVC5HCHNSD1).238 Three compounds have been identified as positive hits capable of binding to PHDVC5HCHNSD1 and blocking its interaction with the C2HRNizp1 (Zinc finger domain) of the transcriptional repressor NSD1-interacting Zn-finger protein (Nizp1), including a type II topoisomerase inhibitor mitoxantrone dihydrochloride (43) that has been used for treating AML, and two antimalarial drugs chloroquine diphosphate (44) and quinacrine dihydrochloride (45) that have been repositioned for treating cancer (Figure 14). Compounds 43–45 showed weak interactions with PHDVC5HCHNSD1 with Kd values of 1.2 ± 0.4 mM, 4.7 ± 1.4 mM, and 1.4 ± 0.3 mM, respectively, as determined by NMR. This study supports the feasibility of disrupting the finger-finger interactions between PHDVC5HCHNSD1 and C2HRNizp1 by small molecules to develop potent inhibitors modulating these protein-protein interactions. These findings suggest that targeting the PHD domain may be a potential strategy for developing potent and selective NSD1 inhibitors to treat NSD1-related diseases. Given the high structural homology among three NSD members, it is reasonable to believe that this strategy may also be applicable for developing selective NSD2 or NSD3 inhibitors in the future.
Figure 14.

Chemical structures of compounds 43–45.
Targeting the PWWP1 Domain of NSD2.
As we mentioned earlier, NSD2 is a promising therapeutic target, and its translocation, mutations, and overexpression are closely associated with the proliferation, migration, and invasion of cancer cells.11, 21–25 However, developing small molecule inhibitors targeting the catalytic SET domain has yielded little success.24, 25 Recently, a selective covalent NSD1 inhibitor 30 (BT5) and two specific NSD2 inhibitors 11 (LEM-14) and KTX-1001 (ClinicalTrials.gov Identifier: NCT05651932) that targeting the catalytic SET domain have been identified.210, 225 Among previously reported NSD inhibitors, 30 (BT5) and 11 (LEM-14) are the first inhibitors selectively targeting NSD1 and NSD2, respectively. Other than the catalytic SET domain, NSD2 also possesses multiple PPI domains, including PHD domains and PWWP domains (PWWP1 and PWWP2),39, 239 which may be clinically relevant and selectively blocked by small molecules (Figure 15). NSD2-PWWP1 domain binds to H3K36me2 through cation-π and hydrophobic interactions with the ammonium group of the methylated lysine, provided by a conserved aromatic cage formed by three orthogonally positioned aromatic side chains (Y233, W236, and F266).49 Importantly, the F266A mutation occurring at the aromatic cage disrupts the interactions between H3K36me2 and full-length NSD2 without obviously affecting the level of H3K36me2, thereby leading to the proliferation suppression of cancer cells.39 Therefore, developing small molecules targeting the PWWP1 domain of NSD2 to block its interactions with H3K36me2 may offer a potential therapeutic strategy for treating NSD2-driven diseases such as various cancers.39
Figure 15.

NSD2 PWWP domain modulates the functions of NSD2 through recognizing H3K36me2 and may serve as a drug target domain to be blocked by small molecule inhibitors.
In 2019, compound BI-9321 (46, Figure 16) was identified as the first potent and selective NSD3-PWWP1 antagonist (IC50 = 0.2 μM), which can disrupt the interactions between NSD3-PWWP1 and H3K36me2, leading to a significant reduction of MYC mRNA levels and suppressing the proliferation of leukemia cell lines.240 In 2021, to develop a selective NSD2 chemical probe, especially targeting the NSD2-PWWP1 domain, virtual screening and experimental validation were conducted.241 Initially, the structure of the PWWP domain of NSD2 was not available in Protein Data Bank (PDB), and the chromatin factor ZMYND11A with a methyl-lysine binding pocket was determined as a target for virtual screening. A virtual screen on a commercial library of ~2 million compounds was conducted against the PWWP domain of ZMYND11A. 39 compounds were identified as hits, and their activities were experimentally evaluated utilizing differential static light scattering (DSLS). While none of them was confirmed active for ZMYND11A, the other members of the PWWP family were then chosen for screening. Excitingly, compound 47 (Figure 16) exerted a stabilization effect on the PWWP domain of NSD2, increasing the thermal shift by 4 °C at 400 μM (ΔTagg = 4 °C). The interaction between 47 and the PWWP1 domain of NSD2 was further validated by surface plasmon resonance (SPR) assay, displaying a dissociation constant (Kd) value of 41 ± 8 μM. Since the solubility of 47 was poor, a reverse ITC titration was conducted to further validate the interaction between 47 and NSD2-PWWP1 through titrating a concentrated NSD2-PWWP1 protein solution (1 mM) into the sample cell filled with a solution of 47 at 40 μM, with a Kd value of 8.9 μM. DSLS results showed that compound 47 at 400 μM selectively binds to NSD2-PWWP1, showing almost no interaction with six other PWWP-containing proteins. To further improve the potency of 47, a series of derivatives of 47 was designed based on the scaffold-hopping strategy, and 24 analogs were experimentally evaluated. However, only compound 48 (Figure 16) at 500 μM showed a modest stabilization effect on NSD2-PWWP1 (ΔTagg = 1.9 °C). SPR further confirmed the binding affinity of 48 with NSD2-PWWP1 with a Kd value of 175 μM. Despite the binding affinity of 48 (Kd = 175 μM) was weaker than that of 47 (Kd = 41 ± 8 μM), its new scaffold provides a wide chemical space for further SAR studies with many more analogs. The preliminary SAR study suggests that the nitrile group provides a critical interaction with NSD2-PWWP1, presumably as a hydrogen bond acceptor.
Figure 16.

Chemical structures of compounds 46–50.
Structural optimization was performed to identify more potent compounds by maintaining two phenyl groups and the cyano of 48. Encouragingly, compound 49 (Figure 16) has been obtained to be 6- and 25-times more potent than 47 and 48, respectively. Compound 49 at 500 μM shows a significantly improved binding affinity to NSD2-PWWP1 with a Kd value of 7 μM determined by SPR and it displays a strong protein stabilization effect on NSD2-PWWP1 (ΔTagg = 5.7 °C) determined by DSLS. To further explore the SAR for compounds with higher binding affinities, 40 analogs were synthesized or purchased to test their binding capability with NSD2-PWWP1. The compounds without cyclopropyl, either larger or smaller groups, showed much weaker binding or a complete loss of activity, suggesting that cyclopropyl is essential for maintaining high protein binding affinity. Compound MR837 (50, Figure 16) showed the best binding affinity towards NSD2-PWWP1 (SPR Kd = 7 ± 3 μM and DSLS ΔTagg = 6.2 °C), which was about 6-fold more potent than that of 47 (SPR Kd = 41 ± 8 μM). Given that high dimethyl sulfoxide (DMSO) concentration can limit the potency of PWWP ligands, the binding affinity 50 was measured with 0.5% DMSO in an SPR experiment, and the Kd value was confirmed as 3.4 ± 0.4 μM. In addition, 50 showed high binding selectivity against NSD2-PWWP1 over the other nine PWWP domains. It was found that the binding affinity to NSD2-PWWP1 was lost entirely when residues Y233 or F266 located at the aromatic cage were mutated to alanine, indicating that it binds to NSD2 at the methyl-lysine binding site.
The crystal structure of 50 in complex with NSD2-PWWP1 (Figure 17, PDB ID: 6UE6) revealed that 50 is located at a Kme reader aromatic cage, which provides a strong molecular basis to support its high binding capability with NSD2-PWWP1.241 The cyclopropyl sits in a hydrophobic pocket formed by the residues Y233, W236, F266, and V230, and exerts hydrophobic interactions with the residues, validating the importance of cyclopropyl group. As the pocket was narrow and lipophilic, smaller groups such as methyl and hydrogen atom may impair or lose hydrophobic interactions. In comparison, larger groups such as cyclohexyl may clash with the amino acid residues within the aromatic cage formed by (Y233, W236, F266), indicating that cyclopropyl may be the optimal group at this site. This observation is in line with the initial SAR results. An extremely coordinated water molecule is located at the bottom of the pocket. Moreover, the cyanophenyl group could reach a pocket formed by amino acid residues W236, G268, D269, A270, E272, L318, and Q321. The nitrogen of the cyano group plays a role as a hydrogen bond acceptor to form key hydrogen bonding with the amide nitrogen of A270, indicating that compounds without cyano display weak potency or no activity. Additionally, the carbonyl oxygen, a hydrogen bond acceptor, interacts with the side chain of Y233 through a key hydrogen bond interaction. Furthermore, the thiophene ring sits in a hydrophobic pocket, defined by V230 and A274, and is partially exposed to the solvent, suggesting that structural modifications at this site appear tolerable. Compared to the structure of NSD2-PWWP1 in complex with DNA (Figure 4b, PBD: 5VC8), 50 induces a significant conformational change of Y233 and E272, leading to opening-up of the aromatic cage that is closed in apo structure. Excitingly, 50 disengages NSD2 PWWP1 but not NSD3-PWWP1 from histone H3 in cells in a dose-dependent manner (IC50 = 17.3 μM), indicating its selective interactions with NSD2-PWWP1 in cells. Overall, 50 can effectively block the interactions between H3K36me2 and NSD2-PWWP1 domain in cells by selectively binding to NSD2-PWWP1 and may be further developed into a useful chemical probe through more extensive structure-based optimization to better understand the biological role and functions of NSD2 in cells and even human body.
Figure 17.

Crystal structure of compound 50 (MR837) in complex with NSD2-PWWP1 domain (PDB ID: 6UE6). Hydrogen bonds formed between 50 and the key residues in the NSD2-PWWP1 domain are highlighted by red dashed lines. Compound 50 is shown as yellow sticks. Key residues ALA-270, TYR-233, TRP-236, PHE-266, and VAL-230 in the NSD2-PWWP1 domain are shown as green sticks.
In 2022,based on the crystal structure of 50 (MR837) and NSD2-PWWP1 (Figure 17, PDB ID: 6UE6), molecular docking simulations were carried out, indicating that a benzoxazinone bicyclic group may favorably replace the cyanophenyl group in 50.242 Compound MRT866 (51, Figure 18) was then designed and evaluated against NSD2-PWWP1, and it was found to display a significantly enhanced binding to NSD2-PWWP1 (Kd = 349 ± 19 nM), compared to that of compound 50 (Kd = 3.4 ± 0.4 μM). Furthermore, X-ray crystallography (Figure 19a, PDB ID: 7MDN) revealed that 50 and 51 bind to NSD2-PWWP1 in a similar mode, and both occupy the aromatic cage of NSD2-PWWP1. In addition, compared to 50, 51 displays more potent binding activity towards NSD2-PWWP1 through more extensive van der Waals interactions between the benzoxazinone moiety and NSD2-PWWP1, and an additional hydrogen bond formed between the benzoxazinone moiety and the side chain of Q321.
Figure 18.

Chemical structures of compounds 14 and 50–53.
Figure 19.

(a) Crystal structure of compound 51 (MRT866) in complex with NSD2-PWWP1 domain (PDB ID: 7MDN). Red dash lines highlight the hydrogen bonds between compound 51 and the key residues in NSD2-PWWP1 domain. Compound 51 is shown as yellow sticks. Key residues ALA-270, GLN-321, TYR-233, TRP-236, and PHE-266 in the NSD2-PWWP1 domain are shown as green sticks. (b) Crystal structure of compound 14 (UNC6934) in complex with NSD2-PWWP1 domain (PDB ID: 6XCG). Red dash lines highlight the hydrogen bonds formed between compound 14 and the key residues in NSD2-PWWP1 domain. Compound 14 is shown as yellow sticks. Key residues ALA-270, GLN-321, TYR-233, ARG-273, TRP-236, and PHE-266 in the NSD2-PWWP1 domain are shown as green sticks.
A more effective compound UNC6934 (14, Figure 18), was successfully discovered based on further structure-based optimization focusing on the thiophene ring. Compared to 51 (Kd = 349 ± 19 nM), compound 14 showed about 3-fold stronger binding activity with NSD2-PWWP1 (Kd = 91 ± 8 nM). Interestingly, compound 52 (UNC7145), containing an isopropyl group instead of a cyclopropyl ring, showed poor binding activity towards NSD2-PWWP1 (Kd > 20 μM) and could serve as an ideal negative control. 14 binds to NSD2-PWWP1 through occupying the canonical methyl-lysine binding pocket and blocks the interactions between NSD2-PWWP1 and nucleosomal H3K36me2 in a dose-dependent manner (IC50 = 104 ± 13 nM), and selectively engages endogenous NSD2-PWWP1 in cells (EC50 = 1.23 ± 0.25 μM), while 52 exhibits no measurable inhibitory effect. Moreover, 14 displays high selectivity for NSD2-PWWP1 over 15 other human PWWP domains with no inhibitory effect on a panel of 33 methyltransferase domains, including NSD1, NSD2, NSD3, and SETD2. At the same time, 52 shows no inhibitory effect against all tested PWWP domains and methyltransferases. The binding pocket of NSD3-PWWP1 appears to be structurally closest to that of NSD2-PWWP1, with only three different amino acid side chains between both domains. In NSD2-PWWP1, G268, located at the bottom of a cavity fits well with the benzoxazinone moiety of 14, while a serine in NSD3-PWWP1 impedes the binding of 14. Such observation strongly supports the high binding of 14 with NSD2-PWWP1 but not with NDS3-PWWP1. Importantly, mutations of other NSD2 chromatin reader domains also increase NSD2 nucleolar localization and enhance the effect of 14. PWWP domains have two pockets, a methyl-lysine-binding pocket and a DNA-binding surface. DNA binds to the PWWP domain through direct electrostatic interactions between its phosphate backbone and side chains of K304, K309, and K312 located at a basic surface area (Figure 4b, PDB ID: 5VC8). 14 can be inserted in the methyl-lysine-binding pocket close to the DNA-binding surface. Despite the proximity of these two pockets, 14 does not compete with DNA in binding to NSD2-PWWP1. The crystal structure of NSD2-PWWP1 in complex with 14 (Figure 19b, PDB ID: 6XCG) revealed that the cyclopropyl group tightly occupies the aromatic cage composed of Y233, W236, and F266. UNC7096 (53, Figure 18), a biotin-labeled affinity reagent, was synthesized based on 14 for chemical pulldown experiments to evaluate the selectivity and target engagement of 14 in cells. 53 displays a high binding affinity to NSD2-PWWP (Kd = 46 nM), comparable to 14 (Kd = 91 ± 8 nM). No acute cytotoxic effects were observed in adherent or hematopoietic malignancy cell lines after administration of 14 (5 μM) for more than 3–12 d, suggesting 14 is a safe and suitable pharmacological tool for further biological function studies of NSD2, together with the negative control 52, and compound 53.
In 2022, by analyzing two crystal structures of NSD2-PWWP1 in complex with 50 (MRT837) (Figure 17, PDB ID: 6UE6) and NSD3-PWWP1 in complex with 46 (BI-9321) (Figure 20, PDB: 6G2O), the side chain of Ser314 in NSD3-PWWP1 was found to impede its binding with benzonitrile group in 50, which is consistent with previously disclosed observations.242, 243 Based on the idea of replacing the quinoline ring of 46 with benzonitrile of 50 to improve the NSD2 selectivity over NSD3 and incorporating a methoxyl into the benzene ring of 46 to mimic the role of the amide of 50 in forming a key hydrogen bond with Tyr233, compound 54 was designed, synthesized and evaluated (Figure 21). Encouragingly, 54 exhibits a high binding affinity to NSD2-PWWP1 with an IC50 value of 4.44 μM. The co-crystal structure of NSD2-PWWP1 with 54 (Figure 22, PDB ID: 7VLN) revealed that 54 binds to the NSD2-PWWP1 in a similar mode to that of 46 binding to NSD3-PWWP1. The N-methylimidazole directly inserts into the canonical aromatic cage, the benzylamine motif interacts with residue Glu272, and the benzonitrile fragment forms hydrogen bonds with residues Asp269 and Ala270. The nitrile group of 54 can clash with the Serine residue in NSD3-PWWP1, providing the molecular basis for its selective binding to NSD2-PWWP1.
Figure 20.

Crystal structure of compound 46 (BI-9321) in complex with NSD3-PWWP1 domain (PDB ID: 6G2O). Red dash lines highlight the hydrogen bonds between compound 46 and the key residues in NSD3-PWWP1 domain. A water molecule is shown as a red ball. Compound 46 is shown as yellow sticks. Key residues SER-314 and GLU-318 in the NSD3-PWWP1 domain are shown as green sticks.
Figure 21.
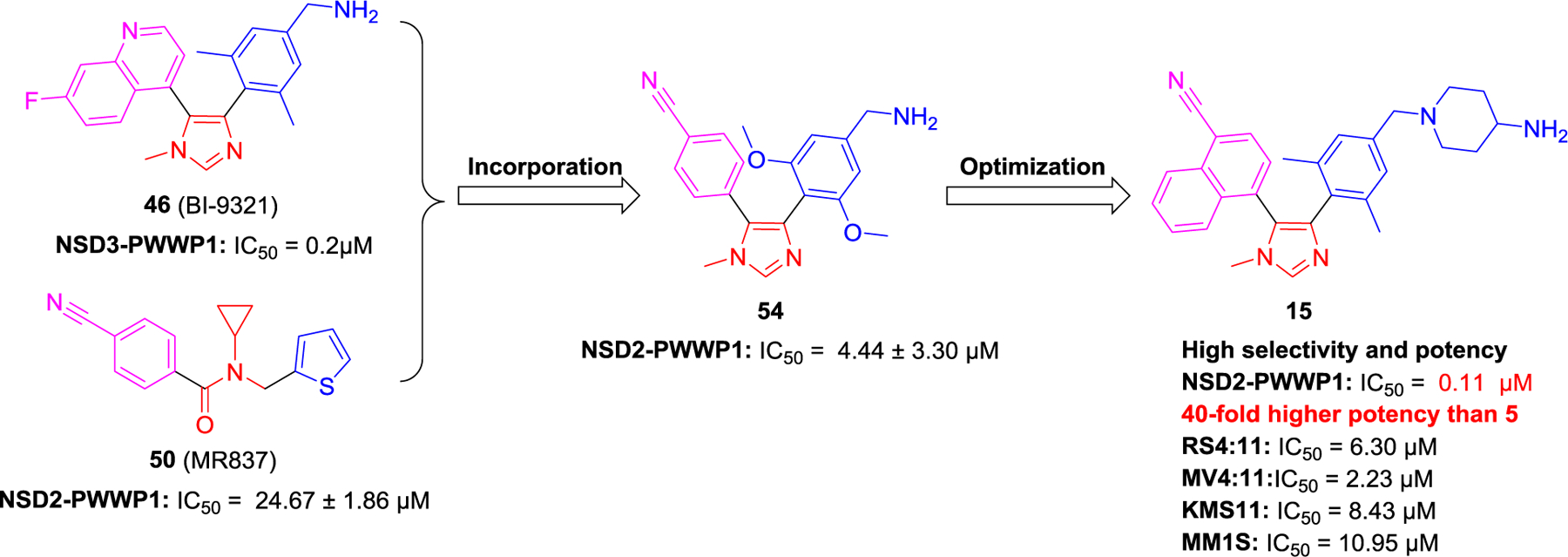
Chemical structures of compounds 15, 46, 50, and 54.
Figure 22.

Crystal structure of compound 54 in complex with NSD2-PWWP1 domain (PDB ID: 7VLN). Hydrogen bonds formed between compound 54 and the key residues in the NSD2-PWWP1 domain are highlighted by red dashed lines. Compound 54 is shown as yellow sticks. Key residues ALA-270, ASP-269, TYR-233, GLU-291, and GLU-272 in the NSD2-PWWP1 domain are shown as green sticks.
Further structure-based optimization focusing on benzylamine and benzonitrile motifs has resulted in the discovery of compound 15 (Figure 21) as the most potent NSD2-PWWP1 antagonist with an IC50 value of 0.11 ± 0.01 μM, displaying 40-fold higher activity than that of 54. Compound 15 directly binds to NSD2-PWWP1 and improves protein stability in vitro. Moreover, 15 displays extremely high binding selectivity to NSD2-PWWP1 over other PWWP domains, including NSD3-PWWP1, DNMT3A-PWWP, and ZCWPW1-PWWP. Importantly, 15 has little effect on the global level of H3K36me2 in cells, suggesting that 15 exhibits no impact on the SET domain or catalytic function after binding to NSD2-PWWP1. Furthermore, 15 at a high concentration (10 μM) can remarkably decrease the levels of NSD2 target genes, including PAK1, RRAS2, TGFA, TEMEL2, and NCAM1. In addition, 15 can suppress the proliferation of some tumor cell lines, including RS4:11 (IC50 = 6.30 μM), MV4:11 (IC50 = 2.23 μM), KMS11 (IC50 = 8.43 μM), and MM1S (IC50 = 10.95 μM). 15 induces the apoptosis of KMS11 and MV4:11 cells in a dose-dependent manner and the cell cycle arrest at G0/G1 phase. Taken together, 15 appears to be a useful chemical probe to understand the specific regulation mode of NSD2 by PWWP1 antagonism, facilitating the discovery of more potent NSD2 antagonists.
Proteolysis Targeting Chimeras (PROTACs) Targeting NSD2.
Over the past decade, the PROTAC technology has been emerging as a novel efficient strategy for the treatment of many diseases by inducing the degradation of target proteins via the ubiquitin-proteasome system (UPS), including those target proteins that were previously described as “undruggable”.244–252 PROTACs, characterized by heterobifunctional small molecules, are composed of a ligand for the protein of interest (POI), a ligand for an E3 ligase, such as von Hippel-Lindau (VHL), cereblon (CRBN), mouse double minute 2 homolog (MDM2), a cellular inhibitor of apoptosis protein-1 (cIAP1), etc. to recruit the UPS and a linker for connecting the two ligands.253–259 PROTACs exert their function commonly through three steps: first, the formation of POI-PROTAC-E3 ligase ternary complexes, then induction of the polyubiquitylation of POIs, followed by the recognition and degradation of polyubiquitylated POIs by the 26S proteasome.260, 261 Compared to traditional small molecule inhibitors, PROTACs possess several advantages244, 250, 253 including that 1) PROTACs can induce the degradation of proteins, even those targets currently undruggable, providing new therapeutic options for various diseases; 2) PROTACs act through an “event-driven” mode instead of an “occupancy-driven” mode, thereby displaying their potency at a catalytic amount with potential to mitigate or avoid the off-target side effects; and 3) PROTACs can improve the selectivity for POIs toward other subtypes or proteins sharing similar active structures, further reducing the off-target toxicity. In recent years, significant advances have been achieved in the field of PROTAC technologies to develop novel degraders induing the degradation of numerous disease-related targets such as AR, ER, BRD4, EZH2, BTK, EGFR, etc.250, 251, 255 More than 18 PROTACs have entered clinical trials, of which, the most advanced is an ER degrader ARV-471 in phase III clinical trials.249, 261
The PROTAC technology has also been applied for inducing NSD3 degradation based on a potent and selective NSD3-PWWP1 antagonist 46 (BI-9321, Figure 16).262, 263 In 2022, compound MS9715 (55, Figure 23) was designed and synthesized, comprising an NSD3-PWWP1 inhibitor 46, an E3 ligase VHL ligand, and an alkyl connecting linker.262 Compared with 46 (Kd = 1.7 ± 0.04 μM), compound 55 (Kd = 1.3 ± 0.17 μM) displays a comparable binding affinity to the NSD3-PWWP1 domain. Compound 55 but not 46 can effectively downregulate the expression level of NSD3 and associated cMyc genes in multiple hematological cancer cells, exerting similar effects with NSD3 knockout mediated by CRISPR-Cas9. Notably, 55 can significantly induce the degradation of NSD3 in MOLM13 cells with a DC50 value of 4.9 ± 0.4 μM and Dmax value of greater than 80% after 48 h treatment. Interestingly, 55, but not 46, can effectively prevent the growth of a panel of NSD-dependent hematological cancer cells, including EOL-1, MM1.S, MOLM13, etc. In addition, 55 causes NSD3 degradation depending on VHL and the ubiquitin-proteasome system. In 2022, based on the PROTAC technology, a series of NSD3 degraders were designed by connecting an NSD3-PWWP1 antagonist 46 and a VHL or CRBN ligase ligand via diverse linkers.263 Compound 56 (Figure 23) can effectively and specifically induce NSD3 degradation with DC50 values of 1.43 and 0.94 μM in lung cancer cells NCI-H1703 and A549, respectively. Furthermore, 56 can suppress the methylation of H3K36, induce apoptosis, and cause cell cycle arrest in lung cancer cells. Moreover, 56 can effectively downregulate the expression of NSD3-associated genes such as CDC25A, ALDH1A1, and IGFBP. Importantly, 56, but not 46, can suppress the proliferation of NCI-H1703 and A549 cells (IC50 = 2.74 and 5.49 μM, respectively). When 56 was administrated alone, the level of NSD3 protein was significantly decreased in lung cancer xenograft models. These results support that targeting NSD3 degradation by PROTACs anchored to the PWWP1 domain to block its function may provide an effective strategy for cancer treatment, showing superiority over NSD3-PWWP1 antagonists.
Figure 23.

Chemical structures of PROTACs 16, and 55–58.
The co-crystal structure of the NSD2-PWWP1 domain in complex with 14 (UNC6934) revealed that the pyrimidine ring (marked by the red circle) points into the solvent-exposed region (Figure 24, PDB ID: 6XCG). Based on such structural information, a biotin-labeled compound 53 (Figure 18) was prepared for pull-down studies by replacing the pyrimidine ring with a phenyl ring and introducing the biotin to the para-position of the phenyl group.242 Excitingly, compound 53 displayed appreciable binding affinity (Kd = 46 nM) to the NSD2-PWWP1 domain. In 2022, according to the insights obtained from the co-crystal structure and binding affinity results of 53, a series of NSD2 PROTACs were designed and synthesized by connecting 14 to VHL or CRBN E3 ligase ligands via diverse linkers.264 These NSD2 degraders contain a phenyl or pyridine group instead of the pyrimidine ring to leave the para-position attached to a linker and E3 ligase ligand. The binding affinities of 14, degrader MS159 (16), and two negative controls 57 and 58 (Figure 23) to the NSD2-PWWP1 domain were evaluated using the ITC assay. Compounds 16 (Kd = 1.1 ± 0.4 μM) and 57 (Kd = 0.9 ± 0.3 μM) display similar binding affinities to the NSD2-PWWP1 domain with that of 14 (Kd = 0.5 ± 0.3 μM), supporting their initial design hypothesis. As expected, 58, designed based on an inactive NSD2-PWWP1 domain antagonist, showed no appreciable binding affinity to the NSD2-PWWP1 domain. Compound 16, capable of binding with NSD2-PWWP1 and E3 ligase simultaneously, displayed the most potent NSD2 degradation activity with a DC50 value of 5.2 ± 0.9 μM and Dmax > 82% in 293FT cells after 48 h treatment. Moreover, 16 can significantly induce the degradation of NSD2 protein in cells in a concentration-, time-, CRBN-, and proteasome-dependent manner. Furthermore, 16 suppresses the growth of MM cell lines (KMS11 and H929) much more effectively than 14, suggesting that NSD2 degradation induced by PROTACs may be developed into an effective and superior therapeutic strategy for blocking the PPI between NSD2-PWWP1 domain and chromatin to treat patients with MM. In addition, 16 can also effectively cause the degradation of CRBN neo-substrates, including IKZF1 and IKZF3, but not GSPT1.
Figure 24.

Crystal structure of compound 14 (UNC6934) in complex with NSD2-PWWP1 domain (PDB ID: 6XCG). Compound 14 is shown as green sticks, and the red dashed circle highlights the pyrimidine ring that points into the solvent-exposed region.
N-degrons, also known as “N-recognins”, mimic the N-terminal residues that a class of E3 ligases has commonly recognized to induce the efficient degradation of different proteins via the N-end rule pathway.265–267 This class of E3 ligases consists of at least four members (UBR1, UBR2, UBR4, and UBR5), and their names were originated by their conserved ubiquitin-recognin box (UBR-box) domain, which can recognize the specific N-terminal protein sequences. Type 1 N-degrons contain basic residues, including Arg, Lys, and His. In 2023, inspired by this novel approach of using N-degron as a UBR E3 ligase recruiting ligand,268 a series of 14 (UNC6934)-based heterobifunctional molecules were designed by replacing the terminal pyrimidine ring of 14 that extends into the solvent exposure region with a phenyl ring.269 Simultaneously, an Arg or His was tethered to the para-position of benzene through alkyl amine linkers with different lengths. Such a design with eight new compounds containing Arg and His was anticipated to induce the degradation of NSD2 through recruiting a UBR E3 ligase into proximity with NSD2. Excitingly, these molecules maintain potent binding affinities to NSD2-PWWP1 with Kd values of 10–60 nM determined by SPR, revealing that the attachment of a linker and basic amino acid has no impact on the binding ability. In-cell western (ICW) results showed that compound UNC7753 (59, Kd = 14 ± 2 nM, Figure 25), containing an Arg group and a six-carbon linker, displayed the most effective NSD2 degradation activity in U2OS cells (Dmax = 81 ± 2% and DC50 = 1.2 ± 0.3 μM).
Figure 25.

Chemical structures of compounds 59 and 60.
Further, SAR explorations around compound 59 were conducted to improve the potency and cell permeability. Interestingly, 59 and its analogs were observed to induce NSD2 degradation through a novel mechanism independent of UBR E3 ubiquitin ligases, which is inconsistent with their initial expectation. Compound UNC8153 (60, Kd = 24 ± 7 nM, Figure 25) without the amino acid moiety emerged as the most potent NSD2 degrader (Dmax = 79 ± 1% and DC50 = 0.35 ± 0.1 μM), which is over 3-fold more potent than 59. Compound 59 can be converted into 60 to induce NSD2 degradation in cells under the proteolysis of cellular proteases, indicating that 59 functions as a prodrug. Moreover, 60-trigged degradation of NSD2 depends on proteasome and neddylation; however, the E3 ligase engaged in this process remains unclear. In addition, 60 displays excellent selectivity, inducing the degradation of only NSD2 but not other NSD members, including NSD1 and NSD3. Furthermore, 60 significantly reduces the level of H3K36me2 in multiple cell lines, including t(4;14)-translocated KMS11 (MM) cells, E1099K-mutated MM1S (MM) and RS411 (ALL) cells, and wild-type U2OS cells. Importantly, 60 treatment can result in mild proliferation suppression in MM1S cells carrying E1099K mutation, while no apparent suppression in t(4;14)-translocated KMS11 cells and other cells containing unaltered, wide-type NSD2 gene, including MDA-MB-231 (breast cancer), U2OS (bone cell), and HEK293 (kidney cancer) cells, despite significant NSD2 degradation. This finding suggests that the growth or survival of MM1S cells harboring E1099K activating mutation depends on the catalytic activity of NSD2. Additionally, 60-mediated NSD2 degradation can exert significant antiadhesive effects in KMS11 cells, which is consistent with the results of NSD2 knockdown. Therefore, 60 and its analogs may serve as useful tools for exploring the role of NSD2 degradation and its therapeutic potential in NSD2-associated cancers.
Combination Use of NSD2 Inhibitors and Other Therapies.
In recent years, developing combination therapy has been gaining increasing attention, especially in the field of cancers, due to its potential superiority to monotherapies, including synergetic therapeutic effects and decreased drug resistance.270–273 Moreover, combination therapy has been universally implemented for epigenetic targets and demonstrated additive and synergetic therapeutic effects.186, 274 Substantial efforts have been made in developing combination therapy for NSD2-associated diseases to improve the therapeutic potential.172, 206, 227, 275–277
Combinatorial use of NSD2 inhibitors, such as small molecules MCTP-39 and its analogs, siRNAs, antibodies, and antisense, with chemotherapeutic agents such as alkaloids, alkylating agents, and antitumor antibiotics, was demonstrated to significantly suppress the proliferation and invasion of many types of cancer cells, including PCa and breast cancer.172 Moreover, they identified H3K36me2 as EZH2 target markers. Furthermore, they found that EZH2 can indirectly affect the level of H3K36me2 by regulating NSD2 and NSD2 knockdown attenuates EZH2-mediated cancer cell invasion, suggesting that NSD2 is a downstream target protein of EZH2. These two proteins play critical synergetic roles in the progression of multiple cancers. Therefore, inhibiting the function of EZH2 and NSD2 simultaneously by combinatorial therapy may display additive or synergetic therapeutic effects. In addition, dual EZH2 and NSD2 inhibitors can also be developed to enhance the anti-tumor activity. Verticillin A was reported to lead to the significant induction of latent virus, indicating that its combination with one or more antiretroviral drugs may enhance the antiviral effects, especially for HIV.206 NSD2 inhibitors, including polypeptides, polynucleotides comprising of antisense molecule, siRNA and shRNA, and small molecules DZNep, MCTP-39, 10 (LEM06), Ku55933 and AZD0516, were disclosed to increase the sensitivity of diverse cancer cells (e.g., PCa, breast cancer, colorectal cancer, pancreatic cancer, gastric cancer) to chemotherapeutic agents such as doxorubicin, etoposide, and PI3 kinase (PI3K) inhibitors BKM120BYL719 and RP6530, indicating that combination use of NSD2 and PI3K inhibitors simultaneously show synergetic antitumor effect.275 Researchers from Epizyme Inc. disclosed a patent that SETD2 inhibitors with a scaffold of substituted indoles or indolines, were used to treat or slow the progression of cancers with NSD2-overexpression, including t(4;14) MM, suggesting that SETD2 inhibitors used as a single agent or in combination with an NSD2 inhibitor might provide a potential therapy for cancers.276 Some compounds described in this patent significantly suppress the growth of KMS-34 cells in a dose-dependent manner. Moreover, these compounds can significantly reduce the level of H3K36me3 while showing no effect on H3K36me2, which was directly associated with its obvious anti-proliferation activity. Notably, they can cause the tumor growth regression in a dose-dependent manner in a KMS11 t(4; 14) xenograft mice model. NSD2 was revealed to be highly overexpressed in breast cancer cells resistant to tamoxifen treatment.170 Moreover, NSD2 inhibitor DZNep can remarkably inhibit the survival of tamoxifen-resistant breast cancer cells and induce cell apoptosis both in vitro and in vivo, suggesting that combination use of tamoxifen and NSD2 inhibitor DZNep can exhibit synergetic anti-tumor effects. Researchers from Novartis Inc. also disclosed the combination use of an NSD2 inhibitor and therapeutic agent to treat or prevent various diseases or disorders, especially for cancers, including MM and thyroid carcinoma.227 It was reported that the combined use of an NSD2 inhibitor, including small molecules, antibodies, aptamers (oligonucleotides or peptides), siRNAs, and antisense oligonucleotides, with conventional therapies such as surgical, immunotherapy (checkpoint inhibitors), chemotherapy, radiotherapy, may provide additive or synergistic effects for treating MM patients, including those with t(4;14) translocation.277 Overall, accumulating evidence supports that combinatorial use of NSD2 inhibitors and other therapies may exert additive or synergetic therapeutic effects, providing potential new therapeutic alternatives for NSD2-driven diseases.
CONCLUSIONS AND FUTURE PERSPECTIVES
NSD family proteins (NSD1, NSD2, and NSD3) are important members of HMTases and are well known to catalyze the mono- and di-methylation of H3K36 (H3K36me1 and H3K36me2). The aberrant expression, translocations, or mutations of NSDs have been widely considered the driving force for various human diseases such as cancers, inflammations, and infectious diseases. As one key member of the NSD family, NSD2 roles as an epigenetic modifier, and its aberrant functions caused by overexpression, mutation, or translocation, are widely implicated in various diseases mentioned above. Moreover, knockout or knockdown of NSD2 has been proven to effectively suppress the growth of cancer cells, induce cell apoptosis, and delay cell-cycle progression. Therefore, inhibiting the hyperactivity of NSD2 by therapeutic agents, especially small molecule inhibitors, may provide new therapeutic options for patients with NSD2-driven conditions. In this review, we briefly introduced the structures and functions of NSDs, distinctly described the role of NSD2 in various cancers and other diseases, and comprehensively summarized the recent advances in approaches targeting NSD2 inhibition, including the development of potent and selective small molecule inhibitors, PROTACs protein degraders, and combinatorial therapies.
Given its critical pharmacological role in various diseases or disorders, NSD2 as an emerging drug target has attracted the increasing attention of researchers from both academic and industrial settings. Through continuous efforts, several NSD2 inhibitors have been discovered and may serve as important pharmacological tools for exploring the functions of NSD2 in diverse diseases. The available findings also demonstrate the feasibility of targeting the inhibition of NSD2 by small molecules that may pave the way for developing potent and specific NSD2 inhibitors as potential therapeutics. Encouragingly, KTX-1001, a potent and selective NSD2 SET domain inhibitor, has successfully entered phase I clinical trials. Nevertheless, most currently available NSD2 inhibitors remain in very early stage of development, suffering issues such as weak potency, especially in cells and animal models, poor subtype selectivity, or limited druglike properties. Hence, developing more potent and selective NSD2 inhibitors and protein degraders with favorable DMPK profiles are urgently needed for powerful pharmacological tools and potential drug candidates.
Although NSDs proteins have been studied for over two decades since their initial identification, several challenges, as described earlier, significantly hinder the drug development of novel specific NSD2 inhibitors or even pan-inhibitors for the whole NSD family proteins. Meanwhile, ongoing studies and technology innovation also bring new opportunities for discovering and developing novel molecules targeting NSD2. Opportunities include: 1) The currently available NSD2 inhibitors provide useful chemical leads and meaningful SAR information that may pave the way for further optimization of more potent and selective NSD2 inhibitors; 2) The disclosure of several co-crystal structures of NSD2 in complexed with small molecule NSD2 inhibitors may facilitate the virtual screening to identify new hits as well as the structure-based rational drug design; 3) The development and optimization of various bioassays for evaluation of NSD2 inhibitory activities may also facilitate the HTS, accelerate the screen speed and improve the accuracy of activities; 4) Other than the catalytic SET domain, the PHD and PWWP domains are also emerging as promising targets for discovery of selective NSD2 inhibitors; 5) The rapid advances in PROTAC technologies may also open new avenues for the development of novel therapeutics targeting NSD2, or even those drug targets that are currently undruggable and readily mutated proteins. PROTAC technology has been applied for degrading NSD2 and NSD3 proteins as proof-of-concept studies, displaying better efficacy than corresponding inhibitors. It is reasonable to believe that more potent PROTACs selectively targeting NSDs will be discovered through structural optimization and exploration of more E3 ligase ligands; and 6) The combinatorial use of NSD2 inhibitors and other chemotherapeutic agents has been proven to exhibit synergetic or additive effects, offering a powerful alternative approach for treating NSD2-associated diseases. Moreover, the widely recognized success of combinatorial therapies provides a solid basis for the rational design of dual-targeting inhibitors to enhance efficacy or overcome drug-resistance.
To date, several NSD2 inhibitors have been discovered and may serve as useful tools for exploring the role and functions of NSD2 in various human conditions. However, most of these available molecules’ potency, selectivity, and druglike properties are relatively poor, robustly limiting the exploration of their biological functions and hampering their further development as viable therapeutic agents. Current strategies targeting NSD2 inhibition mainly include small molecule inhibitors, PROTACs, and combinatorial therapies, while most small molecule inhibitors focus on targeting the catalytic SET domain. Currently, several cocrystal structures of small molecule inhibitors in complex with NSD2 and some screening bioassays with high accuracy and stability are available. This may significantly facilitate the virtual screening and HTS to identify more novel hits for further lead optimization to achieve promising candidates with high potency, distinguished selectivity, and excellent ADMET properties. With the emergence of compounds 14, 15 and 43–45, it appears that other than the SET domain, PWWPs and PHDsare also promising domains that small molecules can successfully target. Moreover, inhibitors targeting PWWP and PHD domains tend to show higher selectivity than those targeting SET domains, indicating that targeting PWWP and PHD domains may be a promising approach to improve selectivity. We believe that more potent and selective NSD2 inhibitors targeting PWWP and PHD domains will likely be discovered soon. The SET, PWWP, and PHD domains of NSD2 have diverse biological functions in transcriptional modulation, while the PWWP and PHD domains of NSDs show lower structural similarity than SET domain. Therefore, other than monovalent inhibitors, bivalent molecules, composed of two same or different warheads and a connecting linker, can also be developed to enhance enzyme inhibitory activity and improve subtype selectivity. These bivalent molecules can interact with two same or different domains, such as SET-PWW1 inhibitors, PWWP1-PWWP1 inhibitors, and SET-PHD inhibitors.
In recent years, with the advent and booming of target protein degradation (TPD) technologies, many PROTACs have successfully entered clinical trials, bringing new alternatives for clinical uses. The most advanced degrader is ARV-471 (an ER degrader) in phase III clinical trials for treating advanced-stage ER+HER2− breast cancer (ClinicalTrials.gov Identifier: NCT01564797). Moreover, the proof-of-concept studies of PROTACs for targeting NSD3 and NSD2 have been established with the discovery of compounds 16, 55, 56, and 60. Furthermore, other emerging TPD strategies have been developed,252 such as Molecule Glue (MG), AUtophagy-TArgeting Chimera (AUTAC), AUTOphagy-TArgeting Chimera (AUTOTAC), etc., which may also be applied for designing novel NSD2 degraders. We believe that more effective NSD2-based degraders will be achieved through further structural optimization, new E3 ligase ligands exploration, and the application of novel TPD strategies. In addition to TPD technologies, other emerging novel approaches, such as regulated induced proximity targeting chimera (RIPTAC),278 may also be applied for designing NSD2-targeted therapeutic agents. RIPTACs are a class of heterobifunctional molecules comprising a Target Protein (TP)-ligand selectively expressed in cancer cells, a pan-essential Effector Protein-ligand (EP) required for survival, and a linker connecting these two different components. RIPTACs can selectively kill the cancer cells expressing the TP, while sparing the normal cells by forming a positive, cooperative ternary complex with the TP and EP (TP-RIPTAC-EP). The formation of the ternary complex can enhance the protein-protein PPIs between the TP and EP, abrogating the function of the EP and leading to cell death. NSD2 protein, especially the PWWP1 and PHD domains, can be used as the TP that is selectively overexpressed in tumor cells, and the EPs can be chosen from BRD4, PLK1, and CDKs essential for cell survival. We anticipate that NSD2-targeted RIPTACs may be developed to achieve the goal of increasing the potency, improving the selectivity, and overcoming the mutation-trigged resistance.
Combinatorial use of NSD2 inhibitors and other therapeutic agents has been validated to exhibit synergetic or additive effects, showing great potential to overcome drug resistance, supporting that combinatorial therapy may be a promising strategy for the development of NSD2 inhibitors. Importantly, this also provides a critical basis for the rational design of NSD2-based dual-target inhibitors to solve the problems (e.g., insufficient efficacy) that appear in developing NSD2 individual inhibitors or improve the clinical outcomes of other drugs (e.g., overcoming drug resistance). Therefore, developing dual-target inhibitors may offer a novel potential strategy targeting NSD2 with superiority. NSD2 has been demonstrated to be a downstream target of EZH2, and overexpression of NSD2 and EZH2 exerts a synergic effect in many tumors. Thus, inhibiting the function of both NSD2 and EZH2 through the combination use of these target inhibitors or dual inhibitors may exhibit synergic or additive effects and overcome drug resistance. We hypothesize that blocking the function of NSD2 and other epigenetic modulators (e.g., DOT1L, BRD4, and HDACs) through combinatorial therapy or dual inhibitors can also be developed as a potential therapeutic strategy for the treatment of cancers and other diseases. Furthermore, combination use of NSD2 inhibitor and cell apoptosis protein (e.g., IAP and Bcl-2) inhibitors, cell cycle regulatory proteins (e.g., CDKs and PLK1) inhibitors, and DNA damage repair proteins (e.g., PARPs) inhibitors, or developing dual inhibitors, may enhance the therapeutic effects and combat drug resistance, ultimately benefiting more clinical patients.
In conclusion, NSD2 plays a driving role in the pathology of many diseases, especially cancers, and has been considered a promising therapeutic target. Developing potent and specific NSD2 inhibitors may offer novel therapies for NSD2-associated diseases. In this review, the recent advance in developing NSD2 inhibitors and degraders were comprehensively summarized, and the challenges and opportunities, as well as future directions, were also discussed. Some potential strategies have been presented for developing NSD2-based therapies, such as combinatorial therapy, dual inhibitors, bivalent inhibitors, TPDs, and RIPTACs. However, more proof-of-concept studies are needed to validate the feasibility and effectiveness of these novel approaches toward viable therapeutics. Most importantly, KVX-1001 becomes the first NSD2 inhibitor entering clinical trials, demonstrating that inhibiting the function of NSD2 by small molecule inhibitors may offer a promising and viable therapeutic strategy for cancer treatment. With the ongoing rapid revolution of drug discovery and development technologies and the unremitting efforts of researchers, there is no doubt that more and more potent and selective NSD2 inhibitors and degraders with desirable druglike profiles will be developed as epigenetic therapeutics for clinical trials, eventually benefiting patients with NSD2-driven or -associated diseases, especially various cancers.
SIGNIFICANCE.
Small molecule NSD2 inhibitors and degraders show therapeutic promise for NSD2-driven and -associated diseases, especially cancers.
The recent advances in drug discovery efforts targeting NSD2 are systematically summarized.
Current challenges and opportunities as well as future directions for developing NSD2 inhibitors and degraders are discussed from the medicinal chemistry perspective.
This perspective provides insights into future drug discovery strategies targeting NSD2.
ACKNOWLEGMENTS
This work was partially supported by grants from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP210062) and the Leukemia & Lymphoma Society (MCL7003-24), the John D. Stobo, M.D. Distinguished Chair Endowment, and the Edith & Robert Zinn Chair Endowment in Drug Discovery.
HC and JZ are partially supported by the pharmaceutical industry MapLight Therapeutics, Inc. through an industry-funded collaborative research project, which is not directly related to this manuscript. Other than that, the authors declare no competing financial interest.
ABBREVATIONS USED
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- Bcl-2
B-cell lymphoma-2
- 53BP1
p53-binding protein 1
- BRD4
bromodomain-containing protein 4
- CC
cervical cancer
- ccRCC
clear cell renal cell carcinoma
- CDKs
cyclin-dependent kinases
- cIAP1
cellular inhibitor of apoptosis protein-1
- CML
chronic myeloid leukemia
- CNS
central nervous system
- CRBN
cereblon
- CRC
colorectal cancer
- CRL4Cdt2
cullin-ring ligase 4-Cdt2
- CSCs
cancer stem cells
- DDR
DNA damage response
- DLBCL
diffuse large B-cell lymphoma
- DMPK
drug metabolism and pharmacokinetics
- DOT1L
disruptor of telomeric silencing 1-like
- DSBs
double-strand breaks
- DSLS
differential static light scattering
- EMT
epithelial-mesenchymal transition
- Erα
estrogen receptor alpha
- EZH2
enhancer of zeste homolog 2
- FLT3
FMS-like tyrosine kinase 3
- GLUT1
glucose transporter 1
- G6PD
glucose-6-phosphate dehydrogenase
- GSH
glutathione
- HCC
hepatocellular carcinoma
- HDACs
histone deacetylases
- HK2
hexokinase 2
- H3K36
histone 3 lysine 36
- H3K36me1
momo-methylated H3K36
- H3K36me2
di-methylated H3K36
- H3K36me3
tri-methylated H3K36
- HKMTases
histone lysine methyltransferases
- HMG-box
high mobility group box
- HNSCC
head and neck squamous cell carcinoma
- IAP
inhibitor of apoptosis protein
- IL-6
interleukin-6
- ITC
isothermal titration calorimetry
- KDM
lysine demethylase
- KMT
lysine methyltransferase
- MCL
mantle cell lymphoma
- MDM2
mouse double minute 2 homolog
- MM
multiple myeloma
- MMSET
multiple myeloma SET domain
- mRCC
metastatic renal cell carcinoma
- NADPH
nicotinamide adenine dinucleotide phosphate
- NEK7
NIMA-related kinase 7
- NSD
nuclear receptor binding SET domain protein
- NSD2
nuclear receptor binding SET domain protein 2
- NUP98
nucleoporin-98
- OCCC
ovarian clear cell carcinoma
- OS
osteosarcoma
- PARPs
poly (ADP-ribose) polymerases
- PCa
prostate cancer
- PD
pharmacodynamic
- PHD
plant homeodomain
- PK
pharmacokinetics
- PKMT
protein lysine methyltransferase
- PLK1
polo-like kinase 1
- POI
protein of interest
- PPI
protein-protein interaction
- PPP
pentose phosphate pathway
- pre-RC
pre-replication complex
- PROTAC
proteolysis protein chimera
- PTEN
phosphatase and tesin homolog deleted on chromosome 10
- PWWP
proline-tryptophan-tryptophan-proline
- RCC
renal cell carcinoma
- RIPTAC
regulated induced proximity targeting chimera
- r-MDS
radiation-associated myelodysplastic syndrome
- ROS
reactive oxygen species
- SAH
S-adenosyl-L-homocysteine
- SAM
S-adenosyl-L-methionine
- SCC
skin squamous cell carcinoma
- SPR
surface plasmon resonance
- TGF-β1
transforming growth factor-β1
- TIGAR
TP53-induced glycolysis and apoptosis regulator
- t-MDS
therapy-related myelodysplastic syndrome
- TNBC
triple-negative breast cancer
- TNF-α
tumor necrosis factor alpha
- TPD
target protein degradation
- TWIST
twist family bHLH transcription factor
- UPS
ubiquitin-proteasome system
- VEGFA
vascular endothelial growth factor A
- VHL
von Hippel-Lindau
- WHSC1
Wolfe-Hirschhorn syndrome candidate 1
- WHSC1L1
Wolf-Hirschhorn syndrome candidate 1-like 1
Biographies
Zonghui Ma received his Ph.D. in Medicinal Chemistry from China Pharmaceutical University (CPU) in 2019 under the supervision of Professor Haiying Sun. After graduation, he continued his research work as a Research Associate in CPU and a Senior Scientist in a pharmaceutical company for three years. Dr. Ma is currently pursuing his postdoctoral training in Professor Jia Zhou’s Chemical Biology Program at the University of Texas Medical Branch (UTMB). His research interests focus on the rational design and synthesis of target-based small molecule modulators as promising pharmacological probes and therapeutic agents for treating cancers, inflammatory disorders, and infectious diseases.
Andrew A. Bolinger did his undergraduate research under the supervision of Dr. Joseph L. Richards and completed his BS degree in 2013 at Colorado Mesa University. He then joined Dr. Don Coltart’s group in 2014, where he studied organic chemistry methodologies related to umpolung alkylation via azo compounds at the University of Houston. He later completed his Ph.D. with a rotation in Dr. Jeremy May’s lab. After graduating in 2019, he did a one-year postdoc at Rutgers - Newark in the Freundlich lab during the COVID-19 pandemic before joining Dr. Jia Zhou’s group at UTMB. His current research is focused on the rational design and synthesis of novel small molecules that target GPCR receptors related to substance use disorders and other human diseases.
Haiying Chen obtained her BSc degree in Engineering from Tianjin University in 1995. She then worked at Tianjin Research Institute of Construction Machinery as an engineer focusing on designing and programming computer testing systems. Since 2014, she has been conducting drug discovery and development research in Dr. Jia Zhou’s Chemical Biology Program as a Senior Research Associate by utilizing the computer‐assisted rational drug design of novel target‐based small molecules and the understanding of key interactions and binding modes of ligands with various drug targets for the treatment of human cancers, CNS disorders, and infectious diseases.
Jia Zhou obtained his Ph.D. in organic chemistry from Nankai University, and pursued his postdoctoral training in organic and bioorganic chemistry at the University of Virginia, as well as medicinal chemistry and drug discovery at Georgetown University Medical Center. With 7-year industrial research experience, he is currently a tenured professor at UTMB, John D. Stobo, M.D. Distinguished Chair, and a distinguished inaugural holder of Edith & Robert Zinn Chair in Drug Discovery. He is an author of more than 230 peer‐reviewed articles and eight book chapters as well as an inventor of 32 patents. Dr. Zhou is a National Academy of Inventors (NAI) Fellow, and the Editor‐in‐Chief of Current Topics in Medicinal Chemistry, as well as the Editorial Advisory Board of Journal of Medicinal Chemistry.
References:
- (1).Husmann D; Gozani O Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol 2019, 26, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhat KP; Umit Kaniskan H; Jin J; Gozani O Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov 2021, 20, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Martin C; Zhang Y The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol 2005, 6, 838–849. [DOI] [PubMed] [Google Scholar]
- (4).Kuo AJ; Cheung P; Chen K; Zee BM; Kioi M; Lauring J; Xi Y; Park BH; Shi X; Garcia BA; Li W; Gozani O NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wagner EJ; Carpenter PB Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol 2012, 13, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li J; Ahn JH; Wang GG Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell Mol. Life Sci 2019, 76, 2899–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kaniskan HU; Martini ML; Jin J Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev 2018, 118, 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Morishita M; di Luccio E Cancers and the NSD family of histone lysine methyltransferases. Biochim. Biophys. Acta 2011, 1816, 158–163. [DOI] [PubMed] [Google Scholar]
- (9).Zhang X; Wen H; Shi X Lysine methylation: beyond histones. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 14–27. [DOI] [PubMed] [Google Scholar]
- (10).Bennett RL; Swaroop A; Troche C; Licht JD The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med 2017, 7, a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Vougiouklakis T; Hamamoto R; Nakamura Y; Saloura V The NSD family of protein methyltransferases in human cancer. Epigenomics 2015, 7, 863–874. [DOI] [PubMed] [Google Scholar]
- (12).Kassambara A; Klein B; Moreaux J MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem. Biophys. Res. Commun 2009, 379, 840–845. [DOI] [PubMed] [Google Scholar]
- (13).Hudlebusch HR; Santoni-Rugiu E; Simon R; Ralfkiaer E; Rossing HH; Johansen JV; Jorgensen M; Sauter G; Helin K The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin. Cancer Res 2011, 17, 2919–2933. [DOI] [PubMed] [Google Scholar]
- (14).Keats JJ; Reiman T; Maxwell CA; Taylor BJ; Larratt LM; Mant MJ; Belch AR; Pilarski LM In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [DOI] [PubMed] [Google Scholar]
- (15).Cheong CM; Mrozik KM; Hewett DR; Bell E; Panagopoulos V; Noll JE; Licht JD; Gronthos S; Zannettino ACW; Vandyke K Twist-1 is upregulated by NSD2 and contributes to tumour dissemination and an epithelial-mesenchymal transition-like gene expression signature in t(4;14)-positive multiple myeloma. Cancer Lett. 2020, 475, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mirabella F; Wu P; Wardell CP; Kaiser MF; Walker BA; Johnson DC; Morgan GJ MMSET is the key molecular target in t(4;14) myeloma. Blood Cancer J. 2013, 3, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jaffe JD; Wang Y; Chan HM; Zhang J; Huether R; Kryukov GV; Bhang HE; Taylor JE; Hu M; Englund NP; Yan F; Wang Z; Robert McDonald E 3rd; Wei L; Ma J; Easton J; Yu Z; deBeaumount R; Gibaja V; Venkatesan K; Schlegel R; Sellers WR; Keen N; Liu J; Caponigro G; Barretina J; Cooke VG; Mullighan C; Carr SA; Downing JR; Garraway LA; Stegmeier F Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet 2013, 45, 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Swaroop A; Oyer JA; Will CM; Huang X; Yu W; Troche C; Bulic M; Durham BH; Wen QJ; Crispino JD; MacKerell AD Jr.; Bennett RL; Kelleher NL; Licht JD An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 2019, 38, 671–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Oyer JA; Huang X; Zheng Y; Shim J; Ezponda T; Carpenter Z; Allegretta M; Okot-Kotber CI; Patel JP; Melnick A; Levine RL; Ferrando A; Mackerell AD Jr.; Kelleher NL; Licht JD; Popovic R Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014, 28, 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pierro J; Saliba J; Narang S; Sethia G; Saint Fleur-Lominy S; Chowdhury A; Qualls A; Fay H; Kilberg HL; Moriyama T; Fuller TJ; Teachey DT; Schmiegelow K; Yang JJ; Loh ML; Brown PA; Zhang J; Ma X; Tsirigos A; Evensen NA; Carroll WL The NSD2 p.E1099K Mutation Is Enriched at Relapse and Confers Drug Resistance in a Cell Context-Dependent Manner in Pediatric Acute Lymphoblastic Leukemia. Mol. Cancer Res 2020, 18, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Topchu I; Pangeni RP; Bychkov I; Miller SA; Izumchenko E; Yu J; Golemis E; Karanicolas J; Boumber Y The role of NSD1, NSD2, and NSD3 histone methyltransferases in solid tumors. Cell Mol. Life Sci 2022, 79, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Azagra A; Cobaleda C NSD2 as a Promising Target in Hematological Disorders. Int. J. Mol. Sci 2022, 23, 11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen R; Chen Y; Zhao W; Fang C; Zhou W; Yang X; Ji M The Role of Methyltransferase NSD2 as a Potential Oncogene in Human Solid Tumors. Onco. Targets Ther 2020, 13, 6837–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shrestha A; Kim N; Lee SJ; Jeon YH; Song JJ; An H; Cho SJ; Kadayat TM; Chin J Targeting the Nuclear Receptor-Binding SET Domain Family of Histone Lysine Methyltransferases for Cancer Therapy: Recent Progress and Perspectives. J. Med. Chem 2021, 64, 14913–14929. [DOI] [PubMed] [Google Scholar]
- (25).Zhang L; Zha X Recent advances in nuclear receptor-binding SET domain 2 (NSD2) inhibitors: An update and perspectives. Eur. J. Med. Chem 2023, 250, 115232. [DOI] [PubMed] [Google Scholar]
- (26).Rothbart SB; Baylin SB Epigenetic Therapy for Epithelioid Sarcoma. Cell 2020, 181, 211. [DOI] [PubMed] [Google Scholar]
- (27).Stein EM; Garcia-Manero G; Rizzieri DA; Tibes R; Berdeja JG; Savona MR; Jongen-Lavrenic M; Altman JK; Thomson B; Blakemore SJ; Daigle SR; Waters NJ; Suttle AB; Clawson A; Pollock R; Krivtsov A; Armstrong SA; DiMartino J; Hedrick E; Lowenberg B; Tallman MS The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shukla N; Wetmore C; O’Brien MM; Silverman LB; Brown P; Cooper TM; Thomson B; Blakemore SJ; Daigle S; Suttle B; Waters NJ; Krivstov AV; Armstrong SA; Ho PT; Gore L Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood 2016, 128, 2780–2780. [Google Scholar]
- (29).Liu F; Barsyte-Lovejoy D; Li F; Xiong Y; Korboukh V; Huang XP; Allali-Hassani A; Janzen WP; Roth BL; Frye SV; Arrowsmith CH; Brown PJ; Vedadi M; Jin J Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem 2013, 56, 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ma A; Yu W; Li F; Bleich RM; Herold JM; Butler KV; Norris JL; Korboukh V; Tripathy A; Janzen WP; Arrowsmith CH; Frye SV; Vedadi M; Brown PJ; Jin J Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem 2014, 57, 6822–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ma A; Yu W; Xiong Y; Butler KV; Brown PJ; Jin J Structure-activity relationship studies of SETD8 inhibitors. MedChemComm 2014, 5, 1892–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Barsyte-Lovejoy D; Li F; Oudhoff MJ; Tatlock JH; Dong A; Zeng H; Wu H; Freeman SA; Schapira M; Senisterra GA; Kuznetsova E; Marcellus R; Allali-Hassani A; Kennedy S; Lambert JP; Couzens AL; Aman A; Gingras AC; Al-Awar R; Fish PV; Gerstenberger BS; Roberts L; Benn CL; Grimley RL; Braam MJ; Rossi FM; Sudol M; Brown PJ; Bunnage ME; Owen DR; Zaph C; Vedadi M; Arrowsmith CH (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 12853–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Eggert E; Hillig RC; Koehr S; Stockigt D; Weiske J; Barak N; Mowat J; Brumby T; Christ CD; Ter Laak A; Lang T; Fernandez-Montalvan AE; Badock V; Weinmann H; Hartung IV; Barsyte-Lovejoy D; Szewczyk M; Kennedy S; Li F; Vedadi M; Brown PJ; Santhakumar V; Arrowsmith CH; Stellfeld T; Stresemann C Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem 2016, 59, 4578–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Thomenius MJ; Totman J; Harvey D; Mitchell LH; Riera TV; Cosmopoulos K; Grassian AR; Klaus C; Foley M; Admirand EA; Jahic H; Majer C; Wigle T; Jacques SL; Gureasko J; Brach D; Lingaraj T; West K; Smith S; Rioux N; Waters NJ; Tang C; Raimondi A; Munchhof M; Mills JE; Ribich S; Porter Scott M; Kuntz KW; Janzen WP; Moyer M; Smith JJ; Chesworth R; Copeland RA; Boriack-Sjodin PA Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS One 2018, 13, e0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bromberg KD; Mitchell TR; Upadhyay AK; Jakob CG; Jhala MA; Comess KM; Lasko LM; Li C; Tuzon CT; Dai Y; Li F; Eram MS; Nuber A; Soni NB; Manaves V; Algire MA; Sweis RF; Torrent M; Schotta G; Sun C; Michaelides MR; Shoemaker AR; Arrowsmith CH; Brown PJ; Santhakumar V; Martin A; Rice JC; Chiang GG; Vedadi M; Barsyte-Lovejoy D; Pappano WN The SUV4–20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol 2017, 13, 317–324. [DOI] [PubMed] [Google Scholar]
- (36).Allali-Hassani A; Szewczyk MM; Ivanochko D; Organ SL; Bok J; Ho JSY; Gay FPH; Li F; Blazer L; Eram MS; Halabelian L; Dilworth D; Luciani GM; Lima-Fernandes E; Wu Q; Loppnau P; Palmer N; Talib SZA; Brown PJ; Schapira M; Kaldis P; O’Hagan RC; Guccione E; Barsyte-Lovejoy D; Arrowsmith CH; Sanders JM; Kattar SD; Bennett DJ; Nicholson B; Vedadi M Discovery of a chemical probe for PRDM9. Nat. Commun 2019, 10, 5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dillon SC; Zhang X; Trievel RC; Cheng X The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Herz HM; Garruss A; Shilatifard A SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci 2013, 38, 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sankaran SM; Wilkinson AW; Elias JE; Gozani O A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. J. Biol. Chem 2016, 291, 8465–8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Pasillas MP; Shah M; Kamps MP NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum. Mutat 2011, 32, 292–298. [DOI] [PubMed] [Google Scholar]
- (41).Baker LA; Allis CD; Wang GG PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat. Res 2008, 647, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Angrand PO; Apiou F; Stewart AF; Dutrillaux B; Losson R; Chambon P NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 2001, 74, 79–88. [DOI] [PubMed] [Google Scholar]
- (43).Chesi M; Nardini E; Lim RS; Smith KD; Kuehl WM; Bergsagel PL The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998, 92, 3025–3034. [PubMed] [Google Scholar]
- (44).Morishita M; Mevius D; Shen Y; di Luccio E Recombinant Protein Expression and Purification of the Human HMTase MMSET/NSD2. Curr. Res. Agric. Life Sci 2013, 31, 157–164. [Google Scholar]
- (45).Morishita M; Mevius D; di Luccio E In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L. BMC Struct. Biol 2014, 14, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Rayasam GV; Wendling O; Angrand PO; Mark M; Niederreither K; Song L; Lerouge T; Hager GL; Chambon P; Losson R NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Nimura K; Ura K; Shiratori H; Ikawa M; Okabe M; Schwartz RJ; Kaneda Y A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009, 460, 287–291. [DOI] [PubMed] [Google Scholar]
- (48).Douglas J; Coleman K; Tatton-Brown K; Hughes HE; Temple IK; Cole TR; Rahman N; Childhood Overgrowth C Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur. J. Hum. Genet 2005, 13, 150–153. [DOI] [PubMed] [Google Scholar]
- (49).Qin S; Min J Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci 2014, 39, 536–547. [DOI] [PubMed] [Google Scholar]
- (50).Vermeulen M; Eberl HC; Matarese F; Marks H; Denissov S; Butter F; Lee KK; Olsen JV; Hyman AA; Stunnenberg HG; Mann M Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [DOI] [PubMed] [Google Scholar]
- (51).Zanoni P; Steindl K; Sengupta D; Joset P; Bahr A; Sticht H; Lang-Muritano M; van Ravenswaaij-Arts CMA; Shinawi M; Andrews M; Attie-Bitach T; Maystadt I; Belnap N; Benoit V; Delplancq G; de Vries BBA; Grotto S; Lacombe D; Larson A; Mourmans J; Ounap K; Petrilli G; Pfundt R; Ramsey K; Blok LS; Tsatsaris V; Vitobello A; Faivre L; Wheeler PG; Wevers MR; Wojcik M; Zweier M; Gozani O; Rauch A Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet. Med 2021, 23, 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hirschhorn K; Cooper HL; Firschein IL Deletion of short arms of chromosome 4–5 in a child with defects of midline fusion. Humangenetik 1965, 1, 479–482. [DOI] [PubMed] [Google Scholar]
- (53).Wright TJ; Clemens M; Quarrell O; Altherr MR Wolf-Hirschhorn and Pitt-Rogers-Danks syndromes caused by overlapping 4p deletions. Am. J. Med. Genet 1998, 75, 345–350. [DOI] [PubMed] [Google Scholar]
- (54).Wright TJ; Ricke DO; Denison K; Abmayr S; Cotter PD; Hirschhorn K; Keinanen M; McDonald-McGinn D; Somer M; Spinner N; Yang-Feng T; Zackai E; Altherr MR A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum. Mol. Genet 1997, 6, 317–324. [DOI] [PubMed] [Google Scholar]
- (55).Stec I; Wright TJ; van Ommen GJ; de Boer PA; van Haeringen A; Moorman AF; Altherr MR; den Dunnen JT WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet 1998, 7, 1071–1082. [DOI] [PubMed] [Google Scholar]
- (56).Bergemann AD; Cole F; Hirschhorn K The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005, 21, 188–195. [DOI] [PubMed] [Google Scholar]
- (57).Evans DL; Zhang H; Ham H; Pei H; Lee S; Kim J; Billadeau DD; Lou Z MMSET is dynamically regulated during cell-cycle progression and promotes normal DNA replication. Cell Cycle 2016, 15, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Fnu S; Williamson EA; De Haro LP; Brenneman M; Wray J; Shaheen M; Radhakrishnan K; Lee SH; Nickoloff JA; Hromas R Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Pei H; Zhang L; Luo K; Qin Y; Chesi M; Fei F; Bergsagel PL; Wang L; You Z; Lou Z MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhang J; Lee YR; Dang F; Gan W; Menon AV; Katon JM; Hsu CH; Asara JM; Tibarewal P; Leslie NR; Shi Y; Pandolfi PP; Wei W PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Discov. 2019, 9, 1306–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Shah MY; Martinez-Garcia E; Phillip JM; Chambliss AB; Popovic R; Ezponda T; Small EC; Will C; Phillip MP; Neri P; Bahlis NJ; Wirtz D; Licht JD MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).FitzGerald JE; Grenon M; Lowndes NF 53BP1: function and mechanisms of focal recruitment. Biochem. Soc. Trans 2009, 37, 897–904. [DOI] [PubMed] [Google Scholar]
- (63).Botuyan MV; Lee J; Ward IM; Kim JE; Thompson JR; Chen J; Mer G Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Sanders SL; Portoso M; Mata J; Bahler J; Allshire RC; Kouzarides T Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 2004, 119, 603–614. [DOI] [PubMed] [Google Scholar]
- (65).Fang J; Feng Q; Ketel CS; Wang H; Cao R; Xia L; Erdjument-Bromage H; Tempst P; Simon JA; Zhang Y Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol 2002, 12, 1086–1099. [DOI] [PubMed] [Google Scholar]
- (66).Nishioka K; Rice JC; Sarma K; Erdjument-Bromage H; Werner J; Wang Y; Chuikov S; Valenzuela P; Tempst P; Steward R; Lis JT; Allis CD; Reinberg D PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 2002, 9, 1201–1213. [DOI] [PubMed] [Google Scholar]
- (67).Schotta G; Lachner M; Sarma K; Ebert A; Sengupta R; Reuter G; Reinberg D; Jenuwein T A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Schotta G; Sengupta R; Kubicek S; Malin S; Kauer M; Callen E; Celeste A; Pagani M; Opravil S; De La Rosa-Velazquez IA; Espejo A; Bedford MT; Nussenzweig A; Busslinger M; Jenuwein T A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008, 22, 2048–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Fidler IJ The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [DOI] [PubMed] [Google Scholar]
- (70).Yang J; Mani SA; Donaher JL; Ramaswamy S; Itzykson RA; Come C; Savagner P; Gitelman I; Richardson A; Weinberg RA Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [DOI] [PubMed] [Google Scholar]
- (71).Kwok WK; Ling MT; Lee TW; Lau TC; Zhou C; Zhang X; Chua CW; Chan KW; Chan FL; Glackin C; Wong YC; Wang X Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005, 65, 5153–5162. [DOI] [PubMed] [Google Scholar]
- (72).Brabletz T; Jung A; Spaderna S; Hlubek F; Kirchner T Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [DOI] [PubMed] [Google Scholar]
- (73).Acloque H; Adams MS; Fishwick K; Bronner-Fraser M; Nieto MA Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Invest 2009, 119, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Han X; Piao L; Yuan X; Wang L; Liu Z; He X Knockdown of NSD2 Suppresses Renal Cell Carcinoma Metastasis by Inhibiting Epithelial-Mesenchymal Transition. Int. J. Med. Sci 2019, 16, 1404–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ezponda T; Popovic R; Shah MY; Martinez-Garcia E; Zheng Y; Min DJ; Will C; Neri A; Kelleher NL; Yu J; Licht JD The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013, 32, 2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Dring AM; Davies FE; Fenton JA; Roddam PL; Scott K; Gonzalez D; Rollinson S; Rawstron AC; Rees-Unwin KS; Li C; Munshi NC; Anderson KC; Morgan GJ A global expression-based analysis of the consequences of the t(4;14) translocation in myeloma. Clin. Cancer Res 2004, 10, 5692–5701. [DOI] [PubMed] [Google Scholar]
- (77).Groen RW; de Rooij MF; Kocemba KA; Reijmers RM; de Haan-Kramer A; Overdijk MB; Aalders L; Rozemuller H; Martens AC; Bergsagel PL; Kersten MJ; Pals ST; Spaargaren M N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica 2011, 96, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Martin MC; Zeng G; Yu J; Schiltz GE Small Molecule Approaches for Targeting the Polycomb Repressive Complex 2 (PRC2) in Cancer. J. Med. Chem 2020, 63, 15344–15370. [DOI] [PubMed] [Google Scholar]
- (79).Tomassi S; Romanelli A; Zwergel C; Valente S; Mai A Polycomb Repressive Complex 2 Modulation through the Development of EZH2-EED Interaction Inhibitors and EED Binders. J. Med. Chem 2021, 64, 11774–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Jin B; Zhang P; Zou H; Ye H; Wang Y; Zhang J; Yang H; Pan J Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol. Cancer 2020, 19, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kojima M; Sone K; Oda K; Hamamoto R; Kaneko S; Oki S; Kukita A; Machino H; Honjoh H; Kawata Y; Kashiyama T; Asada K; Tanikawa M; Mori-Uchino M; Tsuruga T; Nagasaka K; Matsumoto Y; Wada-Hiraike O; Osuga Y; Fujii T The histone methyltransferase WHSC1 is regulated by EZH2 and is important for ovarian clear cell carcinoma cell proliferation. BMC Cancer 2019, 19, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Popovic R; Martinez-Garcia E; Giannopoulou EG; Zhang Q; Zhang Q; Ezponda T; Shah MY; Zheng Y; Will CM; Small EC; Hua Y; Bulic M; Jiang Y; Carrara M; Calogero RA; Kath WL; Kelleher NL; Wang JP; Elemento O; Licht JD Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Gao B; Liu X; Li Z; Zhao L; Pan Y Overexpression of EZH2/NSD2 Histone Methyltransferase Axis Predicts Poor Prognosis and Accelerates Tumor Progression in Triple-Negative Breast Cancer. Front. Oncol 2020, 10, 600514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Asangani IA; Ateeq B; Cao Q; Dodson L; Pandhi M; Kunju LP; Mehra R; Lonigro RJ; Siddiqui J; Palanisamy N; Wu YM; Cao X; Kim JH; Zhao M; Qin ZS; Iyer MK; Maher CA; Kumar-Sinha C; Varambally S; Chinnaiyan AM Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell 2013, 49, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Toyokawa G; Cho HS; Masuda K; Yamane Y; Yoshimatsu M; Hayami S; Takawa M; Iwai Y; Daigo Y; Tsuchiya E; Tsunoda T; Field HI; Kelly JD; Neal DE; Maehara Y; Ponder BA; Nakamura Y; Hamamoto R Histone lysine methyltransferase Wolf-Hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia 2011, 13, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Yang P; Guo L; Duan ZJ; Tepper CG; Xue L; Chen X; Kung HJ; Gao AC; Zou JX; Chen HW Histone methyltransferase NSD2/MMSET mediates constitutive NF-kappaB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell Biol 2012, 32, 3121–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Brito JL; Walker B; Jenner M; Dickens NJ; Brown NJ; Ross FM; Avramidou A; Irving JA; Gonzalez D; Davies FE; Morgan GJ MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009, 94, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Martinez-Garcia E; Popovic R; Min DJ; Sweet SM; Thomas PM; Zamdborg L; Heffner A; Will C; Lamy L; Staudt LM; Levens DL; Kelleher NL; Licht JD The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Li W; Tian W; Yuan G; Deng P; Sengupta D; Cheng Z; Cao Y; Ren J; Qin Y; Zhou Y; Jia Y; Gozani O; Patel DJ; Wang Z Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 2021, 590, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Bea S; Valdes-Mas R; Navarro A; Salaverria I; Martin-Garcia D; Jares P; Gine E; Pinyol M; Royo C; Nadeu F; Conde L; Juan M; Clot G; Vizan P; Di Croce L; Puente DA; Lopez-Guerra M; Moros A; Roue G; Aymerich M; Villamor N; Colomo L; Martinez A; Valera A; Martin-Subero JI; Amador V; Hernandez L; Rozman M; Enjuanes A; Forcada P; Muntanola A; Hartmann EM; Calasanz MJ; Rosenwald A; Ott G; Hernandez-Rivas JM; Klapper W; Siebert R; Wiestner A; Wilson WH; Colomer D; Lopez-Guillermo A; Lopez-Otin C; Puente XS; Campo E Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 18250–18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Jaju RJ; Fidler C; Haas OA; Strickson AJ; Watkins F; Clark K; Cross NC; Cheng JF; Aplan PD; Kearney L; Boultwood J; Wainscoat JS A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 2001, 98, 1264–1267. [DOI] [PubMed] [Google Scholar]
- (92).Wang GG; Cai L; Pasillas MP; Kamps MP NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol 2007, 9, 804–812. [DOI] [PubMed] [Google Scholar]
- (93).Leonards K; Almosailleakh M; Tauchmann S; Bagger FO; Thirant C; Juge S; Bock T; Mereau H; Bezerra MF; Tzankov A; Ivanek R; Losson R; Peters A; Mercher T; Schwaller J Nuclear interacting SET domain protein 1 inactivation impairs GATA1-regulated erythroid differentiation and causes erythroleukemia. Nat. Commun 2020, 11, 2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Impelman K; Stitzlein L; Giuliani V; Chandra J Evaluation of a Nuclear Receptor-Binding SET Domain 2 (NSD2) Inhibitor for the Treatment of Leukemia & Brain Tumors. Summer Program in Cancer Research (SPCR) 2022. [Google Scholar]
- (95).Rosati R; La Starza R; Veronese A; Aventin A; Schwienbacher C; Vallespi T; Negrini M; Martelli MF; Mecucci C NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002, 99, 3857–3860. [DOI] [PubMed] [Google Scholar]
- (96).Consortium APG AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Wang JJ; Zou JX; Wang H; Duan ZJ; Wang HB; Chen P; Liu PQ; Xu JZ; Chen HW Histone methyltransferase NSD2 mediates the survival and invasion of triple-negative breast cancer cells via stimulating ADAM9-EGFR-AKT signaling. Acta Pharmacol. Sin 2019, 40, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Zhao X; Xie T; Zhao W; Cai W; Su X Downregulation of MMSET impairs breast cancer proliferation and metastasis through inhibiting Wnt/beta-catenin signaling. Onco Targets Ther. 2019, 12, 1965–1977. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (99).Feng Q; Zhang Z; Shea MJ; Creighton CJ; Coarfa C; Hilsenbeck SG; Lanz R; He B; Wang L; Fu X; Nardone A; Song Y; Bradner J; Mitsiades N; Mitsiades CS; Osborne CK; Schiff R; O’Malley BW An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Wang J; Duan Z; Nugent Z; Zou JX; Borowsky AD; Zhang Y; Tepper CG; Li JJ; Fiehn O; Xu J; Kung HJ; Murphy LC; Chen HW Reprogramming metabolism by histone methyltransferase NSD2 drives endocrine resistance via coordinated activation of pentose phosphate pathway enzymes. Cancer Lett. 2016, 378, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Yang ZQ; Liu G; Bollig-Fischer A; Giroux CN; Ethier SP Transforming properties of 8p11–12 amplified genes in human breast cancer. Cancer Res. 2010, 70, 8487–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Bianco-Miotto T; Chiam K; Buchanan G; Jindal S; Day TK; Thomas M; Pickering MA; O’Loughlin MA; Ryan NK; Raymond WA; Horvath LG; Kench JG; Stricker PD; Marshall VR; Sutherland RL; Henshall SM; Gerald WL; Scher HI; Risbridger GP; Clements JA; Butler LM; Tilley WD; Horsfall DJ; Ricciardelli C; Australian Prostate Cancer B Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomarkers Prev 2010, 19, 2611–2622. [DOI] [PubMed] [Google Scholar]
- (103).Li N; Xue W; Yuan H; Dong B; Ding Y; Liu Y; Jiang M; Kan S; Sun T; Ren J; Pan Q; Li X; Zhang P; Hu G; Wang Y; Wang X; Li Q; Qin J AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Invest 2017, 127, 1284–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Aytes A; Giacobbe A; Mitrofanova A; Ruggero K; Cyrta J; Arriaga J; Palomero L; Farran-Matas S; Rubin MA; Shen MM; Califano A; Abate-Shen C NSD2 is a conserved driver of metastatic prostate cancer progression. Nat. Commun 2018, 9, 5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Xiao M; Yang S; Chen J; Ning X; Guo L; Huang K; Sui L Overexpression of MMSET in endometrial cancer: a clinicopathologic study. J. Surg. Oncol 2013, 107, 428–432. [DOI] [PubMed] [Google Scholar]
- (106).Wu J; Luo M; Duan Z; Jia Y; Linghu H; Tian P; Qi H WHSC1 acts as a prognostic indicator and functions as an oncogene in cervical cancer. Onco Targets Ther. 2019, 12, 4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Yin Z; Sun Y; Ge S; Sun J Epigenetic activation of WHSC1 functions as an oncogene and is associated with poor prognosis in cervical cancer. Oncol. Rep 2017, 37, 2286–2294. [DOI] [PubMed] [Google Scholar]
- (108).Zhu L; Yu CL; Zheng Y NSD2 inhibition suppresses metastasis in cervical cancer by promoting TGF-beta/TGF-betaRI/SMADs signaling. Biochem. Biophys. Res. Commun 2019, 519, 489–496. [DOI] [PubMed] [Google Scholar]
- (109).Brennan K; Shin JH; Tay JK; Prunello M; Gentles AJ; Sunwoo JB; Gevaert O NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci. Rep 2017, 7, 17064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Garcia-Carpizo V; Sarmentero J; Han B; Grana O; Ruiz-Llorente S; Pisano DG; Serrano M; Brooks HB; Campbell RM; Barrero MJ NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci. Rep 2016, 6, 32952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Li Z; Ivanov AA; Su R; Gonzalez-Pecchi V; Qi Q; Liu S; Webber P; McMillan E; Rusnak L; Pham C; Chen X; Mo X; Revennaugh B; Zhou W; Marcus A; Harati S; Chen X; Johns MA; White MA; Moreno C; Cooper LA; Du Y; Khuri FR; Fu H The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun 2017, 8, 14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Yuan G; Flores NM; Hausmann S; Lofgren SM; Kharchenko V; Angulo-Ibanez M; Sengupta D; Lu X; Czaban I; Azhibek D; Vicent S; Fischle W; Jaremko M; Fang B; Wistuba II; Chua KF; Roth JA; Minna JD; Shao NY; Jaremko L; Mazur PK; Gozani O Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 2021, 590, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Lu MH; Fan MF; Yu XD NSD2 promotes osteosarcoma cell proliferation and metastasis by inhibiting E-cadherin expression. Eur. Rev. Med. Pharmacol. Sci 2017, 21, 928–936. [PubMed] [Google Scholar]
- (114).He C; Liu C; Wang L; Sun Y; Jiang Y; Hao Y Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis. 2019, 10, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Liu Z; Piao L; Zhuang M; Qiu X; Xu X; Zhang D; Liu M; Ren D Silencing of histone methyltransferase NSD3 reduces cell viability in osteosarcoma with induction of apoptosis. Oncol. Rep 2017, 38, 2796–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Zhou P; Wu LL; Wu KM; Jiang W; Li JD; Zhou LD; Li XY; Chang S; Huang Y; Tan H; Zhang GW; He F; Wang ZM Overexpression of MMSET is correlation with poor prognosis in hepatocellular carcinoma. Pathol. Oncol. Res 2013, 19, 303–309. [DOI] [PubMed] [Google Scholar]
- (117).Seiwert TY; Zuo Z; Keck MK; Khattri A; Pedamallu CS; Stricker T; Brown C; Pugh TJ; Stojanov P; Cho J; Lawrence MS; Getz G; Bragelmann J; DeBoer R; Weichselbaum RR; Langerman A; Portugal L; Blair E; Stenson K; Lingen MW; Cohen EE; Vokes EE; White KP; Hammerman PS Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res 2015, 21, 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Saloura V; Cho HS; Kiyotani K; Alachkar H; Zuo Z; Nakakido M; Tsunoda T; Seiwert T; Lingen M; Licht J; Nakamura Y; Hamamoto R WHSC1 promotes oncogenesis through regulation of NIMA-related kinase-7 in squamous cell carcinoma of the head and neck. Mol. Cancer Res 2015, 13, 293–304. [DOI] [PubMed] [Google Scholar]
- (119).Saloura V; Vougiouklakis T; Zewde M; Kiyotani K; Park JH; Gao G; Karrison T; Lingen M; Nakamura Y; Hamamoto R WHSC1L1 drives cell cycle progression through transcriptional regulation of CDC6 and CDK2 in squamous cell carcinoma of the head and neck. Oncotarget 2016, 7, 42527–42538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Saloura V; Vougiouklakis T; Zewde M; Deng X; Kiyotani K; Park JH; Matsuo Y; Lingen M; Suzuki T; Dohmae N; Hamamoto R; Nakamura Y WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci. Rep 2017, 7, 40664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Chen HQ; Gao D Inhibitory effect of microRNA-154 targeting WHSC1 on cell proliferation of human skin squamous cell carcinoma through mediating the P53 signaling pathway. Int. J. Biochem. Cell Biol 2018, 100, 22–29. [DOI] [PubMed] [Google Scholar]
- (122).Berdasco M; Ropero S; Setien F; Fraga MF; Lapunzina P; Losson R; Alaminos M; Cheung NK; Rahman N; Esteller M Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 21830–21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Hudlebusch HR; Skotte J; Santoni-Rugiu E; Zimling ZG; Lees MJ; Simon R; Sauter G; Rota R; De Ioris MA; Quarto M; Johansen JV; Jorgensen M; Rechnitzer C; Maroun LL; Schroder H; Petersen BL; Helin K MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res. 2011, 71, 4226–4235. [DOI] [PubMed] [Google Scholar]
- (124).Li J; Yin C; Okamoto H; Mushlin H; Balgley BM; Lee CS; Yuan K; Ikejiri B; Glasker S; Vortmeyer AO; Oldfield EH; Weil RJ; Zhuang Z Identification of a novel proliferation-related protein, WHSC1 4a, in human gliomas. Neuro. Oncol 2008, 10, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Lu T; Jackson MW; Wang B; Yang M; Chance MR; Miyagi M; Gudkov AV; Stark GR Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Wang Y; Zhu L; Guo M; Sun G; Zhou K; Pang W; Cao D; Tang X; Meng X Histone methyltransferase WHSC1 inhibits colorectal cancer cell apoptosis via targeting anti-apoptotic BCL2. Cell Death Discov. 2021, 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zhao LH; Li Q; Huang ZJ; Sun MX; Lu JJ; Zhang XH; Li G; Wu F Identification of histone methyltransferase NSD2 as an important oncogenic gene in colorectal cancer. Cell Death Dis. 2021, 12, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).D’Afonseca V; Gonzalez G; Salazar M; Arencibia AD Computational analyses on genetic alterations in the NSD genes family and the implications for colorectal cancer development. Ecancermedicalscience 2020, 14, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Sun Y; Xie J; Cai S; Wang Q; Feng Z; Li Y; Lu JJ; Chen W; Ye Z Elevated expression of nuclear receptor-binding SET domain 3 promotes pancreatic cancer cell growth. Cell Death Dis. 2021, 12, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Kang D; Cho HS; Toyokawa G; Kogure M; Yamane Y; Iwai Y; Hayami S; Tsunoda T; Field HI; Matsuda K; Neal DE; Ponder BA; Maehara Y; Nakamura Y; Hamamoto R The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer 2013, 52, 126–139. [DOI] [PubMed] [Google Scholar]
- (131).French CA; Rahman S; Walsh EM; Kuhnle S; Grayson AR; Lemieux ME; Grunfeld N; Rubin BP; Antonescu CR; Zhang S; Venkatramani R; Dal Cin P; Howley PM NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 2014, 4, 928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Jones DH; Lin DI Amplification of the NSD3-BRD4-CHD8 pathway in pelvic high-grade serous carcinomas of tubo-ovarian and endometrial origin. Mol. Clin. Oncol 2017, 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Terwilliger T; Abdul-Hay M Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Cobaleda C; Vicente-Duenas C; Sanchez-Garcia I Infectious triggers and novel therapeutic opportunities in childhood B cell leukaemia. Nat. Rev. Immunol 2021, 21, 570–581. [DOI] [PubMed] [Google Scholar]
- (135).Nguyen K; Devidas M; Cheng SC; La M; Raetz EA; Carroll WL; Winick NJ; Hunger SP; Gaynon PS; Loh ML; Children’s Oncology G Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Carroll WL; Raetz EA Clinical and laboratory biology of childhood acute lymphoblastic leukemia. J. Pediatr 2012, 160, 10–18. [DOI] [PubMed] [Google Scholar]
- (137).Sato K; Kumar A; Hamada K; Okada C; Oguni A; Machiyama A; Sakuraba S; Nishizawa T; Nureki O; Kono H; Ogata K; Sengoku T Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat. Commun 2021, 12, 6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Li J; Hlavka-Zhang J; Shrimp JH; Piper C; Dupere-Richer D; Roth JS; Jing D; Casellas Roman HL; Troche C; Swaroop A; Kulis M; Oyer JA; Will CM; Shen M; Riva A; Bennett RL; Ferrando AA; Hall MD; Lock RB; Licht JD PRC2 Inhibitors Overcome Glucocorticoid Resistance Driven by NSD2 Mutation in Pediatric Acute Lymphoblastic Leukemia. Cancer Discov. 2022, 12, 186–203. [DOI] [PubMed] [Google Scholar]
- (139).Rajkumar SV; Dimopoulos MA; Palumbo A; Blade J; Merlini G; Mateos MV; Kumar S; Hillengass J; Kastritis E; Richardson P; Landgren O; Paiva B; Dispenzieri A; Weiss B; LeLeu X; Zweegman S; Lonial S; Rosinol L; Zamagni E; Jagannath S; Sezer O; Kristinsson SY; Caers J; Usmani SZ; Lahuerta JJ; Johnsen HE; Beksac M; Cavo M; Goldschmidt H; Terpos E; Kyle RA; Anderson KC; Durie BG; Miguel JF International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–548. [DOI] [PubMed] [Google Scholar]
- (140).Vincent Rajkumar S Multiple myeloma: 2014 Update on diagnosis, risk-stratification, and management. Am. J. Hematol 2014, 89, 999–1009. [DOI] [PubMed] [Google Scholar]
- (141).Alzrigat M; Parraga AA; Jernberg-Wiklund H Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol 2018, 51, 101–115. [DOI] [PubMed] [Google Scholar]
- (142).Siegel RL; Miller KD; Fuchs HE; Jemal A Cancer statistics, 2022. CA Cancer J. Clin 2022, 72, 7–33. [DOI] [PubMed] [Google Scholar]
- (143).Landgren O; Graubard BI; Katzmann JA; Kyle RA; Ahmadizadeh I; Clark R; Kumar SK; Dispenzieri A; Greenberg AJ; Therneau TM; Melton LJ 3rd; Caporaso N; Korde N; Roschewski M; Costello R; McQuillan GM; Rajkumar SV Racial disparities in the prevalence of monoclonal gammopathies: a population-based study of 12,482 persons from the National Health and Nutritional Examination Survey. Leukemia 2014, 28, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Rajkumar SV Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol 2016, 91, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Lauring J; Abukhdeir AM; Konishi H; Garay JP; Gustin JP; Wang Q; Arceci RJ; Matsui W; Park BH The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 2008, 111, 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Hajdu I; Ciccia A; Lewis SM; Elledge SJ Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 13130–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).De Smedt E; Lui H; Maes K; De Veirman K; Menu E; Vanderkerken K; De Bruyne E The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol 2018, 8, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Min DJ; Ezponda T; Kim MK; Will CM; Martinez-Garcia E; Popovic R; Basrur V; Elenitoba-Johnson KS; Licht JD MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia 2013, 27, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Park JW; Chae YC; Kim JY; Oh H; Seo SB Methylation of Aurora kinase A by MMSET reduces p53 stability and regulates cell proliferation and apoptosis. Oncogene 2018, 37, 6212–6224. [DOI] [PubMed] [Google Scholar]
- (150).Wu SP; Pfeiffer RM; Ahn IE; Mailankody S; Sonneveld P; van Duin M; Munshi NC; Walker BA; Morgan G; Landgren O Impact of Genes Highly Correlated with MMSET Myeloma on the Survival of Non-MMSET Myeloma Patients. Clin. Cancer Res 2016, 22, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Jain P; Wang M Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am. J. Hematol 2019, 94, 710–725. [DOI] [PubMed] [Google Scholar]
- (152).Hill HA; Qi X; Jain P; Nomie K; Wang Y; Zhou S; Wang ML Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Jain P; Kanagal-Shamanna R; Zhang S; Ahmed M; Ghorab A; Zhang L; Ok CY; Li S; Hagemeister F; Zeng D; Gong T; Chen W; Badillo M; Nomie K; Fayad L; Medeiros LJ; Neelapu S; Fowler N; Romaguera J; Champlin R; Wang L; Wang ML Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br. J. Haematol 2018, 183, 578–587. [DOI] [PubMed] [Google Scholar]
- (154).Agarwal R; Chan YC; Tam CS; Hunter T; Vassiliadis D; Teh CE; Thijssen R; Yeh P; Wong SQ; Ftouni S; Lam EYN; Anderson MA; Pott C; Gilan O; Bell CC; Knezevic K; Blombery P; Rayeroux K; Zordan A; Li J; Huang DCS; Wall M; Seymour JF; Gray DHD; Roberts AW; Dawson MA; Dawson SJ Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med 2019, 25, 119–129. [DOI] [PubMed] [Google Scholar]
- (155).Yang P; Zhang W; Wang J; Liu Y; An R; Jing H Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018, 25, 129–140. [DOI] [PubMed] [Google Scholar]
- (156).Zhang J; Jima D; Moffitt AB; Liu Q; Czader M; Hsi ED; Fedoriw Y; Dunphy CH; Richards KL; Gill JI; Sun Z; Love C; Scotland P; Lock E; Levy S; Hsu DS; Dunson D; Dave SS The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Peng X; Peng Q; Zhong L Targeting H3K36 methyltransferases NSDs: a promising strategy for tumor targeted therapy. Signal Transduct. Target. Ther 2021, 6, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Bain BJ; Bene MC Morphological and Immunophenotypic Clues to the WHO Categories of Acute Myeloid Leukaemia. Acta Haematol. 2019, 141, 232–244. [DOI] [PubMed] [Google Scholar]
- (159).Naymagon L; Marcellino B; Mascarenhas J Eosinophilia in acute myeloid leukemia: Overlooked and underexamined. Blood Rev. 2019, 36, 23–31. [DOI] [PubMed] [Google Scholar]
- (160).Medeiros BC; Chan SM; Daver NG; Jonas BA; Pollyea DA Optimizing survival outcomes with post-remission therapy in acute myeloid leukemia. Am. J. Hematol 2019, 94, 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Vakiti A; Mewawalla P Acute Myeloid Leukemia. StatPearls 2023. [PubMed] [Google Scholar]
- (162).Zhu X; He F; Zeng H; Ling S; Chen A; Wang Y; Yan X; Wei W; Pang Y; Cheng H; Hua C; Zhang Y; Yang X; Lu X; Cao L; Hao L; Dong L; Zou W; Wu J; Li X; Zheng S; Yan J; Zhou J; Zhang L; Mi S; Wang X; Zhang L; Zou Y; Chen Y; Geng Z; Wang J; Zhou J; Liu X; Wang J; Yuan W; Huang G; Cheng T; Wang QF Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat. Genet 2014, 46, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Dong Y; Zhao X; Feng X; Zhou Y; Yan X; Zhang Y; Bu J; Zhan D; Hayashi Y; Zhang Y; Xu Z; Huang R; Wang J; Zhao T; Xiao Z; Ju Z; Andreassen PR; Wang QF; Chen W; Huang G SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia 2019, 33, 2585–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Taketani T; Taki T; Nakamura H; Taniwaki M; Masuda J; Hayashi Y NUP98-NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet. Cytogenet 2009, 190, 108–112. [DOI] [PubMed] [Google Scholar]
- (165).De Deus Wagatsuma VM; Pereira-Martins DA; Do Nascimento MC; Sanchez Mendoza SE; Lucena-Araujo AR; Lima A; Saldanha-Araujo F; Pitella F; Traina F; Madeira MIA; Figueiredo-Pontes LL; Rego EM NSD1 and NSD2 Transcriptional Levels Might Predict Clinical Outcome in AML Patients. Blood 2018, 132, 5257–5257. [Google Scholar]
- (166).Bamford S; Dawson E; Forbes S; Clements J; Pettett R; Dogan A; Flanagan A; Teague J; Futreal PA; Stratton MR; Wooster R The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Sung H; Ferlay J; Siegel RL; Laversanne M; Soerjomataram I; Jemal A; Bray F Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin 2021, 71, 209–249. [DOI] [PubMed] [Google Scholar]
- (168).Giaquinto AN; Sung H; Miller KD; Kramer JL; Newman LA; Minihan A; Jemal A; Siegel RL Breast Cancer Statistics, 2022. CA Cancer J. Clin 2022, 72, 524–541. [DOI] [PubMed] [Google Scholar]
- (169).Arnold M; Morgan E; Rumgay H; Mafra A; Singh D; Laversanne M; Vignat J; Gralow JR; Cardoso F; Siesling S; Soerjomataram I Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Wang Q; Zheng J; Zou JX; Xu J; Han F; Xiang S; Liu P; Chen HW; Wang J S-adenosylhomocysteine (AdoHcy)-dependent methyltransferase inhibitor DZNep overcomes breast cancer tamoxifen resistance via induction of NSD2 degradation and suppression of NSD2-driven redox homeostasis. Chem. Biol. Interact 2020, 317, 108965. [DOI] [PubMed] [Google Scholar]
- (171).Wang L; Lu B; He M; Wang Y; Wang Z; Du L Prostate Cancer Incidence and Mortality: Global Status and Temporal Trends in 89 Countries From 2000 to 2019. Front. Public Health 2022, 10, 811044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Chinnaiyan AM; Lnu S; Cao Q; Asangani I Compositions and methods for treating cancer by inhibiting MMSET protein. WO 2011103028 A2, 2011.
- (173).Want MY; Tsuji T; Singh PK; Thorne JL; Matsuzaki J; Karasik E; Gillard B; Cortes Gomez E; Koya RC; Lugade A; Odunsi K; Battaglia S WHSC1/NSD2 regulates immune infiltration in prostate cancer. J. Immunother. Cancer 2021, 9, e001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Taylor BS; Schultz N; Hieronymus H; Gopalan A; Xiao Y; Carver BS; Arora VK; Kaushik P; Cerami E; Reva B; Antipin Y; Mitsiades N; Landers T; Dolgalev I; Major JE; Wilson M; Socci ND; Lash AE; Heguy A; Eastham JA; Scher HI; Reuter VE; Scardino PT; Sander C; Sawyers CL; Gerald WL Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Wang S; Gao J; Lei Q; Rozengurt N; Pritchard C; Jiao J; Thomas GV; Li G; Roy-Burman P; Nelson PS; Liu X; Wu H Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [DOI] [PubMed] [Google Scholar]
- (176).Denny L; de Sanjose S; Mutebi M; Anderson BO; Kim J; Jeronimo J; Herrero R; Yeates K; Ginsburg O; Sankaranarayanan R Interventions to close the divide for women with breast and cervical cancer between low-income and middle-income countries and high-income countries. Lancet 2017, 389, 861–870. [DOI] [PubMed] [Google Scholar]
- (177).Tonon G; Wong KK; Maulik G; Brennan C; Feng B; Zhang Y; Khatry DB; Protopopov A; You MJ; Aguirre AJ; Martin ES; Yang Z; Ji H; Chin L; Depinho RA High-resolution genomic profiles of human lung cancer. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Mahmood SF; Gruel N; Nicolle R; Chapeaublanc E; Delattre O; Radvanyi F; Bernard-Pierrot I PPAPDC1B and WHSC1L1 are common drivers of the 8p11–12 amplicon, not only in breast tumors but also in pancreatic adenocarcinomas and lung tumors. Am. J. Pathol 2013, 183, 1634–1644. [DOI] [PubMed] [Google Scholar]
- (179).Zhao X; Wu Q; Gong X; Liu J; Ma Y Osteosarcoma: a review of current and future therapeutic approaches. Biomed. Eng. Online 2021, 20, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Kansara M; Teng MW; Smyth MJ; Thomas DM Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [DOI] [PubMed] [Google Scholar]
- (181).Kansara M; Thomas DM Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007, 26, 1–18. [DOI] [PubMed] [Google Scholar]
- (182).Bacci G; Ferrari S; Longhi A; Picci P; Mercuri M; Alvegard TA; Saeter G; Donati D; Manfrini M; Lari S; Briccoli A; Forni C; Italian Sarcoma Group/Scandinavian Sarcoma, G. High dose ifosfamide in combination with high dose methotrexate, adriamycin and cisplatin in the neoadjuvant treatment of extremity osteosarcoma: preliminary results of an Italian Sarcoma Group/Scandinavian Sarcoma Group pilot study. J. Chemother 2002, 14, 198–206. [DOI] [PubMed] [Google Scholar]
- (183).Saloura V; Vougiouklakis T; Sievers C; Burkitt K; Nakamura Y; Hager G; van Waes C The role of protein methyltransferases as potential novel therapeutic targets in squamous cell carcinoma of the head and neck. Oral Oncol. 2018, 81, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Cheng Y; He C; Wang M; Ma X; Mo F; Yang S; Han J; Wei X Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther 2019, 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Yoo CB; Jones PA Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov 2006, 5, 37–50. [DOI] [PubMed] [Google Scholar]
- (186).Kelly TK; De Carvalho DD; Jones PA Epigenetic modifications as therapeutic targets. Nat. Biotechnol 2010, 28, 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Zhang S; Meng Y; Zhou L; Qiu L; Wang H; Su D; Zhang B; Chan KM; Han J Targeting epigenetic regulators for inflammation: Mechanisms and intervention therapy. MedComm 2022, 3, e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Kristie TM The rise of epigenetic targets for the development of novel antivirals. Expert Rev. Anti. Infect. Ther 2012, 10, 1359–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Szyf M Prospects for the development of epigenetic drugs for CNS conditions. Nat. Rev. Drug Discov 2015, 14, 461–474. [DOI] [PubMed] [Google Scholar]
- (190).Liang D; Yu Y; Ma Z Novel strategies targeting bromodomain-containing protein 4 (BRD4) for cancer drug discovery. Eur. J. Med. Chem 2020, 200, 112426. [DOI] [PubMed] [Google Scholar]
- (191).Zhou M; Huang K; Jung KJ; Cho WK; Klase Z; Kashanchi F; Pise-Masison CA; Brady JN Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol 2009, 83, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Lin A; Wang S; Nguyen T; Shire K; Frappier L The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol 2008, 82, 12009–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Gagnon D; Joubert S; Senechal H; Fradet-Turcotte A; Torre S; Archambault J Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol 2009, 83, 4127–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Wu SY; Lee AY; Hou SY; Kemper JK; Erdjument-Bromage H; Tempst P; Chiang CM Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Belkina AC; Denis GV BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Brasier AR; Zhou J Validation of the epigenetic reader bromodomain-containing protein 4 (BRD4) as a therapeutic target for treatment of airway remodeling. Drug Discov Today 2020, 25, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Ma Z; Bolinger AA; Zhou J; Tian B Bromodomain-containing protein 4 (BRD4): a key player in inflammatory bowel disease and potential to inspire epigenetic therapeutics. Expert Opin. Ther. Targets 2023, 27, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Padmanabhan B; Mathur S; Manjula R; Tripathi S Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J. Biosci 2016, 41, 295–311. [DOI] [PubMed] [Google Scholar]
- (199).Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem 2017, 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Liu Z; Tian B; Chen H; Wang P; Brasier AR; Zhou J Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem 2018, 151, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Niu Q; Liu Z; Alamer E; Fan X; Chen H; Endsley J; Gelman BB; Tian B; Kim JH; Michael NL; Robb ML; Ananworanich J; Zhou J; Hu H Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Invest 2019, 129, 3361–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Liu Z; Chen H; Wang P; Li Y; Wold EA; Leonard PG; Joseph S; Brasier AR; Tian B; Zhou J Discovery of Orally Bioavailable Chromone Derivatives as Potent and Selective BRD4 Inhibitors: Scaffold Hopping, Optimization, and Pharmacological Evaluation. J. Med. Chem 2020, 63, 5242–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Liu Z; Li Y; Chen H; Lai HT; Wang P; Wu SY; Wold EA; Leonard PG; Joseph S; Hu H; Chiang CM; Brasier AR; Tian B; Zhou J Discovery, X-ray Crystallography, and Anti-inflammatory Activity of Bromodomain-containing Protein 4 (BRD4) BD1 Inhibitors Targeting a Distinct New Binding Site. J. Med. Chem 2022, 65, 2388–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Tutuncuoglu B; Cakir M; Batra J; Bouhaddou M; Eckhardt M; Gordon DE; Krogan NJ The Landscape of Human Cancer Proteins Targeted by SARS-CoV-2. Cancer Discov. 2020, 10, 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Long X; Zhang L; Zhang Y; Min M; Lin B; Chen J; Ma X; Zhai S; Cai Z; Liu Y; Lu Y; Che N; Tan W; Qin J; Wang X Histone methyltransferase Nsd2 is required for follicular helper T cell differentiation. J. Exp. Med 2020, 217, e20190832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Liu F; Wu P; Liu K Verticilin A inhibition of histone methyltransferases. US 20140161785 A1, 2014.
- (207).Crotty S T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Ueno H; Banchereau J; Vinuesa CG Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol 2015, 16, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Morishita M; Mevius DEHF; Shen Y; Zhao S; di Luccio E BIX-01294 inhibits oncoproteins NSD1, NSD2 and NSD3. Medicinal Chemistry Research 2017, 26, 2038–2047. [Google Scholar]
- (210).Shen Y; Morishita M; Lee D; Kim S; Lee T; Mevius D; Roh Y; di Luccio E Identification of LEM-14 inhibitor of the oncoprotein NSD2. Biochem. Biophys. Res. Commun 2019, 508, 102–108. [DOI] [PubMed] [Google Scholar]
- (211).Richon VM; Johnston D; Sneeringer CJ; Jin L; Majer CR; Elliston K; Jerva LF; Scott MP; Copeland RA Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des 2011, 78, 199–210. [DOI] [PubMed] [Google Scholar]
- (212).Allali-Hassani A; Kuznetsova E; Hajian T; Wu H; Dombrovski L; Li Y; Graslund S; Arrowsmith CH; Schapira M; Vedadi M A Basic Post-SET Extension of NSDs Is Essential for Nucleosome Binding In Vitro. J. Biomol. Screen 2014, 19, 928–935. [DOI] [PubMed] [Google Scholar]
- (213).Zheng W; Ibanez G; Wu H; Blum G; Zeng H; Dong A; Li F; Hajian T; Allali-Hassani A; Amaya MF; Siarheyeva A; Yu W; Brown PJ; Schapira M; Vedadi M; Min J; Luo M Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc 2012, 134, 18004–18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Tisi D; Chiarparin E; Tamanini E; Pathuri P; Coyle JE; Hold A; Holding FP; Amin N; Martin AC; Rich SJ; Berdini V; Yon J; Acklam P; Burke R; Drouin L; Harmer JE; Jeganathan F; van Montfort RL; Newbatt Y; Tortorici M; Westlake M; Wood A; Hoelder S; Heightman TD Structure of the Epigenetic Oncogene MMSET and Inhibition by N-Alkyl Sinefungin Derivatives. ACS Chem. Biol 2016, 11, 3093–3105. [DOI] [PubMed] [Google Scholar]
- (215).Coussens NP; Kales SC; Henderson MJ; Lee OW; Horiuchi KY; Wang Y; Chen Q; Kuznetsova E; Wu J; Chakka S; Cheff DM; Cheng KC; Shinn P; Brimacombe KR; Shen M; Simeonov A; Lal-Nag M; Ma H; Jadhav A; Hall MD High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem 2018, 293, 13750–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Kubicek S; O’Sullivan RJ; August EM; Hickey ER; Zhang Q; Teodoro ML; Rea S; Mechtler K; Kowalski JA; Homon CA; Kelly TA; Jenuwein T Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [DOI] [PubMed] [Google Scholar]
- (217).Chang Y; Zhang X; Horton JR; Upadhyay AK; Spannhoff A; Liu J; Snyder JP; Bedford MT; Cheng X Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol 2009, 16, 312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Vedadi M; Barsyte-Lovejoy D; Liu F; Rival-Gervier S; Allali-Hassani A; Labrie V; Wigle TJ; Dimaggio PA; Wasney GA; Siarheyeva A; Dong A; Tempel W; Wang SC; Chen X; Chau I; Mangano TJ; Huang XP; Simpson CD; Pattenden SG; Norris JL; Kireev DB; Tripathy A; Edwards A; Roth BL; Janzen WP; Garcia BA; Petronis A; Ellis J; Brown PJ; Frye SV; Arrowsmith CH; Jin J A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol 2011, 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Chen P; Yao JF; Huang RF; Zheng FF; Jiang XH; Chen X; Chen J; Li M; Huang HF; Jiang YP; Huang YF; Yang XY Effect of BIX-01294 on H3K9me2 levels and the imprinted gene Snrpn in mouse embryonic fibroblast cells. Biosci. Rep 2015, 35, e00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).di Luccio E Inhibition of Nuclear Receptor Binding SET Domain 2/Multiple Myeloma SET Domain by LEM-06 Implication for Epigenetic Cancer Therapies. J. Cancer Prev 2015, 20, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).di Luccio E; Koehl P A quality metric for homology modeling: the H-factor. BMC Bioinformatics 2011, 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Morrison MJ; Boriack-Sjodin PA; Swinger KK; Wigle TJ; Sadalge D; Kuntz KW; Scott MP; Janzen WP; Chesworth R; Duncan KW; Harvey DM; Lampe JW; Mitchell LH; Copeland RA Identification of a peptide inhibitor for the histone methyltransferase WHSC1. PLoS One 2018, 13, e0197082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Yang S; Li L Small molecule compounds that inhibiting histone lysine methyltransferase NSD2 and their applications therefore. CN 109734677 A, 2019.
- (224).Cierpicki T; Grembecka J; Huang H; Zari S; Cho HJ; Potopnyk M; Dudkin S; Chen W; Adam Y; Howard C; Kim E NSD family inhibitors and methods of treatment therewith. WO 2019113469 A1, 2019.
- (225).Huang H; Howard CA; Zari S; Cho HJ; Shukla S; Li H; Ndoj J; Gonzalez-Alonso P; Nikolaidis C; Abbott J; Rogawski DS; Potopnyk MA; Kempinska K; Miao H; Purohit T; Henderson A; Mapp A; Sulis ML; Ferrando A; Grembecka J; Cierpicki T Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol 2020, 16, 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Wang S; Yang H; Su M; Lian F; Cong Z; Wei R; Zhou Y; Li X; Zheng X; Li C; Fu X; Han X; Shi Q; Li C; Zhang N; Geng M; Liu H; Li J; Huang X; Wang J 5-Aminonaphthalene derivatives as selective nonnucleoside nuclear receptor binding SET domain-protein 2 (NSD2) inhibitors for the treatment of multiple myeloma. Eur. J. Med. Chem 2021, 222, 113592. [DOI] [PubMed] [Google Scholar]
- (227).Deng H; Liu J; Oyang C; Wang C; Xiao Q; Xun G; Zeng H Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agents. WO 2021026803 A1, 2021.
- (228).Tang H; Yu A; Xing L; Chen X; Ding H; Yang H; Song Z; Shi Q; Geng M; Huang X; Zhang A Structural Modification and Pharmacological Evaluation of Substituted Quinoline-5,8-diones as Potent NSD2 Inhibitors. J. Med. Chem 2023, 66, 1634–1651. [DOI] [PubMed] [Google Scholar]
- (229).Liu L; Zhen XT; Denton E; Marsden BD; Schapira M ChromoHub: a data hub for navigators of chromatin-mediated signalling. Bioinformatics 2012, 28, 2205–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Hyun K; Jeon J; Park K; Kim J Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 2017, 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Musselman CA; Kutateladze TG Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011, 39, 9061–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Sanchez R; Zhou MM The PHD finger: a versatile epigenome reader. Trends Biochem. Sci 2011, 36, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Musselman CA; Kutateladze TG PHD fingers: epigenetic effectors and potential drug targets. Mol. Interv 2009, 9, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Amato A; Lucas X; Bortoluzzi A; Wright D; Ciulli A Targeting Ligandable Pockets on Plant Homeodomain (PHD) Zinc Finger Domains by a Fragment-Based Approach. ACS Chem. Biol 2018, 13, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Miller TC; Rutherford TJ; Birchall K; Chugh J; Fiedler M; Bienz M Competitive binding of a benzimidazole to the histone-binding pocket of the Pygo PHD finger. ACS Chem. Biol 2014, 9, 2864–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Wagner EK; Nath N; Flemming R; Feltenberger JB; Denu JM Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry 2012, 51, 8293–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Arrowsmith CH; Schapira M Targeting non-bromodomain chromatin readers. Nat. Struct. Mol. Biol 2019, 26, 863–869. [DOI] [PubMed] [Google Scholar]
- (238).Berardi A; Ghitti M; Quilici G; Musco G In silico derived small molecules targeting the finger-finger interaction between the histone lysine methyltransferase NSD1 and Nizp1 repressor. Comput. Struct. Biotechnol. J 2020, 18, 4082–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Huang Z; Wu H; Chuai S; Xu F; Yan F; Englund N; Wang Z; Zhang H; Fang M; Wang Y; Gu J; Zhang M; Yang T; Zhao K; Yu Y; Dai J; Yi W; Zhou S; Li Q; Wu J; Liu J; Wu X; Chan H; Lu C; Atadja P; Li E; Wang Y; Hu M NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 2013, 73, 6277–6288. [DOI] [PubMed] [Google Scholar]
- (240).Bottcher J; Dilworth D; Reiser U; Neumuller RA; Schleicher M; Petronczki M; Zeeb M; Mischerikow N; Allali-Hassani A; Szewczyk MM; Li F; Kennedy S; Vedadi M; Barsyte-Lovejoy D; Brown PJ; Huber KVM; Rogers CM; Wells CI; Fedorov O; Rumpel K; Zoephel A; Mayer M; Wunberg T; Bose D; Zahn S; Arnhof H; Berger H; Reiser C; Hormann A; Krammer T; Corcokovic M; Sharps B; Winkler S; Haring D; Cockcroft XL; Fuchs JE; Mullauer B; Weiss-Puxbaum A; Gerstberger T; Boehmelt G; Vakoc CR; Arrowsmith CH; Pearson M; McConnell DB Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol 2019, 15, 822–829. [DOI] [PubMed] [Google Scholar]
- (241).Ferreira de Freitas R; Liu Y; Szewczyk MM; Mehta N; Li F; McLeod D; Zepeda-Velazquez C; Dilworth D; Hanley RP; Gibson E; Brown PJ; Al-Awar R; James LI; Arrowsmith CH; Barsyte-Lovejoy D; Min J; Vedadi M; Schapira M; Allali-Hassani A Discovery of Small-Molecule Antagonists of the PWWP Domain of NSD2. J. Med. Chem 2021, 64, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Dilworth D; Hanley RP; Ferreira de Freitas R; Allali-Hassani A; Zhou M; Mehta N; Marunde MR; Ackloo S; Carvalho Machado RA; Khalili Yazdi A; Owens DDG; Vu V; Nie DY; Alqazzaz M; Marcon E; Li F; Chau I; Bolotokova A; Qin S; Lei M; Liu Y; Szewczyk MM; Dong A; Kazemzadeh S; Abramyan T; Popova IK; Hall NW; Meiners MJ; Cheek MA; Gibson E; Kireev D; Greenblatt JF; Keogh MC; Min J; Brown PJ; Vedadi M; Arrowsmith CH; Barsyte-Lovejoy D; James LI; Schapira M A chemical probe targeting the PWWP domain alters NSD2 nucleolar localization. Nat. Chem. Biol 2022, 18, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Li N; Yang H; Liu K; Zhou L; Huang Y; Cao D; Li Y; Sun Y; Yu A; Du Z; Yu F; Zhang Y; Wang B; Geng M; Li J; Xiong B; Xu S; Huang X; Liu T Structure-Based Discovery of a Series of NSD2-PWWP1 Inhibitors. J. Med. Chem 2022, 65, 9459–9477. [DOI] [PubMed] [Google Scholar]
- (244).Toure M; Crews CM Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. Engl 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- (245).Crews CM; Georg G; Wang S Inducing Protein Degradation as a Therapeutic Strategy. J. Med. Chem 2016, 59, 5129–5130. [DOI] [PubMed] [Google Scholar]
- (246).Crews CM Inducing Protein Degradation as a Therapeutic Strategy. J. Med. Chem 2018, 61, 403–404. [DOI] [PubMed] [Google Scholar]
- (247).Wang P; Zhou J Proteolysis Targeting Chimera (PROTAC): A Paradigm-Shifting Approach in Small Molecule Drug Discovery. Curr. Top. Med. Chem 2018, 18, 1354–1356. [DOI] [PubMed] [Google Scholar]
- (248).Bekes M; Langley DR; Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Chirnomas D; Hornberger KR; Crews CM Protein degraders enter the clinic - a new approach to cancer therapy. Nat. Rev. Clin. Oncol 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).He M; Cao C; Ni Z; Liu Y; Song P; Hao S; He Y; Sun X; Rao Y PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target. Ther 2022, 7, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Li K; Crews CM PROTACs: past, present and future. Chem. Soc. Rev 2022, 51, 5214–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Xue Y; Bolinger AA; Zhou J Novel approaches to targeted protein degradation technologies in drug discovery. Expert Opin. Drug Discov 2023, 18, 467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Gao H; Sun X; Rao Y PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett 2020, 11, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Liu Z; Hu M; Yang Y; Du C; Zhou H; Liu C; Chen Y; Fan L; Ma H; Gong Y; Xie Y An overview of PROTACs: a promising drug discovery paradigm. Mol. Biomed 2022, 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Cao C; He M; Wang L; He Y; Rao Y Chemistries of bifunctional PROTAC degraders. Chem. Soc. Rev 2022, 51, 7066–7114. [DOI] [PubMed] [Google Scholar]
- (256).Lee J; Lee Y; Jung YM; Park JH; Yoo HS; Park J Discovery of E3 Ligase Ligands for Target Protein Degradation. Molecules 2022, 27, 6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Ishida T; Ciulli A E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov. 2021, 26, 484–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Zhao C; Dekker FJ Novel Design Strategies to Enhance the Efficiency of Proteolysis Targeting Chimeras. ACS Pharmacol. Transl. Sci 2022, 5, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Ma Z; Ji Y; Yu Y; Liang D Specific non-genetic IAP-based protein erasers (SNIPERs) as a potential therapeutic strategy. Eur. J. Med. Chem 2021, 216, 113247. [DOI] [PubMed] [Google Scholar]
- (260).Dale B; Cheng M; Park KS; Kaniskan HU; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Li M; Zhi Y; Liu B; Yao Q Advancing Strategies for Proteolysis-Targeting Chimera Design. J. Med. Chem 2023, 66, 2308–2329. [DOI] [PubMed] [Google Scholar]
- (262).Xu C; Meng F; Park KS; Storey AJ; Gong W; Tsai YH; Gibson E; Byrum SD; Li D; Edmondson RD; Mackintosh SG; Vedadi M; Cai L; Tackett AJ; Kaniskan HU; Jin J; Wang GG A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol 2022, 29, 386–397.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Sun Y; Zhang Y; Chen X; Yu A; Du W; Huang Y; Wu F; Yu L; Li J; Wen C; Yang H; Shi Q; Geng M; Huang X; Xu S Discovery of a potent and selective proteolysis targeting chimera (PROTAC) degrader of NSD3 histone methyltransferase. Eur. J. Med. Chem 2022, 239, 114528. [DOI] [PubMed] [Google Scholar]
- (264).Meng F; Xu C; Park KS; Kaniskan HU; Wang GG; Jin J Discovery of a First-in-Class Degrader for Nuclear Receptor Binding SET Domain Protein 2 (NSD2) and Ikaros/Aiolos. J. Med. Chem 2022, 65, 10611–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Ryan A; Liu J; Deiters A Targeted Protein Degradation through Fast Optogenetic Activation and Its Application to the Control of Cell Signaling. J. Am. Chem. Soc 2021, 143, 9222–9229. [DOI] [PubMed] [Google Scholar]
- (266).Matta-Camacho E; Kozlov G; Li FF; Gehring K Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat. Struct. Mol. Biol 2010, 17, 1182–1187. [DOI] [PubMed] [Google Scholar]
- (267).Bachmair A; Finley D; Varshavsky A In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [DOI] [PubMed] [Google Scholar]
- (268).Shanmugasundaram K; Shao P; Chen H; Campos B; McHardy SF; Luo T; Rao H A modular PROTAC design for target destruction using a degradation signal based on a single amino acid. J. Biol. Chem 2019, 294, 15172–15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Hanley RP; Nie DY; Tabor JR; Li F; Sobh A; Xu C; Barker NK; Dilworth D; Hajian T; Gibson E; Szewczyk MM; Brown PJ; Barsyte-Lovejoy D; Herring LE; Wang GG; Licht JD; Vedadi M; Arrowsmith CH; James LI Discovery of a Potent and Selective Targeted NSD2 Degrader for the Reduction of H3K36me2. J. Am. Chem. Soc 2023, 145, 8176–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Yap TA; Omlin A; de Bono JS Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol 2013, 31, 1592–1605. [DOI] [PubMed] [Google Scholar]
- (271).Bayat Mokhtari R; Homayouni TS; Baluch N; Morgatskaya E; Kumar S; Das B; Yeger H Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Rationalizing combination therapies. Nat. Med 2017, 23, 1113. [DOI] [PubMed] [Google Scholar]
- (273).Plana D; Palmer AC; Sorger PK Independent Drug Action in Combination Therapy: Implications for Precision Oncology. Cancer Discov. 2022, 12, 606–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Majchrzak-Celinska A; Warych A; Szoszkiewicz M Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Wei W Compositions and methods for treating cancer. WO 2018044906 A1, 2018.
- (276).Thomenius M; Cosmopoulos KL; Totman JA Methods of treating WHSC1-overexpressing cancers by inhibiting SETD2. WO 2020112872 A1, 2020.
- (277).Stong N; Hagner PR; Pierceall WE; Thakurta A; Flynt JE; Ortiz-Estevez M Methods for treating multiple myeloma. WO 2022182953 A1, 2022.
- (278).Raina K; Forbes CD; Stronk R; Rappi JP; Eastman KJ; Gerritz SW; Yu X; Li H; Bhardwaj A; Forgione M; Hundt A; King MP; Posner ZM; Denny A; McGovern A; Puleo DE; Garvin E; Chenard R; Zaware N; Mousseau JJ; Macaluso J; Martin M; Bassoli K; Jones K; Garcia M; Howard K; Smith LM; Chen JM; De Leon CA; Hines J; Kayser-Bricker KJ; Crews CM Regulated Induced Proximity Targeting Chimeras (RIPTACs): a Novel Heterobifunctional Small Molecule Therapeutic Strategy for Killing Cancer Cells Selectively. bioRxiv 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]