

Abstract
Ethanol produces intoxication through actions on numerous molecular and cellular targets. Adaptations involving these and other targets contribute to chronic drug actions that underlie continued and problematic drinking. Among the mechanisms involved in these ethanol actions are alterations in presynaptic mechanisms of synaptic transmission, including presynaptic protein function and excitation-secretion coupling. At synapses in the central nervous system (CNS), excitation-secretion coupling involves ion channel activation followed by vesicle fusion and neurotransmitter release. These mechanisms are altered by presynaptic neurotransmitter receptors, and prominently by G-protein coupled receptors (GPCRs). Studies over the last 20–25 years have revealed that acute ethanol exposure alters neurotransmitter secretion, with especially robust effects on synapses that use the neurotransmitter gamma-aminobutyric acid (GABA). Intracellular signaling pathways involving second messengers such as cyclic AMP and calcium are implicated in these acute ethanol actions. Ethanol-induced release of neuropeptides and small molecule neurotransmitters that act on presynaptic GPCRs also contribute to presynaptic potentiation at synapses in amygdala and hippocampus, and inhibition of GABA release in the striatum. Prolonged exposure to ethanol alters neurotransmitter release at many CNS GABAergic and glutamatergic synapses, and changes in GPCR function are implicated in many of these neuroadaptations. These presynaptic neuroadaptations appear to involve compensation for acute drug effects at some synapses, but “allostatic” effects that result in long-term resetting of synaptic efficacy occur at others. Current investigations are determining how presynaptic neuroadaptations contribute to behavioral changes at different stages of alcohol drinking, with increasing focus on circuit adaptations underlying these behaviors. This chapter will discuss the acute and chronic presynaptic effects of ethanol in the CNS, as well as some of the consequences of these effects in amygdala and corticostriatal circuits that are related to excessive seeking/drinking and ethanol abuse.
Keywords: Alcohol, Addiction, Synaptic transmission, GABA, Glutamate, Endocannabinoid, Synaptic Plasticity, Amygdala, Cortex, Striatum, Long-term depression
Excitation-Secretion Coupling and Modulation at CNS Synapses
Communication between neurons generally occurs at synapses in which neurotransmitters are stored in and released from vesicles. When a neuronal action potential reaches the presynaptic terminal, the depolarization activates voltage-gated calcium channels (VGCCs) that allow calcium to enter the terminal. The increased intraterminal calcium stimulates vesicle fusion in a process known as excitation-secretion coupling (Catterall and Few 2008). Low rates of vesicle fusion also occur in the absence of excitation-secretion coupling, and this fusion appears to involve vesicle and plasma membrane-associated proteins (Kavalali 2015). Upon release, the neurotransmitter is available to bind to receptor proteins that either directly gate ion flux (ligand-gated ion channels, LGICs) or act through intracellular GTP/GDP-binding proteins (G proteins) that alter signaling processes (G protein-coupled receptors, GPCRs) (Betke et al. 2012; Latek et al. 2012).
While postsynaptic receptors are well known to transduce the signals necessary for anterograde transmission, presynaptic receptors have important roles in feedback alterations in the released neurotransmitter (via “autoreceptors”) or crosstalk to alter release of other neurotransmitters (via “heteroreceptors”). Both LGICs and GPCRs serve as presynaptic receptors. The LGICs directly influence excitability of terminals (Engelman and MacDermott 2004; Pinheiro and Mulle 2008), and although these effects are interesting they will not be considered in any detail in the remainder of this chapter given the relative lack of information on ethanol interaction with these presynaptic receptors. The presynaptic GPCRs work through a variety of heterotrimeric G-proteins and signaling pathways. The heterotrimeric G-proteins consist of obligate α, β and γ subunits, with the latter forming stable β /γ complexes. The G-proteins are generally classified according to the type of α subunit present in the complex, and there are several major α subtypes. In this review the focus will be on three subtypes, Gαi/o, Gαq and Gαs/olf. Upon GPCR activation the heterotrimeric complex separates into free α and β /γ components that then bind to intracellular signaling proteins to alter many aspects of cell biochemistry, gene expression and physiology (Oldham and Hamm 2006).
Activation of GPCRs that couple to Gi/o generally inhibits excitation-secretion coupling and vesicle fusion, and hence neurotransmitter release (Atwood et al. 2014; Miller 1998). The predominant mechanism involved in this modulation is inhibition of the VGCCs that mediate excitation-secretion coupling (Herlitze et al. 1996; Ikeda 1996). However, there is also strong evidence for direct G-protein inhibition of vesicle release (Blackmer et al. 2001). It must be emphasized that the Gβ/γ subunit produces these actions by direct binding to channels and vesicle associated proteins. The Gαi/o subunit inhibits adenylyl cyclase (AC), and thus reduces intracellular cyclic AMP levels (Oldham and Hamm 2006). Inhibition of this enzyme is also implicated in inhibition of neurotransmitter release, especially in long-lasting inhibition (Atwood et al., 2014; Seino and Shibasaki, 2005).
A wide variety of Gi/o-coupled GPCRs exist, with a subtype for almost every major neurotransmitter and neuromodulator. Many of these receptors will be discussed throughout this review, with a strong emphasis on the type 1 cannabinoid receptor (CB1). The CB1 receptor is the target of Δ9-tetrahydrocannabinol, the major psychoactive ingredient in preparations of cannabis sativa. This receptor is normally activated by the endocannabinoid (eCB) fatty acid derivatives produced by hydrolysis of arachidonoyl membrane lipids (namely arachidonoyl ethanolamide, or AEA also known as anandamide, and 2-arachidonoylglycerol, or 2-AG) (Araque et al. 2017). Functions of the gamma-aminobutyric acid B (GABAB) receptor and the metabotropic glutamate receptor type 2 (mGluR2) will also be discussed in some detail.
The GPCRs that couple to Gq-containing GPCRs activate the hydrolysis of membrane phospholipids by phospholipases, a mechanism activated by the Gαq subunit (Oldham and Hamm 2006). The best-known pathway is activation of phospholipase C to catalyze the generation of diacylglycerol (DAG) and inositol phosphates. Among the many responses to Gαq actions are neuronal excitation through inhibition of voltage-gated potassium channels and activation of transient receptor potential (TRP) channels. These effectors may contribute to increased VGCC activation and increased neurotransmitter release (reviewed in Brown and Sihra 2008). However, Gβ/γ liberated by dissociation of heterotrimeric Gαq-containing proteins can inhibit VGCCs and neurotransmitter release (Brown and Sihra 2008). In addition, the DAG liberated by PLC-mediated hydrolysis can be further metabolized to the eCB 2-AG. Activation of Gαs/olf G-proteins leads to stimulation of AC activity and cAMP production, leading to stimulation of protein kinase A (PKA) and the exchange protein activated by cAMP (EPAC) proteins. GPCRs that activate Gαs/olf regulate a diverse array of biochemical, protein trafficking and genetic regulation pathways. The direct physiological consequences of this signaling are not widely known, but it has generally been observed that activation of some Gαs/olf-coupled receptors stimulates neurotransmitter release (reviewed in Brown and Sihra 2008). Forskolin, an AC activator, also increases neurotransmitter release at a variety of synapses, via a mechanism that involves cAMP and PKA activation.
GPCRs, Heterotrimeric G-Proteins, and Synaptic Plasticity
Activation of presynaptic Gi/o-coupled GPCRs can produce either short- or long-lasting decreases in neurotransmitter release (Atwood et al. 2014). Inhibition of VGCCs and vesicle fusion are generally responsible for the short-lasting effects (persisting for seconds-10s of seconds). The longer lasting effects (termed long-term depression or LTD) persist at least for hours, and generally for as long as the preparation survives. It is not entirely clear what mechanisms contribute to Gi/o-LTD, but it is most likely that these mechanisms take place within the presynaptic neuronal elements with the axon terminal being the most likely site of action. LTD is observed in slice preparations in which the presynaptic soma is not present (e.g. at glutamatergic synapses in striatum in slices in which axons have been severed, as in Yin et al. 2006). Within the axon terminal, inhibition of AC is a prominent mechanism implicated in Gi/o-LTD, but long-lasting inhibition of VGCCs may also contribute (Atwood et al. 2014; Pelkey et al. 2008). Inhibition of AC will inhibit the activity of PKA, and this mechanism may also contribute to LTD. One PKA substrate, the Rim1 protein, has been implicated in presynaptic LTD (Heifets and Castillo 2009; Grueter et al. 2010). This phosphoprotein is associated with vesicles and implicated in control of vesicle fusion. Thus, it is thought that reducing PKA-catalyzed phosphorylation of Rim1 leads to a decrease in rates of fusion and neurotransmitter release. Presynaptic protein synthesis via translation also appears to have a key role in some forms of presynaptic Gi/o-LTD (Yin et al. 2006; Younts et al. 2017). An elegant recent study indicates that presynaptic GABAergic terminals in the hippocampus contain ribosomal elements that can mediate protein translation, and this process appears to be necessary for the expression of Gi/o-LTD at these synapses (Younts et al. 2017).
Presynaptic long-term potentiation (LTP) also appears to involve Gs/olf-mediated processes (Evans and Morgan 2003; Waltereit and Weller 2003). For example, increased cAMP and PKA activation are implicated in the increased glutamate release observed during LTP at mossy fiber-CA3 pyramidal neuron synapses in the hippocampus (reviewed in Estratova and Tóth 2014).
Acute Ethanol Effects on Neurotransmitter Release
Ethanol acts through a variety of molecular targets to produce acute intoxication. The stages of intoxication range from euphoria, anxiolysis and enhanced movement (which can be quite variable across individuals) to motor and cognitive impairment, sedation, anesthesia, coma and even death from respiratory depression (Abrahao et al. 2017; Mihic and Harris 2011). The blood and brain ethanol concentrations generally associated with these lower dose effects range from 5–10 mM at the low end, through 18 mM (the legal intoxication level in the USA up to ~100 mM which is the lethal range for average non-tolerant humans). Thus, in understanding the molecular and cellular bases of intoxication it is important to examine effects of these relevant concentrations. The behavioral manifestations of intoxication are driven by effects on neurons (and possibly glia) in a number of brain regions and circuits that control everything from reward and movement to respiratory control (Abrahao et al. 2017). Thus, there is a need to understand actions on different cells in different regions to gain a fuller picture of how intoxication develops. It is also becoming clear that ethanol alters neuronal and synaptic activity via different mechanisms at different sites within the brain, and thus the field can no longer assume that effects involving one molecular target in one brain region will necessarily generalize to other regions (chapters in this volume, including: Anderson et al. 2017; Cannady et al. 2017; Chandler et al. 2017; Coleman and Crews 2017; Cuzon Carlson 2017; Dopico et al. 2017; Finn and Jimenez 2017; Hopf and Mangieri 2017; Klenowski and Tapper 2017; N’Gouemo 2017; Roberto et al. 2017; Schreiber and Gilpin 2017; Siciliano et al. 2017).
In this chapter the focus is on presynaptic ethanol effects. While ethanol has clear actions on targets within the postsynaptic elements of neurons, including a number of ligand-gated ion channels and potassium channels, these effects will not be discussed at present. The reader is referred to recent reviews that cover these subjects in detail (Abrahao et al. 2017; Harris et al. 2008; Lovinger and Roberto 2013; Roberto and Varodayan 2017).
GABA
Ethanol has its clearest acute presynaptic effects at GABAergic synapses in many brain regions. Early neurochemical studies showed both inhibitory and stimulatory effects of acute ethanol on GABA release in synaptosomal and brain slice preparations (Howerton and Collins 1984; Strong and Wood 1984; Seilicovich et al. 1988). It is not clear what accounted for these different findings, but they may be due to differences in the methods for stimulating release (mostly assayed with stimulation of release by increasing extracellular potassium concentrations) or the brain regions examined (e.g. as in Peris et al. 1992). Electrophysiological studies beginning in the 1990s began to establish that ethanol potentiation of GABAergic transmission at intact synapses is one of the clearest acute effects of the drug (Wan et al. 1996; Weiner et al. 1997). However, it was often assumed these ethanol effects only involved changes in GABAA receptor function. The first clear evidence of increased GABA release within particular brain regions came from studies in which the ethanol-induced potentiation was accompanied by decreased paired-pulse facilitation, changes in the frequency of miniature synaptic events, and other signs of presynaptic facilitation (Ariwodola and Weiner 2004; Nie et al. 2004; Roberto et al. 2003). Such effects were first reported at synapses made by GABAergic neurons in the hippocampus (Ariwodola and Weiner 2004; Sanna et al. 2004). Subsequently, similar effects have been observed in the basolateral amygdala, central amygdala, cerebellum, dorsal striatum, nucleus accumbens, spinal cord and ventral tegmental area (Bajo et al. 2008; Criswell et al. 2008; Kelm et al. 2008; Richardson and Rossi 2017; Silberman et al. 2008; Talani and Lovinger 2015; Theile et al. 2008; Wilcox et al. 2014; Ziskind-Conhaim et al. 2003). Ethanol also enhances GABA release onto cerebellar Purkinje neurons, although this effect appears to be due mainly to increased firing of Golgi-type interneurons (Carta et al. 2004). Within the BLA ethanol potentiates GABAergic synapses, with presynaptic mechanisms involved at one population of synapses and adrenergic-dependent postsynaptic mechanisms at another synaptic population (Silberman et al. 2008, 2012). Evidence for ethanol potentiation of glycine release has also been observed (Richardson and Rossi 2017; Ziskind-Conhaim et al. 2003).
Interestingly, some of the earliest reports of the presynaptic GABA release-enhancing ethanol effects also noted that these effects could be reduced by activation of the Gi/o-coupled GABAB-type GPCR (Figure 1A) (Ariwodola and Weiner 2004; Wan et al. 1996). This finding provided one of the first clues about the signaling pathways implicated in ethanol potentiation of GABA release. Subsequent studies have implicated the cyclic adenosine monophosphate (cAMP) intracellular signaling pathway in this ethanol action (Figure 1A). Inhibition of adenylyl cyclase (the enzyme that catalyzes cAMP formation) and protein kinase A (PKA, the cAMP-activated protein kinase) has been shown to prevent this ethanol potentiation (Zhu and Lovinger 2006; Kelm et al. 2008; Talani and Lovinger 2015). The actions of Gi/o-GPCRs that prevent ethanol potentiation likely involve AC inhibition, which is a common consequence of activation of such receptors. Indeed, different Gi/o-GPCRs have now been shown to have this ethanol-inhibiting action at GABAergic synapses in several brain regions (Figure 1A) (Kelm et al. 2008; Roberto et al. 2010; Talani and Lovinger 2015). This raises the possibility that such receptors may be used to alter ethanol effects, and indeed there is evidence that CB1, GABAB and mGluR2 receptor-targeted ligands may be useful in this context (Agabio and Colombo 2014; Meinhardt et al. 2013; Pava and Woodward 2012).
Figure 1.
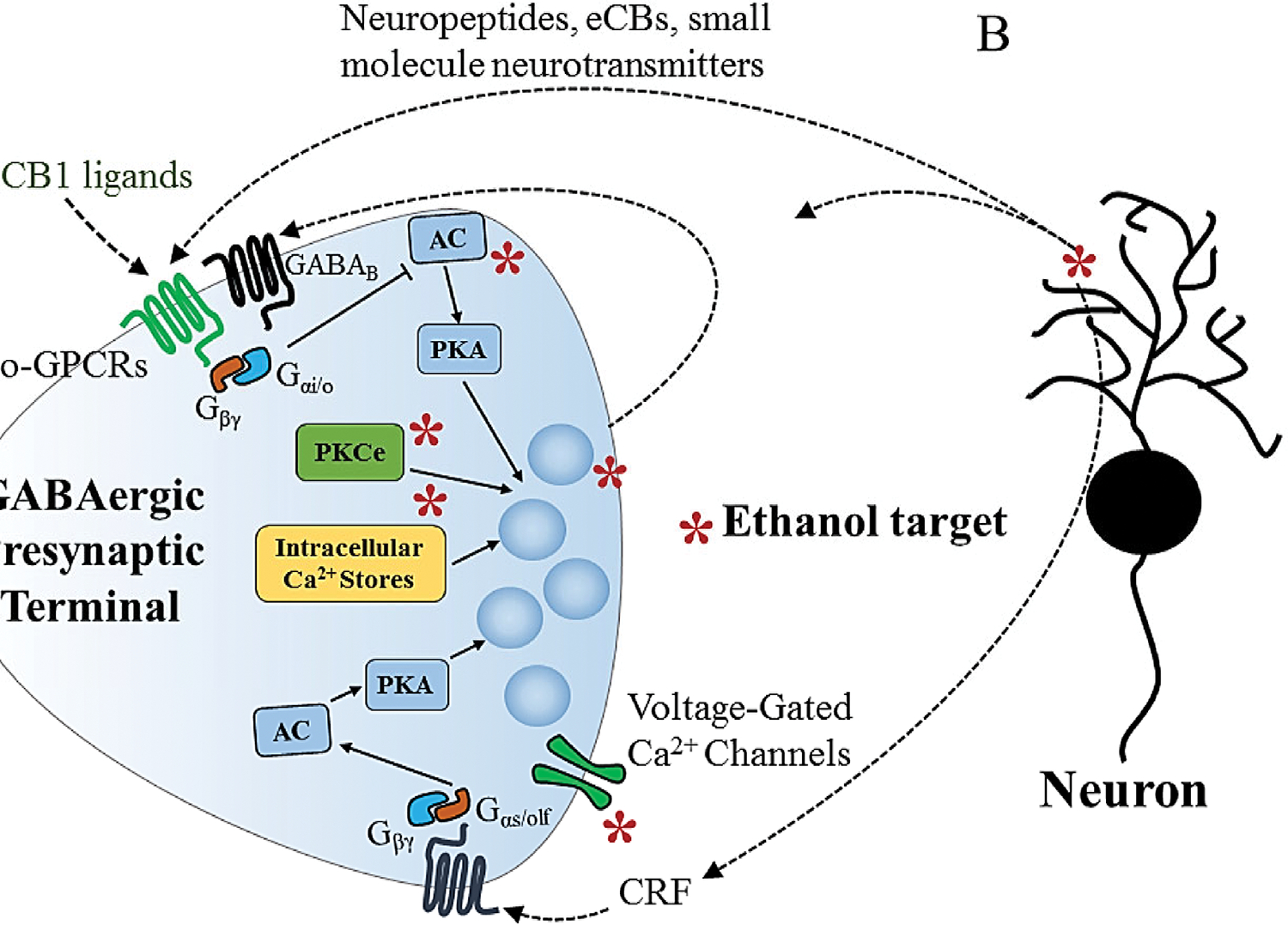
Molecular targets and neuromodulators involved in acute presynaptic ethanol actions at GABAergic synapses. A) Schematic diagram of a presynaptic terminal showing suspected sites of ethanol actions that enhance GABA release (*). The main suspected targets are voltage-gated calcium channels, AC, vesicle fusion, PKCε and intracellular Ca2+ stores. Neuropeptides, including CRF, eCBs and small molecule neurotransmitters (including feedback vesicular GABA release) can contribute to or modulate ethanol actions on presynaptic GABA release through actions on presynaptic GPCRs. Note that ethanol enhances GABA release in many brain regions, but inhibits release in others. B) Ethanol is thought to stimulate release of neuropeptides (including enkephalins and CRF) and eCBs, presumably from neurons, and these neuromodulators act on presynaptic GPCRs to alter GABA release. Arrows indicate stimulation, cross-ended lines indicate inhibition. AC = adenylyl cyclase, CB1 = cannabinoid type 1 receptor, CRF = corticotrophin-releasing factor, eCB = endocannabinoid, GABA = gamma-aminobutyric acid, GPCR = G protein-coupled receptor, Gαi/o = alpha i/o G protein subunit, Gαs/olf = alpha s/olf subunit of G protein, Gβγ = beta/gamma dimer subunit of G protein, PKA = protein kinase A, PKCε = protein kinase C epsilon,
Additional mechanisms may also be involved in the presynaptic GABA-enhancing ethanol action. Knocking out the protein kinase C epsilon (PKCε) isoform appears to prevent ethanol effects in the central amygdala (CeA) (Figure 1A) (Bajo et al. 2008). There may also be a role for stimulation of intracellular calcium release that could enhance excitation/secretion coupling in the cerebellum and VTA (Figure 1A) (Kelm et al. 2007; Theile et al. 2009), and P/Q-type VGCCs appear to be involved in ethanol potentiation in the CeA (Figure 1A) (Varodayan et al. 2017). At several synapses, ethanol has been shown to increase the frequency of action potential- and calcium-entry-independent miniature inhibitory postsynaptic curents (mIPSCs) (Hirono et al. 2009; Kelm et al. 2007; Roberto et al. 2003; Talani et al. 2015; Theile et al. 2008; Zhu and Lovinger 2006), and thus mechanisms downstream of VGCC function are likely involved in this effect (Figure 1A). The function of vesicle- and plasma membrane-associated proteins involved in fusion could be targets for ethanol actions, e.g. through changes in phosphorylation, but this has not yet been examined in detail.
The ethanol-induced increases in GABA release observed in brain slices could involve indirect effects due to release of neuromodulators that stimulate GABAergic terminals (Figure 1A,B). In the CeA, ethanol potentiation of GABA release appears to involve activation of receptors for corticotrophin-releasing factor (CRF), presumably secondary to release of CRF itself (Figure 1A,B) (Nie et al. 2004). Serotonin actions at the 5-HT2C receptor are implicated in ethanol potentiation in VTA (Theile et al. 2009). Application of the nociceptin peptide decreases GABA release in the CeA, and prevents potentiation by ethanol when the peptide is applied before the drug (Roberto and Siggins 2006). Thus, increased release or decreased reuptake of small molecules or neuropeptides may underlie some of these ethanol actions.
However, experiments examining ethanol effects in an isolated “neuron-bouton” preparation provided evidence for a direct effect of ethanol on GABAergic presynaptic terminals (Zhu and Lovinger 2006; Kelm et al. 2007). These neurons are isolated mechanically such that pinched-off presynaptic boutons remain attached to the postsynaptic neuron. These boutons still release GABA and thus spontaneous GABAergic IPSCs (sIPSCs) can be observed independent of the firing of GABAergic neurons and influences of any neurons other than the postsynaptic neuron (Jun et al. 2011). In this preparation, ethanol produces a rapid increase in the frequency of sIPSCs and mIPSCs, indicating a direct effect on GABAergic boutons that appears to be independent of known modulatory or retrograde signals form postsynaptic neurons (Zhu and Lovinger 2006).
It must also be noted that ethanol reduces GABAergic synaptic transmission at some CNS synapses. In the dorsolateral striatum (DLS) acute ethanol application produces such a reduction at synapses onto the medium spiny projection neurons (MSNs) made by both other MSNs and by parvalbumin-positive fast-spiking interneurons (FSIs) (Wilcox et al. 2014; Patton et al. 2016). The inhibition at FSI-MSN synapses appears to involve a presynaptic decrease in GABA release brought about through activation of delta opiate receptors (Figure 1A) (Patton et al. 2016). This finding suggests increased production or release of yet another neuromodulatory peptide by acute ethanol, in this case an enkephalin (Figure 1B). The emerging trend of ethanol modulatory effects through neuropeptide release opens up the possibility that the drug has a variety of actions at different synapses depending on the local peptide expression pattern.
Interactions at GABAergic synapses between the acute presynaptic effects of ethanol and endocannabinoids that act through the CB1 receptor have been especially noteworthy. Within the nervous system eCBs are produced by postsynaptic elements in response to intense neuronal activity. These compounds travel retrogradely across the synaptic cleft to act on presynaptic CB1 receptors, Gi/o-GPCRs that inhibit neurotransmitter release (Figure 1B). At synapses in the CeA and basolateral amygdala (BLA), CB1 activation prevents ethanol potentiation of GABA release (as described previously for other Gi/o-GPCRs) (Figure 1A) (Kelm et al. 2008; Roberto et al. 2010; Talani and Lovinger 2015). There is also evidence that acute ethanol exposure can reduce retrograde eCB signaling at GABAergic synapses in the BLA (Talani and Lovinger 2015). In contrast, acute exposure to ethanol appears to enhance eCB-mediated LTD at glutamatergic synapses in the dorsomedial striatum (Yin et al. 2007). It is not yet clear what mechanisms account for the interaction of ethanol with eCB retrograde signaling. Interactions between ethanol and eCB/CB1 signaling may contribute to the alterations in the in vivo actions of ethanol produced by eCB-targeted drugs (Pava and Woodward 2012), a subject that will be discussed in greater detail in considering the effects of chronic ethanol exposure on the eCB signaling system.
Glutamate and Other Neurotransmitters
Acute ethanol exposure-induced alterations in glutamate release at CNS synapses have not been observed as frequently as effects on GABA release, but a few synapses show some sensitivity, with decreased release being the most common finding (Basavarajappa et al. 2008; Gioia and McCool 2017; Gioia et al. 2017; Li et al. 2013; Maldve et al. 2004; Silberman et al. 2015; Zhu et al. 2007). In the basolateral amygdala (BLA), Ethanol inhibits glutamate release leading to decreased posttetanic potentiation, and reduced synaptic vesicle recycling appears to be the underlying mechanism (Gioia and McCool 2017). The vesicle-associated protein Munc13–2 is implicated in this effect (Gioia et al. 2017). Ethanol decreases glutamatergic synaptic transmission in CeA and this effect is prevented by a CB1 agonist (Kirson et al. 2017), and may also involve N-type VGCCs (Zhu et al. 2007). While it is presumed that this effect involves presynaptic mechanisms there is as yet no direct evidence that this is the case. Potentiation of glutamate release by ethanol has also been reported (e.g. Xiao et al. 2009; Deng et al. 2009), but less frequently than inhibitory actions.
The reasons for the differential effects of ethanol on GABA and glutamate release remain unclear. It is possible that presynaptic molecules that regulate intracellular calcium release and/or vesicle fusion differ at the different synaptic types. In addition, the effects on release secondary to increases in neuromodulator levels and subsequent activation of GPCRs may underlie these differential ethanol actions. This area should be a rich source of important new findings in the future.
There is evidence that presynaptic effects of ethanol alter release of other neurotransmitters, but in many cases it is unclear if these effects involve direct drug actions on presynaptic terminals (Lovinger and Roberto 2013). In the striatum, ethanol inhibits DA release at relatively high concentrations in preparations where DAergic axon terminals are disconnected from their somata (Budygin et al. 2001). While this may still reflect an indirect modulatory action, the findings indicate a local effect on terminal DA release.
Presynaptic Neuroadaptations to Ethanol Exposure and Drinking
Prolonged exposure to ethanol, whether through forced exposure or ethanol drinking, produces neuroadaptations that often compensate for the acute drug actions. However, some adaptations are not always clearly compensatory, and sometimes appear to produce stable alterations that have an “allostatic” effect on neural function.
GABA
Both compensatory and allostatic neuroadaptations to ethanol have been observed at GABAergic synapses in different brain regions. While this chapter focuses on the presynaptic changes at GABAergic synapses, postsynaptic neuroadaptations have also been observed in many brain regions (e.g. Diaz et al. 2011, Abrahao et al. 2017; Roberto and Varodayan 2017). In the CeA, increased GABAergic transmission is observed following prolonged ethanol administration via vapor inhalation (Figure 2) (Roberto et al. 2004a 2010). While there is a prominent postsynaptic component to this neuroadaptation, there is also evidence that the probability of GABA release and/or the number of GABAergic synapses contributes to this effect. Reduced function of GABAB presynaptic autoreceptors is one factor that appears to contribute to this increase in release (Figure 2) (Roberto et al. 2008). Disrupted eCB/CB1 modulation of GABA release may also contribute to the increased release following chronic ethanol exposure (Varodayan et al. 2016). Alterations in CRF levels and function could well play a role in the chronic ethanol actions on CeA GABAergic transmission given the CRF potentiation involved in the acute drug action that was discussed previously (Figures 1, 2). Indeed, CRF levels are increased in the amygdala following withdrawal after chronic ethanol exposure, as measured with in vivo microdialysis (Merlo Pich et al. 1995). The ability of CRF to enhance GABA release in CeA is augmented in ethanol-dependent rats. The acute ethanol-induced potentiation of GABAergic transmission on CeA neurons remains intact following chronic exposure, indicating a lack of tolerance to ethanol. Withdrawal following chronic ethanol intake results in increased extracellular CRF levels in the BNST (Olive et al. 2002), but it is not clear if the increase alters GABAergic transmission in this region. Overall, GABAergic neuroadaptations in the CeA, and perhaps other parts of the extended amygdala, are not compensatory but rather induce a general enhancement of inhibition within CeA that is exacerbated during intoxication.
Figure 2.
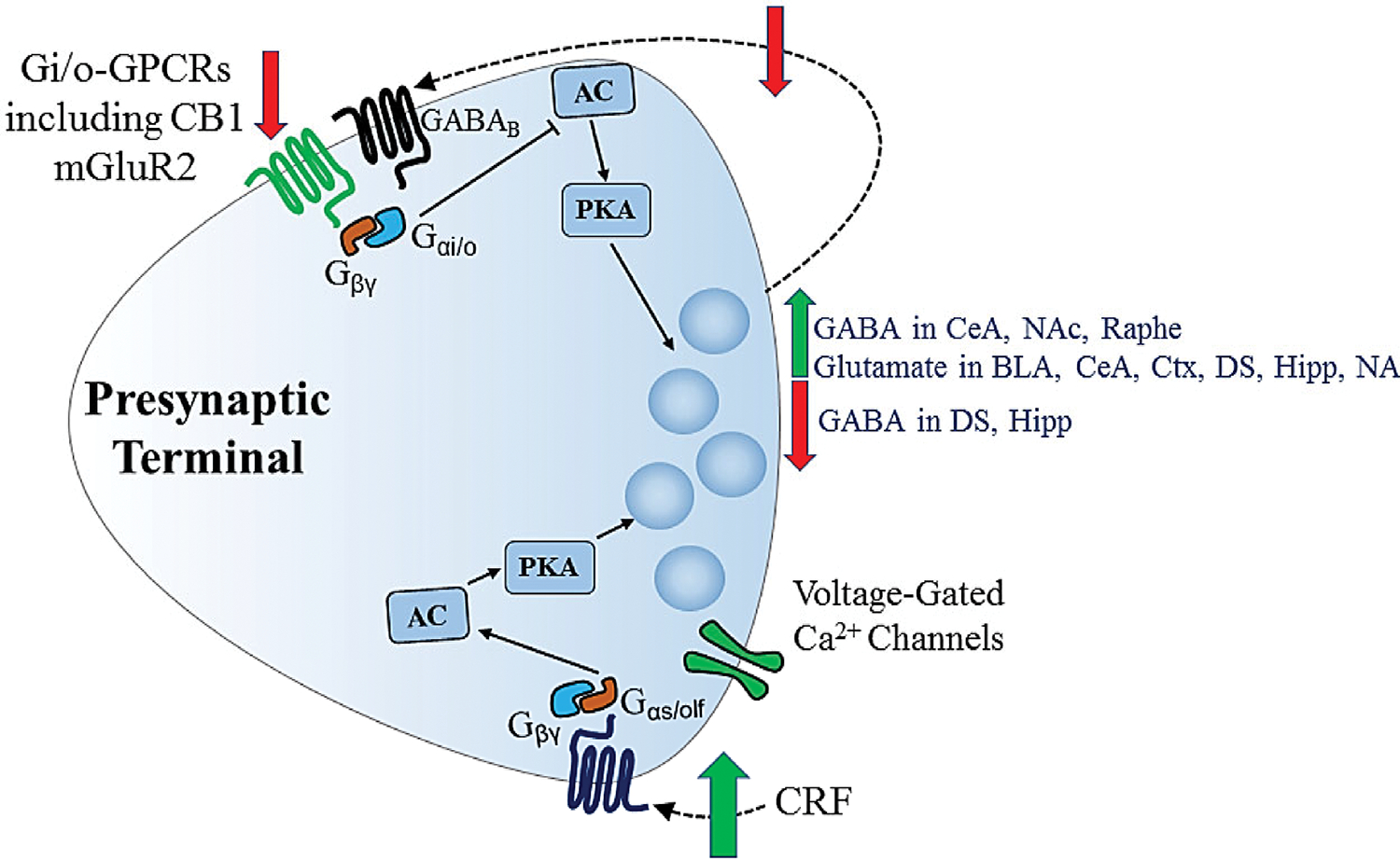
Presynaptic neuroadaptations to chronic ethanol exposure/consumption. Green arrows indicate increases in GABA and glutamate release observed in several brain regions, as well as increased CRF that drives increased GABA release in CeA. Red arrows indicate decreases in Gi/o-GPCR expression/function that occur at both GABAergic and glutamatergic synapses (including decreased GABA/GABABR feedback onto GABAergic terminals), as well as decreased GABA release observed in some brain regions. Decreases in GABA release are thought to compensate for ethanol-induced increases in neurotransmitter release at GABAergic synapses, while activation of GABAB Gi/o-GPCRs participates in compensatory negative feedback that produces tolerance to the direct ethanol action on release (dashed arrow). At glutamatergic synapses, increased neurotransmitter release and decreased Gi/o-GPCR function may compensate for ethanol-induced decreases in glutamatergic transmission. Neuroadaptations that have a more allostatic role include increased GABA release and increased CRF signaling that enhances GABA release. AC = adenylyl cyclase, BLA = basolateral amygdala, CB1 = cannabinoid type 1 receptor, CeA = central amygdala, CRF = corticotrophin-releasing factor, Ctx = cortex, DS = dorsal striatum, GABA = gamma-aminobutyric acid, GABAB = GABA type B receptor, GPCR = G protein-coupled receptor, Gαi/o = alpha i/o G protein subunit, Gαs/olf = alpha s/olf subunit of G protein, Gβγ = beta/gamma dimer subunit of G protein, Hipp = hippocampus, NAc = nucleus accumbens, DRN = dorsal raphe nucleus, PKA = protein kinase A.
GABA release at hippocampal synapses may also be altered through changes in presynaptic function and modulation. In the dentate gyrus hippocampal subfield there is evidence of decreased probability of GABA release following chronic ethanol intake in monkeys (Figure 2) (Weiner et al. 2005). Evidence for decreased GABA release has also been observed in the CA1 subfield (Cagetti et al. 2003). These effects would appear to compensate for the increased GABA release during acute ethanol exposure. However, decreased GABAB receptor function has been implicated in increased GABA release in the CA1 subfield in vivo, and this may be an allostatic type of neuroadaptation (Peris et al. 1997). Decreased GABAergic transmission, involving both pre- and postsynaptic mechanisms has also been observed in the BLA following chronic ethanol drinking, and this neuroadaptation likely contributes to negative affective states that develop during withdrawal (Diaz et al., 2011).
GABAergic synaptic transmission onto serotonergic neurons in the dorsal raphe nucleus is not altered by acute ethanol exposure in naïve mice of the DBA1/J strain. However, following chronic ethanol exposure, acute application of the drug produces a robust enhancement of GABA release (Figure 2) (Lowery-Gionta et al. 2015). This illustrates a case where a change in transmission does not directly compensate for an acute drug effect, but instead chronic exposure induces a hypersensitivity to ethanol that may alter the pattern of intoxication during subsequent encounters with the drug.
In the DLS, long-term changes at GABAergic synapses onto MSNs are mainly allostatic. Decreased frequency of GABAergic mIPSCs has been observed in both mouse DLS and the monkey putamen nucleus (roughly equivalent to rodent DLS) following chronic ethanol drinking protocols (Wilcox et al. 2014; Cuzon Carlson et al. 2011). These findings indicate that the effect of chronic ethanol exposure is similar to that of acute exposure, with the net effect being a loss of inhibition of MSN activity/striatal output (Figure 2). In mouse DLS the effect of acute ethanol is lost after chronic drinking (Wilcox et al. 2014), and thus the effect of chronic ethanol consumption sets a new level of GABAergic inhibition that appears to be stable.
Chronic ethanol drinking leads to depression of DMS GABAergic synapses, i.e. decreased mIPSC frequency, similar to that observed in DLS (Figure 2) (Wilcox et al. 2014). This decrease is accompanied by a change from acute ethanol potentiation of GABA release to a slight depression. In the monkey caudate nucleus GABAergic synaptic transmission exhibits smaller changes following chronic drinking than those observed in the putamen, but decreased mIPSC frequency is the most consistent observation (Cuzon Carlson et al. 2017). Thus, the general effect of ethanol on striatal GABAergic transmission is a decrease that would generally allow for increased striatal output driven by synaptic activation of MSNs.
In the VTA, a single in vivo ethanol exposure appears to produce increased GABA release at synapses on dopaminergic neurons (Melis et al. 2002; Wanat et al. 2009). This potentiation may involve impaired function of GABAB autoreceptors (Melis et al. 2002). However, effects of more prolonged ethanol exposure remain to be determined.
Glutamate and Dopamine
Prolonged ethanol exposure or drinking has generally been proposed to produce an increase in extracellular glutamate levels (Figure 2) (Dahchour and De Witte 1999, 2003; Griffin et al. 2014; Meinhardt et al. 2013; Rossetti and Carboni 1995; Roberto et al. 2004b; Knackstedt and Kalivas 2009). The main evidence supporting this idea comes from microdialysis data demonstrating increases in extracellular glutamate in cortex, dorsal striatum, NAc and other brain regions (Dahchour and De Witte 1999, 2003; Knackstedt and Kalivas 2009; Meinhardt et al. 2013; Rossetti and Carboni 1995). However, it is not clear that the glutamate measured with this approach is of synaptic origin (e.g. Baker et al. 2002). Indeed, changes in the function of the cystine-glutamate transporter accounts for some of this increase (Baker et al. 2002; Knackstedt and Kalivas 2009). Nonetheless, there is evidence for presynaptic changes at glutamatergic synapses that would promote increased glutamate release and direct evidence for increased glutamate release at some brain synapses (Cuzon Carlson et al. 2011; Lack et al. 2007; Lowery-Gionta et al. 2015; Ma et al. 2017; Meinhardt et al. 2013; Zhu et al. 2007; Roberto et al. 2004b). There is also evidence for decreased glutamate uptake in the NAc following chronic ethanol drinking (Melendez et al. 2005). It should also be noted that synaptic glutamate release appears to be decreased in the lateral CeA following chronic ethanol exposure and a 48 hour withdrawal (Pleil et al. 2015). Thus, with some exceptions, it appears that ethanol produces increased glutamate release at synapses in many brain regions.
Another synaptic change that appears to contribute to increased glutamate levels is the loss of regulation of release by presynaptic Gi/o-coupled receptors (Figure 2). In the NAc and DS mGluR2 acts presynaptically as an autoreceptor to reduce glutamate release (Lovinger and McCool 1995; Manzoni et al. 1997). Chronic ethanol exposure decreases mGluR2 expression and function (Meinhardt et al. 2013), and this study supports the idea that loss of the mGluR2 autoreceptor function contributes to enhanced glutamate levels state in NAc. Interestingly, mGluR2 is not expressed by the ethanol-preferring P rats and is prevalent in other rat lines selected for high ethanol drinking preference (Zhou et al. 2013; Wood et al. 2017). The impact of this receptor on ethanol seeking and drinking will be discussed later in this chapter.
Dopamine release in the nucleus accumbens is also altered following chronic ethanol exposure or drinking in rats and mice and in chronic ethanol-consuming rhesus monkeys (Siciliano 2017). Some of these changes appear to reflect direct neuroadaptations in dopamine release mechanisms such as decreased release in brain slices (Karkhanis et al. 2015; Melchior and Jones 2017), while others indicate increased dopamine clearance, most likely due to changes in function of the dopamine transporter (Karkhanis et al. 2015, 2016). It is notable that dopamine release in male monkey NAc slices is increased following chronic drinking (Siciliano et al. 2015), in contrast to the findings in rodent. For a more in-depth discussion of these findings, the reader is referred to the excellent chapter by Siciliano and coworkers (2017) in this volume. Inhibition of dopamine release by the Gi/o-coupled kappa opioid receptor is also enhanced after chronic ethanol exposure in rodent NAc (Karkhanis et al. 2016; Rose et al 2016), contributing to a possible hypodoaminergic state after this exposure. A similar enhancement of kappa receptor function is observed in NAc and caudate nucleus of chronic ethanol consuming monkeys (Siciliano et al 2015, 2016). Overall, several factors contribute to an overall decrease in synaptic dopamine levels following chronic ethanol exposure, particularly during the early stages of abstinence (Hirth et al. 2016). However, increased DA levels have been observed following protracted abstinence in rat, and molecular changes that could contribute to increased extracellular dopamine have been observed in postrmortem tissue from patients with AUD (Hirth et al. 2016). These findings indicate that changes in factors controlling extracellular DA levels may depend on the period of drug withdrawal.
Endocannabinoids and LTD
The CB1 receptor is another presynaptic Gi/o-coupled GPCR whose function is decreased following long-term ethanol exposure (Figure 2) (Xia et al. 2006; Depoy et al. 2013; Adermark et al. 2011a,b). As mentioned previously, retrograde signaling by postsynaptically-released eCBs normally activates presynaptic CB1 receptors inducing either short-term synaptic depression or Gi/o-LTD at GABAergic and glutamatergic synapses throughout the brain (Araque et al. 2017; Heifets and Castillo 2009).
Chronic ethanol exposure or drinking produces decreased CB1 expression and function and loss of the LTD induced by activation of this receptor (Basavarajappa et al. 1998; Xia et al. 2006; Adermark et al. 2011a; DePoy et al. 2013). In the dorsal striatum, depression at glutamatergic synapses induced by a CB1 agonist is lost following chronic ethanol drinking (Adermark et al. 2011a). The loss of CB1-mediated LTD persists for 7 days following the last drug exposure (Xia et al. 2006). At GABAergic synapses, eCB-dependent LTD also occurs, and this synaptic depression indirectly produces a long-lasting increase in neuronal activation by glutamatergic synapses (Adermark et al. 2009). This type of LTD is also impaired following chronic ethanol drinking, facilitating a long-lasting potentiation of striatal output in response to glutamatergic transmission (Adermark et al. 2011b). These changes in eCB-dependent plasticity combine with the decrease in GABAergic transmission to increase striatal output in ethanol-exposed animals.
Roles of Presynaptic Changes in Ethanol-Related Behaviors: Focus on Cortico-Basal Ganglia and Amygdala Circuitry
As the preceding sections indicate we now know a great deal about the acute and chronic ethanol effects on neurotransmitter release as well as presynaptic modulation and plasticity. However, less is known about the roles played by these ethanol actions in the behavioral alterations induced by ethanol. Regarding the consequences of altered GABA release, there are well known interactions between the acute effects of ethanol and many drugs that act at GABAergic synapses (Mihic and Harris 2011). However, these interactions have been mainly ascribed to ethanol effects on GABAA receptors. Thus, it will be important to investigate if presynaptic changes at GABAergic synapses contribute to the drug interactions. This is certainly an important topic, because ethanol drinking in conjunction with drugs that target GABAergic transmission can result in profound acute toxicity, including death.
There has been considerable recent attention on ethanol-induced alterations in presynaptic modulation at synapses in different regions of the striatum. This topic is of interest to investigators examining ethanol seeking and drinking because different cortico-basal ganglia circuits involving specific striatal subregions are implicated in these behaviors. Large regions of the striatum are part of at least three different cortico-basal ganglia circuits, with the DMS/caudate being part of an “associative” circuit, the DLS/putamen participating in the “sensorimotor” circuit, and the NAc being incorporated into the “limbic” circuit (Yin and Knowlton 2006). As discussed in the previous sections of this chapter, ethanol has effects on aspects of presynaptic function in striatal components of all these circuits.
The behavioral consequences of ethanol actions in NAc are widely appreciated (Koob and Volkow 2016). It is clear that this region and the associated limbic circuit have crucial roles in the rewarding effects of the drug (as shown using conditioned place preference and ethanol self-administration procedures). Indeed, alterations within the NAc/limbic circuit are likely to impact affective states, Pavlovian conditioning and Pavlovian-to-instrumental transfer conditioning, as well as responses to stress, withdrawal and other factors that contribute to negative affect that helps to drive relapse to drinking. A decrease in mGluR2 modulation of glutamate release at prefrontal cortical inputs to the NAc contributes to excessive seeking and drinking following chronic ethanol exposure (Meinhardt et al. 2013). Rats and mice lacking mGluR2 also show enhanced ethanol seeking and drinking (Zhou et al. 2013; Wood et al. 2017), although it is clear thatloss of mGluR2 is only one of several genetic and molecular factors that influence these behaviors in alcohol-preferring rats (Zhou et al. 2013). The consequences of mGluR2 absence or hypofunction presumably reflect loss of a crucial feedback control that normally prevents the hyperglutamatergic state thought to drive excessive drinking. Treatment with mGluR2/3 agonists reduces ethanol seeking in rodent models (Backstrom and Hyytia 2005; Rodd et al. 2006; Sidhpura et al. 2010; Zhao et al. 2006). However, considerable additional work is needed to determine the contributions of other presynaptic mechanisms in NAc (e.g. alterations in other presynaptic Gi/o-coupled GPCRs or altered GABA release) to ethanol actions in vivo.
Striatal function within the associative and sensorimotor circuits also has the potential to contribute to a variety of acute and chronic ethanol actions. The major focus of research in this area has been the dissociation of effects on “goal-directed” and “habitual” behaviors, including ethanol seeking and drinking (Lovinger and Alvarez 2017; Corbit and Janak 2016; Gremel and Lovinger 2017). Indeed, the DMS/caudate is implicated in goal-directed behaviors while the DLS has a key role in habit learning, especially in self-paced “free-choice” instrumental tasks and response learning tasks. However, this facile dichotomy has overshadowed important roles of these regions and the larger circuits in behavioral control and ethanol actions.
For example, the associative striatum receives strong synaptic inputs from many regions of frontal cortex, including orbitofrontal and medial prefrontal areas (Haber et al. 2006; Hintiryan et al. 2016; Hunnicutt et al. 2016). These cortical regions show structural and functional alterations following long-term ethanol exposure, both in experimental animals and in humans (reviewed in Barker et al. 2015; Sullivan and Pfefferbaum 2005). The caudate nucleus also shows reduced volume after heavy drinking in adolescents (Squeglia et al. 2014). Thus, the associative circuit is likely to be strongly compromised by this type of ethanol exposure. Given the key role of the DMS/caudate within this circuitry, it is very likely that altered cortical communication to this striatal region contributes to this dysfunction. The evidence that acute and chronic ethanol produce presynaptic alterations in the DMS and caudate has already been discussed. The “hypofrontality” and altered DMS/caudate function induced by ethanol are likely to contribute to deficits in cognitive function and altered decision making induced by ethanol abuse. The loss of conscious executive control during intoxication and following chronic ethanol abuse is likely to contribute to poor decision making and preservation in drinking and other associated maladaptive behaviors. There is a growing literature showing that manipulation of the DMS alters ethanol seeking and drinking (Cheng et al. 2017; Corbit et al. 2012; Nam et al. 2013; Wang et al. 2012), but more work is needed to determine the mechanisms within this striatal region that contribute to this behavioral change.
The sensorimotor circuit has important roles in performance of well-learned actions. Inputs from sensory and motor cortices drive neurons in the DLS/putamen (Haber et al. 2006; Hintiryan et al. 2016; Hunnicutt et al. 2016) allowing for output of automatized movements in appropriate contexts. This circuitry also has key roles in reinforcement-driven “stimulus-response” learning and behavior, especially in self-paced operant tasks that do not include a clear Pavlovian component (Yin and Knowlton 2006). Repeated performance of actions for an outcome in a particular context leads to development of associations between the external context, the internal state of the animal and the action, driven by the history of reinforcement (Dickinson 1985). Indeed, this form of instrumental learning, now sometimes referred to as “habit” learning, received the strongest attention prior to characterization of action-outcome “goal-directed” instrumental learning (Colwill and Rescorla 1990; Dickinson 1985). A number of studies have now shown that chronic ethanol drinking or exposure enhances this type of behavior in both experimental animals and humans (Barker et al. 2010; Corbit et al. 2012; Dickinson et al. 2002; Gladwin and Wiers 2012; Hogarth et al. 2012; Ostlund et al. 2010; Hay et al. 2013; Mangieri et al. 2012; Sjoerds et al. 2013 although see Sebold et al. 2014, 2017), as well as other behaviors that involve the DLS (DePoy et al. 2013).
A number of presynaptic changes in the DLS/putamen have been discussed, including decreased GABA release and decreased Gi/o modulation of cortical/glutamatergic inputs to this striatal subregion. The general consequence of these alterations is to decrease modulatory and inhibitory controls on the activation of MSNs, producing the potential for enhanced DLS/putamen output after chronic ethanol exposure. This would help to foster the learning and performance mediated by the sensorimotor circuit, including increased S-R learning. It remains to be determined what other presynaptic changes occur in other parts of the circuitry that could also contribute to these behavioral changes.
While the focus of the work on ethanol and sensorimotor circuitry has been on how enhanced “habit formation” might contribute to ethanol seeking and drinking, it is important not to lose sight of how the drug effects on sensorimotor circuitry will alter all behaviors related to this circuitry. For example, the fact that ethanol increases S-R learning reinforced by food is part of the pattern of impaired decision making produced by the drug. This effect has consequences across the entire spectrum of behaviors altered by ethanol abuse. In combination with impairment of associative circuit function, enhanced potential for sensorimotor circuit function likely contributes to loss of executive control and behavioral flexibility with enhanced control of behavior by the immediate context. It is worth noting that this effect does not depend on having ethanol as the reinforcer driving learning, as S-R learning is enhanced by forced ethanol exposure when a food reinforcer is used in training (Corbit et al. 2012). Thus, it is unlikely that the enhanced S-R learning is driven by the reinforcement history per se. Rather, it appears to be the effect of ethanol is on the circuitry that influences how reinforcement drives behavior.
The implications of these presynaptic changes in particular circuit changes for ethanol seeking and drinking and other drug-related behaviors can be debated, but there is evidence from studies in both experimental animals and humans that that they have important roles. Multiple circuits contribute to intoxication, binge and excessive ethanol drinking, withdrawal effects, relapse to drinking and excessive drinking following relapse. Acute exposure to ethanol initiates the processes that contribute to escalation of drinking. Presynaptic inhibitory changes in the BLA, CeA and VTA likely contribute to the rewarding effects of ethanol. Enhanced inhibition in the associative circuit may play a part in impairment of cognitive control and executive function that contributes to lack of ability to consciously control drinking as well as poor decisions made under the influence of ethanol. Disinhibition of sensorimotor striatum most likely fosters excessive drinking and poor decision making by fostering more automatized action patterns. Chronic ethanol effects will exacerbate many of these changes, particularly the presynaptic effects on amygdala and sensorimotor circuitry. Presynaptic changes within the limbic circuitry may also begin to have a larger influence with increasing duration and amount of chronic ethanol exposure. Increased inhibition in the hippocampal CA1 region can impair spatial memory and other aspects of episodic learning and memory (Berry et al. 2009; Gibson 1985; Givens 1995; Hunt et al. 2009; Matthews et al. 2002; Ryabinin 1998; Ryabinin et al. 2002). Long-term effects of changes in this limbic region may also underlie the influence of context on relapse to ethanol seeking and taking. Clearly ethanol has strong effects on GABA release in the CeA, with CRF participating in both the acute and chronic drug actions. There is now considerable evidence for participation of these neurotransmitters and this brain region in relapse driven by stress and negative affect (Koob and Volkow 2016). The BLA plays important roles in signaling the relative positive or negative valence of environmental events within the associative and limbic circuits (Johansen et al. 2011; Wassum and Izquierdo 2015). Presynaptic ethanol effects at both GABAergic and glutamatergic synapses likely alter the contribution of this brain region to reward- and punishment-driven behavior, as well as responses to stress. Presynaptic effects of ethanol that alter serotonergic neuronal function will also alter limbic circuit responses to stress, in addition to affecting affective states.
The alterations in all three cortico-basal ganglia circuits, including presynaptic changes, will ultimately participate in a vicious circle of behavioral changes similar to that proposed by Koob and others (Barker et al. 2015; Koob and Volkow 2016). Prolonged ethanol exposure combined with conditioning related to ethanol intake will promote loss of associative circuit-based mechanisms that normally support decisions to limit drinking. Ethanol will alsopromote transition from associative/limbic-based reward-driven actions to sensorimotor reinforcement-based actions that will promote excessive drinking, especially in environments previously associated with heavy drinking. This will drive further neuroadaptations including impairment in prefrontal cortex contributions to associative and limbic circuits leading to compromised executive control and conscious decision making. Parallel changes in other limbic cortical areas will promote excessive responding to negative emotions and stressful/negative environmental events, especially during abstinence. These limbic changes will help to promote relapse. In the proper environmental/social contexts relapse will be fostered by a strengthened sensorimotor circuit, and once drinking has begun, the dominant sensorimotor circuit and impaired associative circuit will likely contribute to continued drinking due to automatization of behavior. Often drinking will then proceed well beyond levels needed to simply overcome negative consequence of abstinence. It will be interesting to determine more about how components of each of these circuits contribute to different stages of alcohol abuse. For example, little is known about how acute and chronic ethanol exposure alters sensory and motor cortex function, and how these actions might contribute to altered circuit function. Even less is known about ethanol actions on the thalamic elements of the three circuits, or effects on basal ganglia regions downstream of the striatum. Presynaptic ethanol actions may occur in many of these brain regions, and discovery of these effects may add to the list of potential targets for treatment of alcohol use disorders.
References
- Abrahao K, Salinas A, Lovinger DM (2017) Alcohol and the Brain: Molecular Targets, Synapses and Circuit. Neuron in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Talani G, Lovinger DM (2009) Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur J Neurosci 29(1):32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Clarke RB, Soderpalm B, Ericson M (2011a). Ethanol-induced modulation of synaptic output from the dorsolateral striatum in rat is regulated by cholinergic interneurons. Neurochem Int 58:693–699 [DOI] [PubMed] [Google Scholar]
- Adermark L, Jonsson S, Ericson M, Soderpalm B (2011b). Intermittent ethanol consumption depresses endocannabinoid-signaling in the dorsolateral striatum of rat. Neuropharmacology 61:1160–1165 [DOI] [PubMed] [Google Scholar]
- Agabio R, Colombo G. (2014) GABAB receptor ligands for the treatment of alcohol use disorder: preclinical and clinical evidence. Front Neurosci. 2014 8:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Moorman DE, Becker HC (2017) Contribution of dynorphin and orexin neuropeptide systems to alcohol reward and motivation. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Castillo PE, Manzoni OJ, Tonini R (2017) Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 124:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariwodola OJ, Weiner JL (2004) Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABA(B) receptors. J Neurosci 24:10679–10686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM, Mathur BN (2014) Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci 37(11):663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom P, Hyytia P (2005) Suppression of alcohol self-administration and cue-induced reinstatement of alcohol seeking by the mGlu2/3 receptor agonist LY379268 and the mGlu8 receptor agonist (S)-3,4-DCPG. Eur J Pharmacol 528:110–118 [DOI] [PubMed] [Google Scholar]
- Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M (2008) Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci USA 105:8410–8415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Shen H, Kalivas PW (2002) Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino Acids. 23(13):161–162 [DOI] [PubMed] [Google Scholar]
- Barker JM, Torregrossa MM, Arnold AP, Taylor JR (2010) Dissociation of genetic and hormonal influences on sex differences in alcoholism-related behaviors. J Neurosci.30(27):9140–9144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JM, Corbit LH, Robinson DL, Gremel CM, Gonzales RA, Chandler LJ (2015) Corticostriatal circuitry and habitual ethanol seeking. Alcohol 49(8):817–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Cooper TB, Hungund BL (1998) Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res. 1998 793(1–2):212–218 [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Ninan I, Arancio O (2008) Acute ethanol suppresses glutamatergic neurotransmission through endocannabinoids in hippocampal neurons. J Neurochem 107, 1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RB, Chandra D, Diaz-Granados JL, Homanics GE, Matthews DB (2009) Investigation of ethanol-induced impairment of spatial memory in gamma2 heterozygous knockout mice. Neurosci Lett 455(2):84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betke KM, Wells CA, Hamm HE (2012) GPCR mediated regulation of synaptic transmission. Progress in Neurobiology 96:304–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE (2001) G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science 292:293–297 [DOI] [PubMed] [Google Scholar]
- Brown DA, Sihra TS (2008) Presynaptic signaling by heterotrimeric G-proteins. In Sudhof TC, Starke K (eds.) Handbook of Experimental Pharmacology, Pharmacology of Neurotransmitter Release (184:207–260 [DOI] [PubMed] [Google Scholar]
- Budygin EA, Phillips PE, Wightman RM, Jones SR (2001) Terminal effects of ethanol on dopamine dynamics in rat nucleus accumbens: an in vitro voltammetric study. Synapse 42:77–79 [DOI] [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW (2003) Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol 63(1):53–64 [DOI] [PubMed] [Google Scholar]
- Cannady R, Rinker JA, Woodward JJ, Mulholland PJ (2017) Chronic alcohol, intrinsic excitability, and potassium channels: Neuroadaptations and drinking behavior. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF (2004) Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci. 24(15):3746–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Few AP (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59:882–901 [DOI] [PubMed] [Google Scholar]
- Chandler CM, Overton JS, Ruedi-Bettschen D, Platt DM (2017) GABAA receptor subtype mechanisms and the abuse-related effects of ethanol. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg [Google Scholar]
- Cheng Y, Huang CCY, Ma T, Wei X, Wang X, Lu J, Wang J (2017) Distinct Synaptic Strengthening of the Striatal Direct and Indirect Pathways Drives Alcohol Consumption. Biol Psychiatry 81:918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Crews FT (2017) Innate immune signaling and alcohol use disorders. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg [Google Scholar]
- Colwill RM, Rescorla RA (1990) Effect of reinforcer devaluation on discriminative control of instrumental behavior. J Exp Psychol Anim Behav Process. 16(1):40–47 [PubMed] [Google Scholar]
- Corbit LH, Nie H, Janak PH (2012) Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry 72(5):389–95. doi: 10.1016/j.biopsych.2012.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit LH, Janak PH (2016) Habitual Alcohol Seeking: Neural Bases and Possible Relations to Alcohol Use Disorders. Alcohol Clin Exp Res 40(7):1380–9. doi: 10.1111/acer.13094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Kelm MK, Breese GR (2008) Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J Pharmacol Exp Ther 326:596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon Carlson VC (2017) GABA and glutamate synaptic co-adaptations to chronic ethanol in the striatum. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg [Google Scholar]
- Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, Daunais J, Alvarez VA, Lovinger DM, Grant KA (2011). Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology 36:2513–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon Carlson VC, Grant KA, Lovinger DM (2017) Synaptic adaptations to ethanol intake in male rhesus monkey dorsal striatum depend on age of drinking onset. Submitted for publication [DOI] [PMC free article] [PubMed]
- Dahchour A, De Witte P (1999) Effect of repeated ethanol withdrawal on glutamate microdialysate in the hippocampus. Alcohol Clin Exp Res 23:1698–1703 [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P (2003) Excitatory and inhibitory amino acid changes during repeated episodes of ethanol withdrawal: an in vivo microdialysis study. Eur J Pharmacol 459:171–178 [DOI] [PubMed] [Google Scholar]
- Deng C, Li KY, Zhou C, Ye JH (2009) Ethanol enhances glutamate transmission by retrograde dopamine signaling in a postsynaptic neuron/synaptic bouton preparation from the ventral tegmental area. Neuropsychopharmacology 34(5):1233–1244. doi: 10.1038/npp.2008.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, Holmes A (2013) Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci USA 110:14783–14788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson A (1985) Actions and habits: the development of behavioural autonomy. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 308:67–78 [Google Scholar]
- Dickinson A, Wood N, Smith JW (2002) Alcohol seeking by rats: action or habit? Q J Exp Psychol B. 55(4):331–348 [DOI] [PubMed] [Google Scholar]
- Diaz MR, Christian DT, Anderson NJ, McCool BA (2011) Chronic ethanol and withdrawal differentially modulate lateral/basolateral amygdala paracapsular and local GABAergic synapses. J Pharmacol Exp Ther. 337(1):162–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopico AM, Bukiya AN, Bettinger JC (2017) Voltage-sensitive potassium channels: BK. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB (2004) Presynaptic ionotropic receptors and control of transmitter release. Nature Reviews Neuroscience 5(2):135–145 [DOI] [PubMed] [Google Scholar]
- Evstratova A, Tóth K (2014) Information processing and synaptic plasticity at hippocampal mossy fiber terminals. Front Cell Neurosci 8:28. doi: 10.3389/fncel.2014.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GJ, Morgan A (2003) Regulation of the exocytotic machinery by cAMP-dependent protein kinase: implications for presynaptic plasticity. Biochem Soc Trans 31(Pt 4):824–827 [DOI] [PubMed] [Google Scholar]
- Finn DA, Jimenez VA (2017) Dynamic adaptation in neurosteroid networks in response to alcohol. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WE (1985) Effects of alcohol on radial maze performance in rats. Physiol Behav 35(6):1003–1005 [DOI] [PubMed] [Google Scholar]
- Givens B (1995) Low doses of ethanol impair spatial working memory and reduce hippocampal theta activity. Alcohol Clin Exp Res 19:763–767 [DOI] [PubMed] [Google Scholar]
- Gioia DA, McCool B (2017) Strain-Dependent Effects of Acute Alcohol on Synaptic Vesicle Recycling and Post-Tetanic Potentiation in Medial Glutamate Inputs to the Mouse Basolateral Amygdala. Alcohol Clin Exp Res 41:735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia DA, Alexander N, McCool BA (2017) Ethanol Mediated Inhibition of Synaptic Vesicle Recycling at Amygdala Glutamate Synapses Is Dependent upon Munc13–2. Front Neurosci 11:424 doi: 10.3389/fnins.2017.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin TE, Wiers RW (2012) Alcohol-related effects on automaticity due to experimentally manipulated conditioning. Alcohol Clin Exp Res 36(5):895–9. doi: 10.1111/j.1530-0277.2011.01687 [DOI] [PubMed] [Google Scholar]
- Gremel CM, Lovinger DM (2017) Associative and sensorimotor cortico-basal ganglia circuit roles in effects of abused drugs. Genes Brain Behav 16(1):71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC 3rd, Haun HL, Hazelbaker CL, Ramachandra VS, Becker HC (2014) Increased extracellular glutamate in the nucleus accumbens promotes excessive ethanol drinking in ethanol dependent mice. Neuropsychopharmacology 39:707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC (2010) Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13(12):1519–1525 doi: 10.1038/nn.2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Kim KS, Mailly P, Calzavara R (2006) Reward-related cortical inputs define a large striatal region in primates that interface with associative cortical connections, providing a substrate for incentive-based learning. Journal of Neuroscience 26:8368–8376. doi: 10.1523/JNEUROSCI.0271-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ (2008) Ethanol’s molecular targets. Sci Signal 1, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay RA, Jennings JH, Zitzman DL, Hodge CW, Robinson DL (2013) Specific and nonspecific effects of naltrexone on goal-directed and habitual models of alcohol seeking and drinking. Alcohol Clin Exp Res 37(7):1100–1110. doi: 10.1111/acer.12081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annual Review of Physiology 71:283–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380:258–262 [DOI] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, Yang XW, Shih JC, Toga AW, Dong HW (2016) The mouse cortico-striatal projectome. Nature Neuroscience. 19:1100–1114. doi: 10.1038/nn.4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M, Yamada M, Obata K (2009) Ethanol enhances both action potential dependent and action potential-independent GABAergic transmission onto cerebellar Purkinje cells. Neuropharmacology 57:109–120 [DOI] [PubMed] [Google Scholar]
- Hirth N, Meinhardt MW, Noori HR, Salgado H, Torres-Ramirez O, Uhrig S, Broccoli L, Vengeliene V, Rossmanith M, Perreau-Lenz S, Kohr G, Sommer WH, Spanagel R, Hansson AC (2016) Convergent evidence from alcohol-dependent humans and rats for a hyperdopaminergic state in protracted abstinence. Proc Natl Acad Sci U S A 113:3024–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth L, Attwood AS, Bate HA, Munafò MR (2012) Acute alcohol impairs human goal-directed action. Biol Psychol. 90(2):154–60. doi: 10.1016/j.biopsycho.2012.02.016 [DOI] [PubMed] [Google Scholar]
- Hopf FW, Mangieri RA (2017) Do alcohol-related AMPA-type glutamate receptor adaptations promote intake? In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PubMed] [Google Scholar]
- Howerton TC, Collins AC (1984) Ethanol-induced inhibition of GABA release from LS and SS mouse brain slices. Alcohol. 1(6):471–477 [DOI] [PubMed] [Google Scholar]
- Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, Mao T (2016) A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife. 5 pii: e19103. doi: 10.7554/eLife.19103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PS, Levillain ME, Spector BM, Kostelnik LA (2009) Post-training ethanol disrupts trace conditioned fear in rats: Effects of timing of ethanol, dose and trace interval duration. Neurobiol Learn Mem 91:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR (1996) Voltage-dependent modulation of N-type Ca2+ channels by G-protein beta gamma subunits. Nature 380:255–258 [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE (2011) Molecular mechanisms of fear learning and memory. Cell 147(3):509–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun SB, Cuzon Carlson V, Ikeda S, Lovinger D (2011) Vibrodissociation of neurons from rodent brain slices to study synaptic transmission and image presynaptic terminals. J Vis Exp (51). pii: 2752. doi: 10.3791/2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis AN, Rose JH, Huggins KN, Konstantopoulos JK, Jones SR (2015) Chronic intermittent ethanol exposure reduces presynaptic dopamine neurotransmission in the mouse nucleus accumbens. Drug Alcohol Depend 150:24–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis AN, Huggins KN, Rose JH, Jones SR (2016) Switch from excitatory to inhibitory actions of ethanol on dopamine levels after chronic exposure: Role of kappa opioid receptors. Neuropharmacology 110:190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET (2015) The mechanisms and function of spontaneous neurotransmitter release. Nature Rev Neurosci 16:5–16 [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR (2007) Calcium release from presynaptic internal stores is required for ethanol to increase spontaneous gamma-aminobutyric acid release onto cerebellum Purkinje neurons. J Pharmacol Exp Ther 323:356–364 [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR (2008) The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J Neurophysiol. 100(6):3417–3428. doi: 10.1152/jn.90970.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirson D, Oleata CS, Parsons LH, Ciccocioppo R, Roberto M (2017) CB1 and ethanol effects on glutamatergic transmission in the central amygdala of male and female msP and Wistar rats. Addict Biol. doi: 10.1111/adb.12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenowski PM, Tapper AR (2017) Molecular, neuronal and behavioral effects of ethanol and nicotine interactions. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW (2009) Glutamate and reinstatement. Curr Opin Pharmacol 9(1):59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2016) Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3:760–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA (2007) Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol 98:3185–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latek D Modzelewska A, Trzaskowski B, Palczewski K, Filipek S (2012) G protein-coupled receptors--recent advances. Acta Biochimica Polonica 59:515–529 [PMC free article] [PubMed] [Google Scholar]
- Li C, McCall NM, Lopez AJ, Kash TL (2013) Alcohol effects on synaptic transmission in periaqueductal gray dopamine neurons. Alcohol. 47(4):279–287. doi: 10.1016/j.alcohol.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Alvarez VA (2017) Alcohol and basal ganglia circuitry: Animal models. Neuropharmacology 122:46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, McCool BA (1995) Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGLuR2 or 3. J. Neurophys 73(03):1076–1083 [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Roberto M (2013) Synaptic effects induced by alcohol. Curr Top Behav Neurosci 13:31–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Marcinkiewcz CA, Kash TL (2015) Functional alterations in the dorsal raphe nucleus following acute and chronic ethanol exposure. Neuropsychopharmacology 40(3):590–600 doi: 10.1038/npp.2014.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Barbee B, Wang X, Wang J (2017) Alcohol induces input-specific aberrant synaptic plasticity in the rat dorsomedial striatum. Neuropharmacology 123:46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldve RE, Chen X, Zhang TA, Morrisett RA (2004) Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: real-time visualization in intact hippocampal slices. Alcohol Clin Exp Res 28:143–152 [DOI] [PubMed] [Google Scholar]
- Mangieri RA, Cofresí RU, Gonzales RA (2012) Ethanol seeking by Long Evans rats is not always a goal-directed behavior. PLoS One. 7(8):e42886. doi: 10.1371/journal.pone.0042886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni O, Michel JM, Bockaert J (1997) Metabotropic glutamate receptors in the rat nucleus accumbens. Eur J Neurosci. 9(7):1514–1523 [DOI] [PubMed] [Google Scholar]
- Matthews DB, Morrow AL, Tokunaga S, McDaniel JR (2002) Acute ethanol administration and acute allopregnanolone administration impair spatial memory in the Morris water task. Alcohol Clin Exp Res 26:1747–1751 [DOI] [PubMed] [Google Scholar]
- Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, Stahlin O, Heilig M, Harper C, Drescher KU, Spanagel R, Sommer WH (2013) Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. J Neurosci 33:2794–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior JR, Jones SR (2017) Chronic ethanol exposure increases inhibition of optically targeted phasic dopamine release in the nucleus accumbens core and medial shell ex vivo. Mol Cell Neurosci 85:93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Hicks MP, Cagle SS, Kalivas PW (2005) Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res 29(3):326–333 [DOI] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A (2002) Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22:2074–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F (1995) Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci 15:5439–5447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic SJ, Harris RA (2011) Hypnotics and sedatives. In Goodman & Gilman’s Pharmacological Basis of Therapeutics. 12th edition (New York: McGraw Hill; ) pp. 457–480 [Google Scholar]
- Miller RJ (1998) Presynaptic receptors. Ann Rev Pharmacol Toxicol 38:201–227 [DOI] [PubMed] [Google Scholar]
- Nam HW, Hinton DJ, Kang NY, Kim T, Lee MR, Oliveros A, Adams C, Ruby CL, Choi DS (2013) Adenosine transporter ENT1 regulates the acquisition of goal-directed behavior and ethanol drinking through A2A receptor in the dorsomedial striatum. J Neurosci. 33(10):4329–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Gouemo P (2017) Voltage-sensitive calcium channels. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR (2004) Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 303:1512–1514 [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE (2006) Structural basis of function in heterotrimeric G proteins. Quarterly reviews of biophysics 39:117–166 [DOI] [PubMed] [Google Scholar]
- Olive MF, Koenig HN, Nannini MA, Hodge CW (2002) Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav 72:213–220 [DOI] [PubMed] [Google Scholar]
- Ostlund SB, Maidment NT, Balleine BW (2010) Alcohol-Paired Contextual Cues Produce an Immediate and Selective Loss of Goal-directed Action in Rats. Front Integr Neurosci. 19;4. pii: 19. doi: 10.3389/fnint.2010.00019. eCollection 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton MH, Roberts BM, Lovinger DM, Mathur BN (2016) Ethanol Disinhibits Dorsolateral Striatal Medium Spiny Neurons Through Activation of A Presynaptic Delta Opioid Receptor. Neuropsychopharmacology 41:1831–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pava MJ, Woodward JJ (2012) A review of the interactions between alcohol and the endocannabinoid system: implications for alcohol dependence and future directions for research. Alcohol 46(3):185–204. doi: 10.1016/j.alcohol.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC, McBain CJ (2008) State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron 60:980–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris J, Coleman-Hardee M, Burry J, Pecins-Thompson M (1992) Selective changes in GABAergic transmission in substantia nigra and superior colliculus caused by ethanol and ethanol withdrawal. Alcohol Clin Exp Res 16(2):311–319 [DOI] [PubMed] [Google Scholar]
- Peris J, Eppler B, Hu M, Walker DW, Hunter BE, Mason K, Anderson KJ (1997) Effects of chronic ethanol exposure on GABA receptors and GABAB receptor modulation of 3H-GABA release in the hippocampus. Alcohol Clin Exp Res 21:1047–1052 [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C (2008) Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nature Reviews Neuroscience 9(6):423–436 [DOI] [PubMed] [Google Scholar]
- Pleil KE, Lowery-Gionta EG, Crowley NA, Li C, Marcinkiewcz CA, Rose JH, McCall NM, Maldonado-Devincci AM, Morrow AL, Jones SR, Kash TL (2015) Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 99:735–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BD, Rossi DJ (2017) Recreational concentrations of alcohol enhance synaptic inhibition of cerebellar unipolar brush cells via pre- and postsynaptic mechanisms. J Neurophysiol 118(1):267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Siggins GR (2006) Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci USA 103(25):9715–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Varodayan FP (2017) Synaptic targets: Chronic alcohol actions. Neuropharmacology 122:85–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR (2003) Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A 100, 2053–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004a) Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci 24:10159–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004b) Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24:1594–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, O’Dell LE, Cruz MT, Morse AC, Siggins GR, Koob GF (2008) Cellular and behavioral interactions of gabapentin with alcohol dependence. J Neurosci 28:5762–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, and Schweitzer P (2010) The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology 35:1962–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Patel RR, Bajo M (2017) Cytokines in the CNS. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg [Google Scholar]
- Rodd ZA, McKinzie DL, Bell RL, McQueen VK, Murphy JM, Schoepp DD, McBride WJ (2006) The metabotropic glutamate 2/3 receptor agonist LY404039 reduces alcohol-seeking but not alcohol self-administration in alcohol-preferring (P) rats. Behav Brain Res 171:207–215 [DOI] [PubMed] [Google Scholar]
- Rose JH, Karkhanis AN, Chen R, Gioia D, Lopez MF, Becker HC, McCool BA, Jones SR (2016) Supersensitive Kappa Opioid Receptors Promotes Ethanol Withdrawal-Related Behaviors and Reduce Dopamine Signaling in the Nucleus Accumbens. Int J Neuropsychopharmacol 19(5):pii: pyv127. doi: 10.1093/ijnp/pyv127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S (1995) Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur J Pharmacol 283:177–183 [DOI] [PubMed] [Google Scholar]
- Ryabinin AE (1998) Role of hippocampus in alcohol-induced memory impairment: implications from behavioral and immediate early gene studies. Psychopharmacology 139:34–43 [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Miller MN, Durrant S (2002) Effects of acute alcohol administration on object recognition learning in C57BL/6J mice. Pharmacology Biochemistry and Behavior 71(1–2):307–312. doi: 10.1016/S0091-3057(01)00661-X [DOI] [PubMed] [Google Scholar]
- Sanna E, Talani F, Busonero MG, Pisu RH, Purdy M, Serra M, Biggio G (2004) Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci 24:6521–6530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber AL, Gilpin NW (2017) Corticotrophin-releasing factor (CRF) neurocircuitry and alcohol drinking. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebold M, Deserno L, Nebe S, Schad DJ, Garbusow M, Hägele C, Keller J, Jünger E, Kathmann N, Smolka MN, Rapp MA, Schlagenhauf F, Heinz A, Huys QJ (2014) Model-based and model-free decisions in alcohol dependence. Neuropsychobiology 70(2):122–131. doi: 10.1159/000362840 [DOI] [PubMed] [Google Scholar]
- Sebold M, Nebe S, Garbusow M, Guggenmos M, Schad DJ, Beck A, Kuitunen-Paul S, Sommer C, Frank R, Neu P, Zimmermann US, Rapp MA, Smolka MN, Huys QJM, Schlagenhauf F, Heinz A (2017) When Habits Are Dangerous: Alcohol Expectancies and Habitual Decision Making Predict Relapse in Alcohol Dependence. Biol Psychiatry pii: S0006–3223(17)31586. doi: 10.1016/j.biopsych.2017.04.019 [DOI] [PubMed] [Google Scholar]
- Seilicovich A, Duvilanski BH, Lasaga M, Debeljuk L, Díaz MC (1988) Effect of ethanol on GABA uptake and release from hypothalamic fragments. Psychopharmacology (Berl). 95(3):418–422 [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiological Reviews 85:1303–1342 [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Cuzon Carlson VC, Helms CM, Lovinger DM, Grant KA, Jones SR (2015) Voluntary ethanol intake predicts kappa-opioid receptor supersensitivity and regionally distinct dopaminergic adaptations in macaques. J Neurosci 35:5959–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Yorgason JT, Lovinger DM, Mateo Y, Jimenez VA, Helms CM, Grant KA and Jones SR (2016) Increased presynaptic regulation of dopamine neurotransmission in the nucleus accumbens core following chronic ethanol self-administration in female macaques. Psychopharmacology (Berl) 233:1435–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Karkhanis A, Holleran KM, Melchior JR, Jones SR (2017) Cross-species alterations in synaptic dopamine regulation after chronic alcohol exposure. In: Grant KA (ed) Handbook of Experimental Pharmacology “Neuropharmacology of Alcohol”. Springer, Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhpura N, Weiss F, Martin-Fardon R (2010) Effects of the mGlu2/3 agonist LY379268 and the mGlu5 antagonist MTEP on ethanol seeking and reinforcement are differentially altered in rats with a history of ethanol dependence. Biol Psychiatry 67:804–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Shi L, Brunso-Bechtold JK, Weiner JL (2008) Distinct mechanisms of ethanol potentiation of local and paracapsular GABAergic synapses in the rat basolateral amygdala.J Pharmacol Exp Ther 324(1):251–260 [DOI] [PubMed] [Google Scholar]
- Silberman Y, Ariwodola OJ, Weiner JL (2012) β1-adrenoceptor activation is required for ethanol enhancement of lateral paracapsular GABAergic synapses in the rat basolateral amygdala. J Pharmacol Exp Ther. 343(2):451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Fetterly TL, Awad EK, Milano EJ, Usdin TB, Winder DG (2015) Ethanol produces corticotropin-releasing factor receptor-dependent enhancement of spontaneous glutamatergic transmission in the mouse central amygdala. Alcohol Clin Exp Res 39:2154–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoerds Z, de Wit S, van den Brink W, Robbins TW, Beekman AT, Penninx BW, Veltman DJ (2013) Behavioral and neuroimaging evidence for overreliance on habit learning in alcohol-dependent patients. Transl Psychiatry 17;3:e337 doi: 10.1038/tp.2013.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squeglia LM, Rinker DA, Bartsch H, Castro N, Chung Y, Dale AM, Jernigan TL, Tapert SF (2014) Brain volume reductions in adolescent heavy drinkers. Dev Cogn Neurosci. 9:117–125. doi: 10.1016/j.dcn.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong R, Wood WG (1984) Membrane properties and aging: in vivo and in vitro effects of ethanol on synaptosomal gamma-aminobutyric acid (GABA) release. J Pharmacol Exp Ther 229(3):726–730 [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A (2005) Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology 180:583–594 [DOI] [PubMed] [Google Scholar]
- Talani G, Lovinger DM (2015) Interactions between ethanol and the endocannabinoid system at GABAergic synapses on basolateral amygdala principal neurons. Alcohol 49:781–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA (2008) Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcohol Clin Exp Res 32:1040–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA (2009) Role of 5-hydroxytryptamine2C receptors in Ca2+-dependent ethanol potentiation of GABA release onto ventral tegmental area dopamine neurons. J Pharmacol Exp Ther 329:625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Soni N, Bajo M, Luu G, Madamba SG, Schweitzer P, Parsons LH, Roberto M (2016) Chronic ethanol exposure decreases CB1 receptor function at GABAergic synapses in the rat central amygdala. Addict Biol 21(4):788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Logrip ML, Roberto M (2017) P/Q-type voltage-gated calcium channels mediate the ethanol and CRF sensitivity of central amygdala GABAergic synapses. Neuropharmacology. 125:197–206. doi: 10.1016/j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Weller M (2003) Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol 27(1):99–106 [DOI] [PubMed] [Google Scholar]
- Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR (1996) Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc Natl Acad Sci USA 93(10):5049–5054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Sparta DR, Hopf FW, Bowers MS, Melis M, Bonci A (2009) Strain specific synaptic modifications on ventral tegmental area dopamine neurons after ethanol exposure. Biol Psychiatry 65:646–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassum KM, Izquierdo A (2015) The basolateral amygdala in reward learning and addiction. Neurosci Biobehav Rev 57:271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Gu C, Dunwiddie TV (1997) Differential ethanol sensitivity of subpopulations of GABAA synapses onto rat hippocampal CA1 pyramidal neurons. J Neurophysiol 77:1306–1312 [DOI] [PubMed] [Google Scholar]
- Weiner JL, Ariwodola OJ, Bates WH, Bryant V, Silberman Y, Daunais JB, Grant KA (2005) Presynaptic mechanisms underlying ethanol actions at GABAergci synapses in rat and monkey hippocampus. Alcohol Clin Exper Res 29:187A (Suppl) [Google Scholar]
- Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, Lovinger DM, Alvarez VA (2014) Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology 39:579–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CM, Nicolas CS, Choi SL, Roman E, Nylander I, Fernandez-Teruel A, Kiianmaa K, Bienkowski P, de Jong TR, Colombo G, Chastagnier D, Wafford KA, Collingridge GL, Wildt SJ, Conway-Campbell BL, Robinson ES, Lodge D (2017) Prevalence and influence of cys407* Grm2 mutation in Hannover-derived Wistar rats: mGlu2 receptor loss links to alcohol intake, risk taking and emotional behaviour. Neuropharmacology 115:128–138 [DOI] [PubMed] [Google Scholar]
- Xia JX, Li J, Zhou R, Zhang XH, Ge YB, Ru Yuan X (2006) Alterations of rat corticostriatal synaptic plasticity after chronic ethanol exposure and withdrawal. Alcohol Clin Exp Res 30:819–824 [DOI] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC 3rd, Li KY, Krnjevic K, Zhou C, Ye JH (2009) Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology 34:307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ (2006) The role of the basal ganglia in habit formation. Nat Rev Neurosci 7(6):464–476 [DOI] [PubMed] [Google Scholar]
- Yin HH, Ronesi J, Davis M, Lovinger DM (2006) The role of protein synthesis in striatal long-term depression. J. Neurosci 26(46):11811–11820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Park BS, Adermark L, Lovinger DM (2007) Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci 25:3226–3232 [DOI] [PubMed] [Google Scholar]
- Younts TJ, Monday HR, Dudok B, Klein ME, Jordan BA, Katona I, Castillo PE (2017) Presynaptic Protein Synthesis Is Required for Long-Term Plasticity of GABA Release. Neuron 92(2):479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Dayas CV, Aujla H, Baptista MA, Martin-Fardon R, Weiss F (2006) Activation of group II metabotropic glutamate receptors attenuates both stress and cue-induced ethanol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci 26:9967–9974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, Yuan Q, Barbier E, Feng A, Flanigan M, Augier E, Enoch MA, Hodgkinson CA, Shen PH, Lovinger DM, Edenberg HJ, Heilig M, Goldman D (2013) Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci USA 110(42):16963–16968. doi: 10.1073/pnas.1309839110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Bie B, Pan ZZ (2007) Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci 27(2):289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM (2006) Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurophysiol 96(1):433–441 [DOI] [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Gao BX, Hinckley C (2003) Ethanol dual modulatory actions on spontaneous postsynaptic currents in spinal motoneurons. J Neurophysiol 89(2):806–813 [DOI] [PubMed] [Google Scholar]