

Abstract
Our body expresses sensors to detect pathogens through the recognition of expressed molecules, including nucleic acids, lipids, and proteins, while immune tolerance prevents an overreaction with self and the development of autoimmune disease. Adenosine (A)-to-inosine (I) RNA editing, catalyzed by adenosine deaminases acting on RNA (ADARs), is a post-transcriptional modification that can potentially occur at over 100 million sites in the human genome, mainly in Alu repetitive elements that preferentially form a double-stranded RNA (dsRNA) structure. A-to-I conversion within dsRNA, which may induce a structural change, is required to escape from the host immune system, given that endogenous dsRNAs transcribed from Alu repetitive elements are potentially recognized by melanoma differentiation-associated protein 5 (MDA5) as non-self. Of note, loss-of-function mutations in the ADAR1 gene cause Aicardi–Goutières syndrome, a congenital autoimmune disease characterized by encephalopathy and a type I interferon (IFN) signature. However, the loss of ADAR1 in cancer cells with an IFN signature induces lethality via the activation of protein kinase R in addition to MDA5. This makes cells more sensitive to immunotherapy, highlighting the opposing immune status of autoimmune diseases (overreaction) and cancer (tolerance). In this review, we provide an overview of insights into two opposing aspects of RNA editing that functions as a modulator of the immune system in autoimmune diseases and cancer.
Keywords: Innate immunity, Adaptive immunity, PD-1, RIG-I, RNase L, SINE
Introduction
The immune system, composed of innate and adaptive immunity, is essential for host defense and protection against foreign agents such as viruses. As the first line of host defense during viral infections, pattern recognition receptors (PRRs), abundantly expressed in innate immune cells such as dendritic cells (DCs) and macrophages, detect viral components and induce anti-viral cytokines, particularly type I interferons (IFNs) [1]. In the second line of defense, adaptive immune cells, such as CD4+ and CD8+ T cells, are activated by viral antigens loaded on major histocompatibility complexes (MHCs) expressed in antigen-presenting cells (APCs), especially DCs, and infected cells [2]. Subsequently, CD8+ T cells directly attack infected cells, whereas CD4+ T cells help antibody production by B cells and activate not only macrophages, but also CD8+ T cells [3].
The immune system simultaneously possesses tolerance to prevent an overreaction to self-antigens. T and B cells mature in the thymus and bone marrow, respectively. Autoreactive cells are eliminated in these organs during their maturation steps, in a process termed central tolerance, while autoreactive cells escaping into peripheral tissues are subjected to peripheral tolerance [4, 5]. Intriguingly, foreign proteins sometimes utilize this tolerance to escape from the host immune system. For instance, programmed cell-death ligand-1 (PD-L1), which is highly expressed in various human cancers, induces tolerance by binding to its receptor, programmed cell death 1 (PD-1), expressed on the surface of T cells [6]. Therefore, blockade of the PD-L1/PD1 pathway with antibodies, an immune checkpoint therapy, induces anticancer immune responses and a marked effect in the treatment of human cancers [7].
Endogenous retrotransposons, considered remnants of past retrovirus integration into the host genome, constitute 43% of the human genome [8]. Their unique retrovirus characteristics can be recognized by the host immune system [9]. Endogenous retrotransposons can be divided into two groups: those with long terminal repeats (LTRs), including endogenous retroviruses (ERVs), make up 8% of the genome, whereas those without LTRs, including long interspersed elements (LINEs) and short interspersed elements (SINEs), account for more than 30% of the human genome [10, 11]. The most common type of SINE in humans is an Alu repetitive element, which can be divided into polymerase (pol) II-transcribed retrotransposition-incompetent elements embedded in mRNAs and pol III-transcribed retrotransposition-competent elements [12, 13]. LINEs are autonomous, because they have two open-reading frames (ORFs) that encode RNA-binding protein, nuclease, and reverse transcriptase, and are required for transposition, whereas SINEs are non-autonomous and utilize LINE transposition machinery [14]. All these retrotransposons can be detected as non-self by nucleic acid sensors under certain conditions.
Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) belong to the RIG-I–like receptor (RLR) family, which is a type of PRR (Fig. 1). Such RLRs are cytosolic sensors for viral dsRNA, the detection of which leads to the recruitment of mitochondrial anti-viral-signaling protein (MAVS) to activate TANK-binding kinase 1 and downstream interferon regulatory factor 3, in turn leading to type I IFN production [15-18]. However, accumulating evidence has shown that endogenous dsRNAs formed by retrotransposon-derived repetitive elements potentially activate these cytosolic dsRNA sensors [19-22]. Therefore, to prevent activation of such sensors, endogenous dsRNAs are simultaneously subjected to chemical modifications such as adenosine (A)-to-inosine (I) RNA editing [23-29].
Fig. 1.

Structural representation of ADARs and RNA-binding proteins involved in dsRNA-sensing pathways. Cytoplasmic adenosine deaminase acting on RNA 1 (ADAR1) p150 comprises two Z-DNA/RNA binding domains (green), three double-stranded (ds)RNA-binding domains (red), and a deaminase domain (dark blue), while nuclear ADAR1 p110 is a truncated isoform that lacks a Z-DNA/RNA-binding domain. A nuclear localization signal (NLS; shown in brown) is present in both p150 and p110 isoforms, whereas a nuclear export signal (NES; shown in yellow) is present in the p150 isoform only. Both ADAR2 and ADAR3 are composed of two dsRNA-binding domains and a deaminase domain, and are located in the nucleus. ADAR3, which contains arginine-rich domain (R; shown in black), has not been shown to have editing activity. Amino acid substitutions resulting from point mutations in the ADAR1 gene, identified in patients with Aicardi–Goutières syndrome (AGS), are also shown. Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) are members of RIG-I-like receptors and comprise two caspase activation and recruitment domains (CARDs; shown in light green), which mediate signal transduction through interaction with the mitochondrial anti-viral-signaling protein (MAVS) with a DExD/H-box RNA helicase domain (orange) and a C-terminal domain (CTD; shown in light blue), both of which are required for RNA binding. Protein kinase R (PKR) is composed of two dsRNA-binding domains (red) and a kinase domain (purple). RNase L comprises nine ankyrin-repeats domain (dark yellow), a kinase-like domain (pink) and an RNase domain (blue). An ankyrin-repeats domain contains the site for binding to 2′,5′-oligoadenylates, which is produced by oligoadenylate synthetase (OAS) proteins
ADAR1-mediated RNA editing prevents MDA5 sensing endogenous dsRNAs
A-to-I RNA editing is a post-transcriptional modification occurring within dsRNA [30, 31]. In mammals, such deamination is catalyzed by adenosine deaminases acting on the RNA (ADAR) protein family [32], composed of ADAR1 [33-36], ADAR2 [37-39], and ADAR3 [40, 41], which all contain dsRNA-binding domains (Figs. 1, 2). ADAR1 is expressed as two isoforms driven by different promoters: interferon (IFN)-inducible full-length ADAR1 p150 that contains a nuclear export signal and is mainly localized in the cytoplasm, and constitutively expressed truncated ADAR1 p110, which is localized in the nucleus [42-45] (Fig. 1). Although both ADAR2 and ADAR3 are located in the nucleus [46, 47], ADAR3 is expressed to a much more limited extent in the brain. Because it is catalytically inactive in vitro, ADAR3 is considered to act as a dominant negative regulator of RNA editing [40, 48, 49]. However, a recent study demonstrated that ADAR3 deficiency in mice does not substantially modulate RNA-editing activity [50]. Therefore, the function of ADAR3 remains undetermined. In contrast, ADAR1 p110 and ADAR2 are active RNA-editing enzymes that are highly expressed in the brain, whereas ADAR1 p150 is especially enriched in the thymus and spleen [51].
Fig. 2.
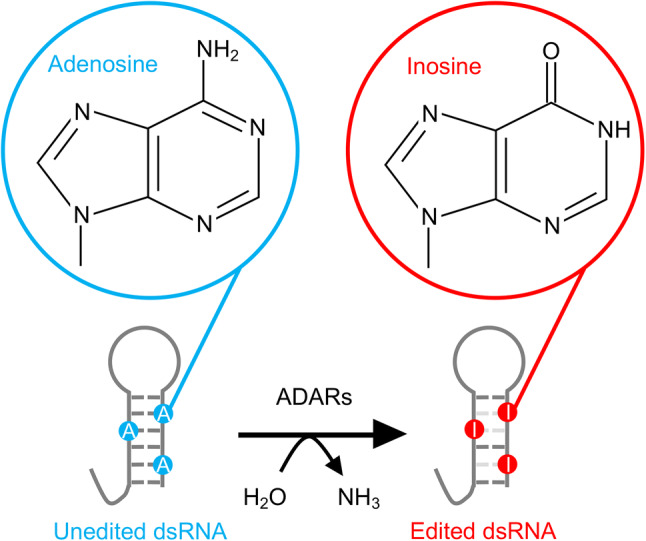
Conversion of adenosine into inosine by ADARs. Adenosine deaminases acting on RNA (ADARs) recognize double-stranded (ds)RNA structures as targets and catalyze the deamination of adenosines in dsRNA into inosine
Because the structure of inosine is very similar to guanosine, the cellular machinery recognizes the inosine within dsRNA as if it were guanosine [52-54]. Therefore, any outcome depends on sites where RNA editing occurs. Although RNA editing in protein coding sequences rarely takes place [55], it can potentially change amino acid sequences, called recoding, and their protein functions [56-62]. It is worth noting that the biological significance of individual recoding events has been demonstrated by introducing edited or unedited versions of the gene of interest in vivo [63-66]. In contrast, of over 100 million sites, approximately 85% of pre-mRNAs are estimated to be edited in humans [67, 68]. This preferentially occurs in the non-coding region of mRNA, especially in the 3′untranslated region (UTR) and introns [69-71]. This is because inverted retrotransposon-derived repetitive elements, which are frequently found in the non-coding region, form intramolecular dsRNA structures targeted by ADARs [67, 72]. Therefore, more than 90% of all RNA-editing events occur within Alu repetitive elements in humans, especially within retrotransposition-incompetent Alu elements embedded in mRNAs but not retrotransposition-competent elements [12]. Although the majority of RNA-editing sites are also found within SINEs in mice, the total number of sites is smaller due to the higher divergence of repeats as compared to humans [73]. It has been reported that RNA editing in the non-coding region of mRNA as well as non-coding RNA modulates splicing patterns [74-78], micro(mi)RNA target specificity [79-81], mRNA stability [82, 83], and circular RNA biogenesis [84-86], although these events are applicable to limited sites only. This indicates that RNA editing in the non-coding region has distinct functions that affect cellular homeostasis in a global manner.
Growing evidence suggests that ADAR1-mediated RNA editing in repetitive elements plays a pivotal role in preventing activation of the host immune system. Given that ADAR1 and ADAR2 have the same preference of U > A > C > G at the nearest 5′ neighbor and a different preference of G > C ~ A > U and G > C > U ~ A at the nearest 3′ neighbor, respectively [87], a rigid motif for RNA editing does not exist. However, a comprehensive study has described how ADAR1 preferentially edits non-coding regions, whereas ADAR2 mainly edits coding regions [49]. Accordingly, Adar1 and Adar2 knockout (KO) mice show different phenotypes: Adar1 KO mice die by embryonic day E12.5, with widespread apoptosis and the overproduction of type I IFN [88-90], whereas Adar2 KO mice show postnatal lethality with progressive seizures [64], which can be rescued by the expression of an edited GRIA2 encoding glutamate receptor subunit GluA2 [59, 64]. Although critical substrates of ADAR1 are unknown, unlike ADAR2, recent studies have reported that several Adar1 mutant mice, such as Adar1 p150-specific KO and Adar1 knock-in (KI) mice that harbor the editing-inactive E861A point mutation (Adar1 E861A KI mice), also show phenotypes similar to those found in Adar1 KO mice; the concurrent deletion of either MDA5 or MAVS rescues embryonic lethality and type I IFN production in these three mutant lines [91-94]. Therefore, it is thought that ADAR1 p150-mediated RNA editing prevents MDA5 sensing endogenous dsRNAs transcribed from repetitive elements as non-self [91-93, 95, 96].
Possible mechanisms underlying the prevention of MDA5-sensing endogenous dsRNAs by RNA editing
Given that RLRs, including RIG-I and MDA5, detect unique structures of viral dsRNA, self-RNAs also possess host-specific molecular markers to prevent recognition by these cytosolic sensors [24] (Fig. 1). Because RIG-I detects short dsRNA with 5′triphosphate blunt ends [97-99], the 5′triphosphate-linked methylguanosine (m7G) cap, only present in eukaryotic organisms, is considered to prevent RIG-I activation. Interestingly, Schuberth–Wagner et al. showed that 2′-O-methylation (2′OMe) at the 5′-terminal nucleotide (N1) completely prevented RIG-I activation, whereas an m7G cap only partially suppressed it [27]. Furthermore, the same group showed that knockdown of the endogenous methyltransferase, MTr1, which is responsible for 2′OMe at N1, caused a loss of RIG-I tolerance to self-RNAs.
In contrast, MDA5 recognizes internal long dsRNA but without characteristic features, unlike RIG-I [100, 101]. Importantly, gain-of-function mutations in IFIH1 that encode MDA5 cause several autoimmune diseases, such as systemic lupus erythematosus (SLE), Singleton–Merten syndrome, and Aicardi–Goutières syndrome (AGS), which is characterized by a childhood-onset autoimmune encephalopathy that shows an excessive expression of type I IFN [102-105]. Although the mechanism underlying mutation-induced MDA5 activation remains controversial, two suggestions have been proposed, which include being constitutively activated in a ligand-independent manner [103, 105], or being activated by self-RNAs due to misrecognition in a ligand-dependent manner [102]. Recently, Ahmad et al. showed that a lack of RNA-binding domain caused by a premature termination single-nucleotide polymorphism failed to activate MDA5 with gain-of-function mutations, supporting the mechanism in a ligand-dependent manner [19]. They further demonstrated that MDA5 activation induced by gain-of-function mutations is caused by the misrecognition of endogenous repetitive elements.
Under physiological conditions, ADAR1-mediated RNA editing prevents MDA5 sensing endogenous dsRNAs transcribed from repetitive elements as non-self [91-93, 95, 96]. However, the mechanisms that underlie escaping MDA5 recognition by A-to-I conversion in dsRNAs remain elusive. Considering the RNA-editing level of each site differs dramatically between developmental stages, organs, and cells [49], one possibility is that edited substrates competitively inhibit MDA5 binding to unedited dsRNAs [91, 106]. It is noteworthy that dsRNAs formed by wobble I–U pairs bind to MDA5, leading to the inhibition of binding of perfect RNA duplexes containing I–C base pairs and the suppression of induced IFN-stimulated genes (ISGs) [106].
In contrast, another possibility is that RNA editing destabilizes A–uridine (U) base pairs by generating multiple I–U mismatches, leading to the prevention of MDA5 recognition [91]. However, when RNA editing occurs at A–C mismatches, which are preferred by ADARs [107], it results in stabilizing a dsRNA structure. Indeed, RNA secondary structure modeling and free energy calculations revealed that a large subset of imperfect RNA duplexes would be stabilized as a consequence of RNA editing [91, 96]. In contrast, a dsRNA structure destabilized by RNA editing can be found in inverted Alu repetitive elements within the 3′UTR of genes involved in vital biological processes in humans. These targets may contain the critical RNA-editing sites required for escaping MDA5 sensing (which needs further study) given that Alu repeats are specific to primates and the inserted position of these repetitive elements is mostly not conserved.
Other dsRNA-sensing pathways regulated by ADAR1
Although concurrent deletion of MDA5 or MAVS extends the survival of Adar1 KO mice until the day of birth, Adar1 E861A KI mice do not show postnatal lethality and survive until adulthood, highlighting the contribution of ADAR1 in alternative signaling pathways [91-93]. Recently, Chung et al. demonstrated that protein kinase R (PKR), a dsRNA sensor, is activated in an ADAR1-deficient human cell line during an IFN response [12]. PKR, encoded by the eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) gene, is a ubiquitously expressed anti-viral protein that is induced by type I IFN [108] (Fig. 1). Once activated by dsRNA, PKR phosphorylates eukaryotic initiation factor 2 alpha (eIF2α), leading to the inhibition of translational initiation [109]. Although it was observed that ADAR1-deficient 293 T cells did not show upregulated expression of type I IFN or ISGs, global translational efficiency and cell proliferation with IFN treatment were impaired in response to PKR activation [12]. It was further demonstrated that the suppression of PKR activation by ADAR1 required dsRNA-binding and catalytic activities. These lines of evidence suggest that upon activation by dsRNAs, PKR has distinct functions that differ from those of MDA5. In addition, the embryonic lethality of Adar1 KO mice could not be rescued by concurrent deletion of PKR [90], which suggests that as-yet-undetermined critical RNA editing targets required for suppressing PKR activation, such as certain Alu elements, may be specific to humans.
In contrast, the lethal phenotype induced by ADAR1 depletion in a human lung adenocarcinoma A549 cell line could be rescued by the concurrent deletion of RNase L, which is involved in another dsRNA-activated anti-viral pathway [110] (Fig. 1). RNase L is a ubiquitously expressed single-stranded RNA–specific ribonuclease that cleaves viral and host RNAs, leading to translational inhibition [111-115]. The molecule, 2′,5′-oligoadenylate, which is produced by oligoadenylate synthetase (OAS) proteins upon dsRNA recognition, binds to monomeric inactive RNase L, leading to catalytically active dimers. Of note, depletion of RNase L restored the lethality of ADAR1-deficient A549 cells in the presence of MDA5, indicating that the OAS–RNase L system is likely the primary pathway activated by ADAR1 depletion, at least in this cell line [110].
A-to-I RNA editing in innate immune cells
Although how RNA editing induces MDA5 tolerance to self-dsRNAs has been clearly demonstrated [91], its role in innate immune cells has not been fully investigated. Recently, Baal et al. reported a role for ADAR1 in the development of DCs using the CD11c-cre transgene, which deletes floxed genes in DCs and alveolar macrophages [116]. CD11c-cre–driven conditional ADAR1 deletion inhibits differentiation and expansion of CD103+ cells among DC subsets, whereas apoptosis, which is generally observed in multiple tissues of Adar1 KO mice, is not induced (Fig. 3). CD103+ DCs mainly contribute to CD8+ T-cell priming via antigen cross-presentation during host defense [117, 118]. In accordance with this, ADAR1-deficient DCs failed to expand CD8+ T cells [116]. Furthermore, CD11c-cre–driven conditional Adar1 KO mice showed the presence of Periodic acid–Schiff (PAS)-positive giant alveolar macrophages; this was also observed when ADAR1 was specifically depleted in macrophages by the LysM-cre transgene, resembling symptoms of pulmonary alveolar proteinosis [119] (Fig. 3). In addition, although the contribution of the MDA5-sensing pathway was not examined, ADAR1-deficient alveolar macrophages showed upregulated ISG expression. Consistently, in humans, knockdown of ADAR1 induces type I IFN responses in primary macrophages differentiated from peripheral blood mononuclear cells (PBMCs) [120]. Given that DCs and macrophages are major type I IFN-producing cells, it is worth investigating how ADAR1 deficiency in these innate immune cells contributes to the pathogenesis of autoimmune diseases such as AGS, caused by loss-of-function mutations in the ADAR1 gene [121].
Fig. 3.

ADAR1-mediated MDA5 tolerance to self-dsRNAs in innate and adaptive immune cells. Melanoma differentiation-associated protein 5 (MDA5) senses viral RNAs upon infection, promoting polymerization of mitochondrial anti-viral-signaling protein (MAVS), and leading to the expression of interferon-stimulated genes (ISGs). To avoid the recognition of self-double-stranded (ds)RNAs by MDA5, adenosine deaminase acting on RNA 1 (ADAR1)-mediated RNA editing is required. Loss of ADAR1 results in activation of the MDA5–MAVS signaling pathway, which impairs homeostasis of innate immune cells, such as dendritic cells (DCs) and macrophages, as well as adaptive immune cells, such as T and B cells
A-to-I RNA editing in adaptive immune cells
Although MDA5 as a specialized molecule for innate immunity has been well studied, given that ADAR1 p150 is especially abundant in lymphoid organs such as the thymus and spleen, the RNA editing/MDA5 axis also has a potential role in lymphocytes. We recently reported a role for RNA editing during T-cell maturation in the thymus [51, 122]. T-cell-specific ADAR1 deficiency reduces mature CD4+ and CD8+ thymocytes due to an impairment of T-cell receptor (TCR) signal transduction (Fig. 3). This comes from the excessive expression of ISGs, given that type I IFN inhibits TCR signal transduction and has an anti-proliferative effect on T cells. Moreover, ADAR1-deficient thymocytes are resistant to negative selection, a process that establishes central tolerance by eliminating autoreactive T cells [123], leading to autoimmunity including intestinal inflammation [51] (Fig. 3). Importantly, this symptom is sometimes observed in patients with AGS [124]. It is worth noting that the concurrent deletion of MDA5 rescues these abnormalities. However, it remains unknown whether the RNA editing/MDA5 axis regulates T-cell functions in peripheral tissues, and this, therefore, requires further study. Another group reported a proviral function of ADAR1 using primary CD4+ T cells isolated from patients with AGS [125]. In accordance with our observation in the mouse, AGS patient-derived primary CD4+ T cells showed the upregulated expression of ISGs, providing a resistant phenotype to infection by HIV-1 [51, 125].
In contrast, Marcu–Malina et al. reported an essential role for ADAR1 in B cells [126]. B-cell-specific ADAR1 deficiency induced by the CD19-cre transgene in mice severely inhibits immature and mature recirculating B cells in the bone marrow (Fig. 3). In agreement with this, peripheral blood and splenic B cells were also reduced in the mutant mice. Importantly, ADAR1-deficient B cells isolated from bone marrow showed upregulated ISG expression and enhanced apoptosis. Of note, Pestal et al. reported that Adar1 p150–MAVS double KO (dKO) mice showed a dramatic reduction in mature B cells, indicating that ADAR1 p150 regulates B-cell homeostasis in a MAVS-independent manner [93]. In contrast, another group reported that both Adar1 KO and Adar1 E861A KI mice on an Ifih1 (encoding MDA5) KO background at the day of birth exhibited a normal proportion of splenic B cells [127], suggesting that aberrantly activated MDA5-dependent signaling, which is caused by ADAR1 deficiency, results in immature B-cell differentiation at this stage. Given that nearly half of Adar1 p150–MAVS dKO mice die around 15–21 days postnatally and that 21-day-old surviving mutant mice affected with several developmental defects were used, the observed reduction in splenic B cells may be derived from an adaptation for survival [93].
Another aspect of RNA editing in adaptive immunity has been reported by Danan–Gotthold et al. [128]. They found abundant RNA-editing events, including recoding in medullary thymic epithelial cells (mTECs), comparable to those of the brain, an organ considered to undergo higher RNA-editing events in the body [129]. During negative selection in the thymus, mTECs present a broad spectrum of self-antigens; autoreactive thymocytes expressing TCRs that strongly react with these cells are eliminated by apoptosis [123]. Therefore, such recoding events are probably important for the elimination of autoreactive thymocytes by the recognition of edited self-antigens, which may be required to prevent their recognition in peripheral tissues. In fact, the same group reported that RNA-editing events, including recoding, were elevated in patients with SLE [130]. Such observations suggest that recoded proteins through RNA editing are processed and loaded on MHCs as recoded self-peptides, which may then be recognized as neo–self-antigens to trigger subsequent autoimmune responses.
A-to-I RNA editing in autoimmune diseases
ADAR1 mutations cause AGS [121] (Fig. 1), a rare autosomal recessive encephalopathy that is characterized by basal ganglia calcification and white matter abnormalities [124]. Intriguingly, other genes found in patients with AGS, such as TREX1 [131], RNASEH2A, RNASEH2B, RNASEH2C [132], SMAHD1 [133], and IFIH1 [102], are all involved in nucleic acid metabolism and signaling [134]. Because patients with AGS show upregulated type I IFN activity and an increased expression of ISGs in the absence of infections, this suggests that a type I IFN signature is likely triggered by impaired metabolism or the sensing of host nucleic acids [9, 135]. ADAR1 mutations in patients with AGS are frequently located in the catalytic domain and decrease the RNA-editing activity of ADAR1 p150, more so than that of ADAR1 p110 [92]. This indicates that the reduced RNA-editing activity of ADAR1 p150 is probably a cause of AGS pathogenesis. In this regard, Adar1 p150-specific KO and Adar1 E861A KI mice show embryonic lethality with a type I IFN signature resembling AGS symptoms [91, 94]. Of note, this lethality was rescued by concurrent deletion of MDA5 [91, 93]. Furthermore, considering that mutations in IFIH1 also cause AGS via the aberrant activation of MDA5 [19, 102, 103, 105], the pathogenesis of AGS caused by ADAR1 mutations is most likely mediated by an activated MDA5-sensing pathway.
In addition, it is worth noting that a P193A mutation, located in the N-terminal Z-DNA/RNA-binding domain of ADAR1 p150, was sometimes observed in patients with AGS [121] (Fig. 1). Z-DNA/RNA form a left-handed double helix in contrast to general right-handed B-DNA/RNA; proline at position 193 is required to interact with Z-DNA and Z-RNA [136]. Intriguingly, dsRNA with Z-RNA is more efficiently edited by ADAR1 p150 than dsRNA without Z-RNA [137]. Therefore, it is worth investigating how Z-RNA modulates RNA-editing activity. Another problem is that AGS pathogenesis has not been fully investigated because of the embryonic lethality of Adar1 KO and Adar1 E861A KI mice that completely lack editing activity [87, 88, 91]. Given that AGS mutations reduce but still retain some ADAR1 editing activity [92] and AGS symptoms appear after birth, KI mice harboring the same Adar1 mutation that is found in patients with AGS may be viable and reflect AGS symptoms more precisely.
In comparison, mutations in the ADAR1 gene are also found in patients with dyschromatosis symmetrica hereditaria (DSH), a pigmentary genodermatosis characterized by hyper- and hypo-pigmented skin lesions [138]. Because several mutations found in patients with DSH are located upstream of the start codon of ADAR1 p110, it is thought that ADAR1 p150 is responsible for pathogenesis [139]. In contrast to AGS mutations, which are generally biallelic except for a G1007R substitution [121], patients with DSH have heterozygous mutations with symptoms obvious only in the skin and that are not fatal, unlike AGS [138]. Importantly, the monoallelic G1007R mutation was identified in DSH patients with neurological symptoms [140], indicating that a mechanism of DSH pathogenesis is at least partially shared with AGS. However, it remains unknown why heterozygotes of Adar1 KO or E861A KI mice do not exhibit skin abnormalities resembling those of DSH, suggesting that further investigation is required.
Dysregulation of RNA editing has also been reported in other autoimmune diseases. ADAR1 p150, but not the p110 isoform, was upregulated in synovium and PBMCs isolated from patients with rheumatoid arthritis (RA) [141]. Accordingly, RNA editing of the 3′UTR of cathepsin S transcripts is increased, and is ameliorated together with decreased ADAR1 p150 expression by anti-rheumatic treatment depending on the clinical response. Stellos et al. previously showed that ADAR1-mediated RNA-editing stabilized cathepsin S transcripts through the recruitment of HuR, an RNA-binding protein [83]. Cathepsin S, a lysosomal cysteine protease, was indispensable for antigen presentation by MHCs [142], autoantibody production [143], and the development of collagen-induced arthritis in a mouse model of RA [144]. Therefore, the stabilization of cathepsin S transcripts by increased ADAR1 p150 expression may contribute to RA pathogenesis.
The upregulation of ADAR1 p150 expression has also been shown in T cells isolated from patients with SLE, which resulted in the increased RNA editing of an α regulatory subunit of type 1 protein kinase A [145]. This may have led to impaired activity of this protein as found in most patients with SLE [146]. The same group further observed changes in RNA-editing efficiency in known and novel RNA-editing sites of ADAR2 transcripts [147]. Consistently, a comprehensive analysis by Rhoth et al. revealed an increase of recoding events in patients with SLE [130], suggesting that recoded self-peptides presented by MHCs potentially behave as neo–self-antigens and contribute to SLE pathogenesis.
A-to-I RNA editing in cancer
Large RNA sequencing data sets obtained from The Cancer Genome Atlas (TCGA) revealed that RNA-editing events and ADAR1 expression were upregulated in most cancers and inversely correlated with patient survival [148, 149]. This upregulation of ADAR1 expression is in response to the increased copy number of chromosome 1q, which contains the ADAR1 gene locus, and a response to type I IFN produced from the chronic inflammatory environment of cancers [150]. However, when we focus on specific RNA-editing sites, the editing efficiency is perturbed, sometimes being upregulated in some sites but downregulated in others (Table 1). These alterations are especially important for RNA recoding events, given that each recoding event alters, in a positive or negative manner, the function of a protein thus regulating cancer progression. For instance, nearly 100% of the RNA editing of GRIA2 transcripts, which leads to changing glutamine (Q) at position 607 of GluA2 to arginine (R), occurs in the brain to regulate Ca2+ permeability of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor [59, 151]. The impairment of such RNA editing was not only found in neurodegenerative diseases such as amyotrophic lateral sclerosis, Huntington’s disease, and Alzheimer’s disease [152-155], but also in glioblastoma multiforme [156]. Overexpression of unedited GluA2 (Q) promotes migration and proliferation of glioblastoma cells [157]. Dysregulated RNA editing of GRIA2 transcripts may be partially attributed to the increased expression of inactive ADAR3, which inhibits the catalytic activity of ADAR2 [48]. In contrast, RNA editing–mediated asparagine (N) to serine (S) substitution at position 136 of Ras Homolog Family Member Q (RHOQ), a member of the Rho family of small GTP-binding proteins that regulates actin-based structures, is increased in patients with colorectal cancer [58, 158]. Such RNA-editing changes the activity of RHOQ and reorganization of the actin cytoskeleton, and promotes an invasive potential. In addition, an amino acid substitution from S to glycine (G) at the position 367 residue of antizyme inhibitor 1 (AZIN1) was increased in hepatocellular carcinoma [57]. This recoding changes the conformation of AZIN1 and its localization from the cytoplasm to the nucleus, and yields gain-of-function phenotypes. Although it is believed that ADAR2 preferentially targets editing sites in coding regions, it is notable that the RNA-editing level of AZIN1 is highly correlated with ADAR1 expression, but not that of ADAR2 [57]. An increase in AZIN1 S/G substitution is also observed in patients with esophageal squamous cell carcinoma and colorectal cancer [159, 160].
Table 1.
RNA-editing-mediated functional alterations found in various cancers
| Gene | Protein | Alteration of amino acid residuea | Function of RNA editing in cancer | RNA-editing level and cancer types | Responsible ADARs | References |
|---|---|---|---|---|---|---|
| AZIN1 | AZIN1 | S367G | Promoting tumor initiation and development | Increase in hepatocellular carcinoma, esophageal squamous cell carcinoma and colorectal cancer | ADAR1 | [57, 159, 160] |
| COPA | COPA | I164V | Not determined | Decrease in hepatocellular carcinoma | ADAR2 | [161] |
| FLNB | Filamin B | M2269V | Not determined | Increase in hepatocellular carcinoma | ADAR1 and ADAR2 | [161] |
| GABRA3 | GABAA receptor subunit α-3 | I342M | Inhibiting cancer metastasis by suppressing AKT pathways | Increase in non-invasive breast cancer | ADAR1 | [162] |
| GRIA2 | AMPA receptor GluA2 subunit | Q607R | Inhibiting migration and proliferation by blocking Ca2+-permeability | Decrease in glioblastoma | ADAR2 | [48, 156, 157] |
| RHOQ | RHOQ | N136S | Promoting an invasive potential by increasing RHOQ protein activity | Increase in colorectal cancer | Not determined | [58] |
| miR-21 and miR-222/221 | - | - | Inhibiting migration and proliferation by preventing microRNA maturation | Increase in glioma cells | ADAR2 | [166] |
| miR-367a* | - | - | Inhibiting invasive properties by altering target transcript | Decrease in glioblastoma multiforme | ADAR2 | [79] |
aAn amino acid substitution due to RNA editing and the position of the residue are shown
The overexpression of ADAR1 and downregulation of ADAR2 can predict a poor clinical outcome for patients with hepatocellular carcinoma [161]. This imbalanced gene expression reflects changes in gene-specific recoding events: an increase in M2269V of FLNB transcripts that encodes filamin B, and a decrease in I164V of coatomer protein complex, subunit α (COPA) transcripts [161]. The other recoding site involved in cancer pathogenesis is type A gamma-aminobutyric acid (GABAA) receptor subunit α-3 (GABRA3), which is edited at a I342M site with nearly 100% efficiency in the adult brain [62]. Intriguingly, such RNA editing of GABRA3 transcripts is detected in non-invasive, but not invasive breast cancers [162]. In this regard, the unedited GABAA receptor shows increased expression on the cell surface and activates AKT pathways, contributing to breast cancer metastasis. Finally, another aspect of recoding function has been reported by Zhang et al. [163]. Using mass spectrometry, they identified several edited peptides as epitopes loaded on the human leukocyte antigen that potentially activate CD8+ T cells. They also showed that edited, but not unedited, cyclin I peptide triggers a cytotoxic response in melanoma cells by CD8+ T cells specific for the edited epitope.
In comparison, RNA-editing events in micro(mi)RNAs were found to be globally downregulated in human cancers and correlated with a poor prognosis in general, given that RNA editing affects miRNA expression and target recognition [80, 164, 165]. For instance, RNA editing of miR-367a* was significantly reduced in human gliomas [79]. Intriguingly, unedited miR-367a* plays a role in glioma cells, which acquire invasive properties, by targeting the tumor suppressor gene, PAP2A, whereas the oncogene, AMFR, a target of edited miR-367a*, failed to be silenced. In contrast, ADAR2-mediated RNA editing inhibits glioma cell proliferation and migration by preventing the maturation of oncogenic miR-21 and miR-222/221 during cleavage steps mediated by DROSHA and DICER [166].
In contrast to the role of ADAR1- and ADAR2-mediated RNA editing at specific sites affecting recoding and miRNA biogenesis, in cancer pathogenesis, the upregulated expression of ADAR1 in most cancers increases RNA-editing frequency in retrotransposon-derived repetitive elements in non-coding regions. This may enhance immune tolerance by preventing activation of dsRNA-sensing pathways associated with MDA5, PKR, and RNase L. Indeed, DNA methyltransferase (DNMT) inhibitors ameliorate hematological and epithelial tumor cells [167]. This clinical effect may be explained by the molecular mechanism in which the hypermethylated promoters of tumor suppressor genes are re-activated by their DNA demethylating function [168, 169]. However, the presence of hypomethylated promoters of tumor suppressor genes observed in patients after DNMT inhibitor treatment is not consistent with the clinical response [170], thus suggesting the involvement of an unknown mechanism in anti-tumor function. Intriguingly, it has been demonstrated that treatment with DNMT inhibitors leads to hypomethylation and subsequent production of self-dsRNAs from ERVs, SINEs, and other repetitive elements [20, 22, 171]. This triggers activation of MDA5, RNase L, and other dsRNA-sensing pathways, thereby contributing to immunotherapy against cancer. Based on these studies, it was expected that ADAR1 depletion may also decrease immune tolerance in cancer cells by activating dsRNA-sensing pathways.
Liu et al. recently reported the existence of ISG signature–positive tumors even without the infiltration of type I IFN-producing immune cells, indicating that they acquire the ability to produce type I IFN [172]. Intriguingly, ISG signature-positive cancer cells are sensitive to ADAR1 depletion. This is in accordance with findings from another group that type I IFN production and lethality induced by ADAR1 depletion in cancer cells could be predicted by the abundance of ISG products, such as MDA5 and PKR [173]. They showed that an increase in type I IFN production was ameliorated by the concurrent deletion of MDA5 or MAVS, whereas the viability of cancer cells was not restored (Fig. 4). In contrast, concurrent deletion of PKR rescued the lethality of cancer cells induced by ADAR1 depletion [173], indicating the different role of MDA5 and PKR in cancer cells. Intriguingly, the overexpression of ADAR1 E861A p150, but not wild-type p110, partially prevented this lethality [173], which is in accordance with a previous finding that the suppression of PKR activation by ADAR1 required its dsRNA-binding and catalytic activities [12]. Of note, Liu et al. showed that an ISG signature was established by the activation of STING, a cytosolic sensor for DNA, and was followed by IFN production, providing a novel cross-talk between DNA- and RNA-sensing pathways [172]. Li et al. further reported that RNase L was activated by ADAR1 deficiency, and induced the death of the human lung adenocarcinoma A549 cell line even in the presence of MDA5 (Fig. 4), suggesting that the OAS–RNase L system is likely the primary pathway activated by ADAR1 depletion [110]. Accordingly, ADAR1 depletion increased DNMT inhibitor-induced cytotoxicity in A549 cells via an OAS–RNase L signaling pathway [171]. Further investigation is required whether ADAR1 depletion exerts the same effect on other cancer cells via activation of RNase L.
Fig. 4.

Depletion of ADAR1 induces vulnerability in cancer cells. RNA-editing efficiency and the expression of adenosine deaminase acting on RNA 1 (ADAR1) are generally upregulated in most cancers. Loss of RNA editing followed by ADAR1 depletion activates multiple double-stranded (ds)RNA-sensing pathways mediated by melanoma differentiation-associated protein 5 (MDA5), protein kinase R (PKR), and oligoadenylate synthetase (OAS), which produces 2′,5′-oligoadenylate (2-5A) resulting in activation of RNase L. The activation of these pathways leads to type I interferon (IFN) production and translational arrest, which make cancer cells vulnerable. In addition, ADAR1 depletion causes cancer cells to be more sensitive to cancer immunotherapy involving, for instance, anti–programmed cell death 1 (PD-1) antibodies
Ishizuka et al. recently reported that the loss of ADAR1 in cancer cells sensitized these to immunotherapy, overcoming resistance to immune checkpoint blockade [174] (Fig. 4). They showed that tumor sizes of implanted ADAR1-deficient B16 melanoma cells were smaller and more sensitive to anti–PD-1 antibodies. Single-cell RNA sequencing analysis revealed an increase in CD8+ T cells and a decrease in M2 macrophages and myeloid-derived suppressor cells, which have a pro-tumor phenotype, in an ADAR1-deficient tumor microenvironment. Moreover, the vulnerability of ADAR1-deficient B16 melanoma cells was canceled by the concurrent deletion of both MDA5 and PKR, but not either alone, suggesting that either PKR or MDA5 is sufficient to sensitize ADAR1-deficient tumor cells to immunotherapy. Importantly, it was shown that the sensitive phenotype of ADAR1-deficient tumor cells to immunotherapy was still observed with the concurrent deletion of beta 2-microglobin, which disrupted MHC-I expression and thus prevented CD8+ T-cell attack. This overcoming of tumor resistance is accompanied by a significant increase in non-MHC-I restricted cytotoxic cells. Taken together, ADAR1 depletion decreases immune tolerance in cancer cells, which is beneficial for cancer therapy.
Concluding remarks
In this review, we summarized RNA-editing function in the immune system and its implication for autoimmune diseases and cancer. The classical functions of RNA editing, which can modulate amino acid sequences, splicing patterns, miRNA target specificity, mRNA stability, and circular RNA biogenesis, have been well investigated. However, recent studies have uncovered the novel role of RNA editing in the immune system in inhibiting the activation of MDA5 and PKR, in addition to the OAS–RNase L system, by preventing their recognition of endogenous dsRNAs formed by retrotransposon-derived repetitive elements. Although the mechanisms underlying the prevention of MDA5-sensing endogenous dsRNAs by RNA editing remain unresolved, the RNA editing/MDA5 axis is not only indispensable for innate immune cells, but is also required for the development and homeostasis of adaptive immune cells. Furthermore, impairment of the RNA-editing/MDA5 axis is most likely a cause of autoimmune disease, given that Adar1 E861A KI mice show a type I IFN signature resembling AGS symptoms and can be rescued by the concurrent deletion of MDA5. Collectively, we expect to determine the critical editing substrates essential for suppressing MDA5 activation as the next step to establishing a strategy for AGS treatment. In contrast, ADAR1 also inhibits the activation of another dsRNA sensor, PKR, through its RNA editing and binding functions. Although several studies reported that PKR activation by a loss of ADAR1 sensitized cancer cells to immunotherapy, this appears to be limited in humans in which dependence may come from a different proportion of SINE repetitive elements between the human and mouse genome [175]. The mechanism of how ADAR1 prevents PKR activation and the critical editing substrates required for preventing PKR activation also requires further investigation. In addition, although recent studies showed that the ADAR1/PKR axis is critical for tumor vulnerability [172-174], it is worth investigating its role in AGS pathogenesis.
Acknowledgements
This work was supported by Grants-in-Aid (KAKENHI; 19K22580 and 19H04210 to Y. K., and 18K15186 and 15K19126 to T. N.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, by grants from the Takeda Science Foundation (to Y. K. and T. N.), the Osaka Nanbyou Zaidan, The Naito Foundation, the Astellas Foundation for Research on Metabolic Disorders and The Mochida Memorial Foundation for Medical and Pharmaceutical Research (to T. N.), and by a Novartis Research Grant (to T. N.).
Abbreviations
- A
Adenosine
- ADAR
Adenosine deaminase acting on RNA
- AGS
Aicardi-Goutières syndrome
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- APC
Antigen-presenting cell
- AZIN1
Antizyme inhibitor 1
- COPA
Coatomer protein complex, subunit alpha
- dsRNA
Double-stranded RNA
- DC
Dendritic cell
- DNMT
DNA methyltransferase
- DSH
Dyschromatosis symmetrica hereditaria
- eIF2α
Eukaryotic initiation factor 2 alpha
- EIF2AK2
Eukaryotic translation initiation factor 2 alpha kinase 2
- ERV
Endogenous retroviruse
- G
Glycine
- GABAA
Type A gamma-aminobutyric acid
- GABRA3
GABAA receptor subunit α-3
- I
Inosine
- IFN
Interferon
- ISG
IFN-stimulated gene
- KI
Knock-in
- KO
Knockout
- LINE
Long interspersed element
- LTR
Long terminal repeat
- MAVS
Mitochondrial anti-viral-signaling protein
- MDA5
Melanoma differentiation-associated protein 5
- MHC
Major histocompatibility complex
- miRNA
MicroRNA
- mTEC
Medullary thymic epithelial cell
- m7G
5′Triphosphate-linked methylguanosine
- N
Asparagine
- N1
5′-Terminal nucleotide
- OAS
Oligoadenylate synthetase
- ORF
Open-reading frame
- PAS
Periodic acid-Schiff
- PBMC
Peripheral blood mononuclear cell
- PD-L1
Programmed cell-death ligand-1
- PD-1
Programmed cell death 1
- PKR
Protein kinase R
- Pol
Polymerase
- Q
Glutamine
- R
Arginine
- RA
Rheumatoid arthritis
- RHOQ
Ras Homolog Family Member Q
- RIG-I
Retinoic acid-inducible gene I
- RLR
RIG-I-like receptor
- PRR
Pattern recognition receptor
- S
Serine
- SINE
Short interspersed element
- SLE
Systemic lupus erythematosus
- TCGA
The Cancer Genome Atlas
- TCR
T-cell receptor
- UTR
Untranslated region
- U
Uridine
- 2′OMe
2′-O-Methylation
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S33–40. doi: 10.1016/j.jaci.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol. 2012 doi: 10.1101/cshperspect.a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28(1):18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19(7):813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 8.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J, International Human Genome Sequencing C Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 9.Volkman HE, Stetson DB. The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol. 2014;15(5):415–422. doi: 10.1038/ni.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedli M, Trono D. The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol. 2015;31:429–451. doi: 10.1146/annurev-cellbio-100814-125514. [DOI] [PubMed] [Google Scholar]
- 12.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, Rice CM. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172(4):811–824. doi: 10.1016/j.cell.2017.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conti A, Carnevali D, Bollati V, Fustinoni S, Pellegrini M, Dieci G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015;43(2):817–835. doi: 10.1093/nar/gku1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14572–14579. doi: 10.1073/pnas.0404838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19(6):727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 18.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, Hur S. Breaching self-tolerance to alu duplex RNA underlies MDA5-mediated inflammation. Cell. 2018;172(4):797–810. doi: 10.1016/j.cell.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Budhu S, Slamon DJ, Wolchok JD, Pardoll DM, Beckmann MW, Zahnow CA, Merghoub T, Chan TA, Baylin SB, Strick R. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, Akira S, Weichselbaum RR, Khodarev NN. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget. 2016;7(18):26496–26515. doi: 10.18632/oncotarget.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, O'Brien C, De Carvalho DD. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, Patel SS, Marcotrigiano J. Structural basis for m7G recognition and 2'-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci USA. 2016;113(3):596–601. doi: 10.1073/pnas.1515152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lassig C, Hopfner KP. Discrimination of cytosolic self and non-self RNA by RIG-I-like receptors. J Biol Chem. 2017;292(22):9000–9009. doi: 10.1074/jbc.R117.788398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24(5):559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- 26.Reich DP, Bass BL. Mapping the dsRNA world. Cold Spring Harb Perspect Biol. 2019 doi: 10.1101/cshperspect.a035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuberth-Wagner C, Ludwig J, Bruder AK, Herzner AM, Zillinger T, Goldeck M, Schmidt T, Schmid-Burgk JL, Kerber R, Wolter S, Stumpel JP, Roth A, Bartok E, Drosten C, Coch C, Hornung V, Barchet W, Kummerer BM, Hartmann G, Schlee M. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1–2'O-methylated self RNA. Immunity. 2015;43(1):41–51. doi: 10.1016/j.immuni.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walkley CR, Li JB. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017;18(1):205. doi: 10.1186/s13059-017-1347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. Ribose 2'-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12(2):137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samuel CE. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J Biol Chem. 2019;294(5):1710–1720. doi: 10.1074/jbc.TM118.004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim U, Garner TL, Sanford T, Speicher D, Murray JM, Nishikura K. Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J Biol Chem. 1994;269(18):13480–13489. [PubMed] [Google Scholar]
- 34.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci USA. 1994;91(24):11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Connell MA, Keller W. Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc Natl Acad Sci USA. 1994;91(22):10596–10600. doi: 10.1073/pnas.91.22.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol. 1995;15(3):1389–1397. doi: 10.1128/mcb.15.3.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber A, O'Connell MA, Keller W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA. 1997;3(5):453–463. [PMC free article] [PubMed] [Google Scholar]
- 38.Lai F, Chen CX, Carter KC, Nishikura K. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol. 1997;17(5):2413–2424. doi: 10.1128/mcb.17.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379(6564):460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 40.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6(5):755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271(50):31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 42.Desterro JM, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116(Pt 9):1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 43.Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol Biol Cell. 2001;12(7):1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15(10):5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol Cell Biol. 2001;21(22):7862–7871. doi: 10.1128/MCB.21.22.7862-7871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maas S, Gommans WM. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009;37(17):5822–5829. doi: 10.1093/nar/gkp599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci USA. 2003;100(24):14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oakes E, Anderson A, Cohen-Gadol A, Hundley HA. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B Pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem. 2017;292(10):4326–4335. doi: 10.1074/jbc.M117.779868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, Gupte A, Keegan LP, George CX, Ramu A, Huang N, Pollina EA, Leeman DS, Rustighi A, Goh YPS, Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, Biospecimen Collection Source Site N, Biospecimen Collection Source Site R, Biospecimen Core Resource V, Brain Bank Repository-University of Miami Brain Endowment B, Leidos Biomedical-Project M, Study E, Genome Browser Data I, Visualization EBI, Genome Browser Data I, Visualization-Ucsc Genomics Institute UoCSC, Chawla A, Del Sal G, Peltz G, Brunet A, Conrad DF, Samuel CE, O'Connell MA, Walkley CR, Nishikura K, Li JB Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550(7675):249–254. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mladenova D, Barry G, Konen LM, Pineda SS, Guennewig B, Avesson L, Zinn R, Schonrock N, Bitar M, Jonkhout N, Crumlish L, Kaczorowski DC, Gong A, Pinese M, Franco GR, Walkley CR, Vissel B, Mattick JS. Adar3 Is Involved in Learning and Memory in Mice. Front Neurosci. 2018;12:243. doi: 10.3389/fnins.2018.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakahama T, Kato Y, Kim JI, Vongpipatana T, Suzuki Y, Walkley CR, Kawahara Y. ADAR1-mediated RNA editing is required for thymic self-tolerance and inhibition of autoimmunity. EMBO Rep. 2018 doi: 10.15252/embr.201846303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S. Synthetic polynucleotides and the amino acid code. V. Proc Natl Acad Sci USA. 1962;48:613–616. doi: 10.1073/pnas.48.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Licht K, Hartl M, Amman F, Anrather D, Janisiw MP, Jantsch MF. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 2019;47(1):3–14. doi: 10.1093/nar/gky1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisenberg E, Levanon EY. A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet. 2018;19(8):473–490. doi: 10.1038/s41576-018-0006-1. [DOI] [PubMed] [Google Scholar]
- 55.Porath HT, Knisbacher BA, Eisenberg E, Levanon EY. Massive A-to-I RNA editing is common across the Metazoa and correlates with dsRNA abundance. Genome Biol. 2017;18(1):185. doi: 10.1186/s13059-017-1315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387(6630):303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, Qi L, Li Y, Zhang N, Tong AH, Kwong DL, Man K, Lo CM, Lok S, Tenen DG, Guan XY. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19(2):209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han SW, Kim HP, Shin JY, Jeong EG, Lee WC, Kim KY, Park SY, Lee DW, Won JK, Jeong SY, Park KJ, Park JG, Kang GH, Seo JS, Kim JI, Kim TY. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J Exp Med. 2014;211(4):613–621. doi: 10.1084/jem.20132209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75(7):1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 60.Hoopengardner B, Bhalla T, Staber C, Reenan R. Nervous system targets of RNA editing identified by comparative genomics. Science. 2003;301(5634):832–836. doi: 10.1126/science.1086763. [DOI] [PubMed] [Google Scholar]
- 61.Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Kohr G, Higuchi M, Fatemi-Shariatpanahi H, Harden B, Yue DT, Soong TW. RNA editing of the IQ domain in Ca(v)13 channels modulates their Ca(2)(+)-dependent inactivation. Neuron. 2012;73(2):304–316. doi: 10.1016/j.neuron.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohlson J, Pedersen JS, Haussler D, Ohman M. Editing modifies the GABA(A) receptor subunit alpha3. RNA. 2007;13(5):698–703. doi: 10.1261/rna.349107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2(1):57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 64.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 65.Kawahara Y, Grimberg A, Teegarden S, Mombereau C, Liu S, Bale TL, Blendy JA, Nishikura K. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci. 2008;28(48):12834–12844. doi: 10.1523/JNEUROSCI.3896-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyake K, Ohta T, Nakayama H, Doe N, Terao Y, Oiki E, Nagatomo I, Yamashita Y, Abe T, Nishikura K, Kumanogoh A, Hashimoto K, Kawahara Y. CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep. 2016;17(8):2004–2014. doi: 10.1016/j.celrep.2016.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, Levanon EY. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24(3):365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, Guo J, Dong Z, Liang Y, Bao L, Wang J. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30(3):253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 69.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22(8):1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 70.Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep. 2015;5:14941. doi: 10.1038/srep14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods. 2013;10(2):128–132. doi: 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods. 2012;9(6):579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA. 2006;12(10):1802–1809. doi: 10.1261/rna.165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beghini A, Ripamonti CB, Peterlongo P, Roversi G, Cairoli R, Morra E, Larizza L. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet. 2000;9(15):2297–2304. doi: 10.1093/oxfordjournals.hmg.a018921. [DOI] [PubMed] [Google Scholar]
- 75.Flomen R, Knight J, Sham P, Kerwin R, Makoff A. Evidence that RNA editing modulates splice site selection in the 5-HT2C receptor gene. Nucleic Acids Res. 2004;32(7):2113–2122. doi: 10.1093/nar/gkh536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laurencikiene J, Kallman AM, Fong N, Bentley DL, Ohman M. RNA editing and alternative splicing: the importance of co-transcriptional coordination. EMBO Rep. 2006;7(3):303–307. doi: 10.1038/sj.embor.7400621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lev-Maor G, Sorek R, Levanon EY, Paz N, Eisenberg E, Ast G. RNA-editing-mediated exon evolution. Genome Biol. 2007;8(2):R29. doi: 10.1186/gb-2007-8-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399(6731):75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 79.Choudhury Y, Tay FC, Lam DH, Sandanaraj E, Tang C, Ang BT, Wang S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;122(11):4059–4076. doi: 10.1172/JCI62925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, Mosqueda M, Wen YY, Eterovic AK, Sood AK, Hwu P, Gershenwald JE, Robertson AG, Calin GA, Markel G, Fidler IJ, Bar-Eli M. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;17(3):311–321. doi: 10.1038/ncb3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, Davidson BL. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet. 2009;18(24):4801–4807. doi: 10.1093/hmg/ddp443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stellos K, Gatsiou A, Stamatelopoulos K, Perisic Matic L, John D, Lunella FF, Jae N, Rossbach O, Amrhein C, Sigala F, Boon RA, Furtig B, Manavski Y, You X, Uchida S, Keller T, Boeckel JN, Franco-Cereceda A, Maegdefessel L, Chen W, Schwalbe H, Bindereif A, Eriksson P, Hedin U, Zeiher AM, Dimmeler S. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat Med. 2016;22(10):1140–1150. doi: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 84.Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, Rajewsky N. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10(2):170–177. doi: 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 85.Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, Herzog M, Schreyer L, Papavasileiou P, Ivanov A, Ohman M, Refojo D, Kadener S, Rajewsky N. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 86.Shi L, Yan P, Liang Y, Sun Y, Shen J, Zhou S, Lin H, Liang X, Cai X. Circular RNA expression is suppressed by androgen receptor (AR)-regulated adenosine deaminase that acts on RNA (ADAR1) in human hepatocellular carcinoma. Cell Death Dis. 2017;8(11):e3171. doi: 10.1038/cddis.2017.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279(6):4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- 89.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279(6):4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 91.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349(6252):1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O'Connell MA. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9(4):1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015;43(5):933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci USA. 2011;108(1):331–336. doi: 10.1073/pnas.1017241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.George CX, Ramaswami G, Li JB, Samuel CE. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J Biol Chem. 2016;291(12):6158–6168. doi: 10.1074/jbc.M115.709014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Solomon O, Di Segni A, Cesarkas K, Porath HT, Marcu-Malina V, Mizrahi O, Stern-Ginossar N, Kol N, Farage-Barhom S, Glick-Saar E, Lerenthal Y, Levanon EY, Amariglio N, Unger R, Goldstein I, Eyal E, Rechavi G. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat Commun. 2017;8(1):1440. doi: 10.1038/s41467-017-01458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5'-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 98.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 99.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis e Sousa C. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. 2014;514(7522):372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 101.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 102.Rice GI, Del Toro DY, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR, Lebon P, Li C, Livingston JH, Lourenco CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O'Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46(5):503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, Yasumi T, Kato H, Shirai T, Ohara O, Fujita T, Heike T. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet. 2014;95(1):121–125. doi: 10.1016/j.ajhg.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Eyck L, De Somer L, Pombal D, Bornschein S, Frans G, Humblet-Baron S, Moens L, de Zegher F, Bossuyt X, Wouters C, Liston A. Brief report: IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol. 2015;67(6):1592–1597. doi: 10.1002/art.39110. [DOI] [PubMed] [Google Scholar]
- 105.Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, Minowa O, Yoshida A, Deguchi K, Sato H, Ito S, Shiroishi T, Takeyasu K, Noda T, Fujita T. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40(2):199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 106.Vitali P, Scadden AD. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17(9):1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA. 2008;14(6):1110–1118. doi: 10.1261/rna.923308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80(9):4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, Weiss SR. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife. 2017 doi: 10.7554/eLife.25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA. Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 2009;6(3):305–315. doi: 10.4161/rna.6.3.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res. 2011;31(1):49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brennan-Laun SE, Ezelle HJ, Li XL, Hassel BA. RNase-L control of cellular mRNAs: roles in biologic functions and mechanisms of substrate targeting. J Interferon Cytokine Res. 2014;34(4):275–288. doi: 10.1089/jir.2013.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. Interferon action and apoptosis are defective in mice devoid of 2',5'-oligoadenylate-dependent RNase L. EMBO J. 1997;16(21):6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Donovan J, Rath S, Kolet-Mandrikov D, Korennykh A. Rapid RNase L-driven arrest of protein synthesis in the dsRNA response without degradation of translation machinery. RNA. 2017;23(11):1660–1671. doi: 10.1261/rna.062000.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baal N, Cunningham S, Obermann HL, Thomas J, Lippitsch A, Dietert K, Gruber AD, Kaufmann A, Michel G, Nist A, Stiewe T, Rupp O, Goesmann A, Zukunft S, Fleming I, Bein G, Lohmeyer J, Bauer S, Hackstein H. ADAR1 is required for dendritic cell subset homeostasis and alveolar macrophage function. J Immunol. 2019;202(4):1099–1111. doi: 10.4049/jimmunol.1800269. [DOI] [PubMed] [Google Scholar]
- 117.Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, Brooks AG, Heath WR. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10(5):488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 118.Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, Murphy KM, Garcia-Sastre A, Merad M. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest. 2012;122(11):4037–4047. doi: 10.1172/JCI60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ben-Dov I, Segel MJ. Autoimmune pulmonary alveolar proteinosis: clinical course and diagnostic criteria. Autoimmun Rev. 2014;13(4–5):513–517. doi: 10.1016/j.autrev.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 120.Pujantell M, Riveira-Munoz E, Badia R, Castellvi M, Garcia-Vidal E, Sirera G, Puig T, Ramirez C, Clotet B, Este JA, Ballana E. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci Rep. 2017;7(1):13339. doi: 10.1038/s41598-017-13580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O'Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chung H, Rice CM. T time for ADAR: ADAR1 is required for T cell self-tolerance. EMBO Rep. 2018 doi: 10.15252/embr.201847237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 124.Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-Shalev S, Arellano M, Ariaudo G, Aswani V, Babul-Hirji R, Baildam EM, Bahi-Buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Beresford MW, Bernard G, Bianchi M, Billette de Villemeur T, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-Gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Sierra Corcoles C, Cordeiro NJ, Crichiutti G, Dabydeen L, Dale RC, D'Arrigo S, De Goede CG, De Laet C, De Waele LM, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Fazzi E, Ferrie CD, Figueiredo A, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Kara B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Landrieu P, Lauffer H, Laugel V, La Piana R, Lim MJ, Lin JP, Linnankivi T, Mackay MT, Marom DR, Marques Lourenco C, McKee SA, Moroni I, Morton JE, Moutard ML, Murray K, Nabbout R, Nampoothiri S, Nunez-Enamorado N, Oades PJ, Olivieri I, Ostergaard JR, Perez-Duenas B, Prendiville JS, Ramesh V, Rasmussen M, Regal L, Ricci F, Rio M, Rodriguez D, Roubertie A, Salvatici E, Segers KA, Sinha GP, Soler D, Spiegel R, Stodberg TI, Straussberg R, Swoboda KJ, Suri M, Tacke U, Tan TY, te Water Naude J, Wee Teik K, Thomas MM, Till M, Tonduti D, Valente EM, Van Coster RN, van der Knaap MS, Vassallo G, Vijzelaar R, Vogt J, Wallace GB, Wassmer E, Webb HJ, Whitehouse WP, Whitney RN, Zaki MS, Zuberi SM, Livingston JH, Rozenberg F, Lebon P, Vanderver A, Orcesi S, Rice GI Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A(2):296–312. doi: 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cuadrado E, Booiman T, van Hamme JL, Jansen MH, van Dort KA, Vanderver A, Rice GI, Crow YJ, Kootstra NA, Kuijpers TW. ADAR1 facilitates HIV-1 replication in primary CD4+ T cells. PLoS ONE. 2015;10(12):e0143613. doi: 10.1371/journal.pone.0143613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marcu-Malina V, Goldberg S, Vax E, Amariglio N, Goldstein I, Rechavi G. ADAR1 is vital for B cell lineage development in the mouse bone marrow. Oncotarget. 2016;7(34):54370–54379. doi: 10.18632/oncotarget.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Heraud-Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM, Pang L, Liddicoat BJ, Gupte A, Li JB, Walkley CR. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol. 2017;18(1):166. doi: 10.1186/s13059-017-1301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Danan-Gotthold M, Guyon C, Giraud M, Levanon EY, Abramson J. Extensive RNA editing and splicing increase immune self-representation diversity in medullary thymic epithelial cells. Genome Biol. 2016;17(1):219. doi: 10.1186/s13059-016-1079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paul MS, Bass BL. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998;17(4):1120–1127. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roth SH, Danan-Gotthold M, Ben-Izhak M, Rechavi G, Cohen CJ, Louzoun Y, Levanon EY. Increased RNA editing may provide a source for autoantigens in systemic lupus erythematosus. Cell Rep. 2018;23(1):50–57. doi: 10.1016/j.celrep.2018.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 132.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38(8):910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 133.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41(7):829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 135.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18(R2):R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schade M, Turner CJ, Lowenhaupt K, Rich A, Herbert A. Structure-function analysis of the Z-DNA-binding domain Zalpha of dsRNA adenosine deaminase type I reveals similarity to the (alpha + beta) family of helix-turn-helix proteins. EMBO J. 1999;18(2):470–479. doi: 10.1093/emboj/18.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koeris M, Funke L, Shrestha J, Rich A, Maas S. Modulation of ADAR1 editing activity by Z-RNA in vitro. Nucleic Acids Res. 2005;33(16):5362–5370. doi: 10.1093/nar/gki849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73(3):693–699. doi: 10.1086/378209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, Takama H, Sato K, Ishikawa O, Abe M, Shimizu H, Kawai M, Horikawa T, Yoshida K, Matsumoto K, Terui T, Tsujioka K, Tomita Y. Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J Invest Dermatol. 2005;124(6):1186–1192. doi: 10.1111/j.0022-202X.2005.23732.x. [DOI] [PubMed] [Google Scholar]
- 140.Kondo T, Suzuki T, Ito S, Kono M, Negoro T, Tomita Y. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35(10):662–666. doi: 10.1111/j.1346-8138.2008.00540.x. [DOI] [PubMed] [Google Scholar]
- 141.Vlachogiannis NI, Gatsiou A, Silvestris DA, Stamatelopoulos K, Tektonidou MG, Gallo A, Sfikakis PP, Stellos K. Increased adenosine-to-inosine RNA editing in rheumatoid arthritis. J Autoimmunol. 2019 doi: 10.1016/j.jaut.2019.102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4(4):357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 143.Saegusa K, Ishimaru N, Yanagi K, Arakaki R, Ogawa K, Saito I, Katunuma N, Hayashi Y. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J Clin Invest. 2002;110(3):361–369. doi: 10.1172/JCI14682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, McNeish JD, Eastman SE, Howard ED, Clarke SR, Rosloniec EF, Elliott EA, Rudensky AY. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity. 1999;10(2):207–217. doi: 10.1016/S1074-7613(00)80021-7. [DOI] [PubMed] [Google Scholar]
- 145.Laxminarayana D, Khan IU, Kammer G. Transcript mutations of the alpha regulatory subunit of protein kinase A and up-regulation of the RNA-editing gene transcript in lupus T lymphocytes. Lancet. 2002;360(9336):842–849. doi: 10.1016/s0140-6736(02)09966-x. [DOI] [PubMed] [Google Scholar]
- 146.Kammer GM. High prevalence of T cell type I protein kinase A deficiency in systemic lupus erythematosus. Arthritis Rheum. 1999;42(7):1458–1465. doi: 10.1002/1529-0131(199907)42:7<1458::AID-ANR20>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 147.Laxminarayana D, O'Rourke KS, Maas S, Olorenshaw I. Altered editing in RNA editing adenosine deaminase ADAR2 gene transcripts of systemic lupus erythematosus T lymphocytes. Immunology. 2007;121(3):359–369. doi: 10.1111/j.1365-2567.2007.02582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13(2):267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 149.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HMJ, Eterovic AK, Yuan Y, Li J, Nair N, Minelli R, Tsang YH, Cheung LWT, Jeong KJ, Roszik J, Ju Z, Woodman SE, Lu Y, Scott KL, Li JB, Mills GB, Liang H. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28(4):515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, Larsimont D, Polyak K, Willard-Gallo K, Desmedt C, Piccart M, Abramowicz M, Campbell PJ, Sotiriou C, Detours V. Principles governing A-to-I RNA editing in the breast cancer transcriptome. Cell Rep. 2015;13(2):277–289. doi: 10.1016/j.celrep.2015.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seeburg PH, Hartner J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol. 2003;13(3):279–283. doi: 10.1016/S0959-4388(03)00062-X. [DOI] [PubMed] [Google Scholar]
- 152.Aizawa H, Sawada J, Hideyama T, Yamashita T, Katayama T, Hasebe N, Kimura T, Yahara O, Kwak S. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010;120(1):75–84. doi: 10.1007/s00401-010-0678-x. [DOI] [PubMed] [Google Scholar]
- 153.Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427(6977):801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- 154.Akbarian S, Smith MA, Jones EG. Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res. 1995;699(2):297–304. doi: 10.1016/0006-8993(95)00922-d. [DOI] [PubMed] [Google Scholar]
- 155.Gaisler-Salomon I, Kravitz E, Feiler Y, Safran M, Biegon A, Amariglio N, Rechavi G. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer's disease. Neurobiol Aging. 2014;35(8):1785–1791. doi: 10.1016/j.neurobiolaging.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 156.Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci USA. 2001;98(25):14687–14692. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, Sasaki T, Ozawa S. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8(9):971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 158.Neudauer CL, Joberty G, Tatsis N, Macara IG. Distinct cellular effects and interactions of the Rho-family GTPase TC10. Curr Biol. 1998;8(21):1151–1160. doi: 10.1016/s0960-9822(07)00486-1. [DOI] [PubMed] [Google Scholar]
- 159.Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF, Liu H, Fei J, Li Y, Guan XY, Chen L. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014;74(3):840–851. doi: 10.1158/0008-5472.CAN-13-2545. [DOI] [PubMed] [Google Scholar]
- 160.Shigeyasu K, Okugawa Y, Toden S, Miyoshi J, Toiyama Y, Nagasaka T, Takahashi N, Kusunoki M, Takayama T, Yamada Y, Fujiwara T, Chen L, Goel A. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018 doi: 10.1172/jci.insight.99976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong KJ, Liu M, Song Y, Chow RK, Ng VH, Yuan YF, Tenen DG, Guan XY, Chen L. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63(5):832–843. doi: 10.1136/gutjnl-2012-304037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gumireddy K, Li A, Kossenkov AV, Sakurai M, Yan J, Li Y, Xu H, Wang J, Zhang PJ, Zhang L, Showe LC, Nishikura K, Huang Q. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat Commun. 2016;7:10715. doi: 10.1038/ncomms10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, Tsou CC, Hoffgaard F, Goldfinger V, Schoor O, Talukder A, Forget MA, Haymaker C, Bernatchez C, Han L, Tsang YH, Kong K, Xu X, Scott KL, Singh-Jasuja H, Lizee G, Liang H, Weinschenk T, Mills GB, Hwu P. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun. 2018;9(1):3919. doi: 10.1038/s41467-018-06405-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pinto Y, Buchumenski I, Levanon EY, Eisenberg E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2018;46(1):71–82. doi: 10.1093/nar/gkx1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8(8):763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tomaselli S, Galeano F, Alon S, Raho S, Galardi S, Polito VA, Presutti C, Vincenti S, Eisenberg E, Locatelli F, Gallo A. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015;16:5. doi: 10.1186/s13059-014-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, Brock MV, Stearns V, Feller-Kopman D, Yarmus LB, Lin YC, Welm AL, Issa JP, Minn I, Matsui W, Jang YY, Sharkis SJ, Baylin SB, Zahnow CA. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Navada SC, Steinmann J, Lubbert M, Silverman LR. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124(1):40–46. doi: 10.1172/JCI69739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, Yang X, Liang G, Jones PA. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012;21(5):655–667. doi: 10.1016/j.ccr.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, Licht J, McConnell MJ, Nasrallah C, Kim MK, Zhang W, Sun Y, Murgo A, Espinoza-Delgado I, Oteiza K, Owoeye I, Silverman LR, Gore SD, Carraway HE. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Banerjee S, Gusho E, Gaughan C, Dong B, Gu X, Holvey-Bates E, Talukdar M, Li Y, Weiss SR, Sicheri F, Saunthararajah Y, Stark GR, Silverman RH. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc Natl Acad Sci USA. 2019;116(11):5071–5076. doi: 10.1073/pnas.1815071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, Bullock CP, Jones MD, Kerr G, Li L, Rakiec DP, Schlabach MR, Sovath S, Growney JD, Pagliarini RA, Ruddy DA, MacIsaac KD, Korn JM, McDonald ER., 3rd Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med. 2019;25(1):95–102. doi: 10.1038/s41591-018-0302-5. [DOI] [PubMed] [Google Scholar]
- 173.Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, Cherniack AD, Vazquez F, Tsherniak A, Levanon EY, Hahn WC, Meyerson M. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun. 2018;9(1):5450. doi: 10.1038/s41467-018-07824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, Buchumenski I, Comstock DE, Brown FD, Ayer A, Kohnle IC, Pope HW, Zimmer MD, Sen DR, Lane-Reticker SK, Robitschek EJ, Griffin GK, Collins NB, Long AH, Doench JG, Kozono D, Levanon EY, Haining WN. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565(7737):43–48. doi: 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Deniz O, Frost JM, Branco MR. Regulation of transposable elements by DNA modifications. Nat Rev Genet. 2019;20(7):417–431. doi: 10.1038/s41576-019-0106-6. [DOI] [PubMed] [Google Scholar]