

Abstract
VE-cadherin plays a central role in controlling endothelial barrier function, which is transiently disrupted by proinflammatory cytokines such as tumor necrosis factor (TNFα). Here we show that human endothelial cells compensate VE-cadherin degradation in response to TNFα by inducing VE-cadherin de novo synthesis. This compensation increases adherens junction turnover but maintains surface VE-cadherin levels constant. NF-κB inhibition strongly reduced VE-cadherin expression and provoked endothelial barrier collapse. Bacterial lipopolysaccharide and TNFα upregulated the transcription factor ETS1, in vivo and in vitro, in an NF-κB dependent manner. ETS1 gene silencing specifically reduced VE-cadherin protein expression in response to TNFα and exacerbated TNFα-induced barrier disruption. We propose that TNFα induces not only the expression of genes involved in increasing permeability to small molecules and immune cells, but also a homeostatic transcriptional program in which NF-κB- and ETS1-regulated VE-cadherin expression prevents the irreversible damage of endothelial barriers.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03260-9) contains supplementary material, which is available to authorized users.
Keywords: Endothelial barrier function, Permeability, VE-cadherin, NF-κB, ETS1, LPS, Resolution of inflammation, Adherens junctions, Degradation
Introduction
A major effect of long-term inflammatory stimulation is the progressive and transient disruption of endothelial barrier integrity, which underlies the development of many proinflammatory diseases. Inflammatory cytokines such as TNFα, interferon (IFN)-γ and interleukin (IL)-1β induce actin-dependent morphological remodeling and a transcriptional program to express proteins that increase vascular leakiness to immune cells and small molecules through mechanisms that are not fully understood [1, 2].
Vascular endothelial (VE)-cadherin-dependent adherens junctions (AJs) are the main regulators of endothelial barrier properties in many vascular beds [3, 4]. VE-cadherin is the transmembrane partner of cytoplasmic catenins, which stabilize the junctional complex at the cell border and connect AJs to the underlying actin cytoskeleton. Surface VE-cadherin not only regulates permeability, but also promotes cell survival, controls gene transcription, cytoskeleton remodeling and collective endothelial cell migration [3, 5–7]. This wide range of functions suggests that the expression and localization of VE-cadherin must be finely tuned even in the context in which barrier function is transiently compromised during the inflammatory response. Inflammatory cytokines and other inflammatory mediators activate the src family-dependent tyrosine phosphorylation of the cytoplasmic tail of VE-cadherin, which uncouples p120-catenin from VE-cadherin, causing the disruption of AJs and the subsequent internalization of VE-cadherin [2, 8, 9]. However, the effects of cytokine stimulation on VE-cadherin expression and stability have not yet been clarified. It has been shown that at least TNFα and the bacterial endotoxin lipopolysaccharide (LPS) can induce VE-cadherin shedding or intracellular degradation, although this induction does not necessarily reduce cellular VE-cadherin levels [10]. Increased VE-cadherin degradation has been observed by simultaneous stimulation of vascular endothelial cells with more than two inflammatory cytokines or by a combination of certain cytokines and immune cells [11]. In contrast, several reports indicate that single cytokine stimulation causes barrier dysfunction in many endothelial beds without significantly decreasing VE-cadherin expression levels [8, 9, 12, 13].
Taking TNFα as a paradigm of inflammatory stimulus that progressively disrupts cellular barriers, and combining biochemical surface labeling and high-resolution microscopy, we have comprehensively analyzed the effect of this cytokine on surface VE-cadherin expression and dynamics in primary human endothelial cells. We show that long-term stimulation with TNFα increases endothelial permeability and reduces surface VE-cadherin half-life but not total VE-cadherin levels. TNFα induces the compensatory expression of a VE-cadherin intracellular pool through NF-κB-dependent regulation of the transcription factor ETS1, which compensates VE-cadherin degradation. Indeed, inhibiting NF-κB abrogates VE-cadherin and ETS1 expression and causes the collapse of the endothelial barrier, whereas ETS1 gene silencing specifically regulates VE-cadherin expression, but not other TNFα-regulated genes, and exacerbates TNFα-mediated barrier dysfunction. Thus, our results suggest that endothelial cells implement a transcriptional program, which involves de novo expression of ETS1 and VE-cadherin, to preserve barrier integrity in response to inflammatory cytokines.
Results
De novo VE-cadherin expression compensates for reduction of surface VE-cadherin protein half-life in response to TNFα
TNFα induces a progressive increase of permeability in many cellular barriers [2]. In human umbilical vein endothelial cells (HUVECs), long-term exposure to TNFα reduces TEER from 25 to 40% of initial values (Fig. 1a) [1], remodels the endothelial cytoskeleton (Fig. 1b), alters the crosstalk between VE-cadherin and other cell–cell junction proteins and reduces cell density (Fig. S1A) [1, 13, 14]. This causes VE-cadherin reorganization, decreasing reticular AJs and increasing discontinuous AJs, orthogonal to cell borders and aligned with stress fibers (Fig. 1b) [1, 13]. We set out to quantify the effect of TNFα on surface VE-cadherin half-life by performing pulse and chase experiments of endogenous surface VE-cadherin labeled with sulfo-NHS-biotin. HUVECs were stimulated or not with TNFα for 24 h and their surface proteins were biotinylated for 30 min (Fig. 1c, biotin pulse, time 0 post-biotinylation). Biotin-labeled cells were then incubated at 37 °C and lysed at different times (Fig. 1c, chase). Biotinylated surface proteins were isolated by pull-down assay with NeutrAvidin agarose [15] and endogenous VE-cadherin protein levels in the lysates (Fig. 1c, total) were compared with those in the pull-down fraction (Fig. 1c, surface). As a control, VE-cadherin was not detected in the pull-down fraction from non-biotinylated cells (Fig. 1c). Biotinylated VE-cadherin levels in HUVECs treated with TNFα for 24 h (Fig. 1c) and 8 h (Fig. 1d) decreased faster than in unstimulated HUVECs. From these experiments, we calculated that surface VE-cadherin protein half-life decreased from approximately 6 h in unstimulated cells to 2 h and 3 h in cells previously stimulated with TNFα for 24 h and 8 h, respectively. Treatment with lactacystin or MG132 indicated that proteasome is involved in accelerated biotinylated-VE-cadherin protein decrease in response to TNFα (Fig. S1B). As a complementary approach, we labeled junctional VE-cadherin with a specific antibody that does not alter AJ dynamics [16]. Confocal microscopy analysis showed that VE-cadherin labeling decreased faster in cells pretreated with TNFα for 24 h than in untreated cells (Fig. 1e). Together, pulse and chase and antibody-mediated labeling experiments indicate that TNFα decreases the half-life of surface VE-cadherin located at junctional domains. Dose–response assays showed that TNFα caused a 15% decrease in cell density at 10 ng/ml TNFα, the cytokine concentration used for the pulse-chase experiments (Fig. S1A). This decrease may contribute to surface VE-cadherin instability, but had no effect on PECAM-1 surface levels and stability, suggesting that this concentration of TNFα does not induce a general effect on the half-life of junctional protein complexes (Fig. S1C).
Fig. 1.

TNFα induces endothelial barrier remodeling and decreases surface VE-cadherin half-life. a HUVECs were plated at confluence for 48 h in ECIS arrays pre-coated with fibronectin, then starved for 12 h, stimulated or not with 10 ng/ml TNFα and TEER measured by ECIS. b HUVECs were cultured at confluence for 48 h on coverslips pre-coated with fibronectin, starved for 12 h and then stimulated or not with 10 ng/ml TNFα for 24 h. Cells were fixed and stained to analyze the distribution of VE-cadherin and F-actin. Scale bar, 20 μm. c, d Pulse and chase experiment of surface VE-cadherin. HUVECs were cultured at confluence for at least 48 h on dishes pre-coated with fibronectin, starved for 12 h and then stimulated or not with 10 ng/ml TNFα for 24 h (c) or for 8 h (d). Surface proteins were labeled with sulfo-NHS-biotin (biotin pulse), cells were incubated at 37 oC for the indicated times and lysed (chase). Surface-biotinylated proteins were isolated by pull-down assay with NeutrAvidin-agarose. Surface VE-cadherin levels were detected with a specific antibody from the pull-down fraction and compared with total VE-cadherin levels from the lysate. Immunoblots of ERK are shown as loading controls. Graphs show the mean + SEM from three independent experiments. Discontinuous lines mark surface VE-cadherin half-life for each condition. The t test analyzed differences between surface VE-cadherin levels in unstimulated and TNFα-stimulated cells at the indicated times *p < 0.05; **p < 0.01. e HUVECs were cultured at confluence for at least 48 h on coverslips pre-coated with fibronectin, starved for 12 h and then stimulated or not with 10 ng/ml TNFα for 24 h. Cells were then incubated in the cold with mouse anti-VE-cadherin labeling antibody (surface), as described [16]. HUVECs were then incubated at 37 oC for the indicated times, fixed, permeabilized and stained with a rabbit anti-VE-cadherin antibody (total). Scale bar, 20 μm. The right-hand graph shows the ratio between the staining intensities of total and surface-labeled VE-cadherin antibody. Graph shows the mean + SEM from four independent experiments. *p < 0.05. See also Fig. S1
Although surface VE-cadherin stability, analyzed by pulse and chase experiments, decreased in cells exposed to TNFα, total surface VE-cadherin levels, detected immediately after the pulse with sulfo-NHS-biotin, remained unaltered over the time course of TNFα stimulation (pulse in Fig. 1c, d, time 0 post-biotinylation, and Fig. 2a). Moreover, when lysates were analyzed, a transient increase in total VE-cadherin was detected between 4 and 24 h after TNFα stimulation (Fig. 2a), suggesting that endothelial cells activate mechanisms to express intracellular VE-cadherin molecules that compensate for surface VE-cadherin degradation, yielding constant surface VE-cadherin levels. Confocal analysis of the VE-cadherin distribution revealed a transient accumulation of this protein in an intracellular compartment upon TNFα exposure (Fig. 2b). This intracellular pool colocalized with GM130, a Golgi marker (Fig. 2b, enlarged area), which is compatible with accumulation in the Golgi apparatus of newly synthesized VE-cadherin in response to TNFα. The increase in VE-cadherin was also observed in the HUVEC-derived cell line, EA.hy926 and, to a lesser extent, in human dermal microvascular endothelial cells (HDMVECs) stimulated with TNFα (Fig. 2c). In contrast, other components of AJs, such as catenins, and other vascular cadherins, such as N-cadherin [4, 17], did not change expression in response to TNFα (Fig. S2).
Fig. 2.
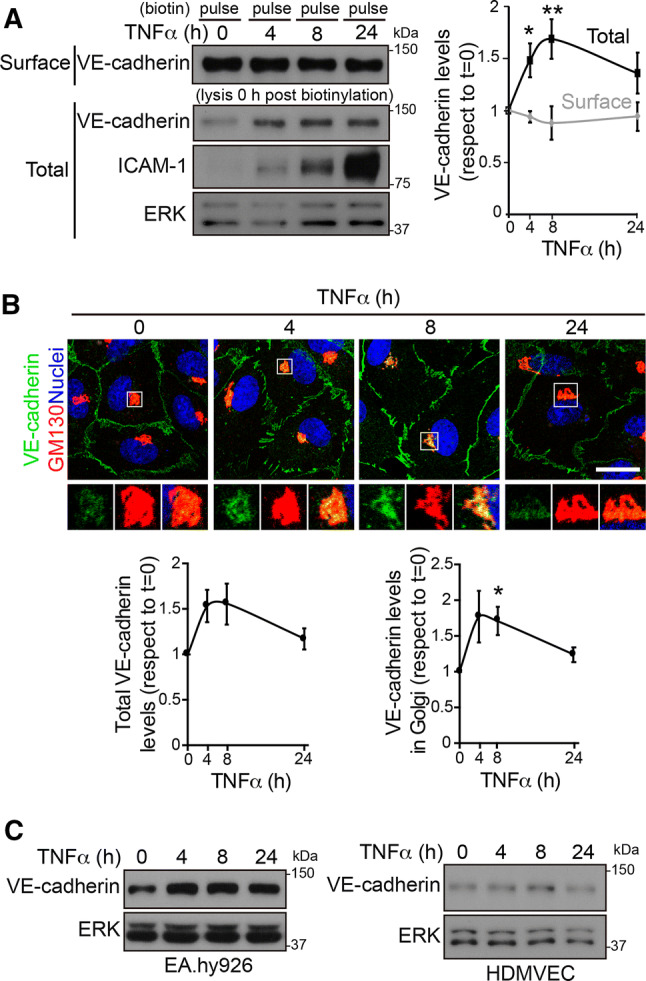
TNFα increases VE-cadherin protein expression but not surface VE-cadherin levels. a HUVECs were cultured at confluence for 48 h on dishes pre-coated with fibronectin, starved for 12 h and then stimulated with 10 ng/ml TNFα for the indicated times. Surface proteins were labeled with sulfo-NHS-biotin, and cells were immediately lysed. Surface-biotinylated proteins were isolated by pull-down assay with NeutrAvidin-agarose. Surface VE-cadherin levels were detected with specific antibodies from the pull-down fraction and compared with the expression levels from the lysates (total). ICAM-1 was immunodetected as a control for TNFα stimulation. ERK was immunoblotted as a loading control. The right-hand graph shows the quantification of the changes in total and surface VE-cadherin expression with respect to values in unstimulated cells. Graphs show the mean + SEM from three independent experiments. The t test compared differences between protein levels at the indicated timepoints of TNFα stimulation with those in unstimulated cells. *p < 0.05; **p < 0.01. b HUVECs were cultured as in (a) and stimulated with TNFα for the indicated times. Cells were fixed, permeabilized and stained with rabbit anti-VE-cadherin against the cytoplasmic VE-cadherin domain, mouse anti-GM130 antibody and DAPI to label nuclei. Scale bar, 20 μm. Bottom graphs show the quantification of changes in response to TNFα of total VE-cadherin staining intensity (left) and VE-cadherin staining intensity in the area positive for the Golgi marker GM130 (right). c Changes in VE-cadherin expression in response to TNFα stimulation in the HUVEC derived cell line EA.hy926 and in HDMVEC. ERK was immunoblotted as a loading control
Intracellular VE-cadherin partially compensates for surface VE-cadherin decrease during acute barrier disruption
In an inflammatory context, endothelial beds are subjected to acute disruption of their integrity by mechanical, thrombotic, and inflammatory factors [2, 14]. We wondered whether endothelial cells maintain VE-cadherin protein levels during acute barrier rupture. TNFα-pretreated primary HUVEC and EA.hy926 cell monolayers were subjected to Ca2+ switch assay, which provides a stimulus-independent mean of synchronizing the disruption, trafficking and reformation of AJs [18–20]. In brief, cells were cultured in the absence of Ca2+ for 20 min to disrupt cell–cell junctions, and then switched to normal medium to replenish normal Ca2+ levels and reform contacts between cells (Fig. 3a). TEER analysis revealed a transient rupture of the endothelial barrier and its rapid recovery upon Ca2+ replenishment (Fig. 3b). This TEER decrease was not caused by mechanical stress induced by sequential buffer and medium replacements in the well containing the electrodes since buffer solution containing Ca2+ did not induce significant changes in endothelial barrier properties (Fig. 3b). TEER results obtained with ECIS can be mathematically modelled and separated into specific values, which reflect the contribution of various physiological parameters to changes in barrier function [21]. Ca2+ depletion specifically decreased Rb values, which measure paracellular permeability, had a moderate effect on alpha values, which reflect cell–matrix interactions, and had almost no effect in cell membrane capacitance (Fig. 3c) [21]. ECIS modelling, therefore, suggested that the main effect of Ca2+ removal is the disruption of cell–cell junctions that control paracellular permeability. Western blot and confocal microscopy revealed that total VE-cadherin significantly decreased when physiological levels of Ca2+ were replenished, even though the cell monolayer recovered its barrier properties 60 min after replenishment (Figs. 3d–f and S3A, B). In contrast, the levels of PECAM-1, another junctional protein, remained unaltered during barrier contraction and reformation (Fig. 3d and S3C). These results suggested the involvement of other junctional components in the initial stages of barrier reformation. Indeed, tight junctions, detected by staining with antibodies specific for ZO-1, were fully reformed 60 min after Ca2+ restoration (Fig. 3g, left). N-cadherin is highly expressed in human endothelial cells and does not localize at mature endothelial cell–cell contacts since it mediates heterotypic interactions between the basal membrane of endothelial cells and vascular smooth muscle cells and pericytes [17, 22, 23]. Sixty min after Ca2+ restoration, N-cadherin partially decorated endothelial cell–cell junctions (Fig. 3g, right), suggesting that junctional complexes other than those mediated by VE-cadherin participate in the early recovery of cell–cell contacts after acute barrier disruption. Alternative Ca2+-switch assays, carried out by adding EDTA to the endothelial medium, also induced a decrease in VE-cadherin expression levels 60 min after Ca2+ replenishment (Fig. S3A, B). In addition, proteasomal inhibition had little effect on total VE-cadherin decrease, but combinations of proteasomal and ADAM-10 inhibitors prevented VE-cadherin decrease (Fig. S3C). Together, these results indicate that barrier repair after acute disruption engages VE-cadherin in a route of degradation and shedding that continues even when cell–cell junctions are being reannealed.
Fig. 3.

An intracellular VE-cadherin pool partially compensates surface VE-cadherin decrease upon acute barrier disruption a, b HUVECs were cultured at confluence for 48 h on dishes pre-coated with fibronectin, starved for 12 h and then stimulated with 10 ng/ml TNFα for 24 h before performing a Ca2+ switch (CS) assay, which induces a transient and acute disruption of the cell monolayer in absence of Ca2+ and a subsequent recovery upon Ca2+ replenishment with EBM-2 endothelial culture medium. b ECIS analysis of the transient effect of Ca2+ switch on TEER. PBS with no Ca2+ induces a strong decrease in TEER, whereas PBS containing 1 mM Ca2+ has no effect. c ECIS mathematical modelling of the TEER analysis in (b) yields three values: Rb values, which reveal changes in paracellular permeability (left), Alpha values, which are proportional to changes in cell–matrix interactions (center), and Cell membrane capacitance (right) [21]. d Analysis of VE-cadherin expression levels during the CS assay in HUVECs and EA.hy926 (EA) detected by western blot. e Graphs show VE-cadherin expression levels with respect to control cells from at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. f VE-cadherin distribution in HUVECs during the CS assay. Scale bar, 20 μm. g ZO-1 and N-cadherin distribution with respect to VE-cadherin staining during the CS assay. Scale bar, 20 μm. h TNFα-pretreated cells were biotinylated before or after a CS assay. Control cells and cells recovering after 90 min of Ca2+ replenishment were lysed and biotinylated proteins isolated by pull-down with NeutrAvidin-agarose beads. Graphs show the quantification of total and surface VE-cadherin levels from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. ERK was immunoblotted as a loading control. Double arrowhead shows differences in surface VE-cadherin during Ca2+ replenishment when cells were biotinylated before and after CS, which correspond to the surface localization, after barrier reformation, of VE-cadherin molecules that were not at the cell surface before the CS assay (right graph). To test whether VE-cadherin localization at the cell surface requires intracellular trafficking, parallel experiments of CS > biotinylation where performed in the presence of Dynasore during Ca2+ replenishment. See also Fig. S3
To establish whether the intracellular pool of VE-cadherin found in TNFα-treated cells (Fig. 2b) participates in the regulation of surface VE-cadherin turnover during the Ca2+ switch assays, we performed surface biotinylation experiments in combination with Ca2+-depletion assays. To better detect intracellular VE-cadherin translocation to the cell surface, barrier recovery was prolonged up to 90 min. First, cells were biotinylated and then subjected to Ca2+ switch assays (Fig. 3h, biotinylation > CS). The reduction of surface and total levels of VE-cadherin during Ca2+ replenishment was compared, revealing that surface and total VE-cadherin disappeared at a similar rate. Then reciprocal experiments were carried out in parallel: cells were subjected or not to Ca2+ switch assays and then biotinylated after Ca2+ replenishment (Fig. 3h, CS > biotinylation). Total VE-cadherin levels decreased at a similar rate in the two types of experiments. However, biotinylated VE-cadherin levels detected during Ca2+ replenishment when biotinylation was performed after Ca2+ switch (CS > biotinylation) were significantly higher than those in cells subjected to biotin labeling before Ca2+ depletion (biotinylation > CS) (Fig. 3h, surface, right graph, double arrowheads). This difference in VE-cadherin biotinylation during Ca2+ replenishment between the two sets of parallel experiments indicates that a significant proportion of surface VE-cadherin detected after barrier reformation was not at the plasma membrane before the Ca2+ switch assay, and thus may correspond to the intracellular pool found in Fig. 2b. Indeed, inhibition of intracellular trafficking with Dynasore reduced total VE-cadherin expression and the presence of VE-cadherin at the cell surface during barrier recovery (Fig. 3h, right graph). In conclusion, our experiments suggest that endothelial cells partially compensate VE-cadherin protein reduction induced by acute disruption by translocating the intracellular pool of VE-cadherin during barrier reformation.
The NF-κB pathway mediates VE-cadherin expression and maintains endothelial barrier integrity
VE-cadherin is fundamental to the preservation of endothelial cell–cell contacts as well as to endothelial cell survival [5]. Reduction of VE-cadherin levels leads to massive collapse of the endothelial barriers in vitro [1, 13] and of some vascular beds in vivo [5]. Therefore, compensation of VE-cadherin degradation through its synthesis must be an essential mechanism for increasing permeability without compromising endothelial barrier integrity, for instance, by a surge in apoptosis [5]. We gained further insight into the TNFα-mediated increase of the intracellular VE-cadherin pool by investigating whether it involves transcriptional activation of the CDH5 gene. First, RT-qPCR analysis of a time course of TNFα stimulation showed an increase in the levels of VE-cadherin mRNA, similar to that observed at the protein level (Fig. 4a). In addition, the use of a panel of inhibitors suggested that TNFα raises VE-cadherin expression through a NF-κB-dependent pathway but not through a JNK-dependent pathway (Fig. 4b, c). Consistent with its inhibitory effect on VE-cadherin expression, treatment with the NF-κB inhibitor BAY11 induced the collapse of the endothelial barrier as measured in TEER experiments and transwell permeability assays, in a similar manner to the barrier disruption induced upon VE-cadherin blockage with specific antibodies (Figs. 4d–g and S4). This collapse was independent of TNFα stimulation and indicated that NF-κB activation is critical not only for the endothelial inflammatory response, but also for endothelial barrier homeostasis.
Fig. 4.

NF-κB is necessary for VE-cadherin expression and endothelial barrier function. a TNFα increases VE-cadherin mRNA levels. RT-qPCR of VE-cadherin mRNA from HUVECs stimulated with TNFα for the indicated times. Graph shows the mean + SEM from three independent experiments performed with two pairs of oligonucleotides. *p < 0.05; **p < 0.01. b The NF-κB inhibitors BAY11 (10 μM) and MG132 (20 μM), but not the JNK inhibitor SP600125, abrogate the expression of VE-cadherin and ICAM-1 (positive control) but not of β-catenin, PECAM-1, tubulin and caspase-3 in HUVECs. Graph shows the mean + SEM from three independent experiments. *p < 0.05. c Decrease of VE-cadherin expression in the presence of the NF-κB inhibitor BAY11 detected by immunofluorescence with a specific antibody. Scale bar, 20 μm. d Effect of Bay11 on endothelial cell morphology detected with anti-β-catenin and anti-PECAM-1 antibodies. Scale bar, 20 μm. e Effect of 20 μg/ml anti-VE-cadherin blocking antibody on endothelial adherens junctions detected with anti-β-catenin antibody. f The NF-κB inhibitor BAY11 induces endothelial barrier collapse. HUVECs were cultured at confluence for 48 h on ECIS electrodes pre-coated with fibronectin, starved for 12 h and then stimulated or not with 10 ng/ml TNFα in the presence or absence of 10 μm BAY11. Graphs show the mean + SEM from at least three independent TEER experiments. ***p < 0.001. g Blocking VE-cadherin with a specific antibody has the same effect than BAY11 on endothelial TEER levels
TNFα and LPS increase ETS1 expression in endothelial cells
The transcription factor ETS1 controls VE-cadherin expression during endothelial cell differentiation [24, 25]. In addition, ETS1 mediates proinflammatory responses in diseases such as rheumatoid arthritis and vascular injury [26–28]. This prompted us to test the effect of TNFα on ETS1 expression in human endothelial cells. TNFα induced transient ETS1 protein expression between 4 and 8 h post-stimulation (Fig. 5a). Furthermore, LPS, an endotoxin that initiates the inflammatory response in vivo by inducing cytokines such as TNFα and IL-1β, induced ETS1 protein expression in HUVECs (Fig. 5b), in murine lung and kidney tissues (Fig. 5c) and in lung endothelial cells isolated from mice intraperitoneally injected with the endotoxin (Fig. 5d). Similar to VE-cadherin expression, ETS1 expression was reduced in the presence of BAY11, suggesting that ETS1 acts downstream of NF-κB (Fig. 5e). We attributed the reduced effect of the MG132 inhibitor to a direct effect on the degradation route of ETS1 itself that prevented its expression from being strongly inhibited.
Fig. 5.

ETS1 expression increases in response to TNFα and LPS in vitro and in vivo a Immunodetection of ETS1 in a time course of TNFα stimulation performed as in Fig. 2. Graph shows the mean + SEM from three independent experiments. ERK was immunoblotted as a loading control. b Immunodetection of ETS1 in HUVECs exposed or not to 1 μg/ml LPS for 17 h. Graphs show the mean + SEM from at least three independent experiments. c Mice were intraperitoneally injected with 1 mg/20 g weight LPS and euthanized 18 h later. Lysates from lung and kidney were subjected to western blot analysis with the indicated antibodies. Graph shows the mean + SEM from five animals per experimental condition. ERK was immunoblotted as a loading control. d An endothelial cell-enriched fraction was isolated as described (see “Materials and methods” section) and immunoblotted for von Willebrand factor (VWF) as an endothelial marker, E-cadherin as an non endothelial marker, ETS1 and β-actin as a loading control. Graph shows the quantification of ETS1 expression levels in endothelial cell-enriched fractions obtained from four mice per experimental condition. e The NF-κB inhibitor BAY11 (10 μM) reduces the expression of ETS1 in HUVECs. ERK was blotted as a loading control. Graphs show the mean + SEM from at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001
ETS1 regulates VE-cadherin expression and paracellular permeability in response to TNFα
siRNA-mediated ETS1 gene silencing reduced VE-cadherin levels, indicating that VE-cadherin upregulation is mediated by this transcription factor (Fig. 6a). Indeed, TNFα induced similar kinetics of VE-cadherin and ETS1 expression increase (Fig. 6a). In contrast, ETS1 knockdown had no effect on ICAM-1 protein expression, a paradigmatic gene induced via NF-κB in response to TNFα (Fig. S5A), suggesting that ETS1 selectively controls VE-cadherin expression but no other NF-κB-regulated genes. In contrast, VE-cadherin knockdown had no effect on ETS1 expression levels (Fig. 6a). Confocal microscopy analysis confirmed that siETS1 reduced nuclear ETS1 and junctional VE-cadherin staining intensities (Fig. 6b). ETS1 nuclear translocation was coincident with the increase of VE-cadherin protein expression (Fig. 6a–c). It is of note that ETS1 depletion also reduced transient VE-cadherin accumulation in the Golgi area (Fig. 6d), although surface labelling with sulfo-NHS-biotin revealed a similar decrease of total and surface VE-cadherin levels (Fig. 6e). Unlike that of NF-κB, ETS1 inhibition by siRNA-mediated knockdown had no significant effect on constitutive permeability in unstimulated HUVECs and a minor effect on TEER reduction upon TNFα stimulation (Fig. 6f, left). However, mathematical modelling suggested that ETS1 gene silencing specifically affects paracellular permeability in response to TNFα since it exacerbated the decrease of Rb values (Fig. 6f, right graph), although it had a minor effect on cell–matrix interactions, measured by alpha values, and on cell membrane capacitance (Fig. S5 B). This also indicates that the remaining VE-cadherin expression upon ETS1 depletion, or other cadherins and junctional proteins unaffected by ETS1 knockdown (not shown) can maintain constitutive permeability in the absence of inflammatory challenges [2, 4, 17]. It also suggests that VE-cadherin is the main junctional protein orchestrating barrier remodeling in response to TNFα. Collectively, our results show that the ETS1 transcription factor is a selective regulator of VE-cadherin expression and endothelial permeability in response to TNFα (Fig. 7).
Fig. 6.

ETS1 expression is required for VE-cadherin expression and reduces TNFα-induced paracellular permeability a siRNA-mediated knockdown of ETS1 (siETS1) reduces VE-cadherin expression, whereas VE-cadherin knockdown (siVE-cadherin) does not affect ETS1 protein levels. TNFα induces similar kinetics of VE-cadherin and ETS1 protein expression. Graphs show quantifications of VE-cadherin and ETS1 levels with respect to siControl after 8 h of TNFα stimulation. Mean + SEM from three independent experiments. *p < 0.05; ***p < 0.001. b Confocal images showing the effect of TNFα stimulation and ETS1 gene silencing on VE-cadherin and ETS1 levels and ETS1 nuclear translocation. Double staining was performed with rabbit anti-ETS1 antibody and mouse anti-VE-cadherin antibody against the extracellular domain of the adhesion receptor. Scale bar, 50 μm. c Quantification of ETS1 nuclear translocation, which occurs mainly at 4 h of TNFα exposure. *p < 0.05. d Effect of ETS1 knockdown on VE-cadherin levels in the Golgi region. Double staining was performed with rabbit anti-VE-cadherin antibody against the cytoplasmic tail and mouse anti-GM130 antibody as a Golgi marker. Scale bar, 20 μm. Graph shows the quantification of VE-cadherin staining in the Golgi area performed as in Fig. 2b. ns, p > 0.05; *p < 0.05. e Effect of ETS1 knockdown on total and surface VE-cadherin levels. HUVECs were transfected with the indicated siRNA oligonucleotides. 72 h post-transfection, cells were labeled with sulfo-NHS-biotin and surface, biotinylated proteins isolated by pull-down with NeutrAvidin agarose as in Fig. 2a. Note that siETS1 has no effect on the total and surface levels of PECAM-1 and on the loading control ERK. f Effect of ETS1 knockdown on TEER decrease (left graph) and Rb value decrease (right) induced by TNFα [21]. Graphs show the mean + SEM from three independent experiments measured at the indicated times of TNFα stimulation. *p < 0.05. See Fig. S5B
Fig. 7.

Endothelial cells compensate for reduction of surface VE-cadherin half-life by expressing VE-cadherin in response to TNFα. Schematic model of the molecular mechanisms by which endothelial cells maintain enough VE-cadherin levels at the plasma membrane when permeability increases in response to TNFα. TNFα induces (1) proteasome-mediated VE-cadherin degradation and, (2) ETS1-mediated VE-cadherin synthesis, which may be caused by ETS1-induced CDH5 gene transcription or by increasing CDH5 mRNA stability
Discussion
Inflammatory cytokines increase endothelial permeability by inducing junctional disassembly, VE-cadherin internalization and degradation, and shedding [9, 10, 29]. Our results suggest that VE-cadherin half-life is strongly reduced from 6 h to less than 3 h by long-term TNFα exposure and that such reduction is ameliorated by proteasomal inhibition. We have also found that a combination of proteasomal and ADAM-10 inhibition clearly prevented VE-cadherin decrease in Ca2+ switch assays, which is consistent with previous observations made in dermal microvascular endothelial cells [10]. These two degradative events may coordinate VE-cadherin decrease after acute barrier disruption in an inflammatory context, similar to the two-step mechanism of calpain-mediated cleavage and intracellular degradation after internalization recently reported for VE-cadherin [30].
Although these molecular mechanisms are critical for the progressive increase of endothelial paracellular permeability, which enables a physiological inflammatory response, the loss of VE-cadherin at the cell surface can be devastating for some endothelial cell barriers. As shown here, a full VE-cadherin inhibition, either by targeting NF-κB or with anti-VE-cadherin-blocking antibodies, completely disrupts the endothelial monolayer. VE-cadherin expression transmits prosurvival signaling that prevents endothelial apoptosis through its interaction with β-catenin [5] or by controlling the expression of survivin, a member of the family of inhibitors of apoptosis (IAP) involved in survival and differentiation that is upregulated in many cancers [31]. Hence, it is likely that endothelial cells, particularly those from large vessels such as HUVECs, establish molecular mechanisms to ensure enough VE-cadherin molecules at their plasma membrane, thus preserving barrier integrity and cell survival even when endothelial permeability is increased and AJs are transiently destabilized in an inflammatory scenario. Our results indicate that the progressive, TNFα-mediated VE-cadherin degradation can be compensated by an increase of VE-cadherin synthesis. This compensation induces VE-cadherin accumulation in an intracellular compartment, which also partially contributes to surface VE-cadherin restoration during acute contraction (Fig. 7). Experiments comparing levels of cell surface VE-cadherin by labeling cells with biotin before and after inducing acute contraction suggest an active vesicular trafficking of VE-cadherin from an intracellular pool towards the plasma membrane during barrier reformation. This is consistent with other studies showing the important role for intracellular VE-cadherin trafficking from endosomal compartments in endothelial barrier remodeling in response to various stimuli [16, 32–34]. Although we found that VE-cadherin accumulates in a Golgi/trans-Golgi network compartment, the identity of the vesicular carriers sorting newly synthesized cadherin towards the endothelial plasma membrane, and their potential connection to recycling endosomes remains to be investigated. It is of note that VE-cadherin is not essential for maintaining endothelial barrier function in all tissues and type of vessels [35, 36]. This suggests that other endothelial junctional complexes, including different cadherin-mediated junctions that are still poorly investigated, may play major roles in maintaining cell–cell contacts since many other cadherin superfamily members are expressed in human endothelial cells [22]. Our observation that complete endothelial barrier reformation occurs while VE-cadherin is still engaged in a degradative route, as well as the detection of newly formed tight junctions and N-cadherin-mediated junctions in early and probably immature cell–cell contacts, is in agreement with the existence of a wider than expected range of endothelial cell–cell junctional complexes that contribute to dynamically regulate vascular and lymphatic barrier function in different physiological and pathological situations.
In addition, we have found that ETS1 selectively mediates compensatory VE-cadherin synthesis, but not the synthesis of other TNFα-regulated genes, such as ICAM-1. Although further transcriptomic analysis on the role of ETS1 in the inflammatory response of human mature endothelium should be carried out, our data indicate that ETS1 may constitute part of the transcriptional hub that controls compensatory responses during NF-κB-mediated inflammatory signaling. ETS1, one of the main transcription factors controlling endothelial differentiation and cell density [24, 37], is strongly augmented by TNFα in human mature endothelial cells. Our results show that endothelial responses to inflammatory signaling share common transcriptional features with endothelial cell differentiation, in which the transcription of genes such as CD31 and VE-cadherin is upregulated. Indeed, a paradigmatic overlapping mediated by TNFα between inflammatory and differentiation signaling is found in macrophage-induced differentiation of stem/progenitor cells into endothelial cells. TNFα secretion facilitates the upregulation of VE-cadherin expression via TNFα-receptor-1 and NF-κB in these progenitor cells. The direct binding of the p65 subunit of NF-κB to the VE-cadherin gene promoter region was identified, indicating that, in addition to ETS1, NF-κB also directly regulates CDH5 gene transcription [38]. Likewise, ETS1 mediates peripheral blood stem cell commitment to the endothelial lineage [24] and two ETS1 binding sites can be found in the proximal region of the CDH5 promoter, necessary for VE-cadherin expression. These two sites bind ETS1 in vitro and are able to mediate the transactivation of a reporter by ETS1 [37]. The role of all these regulatory elements has not been investigated before in the context of the mature endothelial inflammatory response but, collectively, these previous reports and our results clearly indicate that the VE-cadherin promoter is regulated by inflammatory transcription factors, which participate in stem cell–endothelial differentiation and, as shown here, in endothelial barrier function.
The intensity, space and time of the physiological inflammatory response must be finely tuned to induce partial remodeling, but not destruction, of vessels and tissues. The molecular analysis of inflammation has mainly focused on the proteins causing endothelial leakage. It is only more recently that researchers have realized the importance of investigating the mechanisms that counteract such barrier disruption and, in general, the inflammatory response [39]. In addition, the inflammatory response can also be considered as a cellular stress response, upon which cells trigger homeostatic signaling. We propose that the observed upregulation of VE-cadherin and ETS1 expression via NF-κB is part of the transcriptional program that endothelial cells implement to tune vascular remodeling, preserve barrier integrity and recover vascular homeostasis in an inflammatory context.
Materials and methods
Materials
Mouse anti-VE-cadherin (610252), mouse anti-VE-cadherin blocking antibody (610251), mouse anti-p120-catenin (610134), mouse anti-E-cadherin (610181) and rat anti-mouse CD102 (553326) antibodies were purchased from BD. Rabbit anti-ICAM-1 (sc-7891), rabbit anti-ERK-1/2 (sc-94), mouse anti-GM130 (sc-16268), mouse anti-PECAM-1 (sc-365804), mouse anti-tubulin (sc-134241) antibodies were purchased from Santa Cruz Biotechnology, Inc. Rabbit anti-VE-cadherin (D87F2), rabbit anti-JNK (9252), rabbit anti-pJNK (9251) and rabbit caspase-3 (9662) antibodies were obtained from Cell Signaling Technology. Rabbit anti-β-catenin (C-2206) antibody, mouse anti-N-cadherin (C3865) and mouse anti-β-actin (A-5441) antibodies were obtained from Sigma-Aldrich. Mouse anti-VE-cadherin (BV6) antibody was obtained from Enzo Life Sciences. Mouse anti-GAPDH (AB-82-45) and mouse anti-α-catenin (EP1793Y) antibodies were purchased from Abcam. Mouse anti-Plakoglobin (PG-11E4) and rabbit ZO-1 (40-2200) antibodies were obtained from Thermo-Fischer. Phalloidin-Alexa 647 (A-22287) was obtained from Invitrogen. Rabbit anti-von Willebrand Factor antibody (A0082) was purchased from Dako. DAPI (10236276001) was obtained from Roche. Goat-anti-mouse and goat-anti-rabbit secondary antibodies were conjugated to Alexa Fluor-488, -555 and -647 and were purchased from Thermo-Fischer. Western blot secondary antibodies were obtained from Jackson Immunoresearch (donkey–anti-goat, 705-036-147 and donkey–anti-mouse, 715-035-151) and GE Healthcare (anti-rabbit, NA934 V). BAY11 (B5556), MG132 (M8699), SP600125 (S5567), chloroquine phosphate (C6628), ammonium chloride (A9434), GI254023X (SML0789) and LPS (L8274) were purchased from Sigma-Aldrich. Lactacystin (sc-3575) was purchased from Santa Cruz Biotechnology, Inc. Recombinant human TNFα (210-TA/CF) was obtained from R&D Systems.
Plasmids, siRNA, and RT-qPCR analysis
The following siRNA oligonucleotides were obtained from Dharmacon: siControl1 (nontargeting control), D-001210-01, 5′-AUGUAUUGGCCUGUAUUAGUU-3′; siControl3 (nontargeting control) D-001810-01, 5′-UAGCGACUAAACACAUCAA-3′ were used individually or as a pool; siRNA for VE-cadherin (NM_001795) 5′-GGAACCAGAUGCACAUUGA-3′. siRNA for ETS1was a SMARTpool (M-003887J, consisting of a pool of 4 oligonucleotides: 5′-GAUAAAUCCUGUCAGUCUU-3′; 5′- GGACCGUGCUGACCUCAAU-3′; 5′-GGAAUUACUCACUGAUAAA-3′ and 5′-GCAUAGAGAGCUACGAUAG-3′. siRNA oligonucleotides were transfected with Oligofectamine (Invitrogen) as previously described [1]. Oligonucleotides for qPCR were purchased from Sigma-Aldrich. 1 μg RNA from HUVECs was subjected to reverse transcription with the High-Capacity RNA-cDNA kit (Applied Biosystems). RT-qPCR was performed from the resulting cDNA in a CFX 384 thermocycler (Bio-Rad) using the SsoFast EvaGreen Supermix (Bio-Rad) and forward and reverse primers, designed with Probefinder software (Roche). Parallel qPCR for the housekeeping genes β-actin and GAPDH; F: 5′-CAGGCACCAGGGCGTG-3´, R: 5′-GTGAGGATGCCTCTCTTGCTCT-3´, and F: 5′-AGCCACATCGCTCAGACAC-3′, R: 5′-CGCCCAATACGACCAAAT-3′ was performed to normalize data from each point of stimulation. Three replicates of all the samples were run in parallel with non-reverse transcription controls for all the targets, which yielded no detectable amplification. VE-cadherin mRNA levels changes were analyzed with two pairs of primers: F: 5′-GAACCAGATGCACATTGATGA-3′, R: 5′-CGACTCACGCTTGACTTGAT-3′, (intron spanning) and F: 5′-CAAAGACGGTCGGCTGACAU-3′, R: 5′-TCTGTTCCGTTGGACTGCCT-3′ (non-intron spanning) and results were pooled for quantification. Unless otherwise indicated, the quantification cycle (Cq) values fell in the 20–24 range, which denotes medium to high levels of these transcripts. Results were quantified and represented relative to the expression of unstimulated HUVEC (2−ΔCq) using GenEx software, correcting for the efficiency of each pair of primers. The analyses were performed in technical triplicates. The contamination with genomic DNA was negligible. Subsequently, these values were normalized again with respect to the mean values of housekeeping genes used as reference. The stability of the candidate reference genes was probed with geNorm and Normfinder algorithms within GenEx, and a combination of the two most stable genes was chosen against which to normalize the results.
Cell culture and transfection
HUVECs were directly extracted from umbilical cords that were either generously donated by the Clínica Ruber Internacional (Madrid), thanks to Dr. Santiago Lamas (CBMSO, Madrid), or purchased from Lonza. Late-passage HUVECs lost sensitivity to inflammatory stimulation and changes in gene expression, so cells were maintained and used during the first 4 passages and then discarded. EA.hy926 cells were a gift from C.J.S. Edgell (University of North Carolina, Chapel Hill, NC). Adult HDMVECs were purchased from Tebu-Bio. HUVECs were grown in EGM-2 medium from Lonza. EA.hy926 cells were grown in DMEM supplemented with 10% FBS. Adult HDMVEC cells were grown in CADMEC medium from Tebu-Bio. All cells were incubated in plates precoated with 10 µg/ml human Fibronectin (Sigma-Aldrich) at 37 °C in an atmosphere of 5% CO2/95% air.
For siRNA transfection, HUVECs were plated at subconfluence (400,000 cells per 60 mm-dish) in EBM-2 medium (Lonza) with no antibiotics. The next day, cells were transfected by mixing 4 μl of oligofectamine (Invitrogen) with the indicated siRNA to a final concentration of 100 nM. 24 h after transfection, cells were trypsinized and plated at confluence onto different types of dish for parallel assays, such as permeability, adhesion, immunofluorescence or western blotting [14]. Experiments were performed 72 h after transfection.
For DNA transfection, 200,000 HUVECs were seeded on fibronectin-coated coverslips in 24-well dishes and were transfected with a mixture of 4 µg poly-ethylene-imide (PEI, Polysciences, Inc.) and 2 µg of plasmid DNA per coverslip. Medium was replaced 24 h after transfection, and experiments were performed 48 h after transfection.
Calcium switch assay
Interendothelial junctions depend on VE-cadherin homophilic interactions, which, in turn, rely on extracellular Ca2+ availability [6]. To induce massive inter-endothelial junctions rupture and surface protein internalization, we performed transient Ca2+ depletion assays, which consisted of replacing the culture medium with phosphate buffer saline (which does not contain Ca2+) for 20 min, followed by the addition of fresh Ca2+ -enriched culture medium to induce cell–cell contact recovery. When indicated, cells were incubated with 60 μM Dynasore (Calbiochem) during Ca2+ replenishment. Alternatively, Ca2+ depletion was induced by adding 3 mM EDTA to the culture medium.
Biotinylation assays for surface protein detection and western blot
We labeled cells with EZ-Link sulfo-NHS-biotin (Thermo Scientific) to detect surface-associated proteins [15]. In brief, we incubated HUVEC monolayers in 60-mm dishes with a solution of 0.1 mg/ml of sulfo-NHS-biotin in PBS supplemented with 1.5 mM Ca2+ for 30 min. Biotinylation was performed at 4 °C to block membrane trafficking. Subsequently, we washed the excess of sulfo-NHS-biotin with cold PBS supplemented with Ca2+ and incubated the cells with cold DMEM 10% FBS to quench the remaining free reagent, and then washed again. Cells were either directly lysed or, in the pulse-chase experiments, cultured at 37 °C for the indicated times and then lysed. Lysates were prepared by passing the cells through a 0.6-mm, 24G needle and then centrifuged at 14,000 rpm for 5 min. Part of the supernatant was conserved as total lysate and the rest was subjected to preclearing with Sepharose resin, followed by pull-down with NeutrAvidin agarose (Thermo Scientific) to isolate biotinylated proteins. Protein extraction and immunodetection procedures were performed as described previously [40]. For Western blot analysis, equal amounts of proteins were loaded onto polyacrylamide gels (8–12%) under reducing conditions and transferred to Immobilon-P membranes (EMD Millipore). After blocking with 5% nonfat dry milk or 5% BSA, the latter when western blotting of phosphorylated proteins was performed, 0.05% (vol/vol) Tween-20 in PBS, the membranes were incubated with the indicated antibodies, washed with PBS + 0.05% Tween 20, and incubated with the appropriate secondary antibodies coupled to HRP. After extensive washes, blots were developed using an enhanced chemiluminescence Western blotting kit (ECL; GE Healthcare).
Endothelial barrier function assays
Trans-endothelial electric resistance (TEER) assays with an electric cell-substrate impedance sensing system (ECIS 1600R; Applied Biophysics) [41] were performed as described [1, 14, 18]. Unless otherwise indicated, resistance values at 4000 Hz were normalized to a value of 1.0 at the beginning of the experiments. The Rb, alpha, and membrane capacitance values (Cm) were modelled using the ECIS software (Applied Biophysics) running the experiment in the multi-frequency mode, which ranges between 250 and 64000 Hz. This mathematical modelling separates TEER values into basolateral cell adhesion (alpha), which correlates with focal adhesion formation, and paracellular barrier strength (Rb) [21]. Transwell permeability assays were performed as described [14]. Briefly, HUVECs were plated at confluence onto fibronectin-coated transwells (Corning). After 72 h, cells were treated as indicated on the top chamber of the transwell. FITC-dextran (Mr 4000, Sigma-Aldrich) was applied on the top chamber at 0.1 mg/ml and allowed to equilibrate for 90 min before a sample from the medium at the lower chamber was collected to measure fluorescence in a Fusion α-FS fluorimeter.
Antibody-mediated labeling of surface VE-cadherin: confocal and time-lapse microscopy
For confocal microscopy, cells were fixed with 4% paraformaldehyde for 20 min, blocked with TBS (25 mM Tris, pH 7.4, and 150 mM NaCl) plus 10 mM Gly for 10 min, permeabilized for 5 min with TBS containing 0.2% Triton X-100 at 4 °C, blocked with TBS containing 3% BSA for 30 min and incubated at 37 °C with primary antibodies and, after extensive washes, with fluorophore-conjugated secondary antibodies and Alexa Fluor 647-labeled phalloidin and/or DAPI. Surface labeling of VE-cadherin with a specific antibody was performed as described [16]. Confocal laser scanning microscopy was performed with an LSM510 (ZEISS) coupled to an Axiovert 200 M microscope, as previously described [1]. Briefly, we used 63 ×/1.4 oil Plan-Apochromat and 100 ×/1.3 oil Plan-Neofluar objectives to acquire images of subcellular structures. Acquisition software (LSM510 4.2) was provided by Zeiss.
Junctional VE-cadherin intensity was quantified with ImageJ. Briefly, an area containing the whole cell was created and then made 10 pixels smaller to exclude the junctional region. This image was then subtracted from the original region to generate a region representing the fluorescence intensity at the cell periphery. The raw pixel intensity surface VE-cadherin was divided by the corresponding area to yield a parameter that could be compared across the different images.
Intraperitoneal injection of LPS
Five mice per condition were intraperitoneally injected with 50 μg/g LPS or saline. 17 h later, a retro-orbital injection of 150 μl 0.5% w/v Evans Blue (Merck) was performed as a control of LPS effect on tissue circulation. 30 min post-injection, mice were euthanized and 100 mg of lung and kidney were collected from each mouse. Evans blue was extracted with formamide at 55 °C for 48 h, measured spectrophotometrically and normalized to the tissue weight revealing an increase of tissue edema in response to LPS injection (not shown). Simultaneously, protein concentration from tissue lysates was measured with BCA protein assay (ThermoFisher) and subjected to Western blot assays with the indicated antibodies. To isolate an endothelial cell-enriched fraction [42, 43], mouse lungs were minced and digested with 0.1% type I collagenase solution (Gibco) for 1 h at 37 °C. Lung tissue was further disaggregated with a 19.5 G needle and subsequently filtered through a cell strainer. Lung cell suspension was then subjected to positive sorting with an antibody against the endothelial cell marker ICAM-2 conjugated to magnetic Dynabeads Sheep Anti-Rat IgG (11035) from Thermo Fisher, for 30 min at 4 °C. After extensive washes, endothelial cell rich fraction was lysed with Laemmli sample buffer and subjected to western blot analysis.
Statistical analysis
Statistical significance was determined using the paired two-tailed Student t test in PRISM and Microsoft Excel.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The expert technical advice of the Confocal Microscopy Facility and the Genomic Facility are gratefully acknowledged. The work was supported by Grants SAF2017-88187-R and S2017/BMD-3817 TomoXliver (to J.M.), BFU2015–67266-R (to I.C) and Instituto de Salud Carlos III (PI18/01662 to CR, co-funded with European FEDER contribution) and of the Programa de Actividades en Biomedicina de la Comunidad de Madrid-B2017/BMD-3671-INFLAMUNE. S.B.F is supported by Endocornea2, convenio colaboración CSIC, funded by Instituto de Investigación Fundación Jiménez Díaz. An institutional support of Fundación Ramón Areces to the CBMSO is also acknowledged. DGW and CCN are recipients of FPI fellowships from MINECO. NCA and AC are recipients of FPU fellowships from MECD. We also thank Dr. Phil Mason, who provided English language support, and Dr. Miguel A. Alonso, for helpful comments.
Author contributions
DGW, NCA and JM contributed to the conception and experimental design. DGW, NCA, CCN and SBF performed the experiments and acquired the data. AC and CR help carry out in vivo experiments and isolated lung endothelial cells. DGW and NCA analyzed the data, IC and CR provided material support, and reviewed the manuscript, which was written by JM.
Compliance with ethical standards
Conflict of interest
The authors declare that no conflicts of interest exist.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Natalia Colás-Algora and Diego García-Weber contributed equally to this work.
Contributor Information
Diego García-Weber, Email: diego.garcia@inserm.fr.
Jaime Millán, Email: jmillan@cbm.csic.es.
References
- 1.Fernandez-Martin L, Marcos-Ramiro B, Bigarella CL, Graupera M, Cain RJ, Reglero-Real N, Jimenez A, Cernuda-Morollon E, Correas I, Cox S, Ridley AJ, Millan J. Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol. 2012;32:e90–e102. doi: 10.1161/ATVBAHA.112.252080. [DOI] [PubMed] [Google Scholar]
- 2.Marcos-Ramiro B, Garcia-Weber D, Millan J. TNF-induced endothelial barrier disruption: beyond actin and Rho. Thromb Haemost. 2014;112(6):1088–1102. doi: 10.1160/TH14-04-0299. [DOI] [PubMed] [Google Scholar]
- 3.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 4.Colas-Algora N, Millan J. How many cadherins do human endothelial cells express? Cell Mol Life Sci. 2019;76(7):1299–1317. doi: 10.1007/s00018-018-2991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 6.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–454. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Hayer A, Shao L, Chung M, Joubert LM, Yang HW, Tsai FC, Bisaria A, Betzig E, Meyer T. Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat Cell Biol. 2016;18:1311–1323. doi: 10.1038/ncb3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, Passaniti A, Hasday JD, Goldblum SE. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1232–L1245. doi: 10.1152/ajplung.00109.2006. [DOI] [PubMed] [Google Scholar]
- 9.Gong H, Gao X, Feng S, Siddiqui MR, Garcia A, Bonini MG, Komarova Y, Vogel SM, Mehta D, Malik AB. Evidence of a common mechanism of disassembly of adherens junctions through Galpha13 targeting of VE-cadherin. J Exp Med. 2014;211:579–591. doi: 10.1084/jem.20131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flemming S, Burkard N, Renschler M, Vielmuth F, Meir M, Schick MA, Wunder C, Germer CT, Spindler V, Waschke J, Schlegel N. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res. 2015;107:32–44. doi: 10.1093/cvr/cvv144. [DOI] [PubMed] [Google Scholar]
- 11.Seynhaeve AL, Rens JA, Schipper D, Eggermont AM, Ten Hagen TL. Exposing endothelial cells to tumor necrosis factor-alpha and peripheral blood mononuclear cells damage endothelial integrity via interleukin-1ss by degradation of vascular endothelial-cadherin. Surgery. 2014;155:545–553. doi: 10.1016/j.surg.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 12.Cain RJ, Vanhaesebroeck B, Ridley AJ. The PI3 K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J Cell Biol. 2010;188:863–876. doi: 10.1083/jcb.200907135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millan J, Cain RJ, Reglero-Real N, Bigarella C, Marcos-Ramiro B, Fernandez-Martin L, Correas I, Ridley AJ. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 2010;8:11. doi: 10.1186/1741-7007-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcos-Ramiro B, Garcia-Weber D, Barroso S, Feito J, Ortega MC, Cernuda-Morollon E, Reglero-Real N, Fernandez-Martin L, Duran MC, Alonso MA, Correas I, Cox S, Ridley AJ, Millan J. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J Cell Biol. 2016;213:385–402. doi: 10.1083/jcb.201504038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reglero-Real N, Alvarez-Varela A, Cernuda-Morollon E, Feito J, Marcos-Ramiro B, Fernandez-Martin L, Gomez-Lechon MJ, Muntane J, Sandoval P, Majano PL, Correas I, Alonso MA, Millan J. Apicobasal polarity controls lymphocyte adhesion to hepatic epithelial cells. Cell Rep. 2014;8:1879–1893. doi: 10.1016/j.celrep.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 17.Navarro P, Ruco L, Dejana E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J Cell Biol. 1998;140:1475–1484. doi: 10.1083/jcb.140.6.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aranda JF, Reglero-Real N, Marcos-Ramiro B, Ruiz-Saenz A, Fernandez-Martin L, Bernabe-Rubio M, Kremer L, Ridley AJ, Correas I, Alonso MA, Millan J. MYADM controls endothelial barrier function through ERM-dependent regulation of ICAM-1 expression. Mol Biol Cell. 2013;24:483–494. doi: 10.1091/mbc.E11-11-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel DM, Dubash AD, Kreitzer G, Green KJ. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J Cell Biol. 2014;206:779–797. doi: 10.1083/jcb.201312110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toret CP, Collins C, Nelson WJ. An Elmo-Dock complex locally controls Rho GTPases and actin remodeling during cadherin-mediated adhesion. J Cell Biol. 2014;207:577–587. doi: 10.1083/jcb.201406135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci USA. 1991;88:7896–7900. doi: 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colas-Algora N, Millan J. How many cadherins do human endothelial cells express? Cell Mol Life Sci. 2019;76:1299–1317. doi: 10.1007/s00018-018-2991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruse K, Lee QS, Sun Y, Klomp J, Yang X, Huang F, Sun MY, Zhao S, Hong Z, Vogel SM, Shin JW, Leckband DE, Tai LM, Malik AB, Komarova YA. N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J Cell Biol. 2019;218:299–316. doi: 10.1083/jcb.201802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MS, Lee CS, Hur J, Cho HJ, Jun SI, Kim TY, Lee SW, Suh JW, Park KW, Lee HY, Kang HJ, Lee DS, Koh GY, Nakagami H, Morishita R, Park YB, Kim HS. Priming with angiopoietin-1 augments the vasculogenic potential of the peripheral blood stem cells mobilized with granulocyte colony-stimulating factor through a novel Tie2/Ets-1 pathway. Circulation. 2009;120:2240–2250. doi: 10.1161/CIRCULATIONAHA.109.856815. [DOI] [PubMed] [Google Scholar]
- 25.Arderiu G, Pena E, Aledo R, Espinosa S, Badimon L. Ets-1 transcription is required in tissue factor driven microvessel formation and stabilization. Angiogenesis. 2012;15:657–669. doi: 10.1007/s10456-012-9293-x. [DOI] [PubMed] [Google Scholar]
- 26.Feng W, Xing D, Hua P, Zhang Y, Chen YF, Oparil S, Jaimes EA. The transcription factor ETS-1 mediates proinflammatory responses and neointima formation in carotid artery endoluminal vascular injury. Hypertension. 2010;55:1381–1388. doi: 10.1161/HYPERTENSIONAHA.110.150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goetze S, Kintscher U, Kaneshiro K, Meehan WP, Collins A, Fleck E, Hsueh WA, Law RE. TNFalpha induces expression of transcription factors c-fos, Egr-1, and Ets-1 in vascular lesions through extracellular signal-regulated kinases 1/2. Atherosclerosis. 2001;159:93–101. doi: 10.1016/s0021-9150(01)00497-x. [DOI] [PubMed] [Google Scholar]
- 28.Redlich K, Kiener HP, Schett G, Tohidast-Akrad M, Selzer E, Radda I, Stummvoll GH, Steiner CW, Groger M, Bitzan P, Zenz P, Smolen JS, Steiner G. Overexpression of transcription factor Ets-1 in rheumatoid arthritis synovial membrane: regulation of expression and activation by interleukin-1 and tumor necrosis factor alpha. Arthritis Rheum. 2001;44:266–274. doi: 10.1002/1529-0131(200102)44:2<266::AID-ANR43>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 29.Nwariaku FE, Liu Z, Zhu X, Turnage RH, Sarosi GA, Terada LS. Tyrosine phosphorylation of vascular endothelial cadherin and the regulation of microvascular permeability. Surgery. 2002;132:180–185. doi: 10.1067/msy.2002.125305. [DOI] [PubMed] [Google Scholar]
- 30.Su W, Kowalczyk AP. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol Biol Cell. 2017;28:76–84. doi: 10.1091/mbc.E16-09-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iurlaro M, Demontis F, Corada M, Zanetta L, Drake C, Gariboldi M, Peiro S, Cano A, Navarro P, Cattelino A, Tognin S, Marchisio PC, Dejana E. VE-cadherin expression and clustering maintain low levels of survivin in endothelial cells. Am J Pathol. 2004;165:181–189. doi: 10.1016/s0002-9440(10)63287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan Z, Wang ZG, Segev N, Hu S, Minshall RD, Dull RO, Zhang M, Malik AB, Hu G. Rab11a mediates vascular endothelial-cadherin recycling and controls endothelial barrier function. Arterioscler Thromb Vasc Biol. 2016;36:339–349. doi: 10.1161/ATVBAHA.115.306549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Yao W, Qian G, Wei Z, Wu G, Wang G. Rab5-mediated VE-cadherin internalization regulates the barrier function of the lung microvascular endothelium. Cell Mol Life Sci. 2015;72:4849–4866. doi: 10.1007/s00018-015-1973-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chichger H, Duong H, Braza J, Harrington EO. p18, a novel adaptor protein, regulates pulmonary endothelial barrier function via enhanced endocytic recycling of VE-cadherin. FASEB J. 2015;29:868–881. doi: 10.1096/fj.14-257212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frye M, Dierkes M, Kuppers V, Vockel M, Tomm J, Zeuschner D, Rossaint J, Zarbock A, Koh GY, Peters K, Nottebaum AF, Vestweber D. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. 2015;212:2267–2287. doi: 10.1084/jem.20150718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagerling R, Hoppe E, Dierkes C, Stehling M, Makinen T, Butz S, Vestweber D, Kiefer F. Distinct roles of VE-cadherin for development and maintenance of specific lymph vessel beds. EMBO J. 2018;37:e98271. doi: 10.15252/embj.201798271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lelievre E, Mattot V, Huber P, Vandenbunder B, Soncin F. ETS1 lowers capillary endothelial cell density at confluence and induces the expression of VE-cadherin. Oncogene. 2000;19:2438–2446. doi: 10.1038/sj.onc.1203563. [DOI] [PubMed] [Google Scholar]
- 38.Wong MM, Chen Y, Margariti A, Winkler B, Campagnolo P, Potter C, Hu Y, Xu Q. Macrophages control vascular stem/progenitor cell plasticity through tumor necrosis factor-alpha-mediated nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol. 2014;34:635–643. doi: 10.1161/ATVBAHA.113.302568. [DOI] [PubMed] [Google Scholar]
- 39.Winsauer G, de Martin R. Resolution of inflammation: intracellular feedback loops in the endothelium. Thromb Haemost. 2007;97:364–369. [PubMed] [Google Scholar]
- 40.Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci. 2012;4:429–434. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA. 1992;89:7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rivas V, Carmona R, Munoz-Chapuli R, Mendiola M, Nogues L, Reglero C, Miguel-Martin M, Garcia-Escudero R, Dorn GW, 2nd, Hardisson D, Mayor F, Jr, Penela P. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest. 2013;123:4714–4730. doi: 10.1172/JCI67333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, Huang X, Sheppard D, Hynes RO, Hodivala-Dilke KM. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.