

Abstract
The interaction of insulin-degrading enzyme (IDE) with the main intracellular proteasome assemblies (i.e, 30S, 26S and 20S) was analyzed by enzymatic activity, mass spectrometry and native gel electrophoresis. IDE was mainly detected in association with assemblies with at least one free 20S end and biochemical investigations suggest that IDE competes with the 19S in vitro. IDE directly binds the 20S and affects its proteolytic activities in a bimodal fashion, very similar in human and yeast 20S, inhibiting at (IDE) ≤ 30 nM and activating at (IDE) ≥ 30 nM. Only an activating effect is observed in a yeast mutant locked in the “open” conformation (i.e., the α-3ΔN 20S), envisaging a possible role of IDE as modulator of the 20S “open”–”closed” allosteric equilibrium. Protein–protein docking in silico proposes that the interaction between IDE and the 20S could involve the C-term helix of the 20S α-3 subunit which regulates the gate opening of the 20S.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2807-y) contains supplementary material, which is available to authorized users.
Keywords: Insulin-degrading enzyme, IDE-20S proteasome interaction, IDE-20S molecular docking, Open-close 20S equilibrium
Introduction
The insulin-degrading enzyme (IDE) is a Zn2+-dependent peptidase of the M16 family of metalloenzymes highly conserved through evolution and ubiquitously expressed in human tissues and also detectable in blood and cerebrospinal fluid [1–5].
IDE is prevalently localized into the cell cytosol; however, mitochondria, intracellular membranes and the plasma membrane, the nucleus and the extracellular milieu (even though this occurrence has been very recently questioned) have been described as additional sites where the canonical IDE (or IDE isoforms) can be detected [1–6]. IDE was originally identified as the key enzyme for the degradation in vitro of insulin and of several short bioactive peptides (such as β-amyloid, amylin, glucagon, transforming growth factor-α, atrial natriuretic peptides, somatostatin, ABri and ADan), which all share the propensity to form β-sheet-rich amyloid fibrils [2, 7–11]. Furthermore, the cohort of molecules (i.e., ATP, PIP3, long-chain fatty acids, metal ions, somatostatin and dynorphin, among others) identified as allosteric or direct modulators of IDE supports the compelling notion that IDE conformation, activity and distribution are finely tuned depending on the metabolic state of the cell [10–16].
Several studies provide evidence that IDE dysregulation is implicated in the onset of type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) [2, 17–19]. With respect to T2DM, IDE has been long considered a promising target in developing novel therapies aimed at reducing the insulin degradation: however, major concerns regarding the therapeutic efficacy of its inhibition are emerging along with a general reconsidering of the activity on insulin in vivo [20, 21]. With respect to AD, following the identification of IDE as the major β-amyloid degrading enzyme, it has become increasingly clear that dysregulation of IDE expression and function, as a result of either acquired or inherited conditions, facilitates the AD onset [2, 8, 9, 13, 22]. The role of IDE in AD is even more attractive in view of the proposed novel biological roles, which all point to a wider contribution of IDE to the proteostasis, a hallmark of AD onset as well as of other neurodegenerative diseases [2, 22–27].
With respect to this, IDE is supposed to regulate the ubiquitin–proteasome system (UPS) as demonstrated by the extensively documented co-purification with the proteasome and the recently proposed role as a non-canonical ubiquitin-activating enzyme [25–32].
The UPS is responsible for most of the non-lysosomal protein degradation into the cells, thus playing a key role in the protein quality control, maintenance of proteostasis and regulation of cell metabolism [33–37].
The proteasome is a proteolytic complex composed of a 20S core particle (CP), harboring the proteolytic active sites, capped by one or two 19S regulatory particles (RP) (forming the 26S and the 30S proteasome, respectively), which assist the binding, unfolding and translocation of the substrate (i.e., poly-ubiquitinated proteins) into the catalytic core [35, 37–45]. The 20S is a barrel-shaped hollow cylinder formed by four stacked rings, containing seven α-subunits in the two outer rings and seven β-subunits in the two inner rings. The β1, β2 and β5 subunits provide the catalytic sites for the caspase-like, trypsin-like and chymotrypsin-like enzymatic activities, respectively [38–47]. Extensive studies indicate that the structural arrangement of the N-terminal tails of the α-subunits, and in particular that of the α-3 subunit, which protrudes into the catalytic channel, are responsible for the transition from the “closed” to the “open” state (and viceversa) of the 20S, which is a requisite for substrate accommodation into the catalytic chamber [39, 47]. Thus, binding of the 19S, as well as of other RPs identified so far (i.e., PA28, Blm10 in yeast, PA200), is expected to regulate the overall structural topology of the α-ring, inducing the opening of the catalytic channel and promoting the entry and the catalysis of proteasome substrates. Moreover, the binding of the RPs modifies proteasome substrate specificities [43–46, 48–52]. In fact, it is widely recognized that, in the intracellular compartment, the 20S exists also in the uncapped form which is proposed to cope with the ubiquitin-independent degradation of oxidized and unfolded substrates, even though a clear demonstration of this role in vivo is still lacking [53–57]. Nonetheless, there is evidence that, under oxidative stress conditions, capped particles undergo dissociation favoring the increase in the pool of free 20S [55, 57]. However, the mechanisms regulating the substrate recognition and the activity of the 20S are still unexplored.
Herewith, we combined a biochemical and a native spectroscopic approach on cell extracts to carry out a molecular investigation which reveals that IDE regulates in a biphasic fashion the enzymatic activities of the 20S through a direct and high-affinity interaction with the subunits of the α-ring. Notably, it further emerges that IDE is able to work as a competitor of the 19S binding to the 20S in vitro, thus envisaging a role of IDE as one of the modulators of the equilibrium between the different proteasome populations (i.e., 30S, 26S and 20S).
Materials and methods
Cell cultures
SHSY5Y and HEK-293 were grown in high-glucose DMEM supplemented with 10% FBS, l-glutamine (2 mM), penicillin (50 IU/mL), streptomycin (50 μg/mL), sodium pyruvate (1 mM) and ciprofloxacin (0.03 mM) at 37 °C in a humidified 5% CO2 chamber. Peripheral blood from healthy volunteers was harvested in tubes containing 2 mM EDTA and centrifuged at 3500 rpm for 5 min. Then, the cells were washed four times with PBS supplemented with 2 mM EDTA and centrifuged at 3000 rpm for 2 min at 4 °C.
Native gel electrophoresis
The assay was performed by following the methodology described elsewhere [58]. SHSY5Y, HEK-293 and red blood cells (RBCs) (1 × 105) were lysed in detergent-free buffer (250 mM sucrose, 25 mM Tris–HCl, 5 mM MgCl2, 1 mM EDTA, 2 mM ATP, 5% glycerol, pH 7.4) through freeze–thaw cycles for preparation of the crude cell extracts (i.e, the soluble fraction of the cytosol). The soluble fraction was then “squeezed out” through centrifugation for 30 min at 14,000 rpm and 4 °C. The protein concentration was normalized by Bradford assay.
Thereafter, 150 µg of proteins was separated under native conditions in a 3.5% acrylamide gel. Gels were then harvested and soaked in a clean dish in a reaction buffer (50 mM Tris, 5 mM MgCl2, 1 mM ATP, pH 7.5) containing 100 μM of a substrate specific for the chymotrypsin-like activity of proteasome (i.e., Suc-LLVY-7-amino-4-methylcoumarin, hereafter referred to as LLVY-amc). This analysis allows to study the distribution and the activity of the three main proteasome sub-assemblies: the 30S, corresponding to the RP2-CP particle, the 26S, corresponding to the RP-CP, and the 20S, corresponding to the CP.
Proteins were then transferred to a HyBond-ECL nitrocellulose filters (see also below for details) and probed with an antibody specific for IDE (Covance) and for α-7 proteasome subunit (PSMA3) or Rpt2 subunit of the 19S base (PSMC1) (Protein-tech), diluted 1:3000 in 0.02% Tween-PBS fat-free milk and, thereafter, incubated with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody (Bio-Rad, Hercules, CA, USA), diluted 1:50,000 in 0.2% Tween-PBS fat-free milk.
In the case of the analysis involving the purified 20S proteasome (1 µg unless otherwise indicated), we have investigated the human 20S proteasome (Boston Biochem) (h20S), the yeast wild-type 20S proteasome (wt y20S) (a generous gift from prof. Groll) and a yeast mutant (i.e., α-3ΔN, a generous gift from prof. Groll, where the first 9 amino acids of the N-terminus of the α-3 chain have been removed) [47]. All three types of 20S proteasomes were incubated in the presence of different concentrations of a rat recombinant IDE which was > 95% identical to the human IDE (R&D System) (ranging from 30 to 200 nM) for 15 min at 37 °C in the standard activity buffer (25 mM Tris–HCl, pH 7.8). In the case of the assay involving also the affinity purified 19S (h19S) (Boston Biochem), h20S (57 nM) and h19S (250 nM) were incubated in the presence of IDE in 25 mM Tris–HCl, 5 mM MgCl2, 1 mM ATP, 1 mM DTT, 1 mM EDTA and 5% glycerol, pH 7.5, for 15 min at 37 °C.
In these cases, the complexes were run under native conditions and then stained either by Western blotting (WB) as previously indicated or by Coomassie Brilliant Blue (CBB).
Kinetic analysis
The characterization of h20S activities (i.e, chymotrypsin-like, trypsin-like and caspase-like) as a function of IDE concentration was carried out through a fluorimetric approach, employing different fluorogenic substrates (namely, the fluorogenic LLVY-amc substrate for the chymotrypsin activity, the fluorogenic LLR-amc substrate for the trypsin-like activity and the fluorogenic LLE-amc substrate for the caspase-like activity) (Boston Biochem, Boston, USA).
All three types of 20S (namely, human, yeast wt and the mutant α-3ΔN) were diluted to a final concentration of 1 nM, unless otherwise indicated, in the assay buffer (25 mM Tris–HCl, pH 7.8) and the activity on fluorogenic substrates was monitored in the absence and in the presence of different concentrations (ranging from 5 to 100 nM) of IDE. Before the substrate addition, the 20S and IDE were allowed to interact for 10 min at 37 °C. The rate of hydrolysis of the fluorogenic substrate was monitored for 45 min, a time interval over which only a small fraction of the substrate underwent proteolysis, and the relative velocities were extrapolated. In the case of the caspase-like activity (which was measured only for h20S) some additional considerations were required, since IDE was able to cleave the fluorogenic LLE-amc substrate in vitro [24]. Therefore, the caspase-like activity was derived by subtracting the enzymatic kinetics (mol/s), obtained from IDE alone at various concentrations, from the catalytic activity of h20S observed in the presence of the same IDE concentrations. Results are reported as relative velocity in the presence and in the absence of IDE.
Protein–protein docking
The interaction between IDE and h20S was studied using protein–protein molecular docking software Hex-v8.0.0 [59]. The structure of IDE was obtained from the PDB entry 2G47 [60] removing any non-protein element (ligands and water). The missing IDE loop (res 971–978) was modeled and the obtained structure was further optimized using the “Modeller” loops refinement tool [61], which is available on UCSF Chimera (v1.10). The structure of h20S was obtained from the PDB entry 4R3O.
The docking was performed using the “Shape-Electro” correlation with “OPLS Energy” refinement. The “range-angle” sampling method was used with a receptor and ligand angle range of 180° (step size 7.5), an “alpha” angle range of 360° (step size 5.5) and a relative distance range of 40.0 Å (step 0.8). Due to the large dimension of the 20S particle, the “macro-docking” procedure was used to uniformly sample the receptor surface, performing a series of ligand–receptor docking on a well-dispersed set of sites.
The hydrogen bond and electrostatic (salt bridges) interactions between IDE and h20S in the docking poses were analyzed with a homemade script using the MDAnalysis Python module [59].
Proteasome enrichment and quantification
2.5 × 108 HEK cells were washed three times with PBS and stored at − 80 °C. Cells were lysed with 2 mL of lysis buffer per plate [10 mM Hepes pH 7.9, 10 mM KCl, 5 mM MgCl2, 10% glycerol, 10 mM ATP, 1% NP40, protease and phosphatase inhibitor (Roche)] by 15 cycles of 1 min sonication at 4 °C. After centrifugation (10 min at 4000 g), the supernatant was filtered with 0.2 µm filters (Sigma-Aldrich) and concentrated down to 800 µL on an Amicon ultracentrifugal device (100 kDa MWCO, Merck Millipore). Samples were then fractionated by gel filtration on a Superose 6 column (GE Healthcare), previously equilibrated with the lysis buffer. 50 fractions of 500 µL were collected. The protein concentration was determined by a detergent-compatible assay (DC assay; Bio-Rad).
Detailed LC–MS/MS analysis, data search and validation
For each fraction, a volume corresponding to 30 µg of total protein was precipitated with 20% TCA and washed with acetone. Samples were boiled 5 min at 95 °C in Laemmli buffer to denature proteins. Proteins were first alkylated with 100 mM chloro-acetamide for 30 min at room temperature in the dark. Thereafter, they were concentrated in a single band on a 12% acrylamide SDS-PAGE gel and visualized by colloidal CBB. One-shot analysis of the entire mixture was performed. A single band, containing the whole sample, was cut and washed in 50 mM ammonium bicarbonate for 15 min at 37 °C, followed by a second wash in 50 mM ammonium bicarbonate and acetonitrile (1:1) for 15 min at 37 °C. Trypsin (Promega) digestion was performed overnight at 37 °C. The resulting peptides were extracted from the gel by three steps: a first incubation in 50 mM ammonium bicarbonate for 15 min at 37 °C and two incubations in 10% formic acid acetonitrile (1:1) for 15 min at 37 °C. The three collected extractions were pooled with the initial digestion supernatant, dried in a Speed-Vac, and resuspended with 2% acetonitrile and 0.05% trifluoroacetic acid. The peptide mixtures were analyzed by nano-LC–MS/MS using an Ultimate3000 system (Dionex) coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Five microliters of each peptide sample, corresponding to an equivalent initial quantity of 7.5 µg of total proteins, was loaded on a C18 precolumn (300-µm inner diameter × 5 mm; Dionex) at 20 µL/min in 5% acetonitrile and 0.05% trifluoroacetic acid. After 5 min of desalting, the precolumn was switched on line with the analytical C18 column (75 µm inner diameter × 15 cm; PepMap C18, Dionex) equilibrated in 95% solvent A (5% acetonitrile, 0.2% formic acid) and 5% solvent B (80% acetonitrile, 0.2% formic acid). Peptides were eluted using a 5–50% gradient of solvent B during 110 min at a 300 nL/min flow rate. The LTQ-Orbitrap Velos was operated in data-dependent acquisition mode with the Xcalibur software. Survey scan MS spectra were acquired in the Orbitrap on the 350–1800 m/z range with the resolution set to a value of 60,000. The 20 most intense ions per survey scan were selected for CID fragmentation, and the resulting fragments were analyzed in the linear trap (LTQ). Dynamic exclusion was used within 60 s to prevent repetitive selection of the same peptide.
Bioinformatic processing of mass spectrometry data
Raw MS files were processed with MaxQuant software (version 1.5.0) for database searching with the Andromeda search engine and for quantitative analysis. Data were searched against human entries of the Swiss-Prot protein database (UniProtKB/Swiss-Prot Knowledgebase release 2016_01, Homo sapiens taxonomy, 20,194 entries). Carbamidomethylation of cysteines was set as a fixed modification, whereas oxidation of methionine, protein N-terminal acetylation, and phosphorylation of serine, threonine, and tyrosine residues were set as variable modifications. The specificity of trypsin digestion was set for cleavage after lysine (K) or arginine (R), and two missed trypsin cleavage sites were allowed. The precursor mass tolerance was set to 20 parts per million (ppm) for the first search and to 4.5 ppm for the main Andromeda database search. The mass tolerances MS/MS mode was set to 0.5 Daltons. Minimum peptide length was set to seven amino acid residues, and the minimum number of unique peptides was set to one. Andromeda results were validated by the target-decoy approach with a reverse database at both a peptide and protein false discovery rate (FDR) of 1%. For label-free relative quantification of the samples, the “match between runs” option of MaxQuant was enabled with a time window of 0.5 min, to enable the cross-assignment of MS features detected in the different runs. The “LFQ” metric from the MaxQuant “protein group.txt” output was used to assess the abundance of each protein.
Data quantification
Gel filtration profiles were normalized to account for protein concentration differences across elution fractions. Then, for each protein group, abundance values were normalized so that the maximum value measured in all the elution fractions was set at 1. For h20S and the 19S, the elution profiles were obtained by averaging the abundances of the constitutive 20S subunits (α1–α7, β3, β4, β6 and β7) and the abundances of the PSMC1–7, PSMD1–4, 6–8, and 10–14 subunits, respectively.
Statistical analysis
For the experimental procedures requiring a statistical analysis, one-way ANOVA followed by Tukey’s post hoc significance test was performed.
Results
Detection of IDE–proteasome particles in the soluble fractions of various cell lines
We have recently reported that downregulation of IDE expression in SHSY5Y cells, upon delivery of an anti-sense oligonucleotide, brought about an increase of the ATP-dependent proteolytic activity of the proteasome, suggesting that IDE exerted an inhibition of the proteolytic activity of the capped particles (i.e., 26S and 30S). Moreover, the hypothesis of an IDE-dependent negative modulation was further supported by the observation that the cleavage kinetics of fluorogenic substrates (specific for the three proteasome activities) by a purified 26S was slower in the presence of IDE concentrations ranging from 5 to 50 nM [26]. To unravel the mechanistic features of the IDE–proteasome association and to identify the proteasome particles interacting with IDE in cells growing under basal metabolic conditions, we first performed a native gel electrophoresis separation of the crude cell extracts of SHSY5Y and HEK-293 human cell lines, which display the three main intracellular proteasome assemblies (i.e., 30S, 26S and 20S) [62, 63]. The proteasome particles were then probed with the LLVY-amc fluorogenic substrate through an overlay assay and transferred to a nitrocellulose filters to further assess their identities.
By probing the filters with an anti-PSMA3 antibody (i.e., the α-7 subunit of h20S), it came out that in SHSY5Y and HEK-293 cells (Fig. 1a), the 26S was the predominant species (52 ± 4 and 44 ± 4%, respectively), followed by the 30S (26 ± 6%) and the 20S (20 ± 2%) (in the case of SHSY5Y cells), whilst followed by the 20S (33 ± 3%) and the 30S (23 ± 6%) in the case of HEK-293 cells.
Fig. 1.
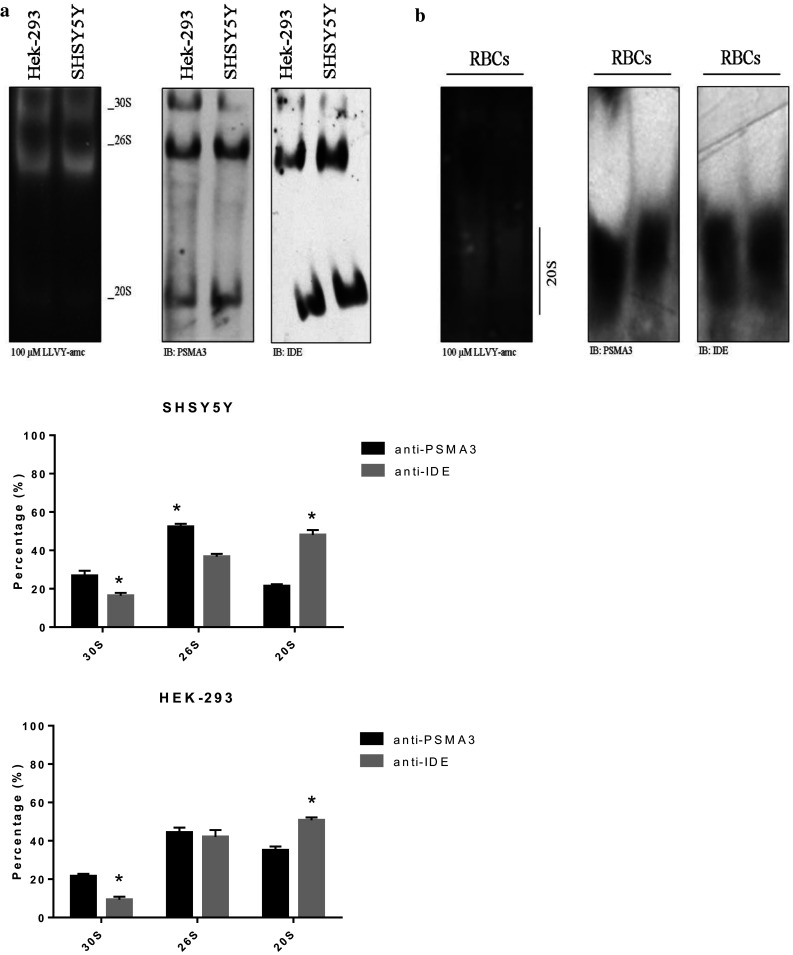
The extracts of the SHSY5Y and HEK-293 cells (left panel) and of the RBCs (right panel) were resolved by native gel electrophoresis and the proteasome particles probed with the LLVY-amc fluorogenic substrate. The proteasome particles were probed with an anti-IDE and anti-PSMA3 antibody by WB. The relative abundance of the three particles was determined through a densitometric analysis of the WB bands (bottom panel). A representative immunoblot is shown. Values reported are the mean ± SE of five independent experiments. *Significantly different from the PSMA3 staining (p < 0.001, one-way ANOVA, followed by Tukey’s test, n = 15)
By probing the filter with the anti-IDE antibody, it emerged that IDE was preferentially bound to the 26S and the 20S (36 ± 4 and 47 ± 7% for SHSY5Y cells, respectively and 50 ± 2 and 41 ± 4% for HEK-293 cells, respectively) (Fig. 1a). Conversely, IDE was faint in correspondence of the 30S (17 ± 2 and 9 ± 3% for SHSY5Y and HEK-293 cells, respectively). Moreover, we could not detect IDE in correspondence of other assemblies (such as the free 19S) over a molecular range comprising between 700 and 2500 kDa. Thus, with the exception of the 26S, the relative abundance of the 30S and the 20S was significantly different upon staining of the filter with the anti-IDE antibody or the anti-PSMA3 antibody (in all cases, p < 0.001).
To further validate the tight interaction between IDE and the 20S, we screened by native gel electrophoresis the terminally differentiated RBCs, wherein the free 20S is abundantly represented [62–64]. In RBCs, the 20S turned out to be the predominant active species and, by further probing the filter with the anti-IDE antibody, it emerged that IDE was strongly immuno-detected in association with the 20S (Fig. 1b).
The preferential binding of IDE to the 20S was further confirmed by nano-liquid chromatography coupled to tandem mass spectrometry (nano-LC–MSMS) showing their co-elution upon size-exclusion chromatography separation (Fig. S1). IDE was associated with 20S assemblies of different sizes; in particular, (1) the first peak (elution volume = 9.5–11 mL) most likely corresponded to 30S, 26S and IDE-bound 26S particles, even though the resolution of the approach did not allow concluding that IDE was bound exclusively to the 26S and not to the 30S, as suggested by the native gel approach; (2) the second peak (elution volume = 12–12.5 mL) most likely contained the 20S singly and doubly capped with IDE, underlying the possibility that one molecule of 20S was able to bind up to two molecules of IDE; (3) finally, the existence of a third peak (elution volume = 14 mL), containing mainly 20S and IDE, suggested the occurrence of IDE associated with hemi-proteasomes (that is, containing only one α-ring and one β-ring). Altogether, this analysis strongly supported the idea that IDE associates with any free side of the 20S assemblies and that the 20S may exist in vivo as a single and doubly IDE-bound form.
IDE directly interacts with the 20S and competes with the 19S
The distribution of IDE between the different proteasome assemblies, and in particular the absence of IDE in association with the 30S, stimulated our interest in addressing two questions, namely: (1) whether a direct association occurs between IDE and the 20S or else this interaction is mediated in cell models by unidentified additional proteasome interacting proteins (PIPs); (2) whether IDE putative binding to the free 20S could interfere with the binding of the canonical 19S RP.
To address the first point, the interaction of an h20S and IDE was investigated by a mobility shift assay (Fig. 2a). The h20S (2 µg, 114 nM) was separated by native gel electrophoresis in the presence and in the absence of 200 nM IDE and probed with the LLVY-amc fluorogenic substrate (Fig. 2a, left panel). In the presence of IDE, the electrophoretic mobility of h20S was clearly delayed with respect to the migration of h20S in the absence of IDE (Fig. 2a, left panel). The particles were then transferred to a nitrocellulose filter and probed with anti-PSMA3 and anti-IDE antibodies to confirm the identity of the species. In the presence of IDE, the band, corresponding to h20S (which displayed a molecular weight higher than that of h20S observed in the absence of IDE), was recognized by both the anti-PSMA3 antibody and the anti-IDE antibody, supporting the occurrence of a direct interaction between the two macromolecules (Fig. 2a, right panel). Interestingly, in the presence of 200 nM IDE, we observed a slight activation of h20S chymotrypsin-like activity, which can be appreciated through the negative stain of the gel (Fig. 2a, left panel).
Fig. 2.

a h20S (114 nM) was incubated in the presence and absence of 200 nM IDE for 10 min at 37 °C. The samples were separated under native conditions and probed with the LLVY-amc fluorogenic substrate (left panel). The identity of the particles was further assessed by WB (right panel). b h20S (57 nM) was allowed to interact with h19S (250 nM) in the presence and in the absence of 200 nM IDE. The complexes were resolved by native gel electrophoresis and the particles probed with 100 µM LLVY-amc. The identity of the complexes was further probed by WB. A representative immunoblot of five independent experiments is shown
To address the second point, h20S (57 nM) was mixed with an average fourfold excess of h19S (250 nM) and separated by native gel electrophoresis. In the absence of IDE, the appearance of three enzymatically active populations, namely (1) h20S, (2) h26S and (3) h30S (Fig. 2b, left panel, left column) was detected. Upon addition of 200 nM IDE, the h26S and the h30S populations seemed to disappear, whereas h20S was left and its activity was markedly enhanced with respect to that of h20S mixed with the h19S but in the absence of IDE (Fig. 2b, left panel, right column). Western blotting analysis of the same experiment showed that, in agreement with the above reported results, IDE was bound to h20S (Fig. 2b, right panel, left column) and the h19S was left in the free state (Fig. 2b, right panel, right column) as a result of an IDE-induced dissociation of the h26S and h30S assemblies.
Notably, the order of addition of the macromolecules was ineffective in altering the experimental outcome (data not shown), indeed, suggesting that the equilibrium situation was attained fast with respect to the observation time. This evidence seemed to indicate that under these conditions, IDE interfered in some way with the binding of h20S particle to the h19S particle, displacing it.
Effect of IDE on the enzymatic activities of human 20S proteasome
The evidence reported above elicited interest in carrying out a functional biochemical characterization of the IDE-dependent modulation of h20S enzymatic activities. The substrate concentration dependence of the enzymatic activity rates follows the Michaelis–Menten mechanism and it has been analyzed according to the double-reciprocal plot:
| 1 |
where [E0] is the total enzyme concentration, ν is the observed rate (expressed in mol/s), [S] is the substrate concentration, Km is the Michaelis–Menten equilibrium constant (referring to the substrate affinity for the enzyme), and kcat is the speed of the rate-limiting step.
Fig. S2A shows the Lineweaver–Burk plot for the three enzymatic activities (i.e., the chymotrypsin-like activity, employing the fluorogenic LLVY-amc substrate, the trypsin-like activity, employing the fluorogenic LLR-amc substrate, and the caspase-like activity, employing the fluorogenic LLE-amc substrate) of the substrate degradation by 1 nM h20S; the catalytic parameters are reported in Table 1. This represents the required starting information to characterize the effect of IDE on the enzymatic activities of h20S.
Table 1.
The catalytic parameters in the absence of IDE at pH 7.8 and 37 °C of the chymotrypsin-like activity (employing the fluorogenic LLVY-amc substrate) for h20S, the wt y20S proteasome and its mutant α-3ΔN
| Human | Yeast | α-3ΔN | |
|---|---|---|---|
| Chymotrypsin-like kcat (s−1) | 12.0 ± 2.7 | 0.95 ± 0.13 | 49.6 ± 6.2 |
| Km (M) | 1.8 (± 0.3) × 10−3 | 1.2 (± 0.3) × 10−4 | 4.1 (± 0.5) × 10−3 |
| Trypsin-like kcat (s−1) | 2.0 ± 0.3 | ||
| Km (M) | 5.3 (± 0.7) × 10−5 | ||
| Caspase-like kcat (s−1) | 1.2 ± 0.3 | ||
| Km (M) | 1.1 (± 0.3) × 10−4 |
For h20S, the catalytic parameters are also reported for the trypsin-like activity (employing the fluorogenic LLR-amc substrate) and for the caspase-like activity (employing the fluorogenic LLE-amc substrate)
Figure 3 shows the dependence on IDE concentration for the relative velocity, which puts in evidence the different IDE-linked effects on the three enzymatic activities of h20S in the presence of 50 μM of either one of the three fluorogenic substrates. A relatively weak IDE-dependent inhibitory effect was observed for the chymotrypsin-like activity and for the trypsin-like activity, while there was a marked inhibitory effect of IDE on the caspase-like activity (see Fig. 3 and Fig. S2B). However, for all three activities, the IDE-linked effect displayed a bimodal behavior, even though to a largely different extent, characterized by an inhibitory effect at lower IDE concentrations, followed by a recovery of the initial activity, which became an activation at higher IDE concentrations that has been fully documented for the chymotrypsin-like activity (see Fig. 3 and Figure S2A).
Fig. 3.

Relative velocity in the presence of 50 μM substrate as a function of IDE concentration for the chymotrypsin-like (circle), trypsin-like (cross) and caspase-like (asterisk) activity of h20S. Continuous lines are the non-linear least-squares global fitting of all three enzymatic activities according to Eq. (2e), employing parameters reported in Tables 1 and 2
This feature indeed suggested the occurrence of (at least) two functionally relevant binding sites for IDE on h20S, which finds a significant correspondence with the native gel and mass spectrometry data (see specific sections), according to Scheme 1, where E is the free enzyme, ES is the enzyme:substrate complex, E(IDE)1 is the complex between the free enzyme and IDE (characterized by the binding affinity constant 1KIDE), ES(IDE)1 is the complex between ES and IDE (characterized by the binding affinity constant α·1KIDE), E(IDE)2 is the complex between the free enzyme and two bound molecules of IDE (the second site being characterized by the binding affinity constant 2KIDE), ES(IDE)2 is the complex between ES and two bound molecules of IDE (the second site being characterized by the binding affinity constant γ·2KIDE), Km and kcat are the catalytic parameters in the absence of IDE, α is the interaction parameter for the affinity to the first site (reflecting the effect of IDE on the substrate affinity and/or the effect of substrate on IDE affinity for the first site, with α < 1 representing a positive interaction, which increases the substrate and/or IDE affinity, with α > 1 representing a negative interaction, which decreases the substrate and/or IDE affinity, and α = 1 no interaction), β is the interaction parameter for the rate-limiting step of h20S with only one binding site occupied by IDE (reflecting the effect of IDE on kcat, with β < 1 representing a negative interaction, which decreases the rate of substrate proteolytic cleavage, with β > 1 representing a positive interaction, which increases the rate of substrate enzymatic processing, and β = 1 no interaction), γ is the interaction parameter for the affinity to the second site (reflecting the effect of IDE on the substrate affinity and/or the effect of substrate on IDE affinity for the second site, with γ < 1 representing a positive interaction, which increases the substrate and/or IDE affinity, with γ > 1 representing a negative interaction, which decreases the substrate and/or IDE affinity, and γ = 1 no interaction), δ is the interaction parameter for the rate-limiting step of h20S with both binding sites occupied by IDE (reflecting the effect of IDE on kcat, with δ < 1 representing a negative interaction, which decreases the rate of substrate proteolytic cleavage, with δ > 1 representing a positive interaction, which increases the rate of substrate enzymatic processing, and δ = 1 no interaction). Therefore, on the basis of Scheme 1, the dependence on IDE concentration of the double-reciprocal plot [see Eq. (1)] becomes:
| 2a |
where
| 2b |
| 2c |
| 2d |
Scheme 1.

Thermodynamic scheme for the representation of the interaction equilibria in the modulation by IDE of the enzymatic activities of h20S (E) toward synthetic substrates (S)
The relative velocity (i.e., vrel) is:
| 2e |
The solid curve in Fig. S2B, describing the inhibitory effect of IDE on the caspase-like activity of h20S, was obtained from Eq. 2e with 1KIDE = 1.35 (± 0.3) × 10−8 M, 2KIDE=8.9 (± 1.3) × 10−8, α = 1.5 ± 0.3, β = 0.01 ± 0.01, γ = 0.46 ± 0.07 and δ = 0.66 ± 0.08, accounting for an about twofold decrease of the enzymatic activity, followed by a partial (at least over the investigated IDE concentration range) restoring (Fig. S2B). The apparent affinity of IDE for these binding sites on h20S (i.e., 1KIDE and 2KIDE) indeed refers to the free enzyme (see Scheme 1) and the value of equilibrium constants is obviously independent of the substrate; therefore, the smaller inhibitory effect of IDE on the chymotrypsin-like and trypsin-like activities (Fig. 3) indicates that IDE binding brings about a much smaller variation of the interaction parameters (see Table 2) for these two enzymatic activities of h20S. This substrate-dependent behavior is completely referable to a much larger IDE-linked inhibitory effect on the rate-limiting proteolytic step of the caspase-like activity after the binding of IDE to the first interaction site, characterized by 1KIDE (i.e., β·kcat, see Scheme 1), underlying a more enhanced sensitivity of the caspase activity to the structural alteration induced by IDE binding to this first site.
Table 2.
Interaction parameters according to Scheme 1 between IDE and h20, the wt y20S proteasome and its mutant α3ΔN
| Human | Yeast | α-3ΔN | |
|---|---|---|---|
| 1KIDE (M) | 1.3 (± 0.3) × 10−8 | 1.6 (± 0.3) × 10−8 | 1.6 (± 0.4) × 10−8a |
| 2KIDE (M) | 8.9 (± 1.3) × 10−8 | 7.4 (± 0.9) × 10−8 | 7.4 (± 0.9) × 10−8a |
| Chymotrypsin-like | |||
| α | 1.5 ± 0.3 | 2.2 ± 0.4 | 1.0a |
| β | 0.79 ± 0.09 | 0.35 ± 0.05 | 1.0a |
| γ | 0.46 ± 0.07 | 0.9 ± 0.2 | 0.64 ± 0.08 |
| δ | 1.2 ± 0.3 | 3.0 ± 0.5 | 1.0 ± 0.2 |
| Trypsin-like | |||
| α | 1.5 ± 0.3 | ||
| β | 0.72 ± 0.09 | ||
| γ | 0.46 ± 0.07 | ||
| δ | 1.15 ± 0.20 | ||
| Caspase-like | |||
| α | 1.5 ± 0.3 | ||
| β | 0.01 ± 0.01 | ||
| γ | 0.46 ± 0.07 | ||
| δ | 0.66 ± 0.08 | ||
aThese values are kept fixed
This inhibitory effect is followed by an activation for all three enzymatic activities as IDE binds the second interaction site, characterized by 2KIDE (see Fig. 3), and, at least for the chymotrypsin-like activity, this effect is essentially completed at 1 μM IDE (see Fig. 4a). The evidence that the IDE-linked effect is exerted on all three enzymatic activities (though to a different extent) indeed suggests that it can be attributed to a conformational change of h20S particle and not to a local effect. This idea appears strengthened by the evidence that the activation is characterized by both an increase of the substrate affinity (as from γ < 1, see Table 2) and of the rate-limiting step velocity (as from δ > 1, see Table 2).
Fig. 4.

a Relative velocity of the chymotrypsin-like activity of h20S in the presence of 50 μM LLVY-amc as a function of IDE concentration at pH 7.8 and 37 °C. The continuous line is the non-linear least-squares global fitting of data according to Eq. (2e), employing parameters reported in Tables 1 and 2. b h20S (114 nM) was allowed to react with the indicated IDE concentrations and separated by native gel electrophoresis (left panel). The particles were probed with 100 µM LLVY-amc (right panel). The identity of the particles was determined by WB. Reaction buffer: 25 mM Tris–HCl, pH 7.8, 37 °C
It is noteworthy that this bimodal behavior as a function of IDE concentration (see Fig. 4a) can be observed also by native gel assay (see Fig. 4b), where different IDE concentrations are employed, clearly demonstrating that such a bimodal feature is highly reproducible and allows to immediately relate the outcome of the enzymatic assay (see Figs. 3, 4a) with the native gel assay (see Fig. 4b).
Interaction of IDE with the wild-type yeast 20S proteasome and its α3ΔN mutant
Starting from these observations and the hypothesis that IDE affects the conformation of h20S, we decided to extend the investigation on the effect of IDE to the chymotrypsin-like activity of another type of 20S, which has been well characterized by the structural viewpoint (namely the wt y20S) [47] and to its α-3ΔN mutant (where the first 9 amino acids of the N-terminus of the α-3 chain have been deleted). Thus, while the former is considered to lie in the “closed” conformation, the mutant α3ΔN particle is thought to be locked in the “open” configuration, since the deletion of the N-terminal sequence of the α-3 subunit was reported to impair a “closed” conformation [47].
To accomplish a comparative analysis for the IDE effect on the chymotrypsin-like activity of human, yeast and α-3ΔN mutant of the 20S, we carried out the characterization of catalytic parameters in the absence of IDE for the three 20S assemblies (see Fig. 5a) and they are reported in Table 1.
Fig. 5.

a Lineweaver–Burk plots of the substrate dependence in the absence of IDE for the chymotrypsin-like activity of humans (circle), yeast wild type (cross) and yeast α-3ΔN mutant (asterisk) at pH 7.8 and 37 °C (buffer: 25 mM Tris–HCl, pH 7.8). Continuous lines were obtained by non-linear least-squares fitting of data according to Eq. (1), employing parameters reported in Table 1. b IDE-dependent modulation of the chymotrypsin-like activity of h20S (circle), as compared to y20S proteasome [wt (cross) and α-3ΔN mutant (asterisk)] on 50 µM LLVY-amc monitored through a fluorimetric assay. Values are reported as relative activity, i.e., the activity of a specific 20S species at each IDE concentration vs that of the same 20S in the absence of IDE. c IDE effect on the wt y20S and α-3ΔN mutant activity was further examined by native gel electrophoresis. The complexes were probed with 100 µM LLVY-amc. The Coomassie Brilliant Blue (CBB) staining of the gel is shown. The relative activity of the 20S activity was quantified by densitometric analysis of the negative stain of the native gel normalized on the CBB staining for each experimental condition. Values reported are the mean ± SE of five independent experiments in the case of the wt y20S and of three independent experiments in the case of the α-3ΔN mutant. *Significantly different from the control (p < 0.001, one-way ANOVA, followed by Tukey’s test, n = 15 for wt y20S and n = 9 for α-3ΔN mutant)
Then, we set up a fluorimetric assay to evaluate the effect of different concentrations of IDE ranging from 5 to 100 nM, on wt y20S and its α-3ΔN mutant (see Fig. 5b). Interestingly, the administration of IDE to wt y20S mirrored the behavior observed for the chymotrypsin-like activity of h20S, characterized by a significant inhibition of the wt y20S activity on the LLVY-amc substrate up to 40 nM IDE, followed by a significant recovery of the activity over the 40–100 nM IDE range (Fig. 5b). Moreover, also in this case, the overall behavior was fully reproducible by native gel electrophoresis (Fig. 5c).
Strikingly, in the case of the α-3ΔN mutant, the IDE-linked inhibitory effect was never observed, not even at very low IDE concentrations (see Fig. 5b), even though a slight mobility shift of the particle was documented allowing to hypothesize that IDE was actually bound. Conversely, an IDE concentration-dependent increase of the mutant α-3ΔN chymotrypsin-like activity on the LLVY-amc substrate was observed throughout the investigated concentration range (Fig. 5b). The experimental outcome was confirmed also by native gel electrophoresis (Fig. 5c).
Protein–protein docking of the interaction of IDE with the 20S
To find a structural explanation to the observed IDE modulation of 20S activity, a set of protein–protein docking in silico experiments was performed. In Fig. 6a, the structures of the top-10 best-scoring pose of IDE on the surface of h20S are reported. A striking majority of the IDE docked poses were found to interact with the α-3 subunit of h20S (only few low scoring poses were found to interact with the α-4 subunit). No direct interactions were observed with the β subunits. A review of the observed residue–residue interactions between IDE and h20S allowed to identify the binding site at the C-term helix of the α-3 subunit, which shows some similarity with the same region of the α-4 subunit.
Fig. 6.

a Top-10 best-scoring poses obtained by docking of IDE on h20S (IDE poses are represented as blue ribbons, proteasome α-3 subunit is shown in magenta). b Molecular surface of IDE colored using electrostatic potential (red: < − 1 eV/kT, blue: > + 1.0 eV/kT). c Close-up of the preferred binding site of IDE (residues showing direct hydrogen bond or salt bridges interaction with the proteasome α-3 subunit are labeled). d Overview of the best-scoring IDE–h20S complex. IDE is indicated by ribbon with orange helices and blue beta-sheets. 20S α-3 subunits are shown in magenta, β-2 subunits (trypsin-like activity) in red, β-5 (chymotrypsin-like activity) in yellow and β-1 (caspase-like activity) in green
This helix contains many charged residues (i.e., Lys237, Lys245, Glu247, Arg248 and Glu249) which can interact with a specific site on the surface of the IDE protein (see Fig. 6b, c). In this site of IDE, a short “crevice” was found with negatively charged residues on the sides (i.e., Glu486, Glu962 and Asp964) and positively charges residues (i.e., Arg674 and Arg 782) at the bottom. These residues were found to form a number of hydrogen bonds and electrostatic (salt-bridges) interactions with the C-term helix of the 20S α-3 subunit.
However, the protein–protein interactions observed in the in silico experiment should be considered with extreme caution. Although the macro-docking protocol ensures an even sampling of the h20S particle, it has to be pointed out that the docking algorithm does not take into account possible changes in secondary (nor tertiary or quaternary) structure of the receptor or the ligand. Therefore, we present the in silico results as a working structural hypothesis for the IDE–h20S interaction, which definitely will need some further experimental confirmation. The molecular model here proposed, if reinforced by additional experimental evidence (e.g., mutational studies, NMR FRET), might constitute the basis for more in silico studies by means of all-atom or coarse-grained molecular dynamics simulations. These studies will permit to acquire more insight into the stability of the complex and the effect on the quaternary structure of 20S at a structural–dynamical level.
The proposed mechanism of IDE interaction can be summarized in Fig. 6d, where IDE is shown to interact with the C-term helix of the h20S α-3 subunit; this interaction indeed is the preferential one, even though IDE seemed to marginally interact also with the α-4 subunit.
Discussion
A functional interrelationship between IDE and proteasome activity on fluorogenic and natural substrates has been already hinted before, putting in evidence that the IDE-linked effect on the proteasome was independent of the enzymatic activity of IDE [26, 28–30]. However, these original studies were addressed to proteasome particles only partially purified from different biological systems and, most notably, without excluding the possibility that they represented indirect effects [26, 28–30]. Moreover, although IDE was reported to be generically associated with 20S and the capped particles, its real distribution among the various proteasome assemblies is unclear [26, 28–30].
In this paper, data obtained through quantitative proteomics and native gel analysis show that IDE predominantly associates with proteasome particles, such as 20S and 26S, which display (at least) one of the two α-rings not involved in the interaction with the 19S particle or other RPs (Fig. 1 and Fig. S1).
In addition, at an IDE concentration of 200 nM, that is when the high-affinity inhibitory site to the α-3 subunit should be fully occupied by IDE and the second activating site is partially saturated (see Fig. 4), IDE likely competes with h19S, enriching the pool of the free h20S. Therefore, the IDE effect appears to induce a stabilization of h20S (see Fig. 2b), even though additional binding sites, such as with the free 19S, even if not detected with the approaches herewith described, cannot be ruled out.
Further, these data demonstrate that IDE directly interacts with h20S, bringing about a modulation of its proteolytic activity which is effective already over the nanomolar range, indicating that this binding occurs under certain physiological conditions.
This modulatory role is exerted through the bimodal behavior of the IDE-linked dependence of h20S activity (Fig. 4b), characterized by a decrease over the 5–30 nM IDE concentration range, followed by an activation at (IDE) ≥ 50 nM (see Figs. 3, 4a).
This bimodal behavior suggests the existence of two binding sites for IDE on h20S, whose occupancy by IDE induces different effects on its activity. A molecular modeling approach has allowed to tentatively localize the higher-affinity IDE binding site for h20S in the α-3 subunit, which plays a crucial role in regulating the opening and the closing of the gate [47]. Therefore, the high affinity of IDE for this binding site (1KIDE= 1.3 (± 0.3) × 10−8 M, see Table 2) indeed strongly supports the hypothesis that IDE could efficiently modulate h20S gating mechanisms.
The bimodal modulation of the chymotrypsin-like activity is also observed in wt y20S (Fig. 5a, b), even though the catalytic parameters as well as the IDE interaction parameters differ between human and yeast 20S (see Fig. 5a, b and Table 2), likely reflecting slight structural differences between the two particles. Besides confirming the remarkable degree of conservation of IDE across evolution [31], the similar IDE-linked behavior between human and yeast 20S proteasome indeed suggests that this feature is correlated to a common type of structural change induced by IDE binding. Conversely, in the yeast α-3ΔN mutant (which has been considered to be “locked” in the “open” state) [47], the first inhibitory effect is not observed (see Fig. 5b) showing only an activation process at (IDE) ≥ 30 nM, with an affinity constant overlapping to that observed in the wt yeast 20S (see Table 2).
The lack of an inhibitory effect on the α-3ΔN mutant cannot be attributed to a lowered affinity of IDE for the C-terminus of the α-3 chain, since this portion has been shown not to be affected by the removal of the N-terminus [47]. Therefore, the lack of the inhibitory effect in the putative presence of IDE binding (as suggested by the slight mobility shift of the 20S in the presence of IDE 80 nM) might rather be tentatively referred to the impossibility of the α-3ΔN mutant to undergo the IDE-linked conformational change toward a more “closed” state, which is instead taking place in both human and yeast wt 20S.
Conversely, all three forms investigated (i.e., the human 20S, the yeast wt 20S and its mutant α-3ΔN) show that (IDE) ≥ 30 nM induces a recovery of (at least) two enzymatic activities of the 20S, suggesting that a second binding pocket of IDE indeed exists, even though at this stage its topology cannot be structurally identified by molecular docking.
However, it is very important to outline that the α-3 and α-4 subunits of the 20S are not directly involved in the interface between the α-ring of h20S and the h19S particles (involved in the formation of the 26S) [46]. Hence, the scenario depicted could be more complex at different IDE concentrations depending on the real localization of the IDE binding sites.
Hence, the research herewith presented well fits in the context of different studies which point to a wider contribution of IDE to the regulation of intracellular proteostasis, a view supported, among the different research topics, by the catalytic activity on amyloidogenic substrates, the “dead-end chaperone” hypothesis and the modulation of the UPS [25–30, 32, 65, 66].
Thus, dysregulation of IDE activity and expression could contribute to protein misfolding diseases in different ways spanning from catalytic to extracatalytic mechanisms.
Therefore, to investigate how IDE modulators alter its biological properties and the interaction with the proteasome possibly represent a step necessary to fulfill the unresolved question regarding IDE biology [2, 22, 67].
Furthermore, it has long been debated whether the 20S has a role in degrading proteins regardless of the ubiquitin tag in vivo. Far from being a demonstration of a role of IDE in regulating the activity and the composition of the 20S in vivo, this study opens to the perspective of a novel mechanism of regulation of the 20S [51–56].
From a more pathophysiological standpoint, it is reasonable to hypothesize that the oxidative stress, as well as other stressors, which is reported both to affect the intracellular abundance and distribution of IDE as well as to induce an adaptation of the relative abundance of the proteasome assemblies (i.e., an increase of the pool of free 20S at the expense of the capped particles) could represent the metabolic conditions during which the IDE-mediated effect on the activity and composition of the proteasome assemblies would be operative in vivo.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Prof. Michael Glickman and Prof. Peter van Endert, for several fruitful discussions. We are grateful to Prof. Michael Groll for the generous gift of wt y20S and α-3ΔN mutant proteasome. The financial support from the Italian Ministry of University and Research (MiUR PRIN20157WZM8 to G.G. and S.M.) is gratefully acknowledged.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
All experiments have been carried out according to the current laws of the country where they have been performed.
Footnotes
Diego Sbardella and Grazia R. Tundo contributed equally to this paper.
References
- 1.Kirschner RJ, Goldberg AL. A high molecular weight metalloendoprotease from the cytosol of mammalian cells. J Biol Chem. 1983;258:967–976. [PubMed] [Google Scholar]
- 2.Kurochkin IV, Guarnera E, Berezovsky IN. Insulin-degrading enzyme in the fight against Alzheimer’s Disease. Trends Pharmacol Sci. 2018;39:49–58. doi: 10.1016/j.tips.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Yfanti C, Mengele K, Gkazepis A, Weirich G, Giersig C, Kuo WL, Tang WJ, Rosner M, Schmitt M. Expression of metalloprotease insulin-degrading enzyme insulysin in normal and malignant humantissues. Int J Mol Med. 2008;4:421–431. [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández-Gamba A, Leal MC, Morelli L, Castaño EM. Insulin-degrading enzyme: structure–function relationship and its possible roles in health and disease. Curr Pharm Des. 2009;15:3644–3655. doi: 10.2174/138161209789271799. [DOI] [PubMed] [Google Scholar]
- 5.Sbardella D, Fasciglione GF, Gioia M, Ciaccio C, Tundo GR, Marini S, Coletta M. Human matrix metalloproteinases: an ubiquitarian class of enzymes involved in several pathological processes. Mol Asp Med. 2012;33:119–208. doi: 10.1016/j.mam.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Song ES, Rodgers DW, Hersh LB. Insulin-degrading enzyme is not secreted from cultured cells. Sci Rep. 2018;8:2335. doi: 10.1038/s41598-018-20597-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19:608–624. doi: 10.1210/edrv.19.5.0349. [DOI] [PubMed] [Google Scholar]
- 8.Kuroichkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci. 2001;26:421–425. doi: 10.1016/S0968-0004(01)01876-X. [DOI] [PubMed] [Google Scholar]
- 9.Kurochkin IV, Goto S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345(1):33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 10.Ciaccio C, Tundo GR, Grasso G, Spoto G, Marasco D, Ruvo M, Gioia M, Rizzarelli E, Coletta M. Somatostatin: a novel substrate and a modulator of insulin-degrading enzyme activity. J Mol Biol. 2009;385:1556–1567. doi: 10.1016/j.jmb.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Ralat LA, Kalas V, Zheng Z, Goldman RD, Sosnick TR, Tang WJ. Ubiquitin is a novel substrate for human insulin-degrading enzyme. J Biol Chem. 2011;286:4670–4679. doi: 10.1074/jbc.M110.173252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grasso G, Salomone F, Tundo GR, Pappalardo G, Ciaccio C, Spoto G, Pietropaolo A, Coletta M, Rizzarelli E. Metal ions affect insulin-degrading enzyme activity. J Inorg Biochem. 2012;117:351–358. doi: 10.1016/j.jinorgbio.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Grasso G, Pietropaolo A, Spoto G, Pappalardo G, Tundo GR, Ciaccio C, Coletta M, Rizzarelli E. Copper(I) and copper(II) inhibit Aβ peptides proteolysis by insulin-degrading enzyme differently: implications for metallostasis alteration in Alzheimer’s disease. Chemistry. 2011;17:2752–2762. doi: 10.1002/chem.201002809. [DOI] [PubMed] [Google Scholar]
- 14.Tundo GR, Ciaccio C, Sbardella D, Boraso MS, Viviani B, Coletta M, Marini S. Somatostatin modulates insulin-degrading-enzyme metabolism: implications for the regulation of microglia activity in AD. PLoS One. 2012;7:e34376. doi: 10.1371/journal.pone.0034376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song ES, Jang H, Guo HF, Juliano MA, Juliano L, Morris AJ, Galperin E, Rodgers DW, Hersh LB. Inositol phosphates and phosphoinositides activate insulin-degrading enzyme, while phosphoinositides also mediate binding to endosomes. Proc Natl Acad Sci USA. 2017;114:E2826–E2835. doi: 10.1073/pnas.1613447114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song ES, Ozbil M, Zhang T, Sheetz M, Lee D, Tran D, Li S, Prabhakar R, Hersh LB, Rodgers DW. An extended polyanion activation surface in insulin degrading enzyme. PLoS One. 2015;10:e0133114. doi: 10.1371/journal.pone.0133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochkina EG, Plesneva SA, Vasilev DS, Zhuravin IA, Turner AJ, Nalivaeva NN. Effects of ageing and experimental diabetes on insulin-degrading enzyme expression in male rat tissues. Biogerontology. 2015;16:473–484. doi: 10.1007/s10522-015-9569-9. [DOI] [PubMed] [Google Scholar]
- 18.Tang WJ. Targeting insulin-degrading enzyme to treat type 2 diabetes mellitus. Trends Endocrinol Metab. 2015;27:24–34. doi: 10.1016/j.tem.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu W, Folstein M. Insulin, insulin-degrading-enzyme and amyloid β-peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–198. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Deprez-Poulain R, Hennuyer N, Bosc D, Liang WG, Enée E, Marechal X, Charton J, Totobenazara J, Berte G, Jahklal J, Verdelet T, Dumont J, Dassonneville S, Woitrain E, Gauriot M, Paquet C, Duplan I, Hermant P, Cantrelle FX, Sevin E, Culot M, Landry V, Herledan A, Piveteau C, Lippens G, Leroux F, Tang WJ, van Endert P, Staels B, Deprez B. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat Commun. 2015;6:8250. doi: 10.1038/ncomms9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durham TB, Toth JL, Kimkovski VJ, Cao JX, Siesky AM, Alexander-Chacko J, Wu GY, Dixon JT, McGee JE, Wang J, Guo SY, Cavitt RN, Schindler J, Thibodeaux SJ, Calvert NA, Coghlan MJ, Sinderlar DK, Christe M, Kiselyov VV, Michael MD, Sloop KW. Dual exosite-binding inhibitors of insulin-degrading enzyme challenge its role as the primary mediator of insulin clearance in vivo. J Biol Chem. 2015;290:20044–20059. doi: 10.1074/jbc.M115.638205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tundo GR, Sbardella D, Ciaccio C, Gioia M, Coletta A, Polticelli F, Di Pierro D, Milardi D, Van Endert P, Marini S, Coletta M. Multiple functions of insulin-degrading enzyme: a metabolic crosslight? Crit Rev Biochem Mol Biol. 2017;21:1–29. doi: 10.1080/10409238.2017.1337707. [DOI] [PubMed] [Google Scholar]
- 23.Parmentier N, Stroobant V, Colau D, de Diesbach P, Morel S, Chapiro J, van Endert P, Van den Heynde B. Production of an antigenic peptide by insulin-degrading enzyme. Nat Immunol. 2010;11:449–454. doi: 10.1038/ni.1862. [DOI] [PubMed] [Google Scholar]
- 24.Radulescu RT, Duckworth WC, Levy JL, Fawcett J. Retinoblastoma protein co-purifies with proteasomal insulin-degrading enzyme: implications for cell proliferation control. Biochem Biophys Res Commun. 2010;395:196–199. doi: 10.1016/j.bbrc.2010.03.157. [DOI] [PubMed] [Google Scholar]
- 25.Tundo GR, Sbardella D, Ciaccio C, Bianculli A, Orlandi A, Desimio MG, Arcuri G, Coletta M, Marini S. Insulin-degrading enzyme (IDE): a novel heat shock-like protein. J Biol Chem. 2013;288:2281–2289. doi: 10.1074/jbc.M112.393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sbardella D, Tundo GR, Sciandra F, Bozzi M, Gioia M, Ciaccio C, Tarantino U, Brancaccio A, Coletta M, Marini S. 26S proteasome activity is affected by fluctuations in insulin-degrading enzyme distribution. PLoS One. 2015;10:e0132455. doi: 10.1371/journal.pone.0132455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tundo GR, Sbardella D, De Pascalis SA, Ciaccio C, Coletta M, Fanizzi FP, Marini S. Novel platinum(II) compounds modulate insulin-degrading enzyme activity and induce cell death in neuroblastoma cells. J Biol Inorg Chem. 2015;20:101–108. doi: 10.1007/s00775-014-1217-3. [DOI] [PubMed] [Google Scholar]
- 28.Hamel FG, Bennett RG, Duckworth WC. Regulation of multicatalytic enzyme activity by insulin and the insulin-degrading enzyme. Endocrinology. 1998;139:4061–4066. doi: 10.1210/endo.139.10.6256. [DOI] [PubMed] [Google Scholar]
- 29.Bennett RG, Hamel FG, Duckworth WC. Insulin inhibits the ubiquitin-dependent degrading activity of the 26S proteasome. Endocrinology. 2000;141:2508–2517. doi: 10.1210/endo.141.7.7575. [DOI] [PubMed] [Google Scholar]
- 30.Bennett RG, Fawcett J, Kruer MC, Duckworth WC, Hamel FG. Insulin inhibition of the proteasome is dependent on degradation of insulin-degrading enzyme. J Endocrinol. 2003;177:399–405. doi: 10.1677/joe.0.1770399. [DOI] [PubMed] [Google Scholar]
- 31.Beuzelin C, Evnouchidou I, Rigolet P, Cauvet-Burgevin A, Girard PM, Dardalhon D, Culina S, Gdoura A, van Endert P, Francesconi S. Deletion of the fission yeast homologue of human insulinase reveals a TORC1-dependent pathway mediating resistance to proteotoxic stress. PLoS One. 2013;8:e67705. doi: 10.1371/journal.pone.0067705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grasso G, Lanza V, Malgieri G, Fattorusso R, Pietropaolo A, Rizzarelli E, Milardi D. The insulin degrading enzyme activates ubiquitin and promotes the formation of K48 and K63 diubiquitin. Chem Commun (Camb) 2015;51:15724–15727. doi: 10.1039/C5CC06786C. [DOI] [PubMed] [Google Scholar]
- 33.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 34.Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 35.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochem Biophys Acta. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Köhler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell. 2011;7:1143–1152. doi: 10.1016/S1097-2765(01)00274-X. [DOI] [PubMed] [Google Scholar]
- 38.Groll M, Nazif T, Huber R, Bogyo M. Probing structural determinants distal to the site of hydrolysis that control substrate specificity of the 20S proteasome. Chem Biol. 2002;9:655–662. doi: 10.1016/S1074-5521(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 39.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008;30:360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009;10:104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- 41.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santoro AM, Cunsolo A, D’Urso A, Sbardella D, Tundo GR, Ciaccio C, Coletta M, Diana D, Fattorusso R, Persico M, Di Dato A, Fattorusso C, Milardi D, Purrello R. Cationic porphyrins are tunable gatekeepers of the 20S proteasome. Chem Sci. 2016;7:1286–1297. doi: 10.1039/C5SC03312H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Förster F, Baumeister W. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci USA. 2016;113:7816–7821. doi: 10.1073/pnas.1608050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sledź P, Förster F, Baumeister W. Allosteric effects in the regulation of 26S proteasome activities. J Mol Biol. 2013;425:1415–1423. doi: 10.1016/j.jmb.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 45.Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.da Fonseca PCA, He J, Morris EP. Molecular model of the human 26S proteasome. Mol Cell. 2012;46:54–66. doi: 10.1016/j.molcel.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 47.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 48.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/S1097-2765(02)00638-X. [DOI] [PubMed] [Google Scholar]
- 50.Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002;21:3516–3525. doi: 10.1093/emboj/cdf333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glickman MH, Raveh D. Proteasome plasticity. FEBS Lett. 2005;579:3214–3223. doi: 10.1016/j.febslet.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 52.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011;41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335:637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/S0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 55.Grune T, Catagol B, Licht A, Ermak G, Pickering AM, Ngo JK, Davies JK. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raynes R, Pomatto LC, Davies KJ. Degradation of oxidized proteins by the proteasome: distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol Asp Med. 2016;50:41–55. doi: 10.1016/j.mam.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livnat-Levanon N, Kevei É, Kleifeld O, Krutauz D, Segref A, Rinaldi T, Erpapazoglou Z, Cohen M, Reis N, Hoppe T, Glickman MH. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014;7:1371–1380. doi: 10.1016/j.celrep.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 58.Elsasser S, Schmidt M, Finley D. Characterization of the proteasome using native gel electrophoresis. Methods Enzymol. 2005;398:353–363. doi: 10.1016/S0076-6879(05)98029-4. [DOI] [PubMed] [Google Scholar]
- 59.Ritchie DW, Venkatraman V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics. 2010;26:2398–2405. doi: 10.1093/bioinformatics/btq444. [DOI] [PubMed] [Google Scholar]
- 60.Shen Y, Joachimiak A, Rosner MR, Tang WJ. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006;443:870–874. doi: 10.1038/nature05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 62.Bousquet-Dubouch MP, Baudelet E, Guerin F, Matondo M, Uttenweller-Joseph S, Burlet-Schiltz O, Monsarrat B. Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol Cell Proteom. 2009;8:1150–1164. doi: 10.1074/mcp.M800193-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fabre B, Lambour T, Garrigues L, Ducoux-Petit M, Amalric F, Monsarrat B, Burlet-Schiltz O, Bousquet-Dubouch MP. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J Proteome Res. 2014;13:3027–3037. doi: 10.1021/pr500193k. [DOI] [PubMed] [Google Scholar]
- 64.Neelam S, Kakhniashvili DG, Wilkens S, Levene SD, Goodman SR. Functional 20S proteasomes in mature human red blood cells. Exp Biol Med. 2011;236:580–591. doi: 10.1258/ebm.2011.010394. [DOI] [PubMed] [Google Scholar]
- 65.de Tullio MB, Castelletto V, Hamley IW, Martino Adami PV, Morelli L, Castaño EM. Proteolytically inactive insulin-degrading enzyme inhibits amyloid formation yielding non-neurotoxic aβ peptide aggregates. PLoS One. 2013;8:e59113. doi: 10.1371/journal.pone.0059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma SK, Chorell E, Steneberg P, Vernersson-Lindahl E, Edlund H, Wittung-Stafshede P. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci Rep. 2015;5:12531. doi: 10.1038/srep12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hersh LB. The insulysin (insulin degrading enzyme) enigma. Cell Mol Life Sci. 2006;63:2432–2434. doi: 10.1007/s00018-006-6238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.