

Abstract
Over 60% of relapsed/refractory (R/R) large B‐cell lymphoma (LBCL) patients who receive chimeric antigen receptor (CAR) T cells will experience disease progression. There is no standard next line of therapy and information in this setting is scarce and heterogeneous. We analyzed 387 R/R LBCL patients who progressed after CAR T cells from July 2018 until March 2022 in Spain and the United Kingdom. Median overall survival (OS) was 5.3 months, with significant differences according to the interval between infusion and progression (<2 months [1.9 months], 2–6 months [5.2 months], and >6 months [not reached]). After progression, 237 (61%) patients received treatment. Focusing on the first subsequent therapy, overall (complete) response rates were 67% (38%) for polatuzumab–bendamustine–rituximab (POLA), 51% (36%) for bispecific antibodies (BsAb), 45% (35%) for radiotherapy (RT), 33% (26%) for immune checkpoint inhibitors (ICIs), 25% (0%) for lenalidomide (LENA), and 25% (14%) for chemotherapy (CT). In terms of survival, 12‐month progression‐free survival and OS was 36.2% and 51.0% for POLA, 32.0% and 50.1% for BsAb, 30.8% and 37.5% for RT, 29.9% and 27.8% for ICI, 7.3% and 20.8% for LENA, and 6.1% and 18.3% for CT. Thirty‐two (14%) patients received an allogeneic hematopoietic cell transplant with median OS not reached after a median follow‐up of 15.1 months. In conclusion, patients with R/R LBCL who progress within the first 2 months after CAR T‐cell therapy have dismal outcomes. Novel targeted agents, such as polatuzumab and BsAbs, can achieve prolonged survival after CAR T‐cell therapy failure.
INTRODUCTION
Chimeric antigen receptor (CAR) T‐cell therapy targeting CD19 has shown durable responses in 30%–40% of patients with relapsed/refractory (R/R) large B‐cell lymphoma (LBCL) in the third or later line setting. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 Therefore, more than 60% of infused patients will progress and require additional treatment options.
There is no standard next line of treatment for patients who progress after CAR T‐cell therapy and long‐term outcomes are dismal. 12 , 13 , 14 Clinical trials are favored in this context, but not all patients meet inclusion criteria nor have access in their local centers. 15 Outside of clinical trials, conventional chemotherapy (CT) has traditionally been the standard option, with very poor results. Recently, novel antibodies and antibody–drug conjugates targeting CD19 (tafasitamab, loncastuximab tesirine), CD79b (polatuzumab vedotin), and CD20/CD3 (epcoritamab, glofitamab), amongst others, have been approved for R/R LBCL patients. 16 , 17 , 18 , 19 , 20 Some of the pivotal trials for these agents included patients with prior CAR T‐cell therapy, with similar outcomes to CAR‐naïve patients. However, the number of CAR‐exposed patients in these trials was low, and real‐world results are scarce and heterogeneous. 12 , 13 , 21 Data on optimal sequencing are warranted to inform decision‐making in this rising patient population, especially in light of the recent approval of CAR T‐cell therapy for second line in refractory or early relapsing LBCL patients. 22 , 23
Here, we report outcomes of R/R LBCL patients who progressed after CD19‐targeted CAR T‐cell therapy in the third or later line setting, and provide a detailed analysis of response and survival after the first‐line subsequent regimen.
METHODS
Patients
We conducted a retrospective, international study including patients from 20 centers in Spain and the United Kingdom with R/R LBCL who experienced disease progression after third or later line treatment with axicabtagene ciloleucel (axi‐cel), tisagenlecleucel (tisa‐cel), or lisocabtagene maraleucel (liso‐cel) from July 2018 until March 2022.
The response was locally assessed according to Lugano criteria 24 and grading of cytokine release syndrome (CRS) and immune effector cell‐associated neurotoxicity syndrome (ICANS) after CAR T‐cell therapy was performed following the American Society for Transplantation and Cellular Therapy criteria. 25 The study was approved by the ethics committee of the Vall d'Hebron University Hospital (PR[AG]404/2020).
Definitions and endpoints
Overall survival (OS1) was defined as the time from disease progression after CAR T‐cell therapy until death from any cause. OS1 was analyzed in the full patient population, according to receipt of treatment after progression (“NT group” [palliative management] vs. “T group” [active treatment]), and depending on the time interval from CAR T‐cell infusion to disease progression (<2, 2–6, and >6 months). A second OS assessment (OS2), restricted to patients from the T group, was defined as the time from the first subsequent treatment start until death from any cause. Progression‐free survival (PFS) was assessed for the T group and defined as the time from the start of the first subsequent treatment until disease progression or death from any cause. Response rates were reported for each regimen, including overall response rate (ORR, complete and partial response) and complete response (CR).
Statistical analyses
A descriptive analysis was performed to examine the baseline variables of all patients who experienced disease progression after CAR T‐cell therapy. Categorical variables were reported with frequencies, and percentages and numerical variables with median and interquartile range (IQR). To determine significant differences between the two cohorts, a logistic regression model and the Wald test were employed to calculate p values for numerical variables, while a test of proportions was used for categorical variables.
Response rates were reported with percentages for each treatment group and a test of proportions was conducted to calculate the p value and identify significant differences between the groups.
Survival endpoints, including PFS and OS, were analyzed with the Kaplan–Meier method and the results were presented with their corresponding 95% confidence intervals (95% CI). Patients who underwent an allogeneic hematopoietic cell transplant (allo‐HCT) were censored at the time of transplant. To assess differences in survival endpoints, univariable and multivariable Cox proportional hazard models were utilized, and hazard ratios with 95% CI and p values were reported.
To address missing values for variables included in multivariable analysis, a Multivariate Imputation via Chained Equations method 26 was employed, which imputed missing values completely at random. This approach was implemented to prevent the omission of cases and ensure a robust analysis. All statistical analyses were conducted using R software version 4.2.2.
RESULTS
Characteristics of the full patient population
The study included 387 R/R LBCL patients with disease progression after CAR T‐cell therapy at a median of 2.6 months (IQR: 1.0–3.3) from infusion. Of these, 237 (61%) patients received subsequent treatment (T group); the remaining 150 (39%) received the best supportive care or palliative therapy such as single‐agent steroids or low‐dose CT (NT group). Baseline variables of the full patient population are summarized in Table 1. In terms of patient and lymphoma characteristics at the time of CAR T‐cell therapy, the NT group was older (63 vs. 58 years; p < 0.01), had a worse performance status (Eastern Cooperative Oncology Group [ECOG] ≥1, 78% vs. 60%; p < 0.01), and was enriched with patients who presented a high‐risk International Prognostic Index score (62% vs. 46%; p < 0.01) and an increased lactate dehydrogenase (82% vs. 66%; p < 0.01), in comparison with the T group. Focusing on CAR T‐cell‐related data, patients in the NT cohort had experienced higher rates of grade ≥3 CRS (13% vs. 4%; p < 0.01), any grade ICANS (37% vs. 23%; p < 0.01), and grade ≥3 ICANS (19% vs. 7%; p < 0.01) in comparison with the T cohort. The former also included an increased rate of early progressors (<2 months, 57% vs. 35%; p < 0.01) compared to the T cohort.
Table 1.
Baseline characteristics of the full patient population.
| All patients, N = 387 | No treatment (NT cohort), N = 150 | Treatment (T cohort), N = 237 | p Value | |
|---|---|---|---|---|
| Variables at the time of CAR T‐cell therapy | ||||
| Median age (years) (IQR) | 60 (50–69) | 63 (54–71) | 58 (48–67) | <0.01 |
| >70 years, n (%) | 77 (20) | 42 (28) | 35 (15) | <0.01 |
| Male gender, n (%) | 253 (65) | 98 (65) | 155 (65) | 1 |
| ECOG ≥ 1, n (%) | 258 (67) | 116 (78) | 142 (60) | <0.01 |
| Histology, n (%) | ||||
| DLBCL | 252 (65) | 97 (65) | 155 (65) | Ref. |
| HGBL | 56 (14) | 21 (14) | 35 (15) | 0.89 |
| PMBL | 19 (5) | 5 (4) | 14 (6) | 0.30 |
| THRLBCL | 14 (4) | 8 (5) | 6 (3) | 0.17 |
| tFL | 37 (10) | 15 (10) | 22 (9) | 0.81 |
| tMZL | 8 (2) | 3 (2) | 5 (2) | 0.95 |
| Primary refractory, n (%) | 228 (60) | 86 (59) | 142 (61) | 0.69 |
| IPI score 3–5, n (%) | 193 (53) | 88 (62) | 105 (46) | <0.01 |
| Bulky (>7.5 cm), n (%) | 125 (33) | 52 (35) | 73 (32) | 0.50 |
| >2 previous lines, n (%) | 165 (44) | 66 (45) | 99 (43) | 0.74 |
| Previous auto‐HCT, n (%) | 80 (21) | 27 (18) | 53 (22) | 0.37 |
| LDH > ULN, n (%) | 254 (72) | 111 (82) | 143 (66) | <0.01 |
| >2 × ULN, n (%) | 101 (29) | 57 (42) | 44 (20) | <0.01 |
| CAR T‐cell‐related characteristics, n (%) | ||||
| Bridging treatment | 338 (88) | 134 (90) | 204 (86) | 0.43 |
| Construct | ||||
| Axi‐cel | 202 (52) | 75 (50) | 127 (54) | Ref. |
| Tisa‐cel | 165 (43) | 69 (46) | 96 (41) | 0.36 |
| Liso‐cel | 19 (5) | 6 (4) | 13 (5) | 0.63 |
| CRS any grade | 306 (79) | 124 (83) | 182 (77) | 0.21 |
| Grade ≥3 | 29 (7) | 19 (13) | 10 (4) | <0.01 |
| ICANS any grade | 110 (28) | 56 (37) | 54 (23) | <0.01 |
| Grade ≥3 | 45 (12) | 28 (19) | 17 (7) | <0.01 |
| Best response to CAR‐T | ||||
| CR/PR | 201 (52) | 59 (39) | 142 (60) | Ref. |
| SD/PD | 186 (48) | 91 (61) | 95 (40) | <0.01 |
| Time from CAR‐T to PD | ||||
| <2 months | 169 (44) | 86 (57) | 83 (35) | <0.01 |
| 2–6 months | 172 (45) | 55 (37) | 120 (51) | <0.01 |
| >6 months | 43 (11) | 9 (6) | 34 (14) | 0.02 |
Note: Missing data in the following variables: Histology (10, 3%), previous lines (10, 3%), primary refractory lymphoma (7, 2%), IPIs (21, 5%), construct (1, 1%), bridging (2, 1%), ECOG (4, 1%), LDH (36, 9%), bulky disease (8, 2%), ICANS grade (1, 1%), time from CAR‐T infusion to progression (1, 1%). Bold values indicate statistically significant results.
Abbreviations: Axi‐cel, axi‐cel; CAR‐T, chimeric antigen receptor T cells; CR, complete response; CRS, cytokine release syndrome; DLBCL, diffuse large B‐cell lymphoma; ECOG, Eastern Cooperative Oncology Group; HCT, hematopoietic cell transplant; HGBL, high‐grade B‐cell lymphoma; ICANS, immune effector cell‐associated neurotoxicity syndrome; IQR, interquartile range; IPI, International Prognostic Index; LDH, lactate dehydrogenase; Liso‐cel, lisocabtagene maraleucel; PD, progressive disease; PMBL, primary mediastinal B‐cell lymphoma; PR, partial response; Ref., reference; SD, stable disease; tFL, transformed follicular lymphoma; THRLBCL, T‐cell/histiocyte‐rich large B‐cell lymphoma; Tisa‐cel, tisagenlecleucel; tMZL, transformed marginal zone lymphoma; ULN, upper limit of normal.
Outcomes of the full patient population
With a median follow‐up of 20.4 months (95% CI: 17.8–23.2) since progression to CAR T‐cell therapy, the median OS1 was 5.3 months (95% CI: 4.4–6.2) (Figure 1A) and the 12‐month OS1 was 31% (95% CI: 26–36). Patients who progressed within 2 months of infusion had a significantly shorter median OS1 (N = 169 [44%], 1.9 months [95% CI: 1.6–2.5]) in comparison to patients who progressed from 2 to 6 months after infusion (N = 175 [45%], 5.2 months [95% CI: 4.3–6.7]) and more than 6 months after infusion (N = 43 [11%], median not reached) (Figure 1B). These survival differences were maintained when the analysis was restricted to the T group (data not shown) and when it was conducted separately for each CAR‐T product (Supporting Information: Figure S1). The cohort of patients who progressed within 2 months of the CAR T‐cell infusion, enriched with tisa‐cel recipients (Supporting Information: Table S1), included a significantly lower proportion of patients receiving subsequent treatment (49% vs. 72%, p < 0.01), in comparison to patients who progressed at a later timepoint.
Figure 1.
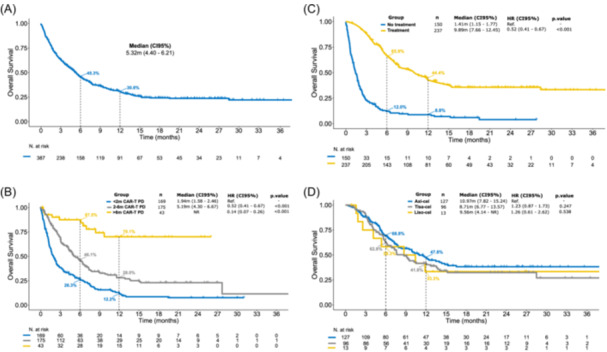
Overall survival (OS) for patients who progressed after chimeric antigen receptor (CAR) T‐cell therapy. (A) Global OS for the full patient population (N = 387). (B) OS depending on the time from CAR T‐cell infusion to disease progression, including patients who progressed within 2 months (N = 175), between 2 and 6 months (N = 169), and beyond 6 months of infusion (N = 43). (C) OS curves for patients who received treatment (N = 237) and for patients who only received palliative treatment or best supportive care (N = 150). (D) OS curves according to CAR T‐cell construct, including axicabtagene ciloleucel (N = 127), tisagenlecleucel (N = 96), and lisocabtagene maraleucel (N = 13); this latter analysis was restricted to patients who received subsequent treatment after progression to CAR T‐cell therapy. CAR‐T‐PD, time interval between CAR T‐cell infusion and disease progression.
Focusing on treatment after CAR T‐cell failure, patients in the NT group had a significantly shorter median OS1 (1.4 months [95% CI: 1.2–1.8]) in comparison to patients in the T group (9.9 months [95% CI: 7.7–12.5]; p < 0.01). The 6‐ and 12‐month OS1 for the NT and T groups was 12% (95% CI: 8–19) versus 66% (95% CI: 60–72) and 9% (95% CI: 5–15) versus 44% (95% CI: 38–52), respectively (Figure 1C). For patients in the T group, median OS1 was comparable across CAR T‐cell constructs (10.97 months for axi‐cel vs. 8.71 months for tisa‐cel vs. 9.56 months for liso‐cel, p = NS [not significant]) (Figure 1D).
First‐line treatment after progression from CAR T‐cell therapy
Descriptive analysis and response rates
Among the 237 patients in the T group, the median number of lines after CAR T‐cell progression was 1 (IQR: 1–2, range: 1–5 lines of therapy) during our study period. The first‐line regimens included polatuzumab–rituximab–bendamustine (POLA) in 67 (28%) patients, bispecific antibodies (BsAb) in 34 (14%), radiotherapy (RT) in 37 (16%), immune checkpoint inhibitors (ICI) in 28 (12%), lenalidomide (LENA)‐containing regimens in 23 (10%; Supporting Information: Table S2), conventional CT in 41 (17%), and other treatments in seven (3%) patients (Supporting Information: Table S3). Further details of the regimens included in each cohort are available in Supporting Information: Figure S2, and the baseline characteristics of each treatment group at the time of CAR T‐cell therapy and at the time of disease progression are summarized in Supporting Information: Table S4. Among the 211 (89%) patients evaluable for disease response after the first‐line subsequent treatment, ORR (CR) was 67% (38%) for POLA, 51% (36%) for BsAb, 45% (35%) for RT, 33% (26%) for ICIs, 25% (0%) for LENA, and 25% (14%) for CT (Figure 2).
Figure 2.
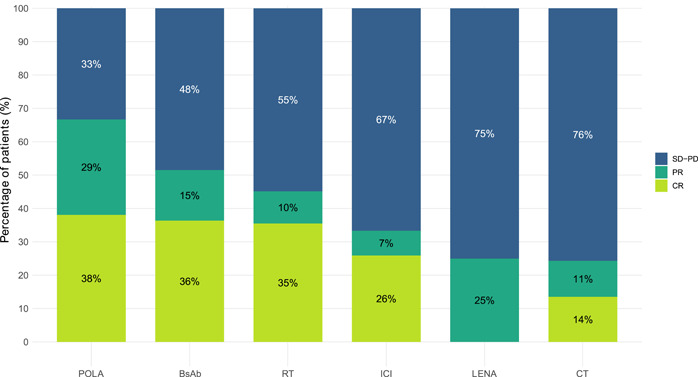
Response rate for the different regimens administered as a first‐line treatment after progression from CAR T‐cell therapy. BsAbs, bispecific antibodies; CR, complete response; CT, chemotherapy; ICI, immune checkpoint inhibitors; LENA, lenalidomide; PD, progressive disease; POLA, polatuzumab–bendamustine–rituximab; PR, partial response; RT, radiotherapy; SD, stable disease.
Survival outcomes after first‐line subsequent therapy
In terms of the T cohort, the median PFS (mPFS) was 3.6 months and the 12‐month PFS was 26% (Figure 3A). Patients who progressed within 2 months of the CAR T‐cell infusion had a significantly shorter mPFS (2.7 months) in comparison to patients who progressed between 2 to 6 months (4.0 months [hazard ratio, HR 0.58, 95% CI: 0.4–0.8]) and later than 6 months (not reached [HR 0.25, 95% CI: 0.1–0.5]) (Supporting Information: Figure S3). Concerning disease histology, patients with high‐grade B‐cell lymphoma had a significantly shorter PFS with the first subsequent treatment in comparison to patients with transformed LBCL (HR 0.53 [95% CI: 0.29–0.98]; p = 0.044) (Supporting Information: Figure S4). Regarding the specific regimens, mPFS (12‐month PFS) was 7.5 months (36.2%) for POLA, 5.3 months (32.0%) for BsAb, 4.8 months (30.8%) for RT, 3.1 months (29.9%) for ICIs, 2.9 months (7.3%) for LENA, and 1.6 months (6.1%) for CT (Figure 3B and Table 2). Focusing on the subgroup of patients who progressed within 2 months of the CAR T‐cell infusion, PFS was comparable across the different subsequent treatment regimens (Supporting Information: Figure S5). In the MVA for all patients from the T cohort, a better pre‐CAR T‐cell therapy performance status (ECOG 0), a longer time interval from CAR T‐cell infusion to disease progression (>6 months), and first‐line subsequent treatment with POLA were associated with a longer PFS (Figure 4A).
Figure 3.
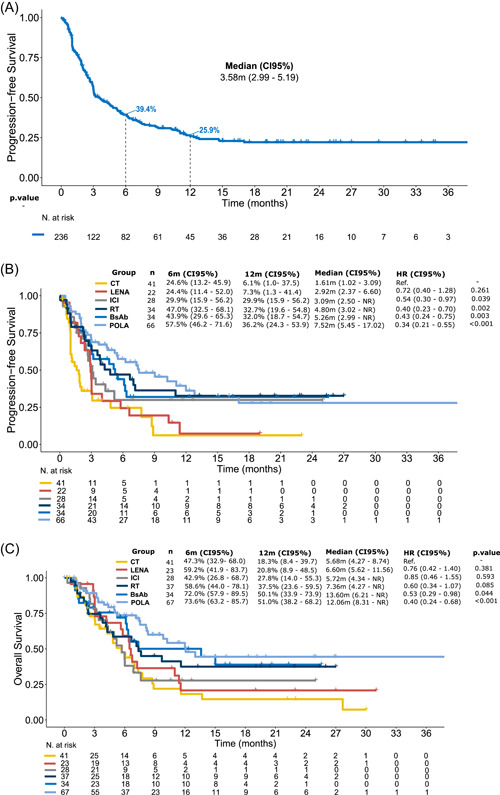
Survival outcomes for patients who received treatment after chimeric antigen receptor T‐cell therapy progression. (A) Progression‐free survival (PFS) for all patients who received treatment (N = 237). (B) PFS according to the type of first subsequent therapy. (C) Overall survival according to the type of first subsequent therapy. BsAbs, bispecific antibodies; CI, confidence interval; CT, chemotherapy; HR, hazard ratio; ICI, immune checkpoint inhibitors; LENA, lenalidomide; POLA, polatuzumab–bendamustine–rituximab; RT, radiotherapy.
Table 2.
Survival outcomes among the different treatment groups.
| Progression‐free survival | Overall survival | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | Median months (95% CI) | HR (95% CI) | p Value | 12‐month PFS (95% CI) | N | Median months (95% CI) | HR (95% CI) | p Value | 12‐month OS (95% CI) | |
| CT | 41 | 1.6 (1.0–3.1) | Ref. | Ref. | 6.1% (1.0–37.5) | 41 | 5.7 (4.3–8.7) | Ref. | Ref. | 18.3% (8.4–39.7) |
| POLA | 66 | 7.5 (5.5–17.0) | 0.3 (0.2–0.6) | <0.01 | 36.2% (24.3–53.9) | 67 | 12.1 (8.3–NR) | 0.4 (0.2–0.7) | <0.01 | 51.0% (38.2–68.2) |
| BsAB | 34 | 5.3 (3.0–NR) | 0.4 (0.2–0.8) | <0.01 | 32.0% (18.7–54.7) | 34 | 13.6 (6.2–NR) | 0.5 (0.3–1.0) | 0.04 | 50.1% (33.9–73.9) |
| RT | 34 | 4.8 (3.0–NR) | 0.4 (0.2–0.7) | <0.01 | 30.8% (18.3–51.9) | 37 | 7.4 (4.3–NR) | 0.6 (0.3–1.1) | 0.09 | 37.5% (23.6–59.5) |
| ICI | 28 | 3.1 (2.5–NR) | 0.5 (0.3–1.0) | 0.04 | 29.9% (15.9–56.2) | 28 | 5.7 (4.3–NR) | 0.9 (0.5–1.6) | 0.59 | 27.8% (14.0–55.3) |
| LEN | 22 | 2.9 (2.4–6.6) | 0.7 (0.4–1.3) | 0.26 | 7.3% (1.3–41.4) | 23 | 6.6 (5.6–11.6) | 0.8 (0.4–1.4) | 0.38 | 20.8% (8.9–48.5) |
Note: Bold values indicate statistically significant results.
Abbreviations: BsAB, bispecific antibodies; CI, confidence interval; CT, chemotherapy; HR, hazard ratio; ICI, immune checkpoint inhibitors; LEN, lenalidomide; N, number of patients evaluable for each survival endpoint; NR, not reported; OS, overall survival; PFS, progression‐free survival; POLA, polatuzumab‐bendamustine‐rituximab; Ref., reference; RT, radiotherapy.
Figure 4.
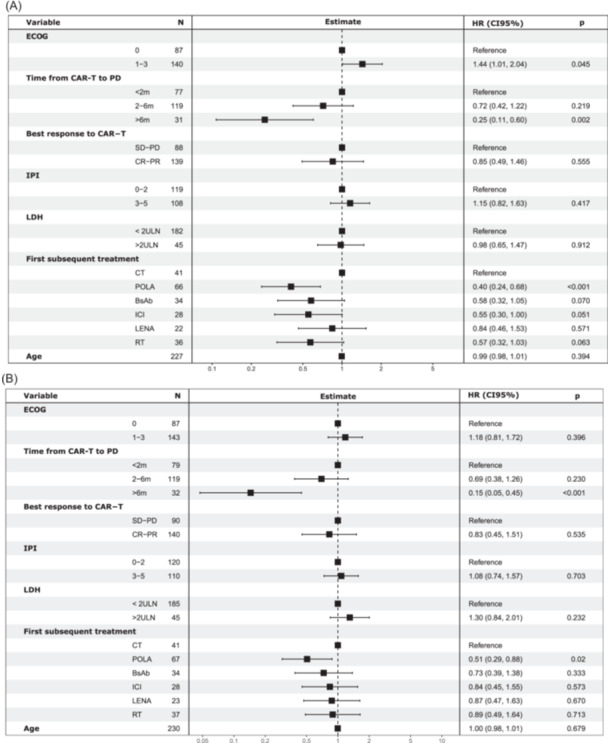
Multivariate analysis (MVA) for patients receiving treatment after CAR T‐cell therapy progression. (A) MVA for progression‐free survival. (B) MVA for overall survival. Footnote shows values of ECOG, LDH, and IPI at the time of CAR T‐cell therapy. BsAbs, bispecific antibodies; CI, confidence interval; CR, complete response; CT, chemotherapy; ECOG, Eastern Cooperative Oncology Group; ICI, immune checkpoint inhibitors; IPI, International Prognostic Index; LDH, lactate dehydrogenase; LENA, lenalidomide; PD, progressive disease; POLA, polatuzumab–bendamustine–rituximab; PR, partial response; RT, radiotherapy; SD, stable disease; ULN, upper limit of normal.
Median OS2 (12‐month OS) was 12.1 months (51.0%) for POLA, 13.6 months (50.1%) for BsAbs, 7.4 months (37.5%) for RT, 5.7 months (27.8%) for ICIs, 6.6 months (20.8%) for LENA, and 5.7 months (18.3%) for CT (Figure 3C and Table 2). In the MVA, a longer time interval from CAR T‐cell infusion to disease progression (>6 months) and subsequent treatment with POLA retained a favorable prognostic impact on OS (Figure 4B).
Second‐line treatment after progression from CAR T‐cell therapy
In our data set, 51 patients (22% among the 237 patients in the T group) received second‐line treatment after CAR‐T failure. Of these, 3 (6%) received BsAb, 16 (31%) CT, 15 (29%) polatuzumab‐containing strategies, 2 (4%) ICIs, 3 (6%) LENA‐based regimens, 3 (6%) RT, and 9 (18%) other treatments. The response rate to the second line of treatment was low and survival was limited for most patients (Supporting Information: Figure S6). Only 14 patients received third‐line or later subsequent treatment (data not shown).
Role of allogeneic stem cell transplant consolidation
Of the 237 patients from the T group, 32 (14%) received an allogeneic stem cell transplant (allo‐HCT) consolidation after subsequent treatment for CAR‐T failure. Patients included in the allo‐HCT cohort were significantly younger (median 48 vs. 60 years; p < 0.01) and had a better performance status (ECOG > 1, 38 vs. 64%; p < 0.01) in comparison with the rest of the T group. Additional baseline characteristics are summarized in Supporting Information: Table S5.
The treatment regimen which served as a bridge to allo‐HCT included POLA (N = 13, 41%), CT (N = 9, 28%), ICIs (N = 4, 12%), BsAbs (N = 3, 9%), and RT (N = 3, 9%). Most patients were in CR (N = 21, 66%) or PR (N = 10, 31%) at the time of the allo‐HCT; 1 (3%) patient was in PD prior to transplant. With a median follow‐up of 15.1 months (95% CI: 13.1–22.3) since the HCT infusion, the median OS was not reached with a 12‐month OS of 84% (Supporting Information: Figure S7). At data cutoff, 7 (22%) patients had died from PD (N = 4), cytomegalovirus encephalitis (N = 1), graft‐versus‐host disease (N = 1), and other causes (N = 1).
DISCUSSION
This is the largest study to date focused on the outcomes of patients with R/R LBCL who progress after CD19‐targeted CAR T‐cell therapy. We describe distinct survival patterns according to the time of disease progression after CAR T‐cell infusion and identify treatment strategies associated with better response rates in this setting.
Our first aim was to analyze survival outcomes in this large, international cohort of patients who progressed after CAR T‐cell therapy. Median OS after progression to CAR T‐cell therapy in our cohort (5.3 months) was similar to previous publications which included this endpoint (5.3–9.6 months). 12 , 21 , 27 , 28 In line with other studies, we confirmed that patients with an early progression after CAR T‐cell therapy had a dismal outcome, irrespective of the subsequent treatment they received. 12 , 21 , 27 However, this was the first study to include a cohort of patients with a late relapse, beyond 6 months of infusion. This group had a particularly good outcome, exceeding the survival reported for patients progressing beyond 90 days from CAR T‐cell infusion in the French study (median OS not reached vs. 9.6 months). 12
In terms of patient management after progression to CAR T‐cell therapy, over one‐third of our cohort did not receive any subsequent treatment, with a median OS of only 1.4 months. This reflects the rapid clinical deterioration in a multiply relapsed patient population and will potentially improve as this cellular therapy moves up in the treatment algorithm of LBCL.
Focusing on the patients who were candidates for subsequent therapy, the POLA subgroup is the largest reported to date in the post‐CAR T‐cell therapy setting. Data in this particular context are essential given the pivotal trial did not include patients with prior CAR T‐cell therapy. 18 , 29 The CR rate observed in our study was similar to multicenter US results (38% vs. 33%–34%) 13 , 21 and the pivotal trial (CRR 39%). 29 Conversely, another retrospective real‐world publication reported a significantly lower CR rate (14% vs. 38%) and a shorter mPFS (2.5 vs. 7.5 months) after CAR T‐cell progression in comparison with our cohort. 30 These differences could have been driven by distinct baseline characteristics, together with the omission of bendamustine in up to 40% of patients in this latter report. Noteworthy, in our study, this regimen was the most common bridge to an allo‐HCT, in line with a recent report focused exclusively on allo‐HCT outcomes after CAR T‐cell therapy progression. 14 Efficacy results in our BsAb cohort were similar to the CAR‐exposed patients in the pivotal trials of glofitamab and epcoritamab. 20 , 31 In comparison to other retrospective reports focusing on post‐CAR T‐cell treatment strategies, 12 , 21 patients who received BsAbs in our study showed a better CR rate (36% vs. 14%–20%) and improved survival outcomes (mPFS of 5.3 vs. 3.7–3.9 months). Interestingly, very few patients from this cohort underwent an allo‐HCT consolidation, possibly linked to the prolonged treatment schedule and durable responses reported after treatment discontinuation. 32 Noteworthy, the patients from this cohort received treatment in the setting of a clinical trial; however, we observed that most baseline characteristics were comparable with the other treatment groups. The favorable response rate observed with RT was potentially associated with a more localized relapse; only 42% had an advanced stage of disease at progression to CAR T‐cell therapy, in comparison to 75% for the other cohorts (Supporting Information: Table S4). Regarding the ICI group, the rationale for this therapy after CAR‐T failure is based on its potential to reactivate exhausted CAR T cells, counteracting an immunosuppressive tumor microenvironment. 33 , 34 Small case series of ICIs as subsequent treatment after CAR T‐cells 35 and clinical trials combining CAR‐T with ICIs have reported modest clinical efficacy. 36 , 37 In terms of our ICI cohort, the CR rate was similar to other reports in the same setting (26% vs. 25%) 21 and higher than the large US multicenter study recently published (N = 96, ORR 19%, CR 10%). 38 Noteworthy, even though this group had a comparable PFS to the POLA and BsAb patients, OS was significantly shorter. Patients who received ICI had negative prognostic features in comparison to other treatment groups, such as a shorter time interval from CAR T‐cell infusion to disease progression, possibly precluding additional treatment after ICI failure. Finally, patients receiving CT and LENA‐based regimens as the next line of therapy had low response rates and short survival. Two retrospective US studies in the post‐CAR‐T setting showed encouraging CR rates (29%–33%) with LENA as subsequent therapy, 13 , 21 supported by preclinical data suggesting immunomodulatory activity which could mitigate CAR T‐cell exhaustion. 39 , 40 However, the French study included the largest cohort to date with this agent (N = 59) and also reported low response rates (ORR 11%, CR 7%). Considering the limited survival in the ICIs, LENA, and CT cohorts, patients who achieve a response with these agents and meet additional required criteria could be considered for an allo‐HCT, taking into account the favorable long‐term outcomes observed in this setting. 14
The main limitation of this study is derived from its retrospective nature, preventing a completely unbiased distribution of subsequent treatment selection at the different participating centers. Also, some clinical and laboratory variables at the time of disease progression after CAR T‐cell therapy were not available. Longer follow‐up is warranted to inform on response duration for each treatment in this particular setting.
In conclusion, this large multicenter cohort demonstrates the poor outcomes of patients with LBCL progressing after CAR T‐cell therapy, particularly those with very early progression. Treatment with rituximab–bendamustine–polatuzumab and BsAbs can achieve prolonged survival after CAR‐T failure. For selected patients achieving a response to subsequent therapy, consolidation with an all‐HCT could be considered.
AUTHOR CONTRIBUTIONS
Conception and design: Gloria Iacoboni, Pau Abrisqueta, and Andrea Kuhnl. Provision of study patients: Gloria Iacoboni, Maeve O'Reilly, Tobias Menne, Mi Kwon, Ana África Martín‐López, Sridhar Chaganti, Javier Delgado, Claire Roddie, Ariadna Pérez, Jane Norman, Manuel Guerreiro, Adam Gibb, Ana Carolina Caballero, Caroline Besley, Nuria Martínez‐Cibrián, Alberto Mussetti, Robin Sanderson, Hugo Luzardo, Sunil Iyengar, Jose Maria Sánchez, Ceri Jones, Juan‐Manuel Sancho, Pere Barba, Anne‐Louise Latif, Lucia López‐Corral, Rafael Hernani, Juan Luis Reguera, Anna Sureda, Alejandro Martin Garcia‐Sancho, Mariana Bastos, Pau Abrisqueta, and Andrea Kuhnl. Data collection and analysis: Gloria Iacoboni, Josu Iraola‐Truchuelo, Víctor Navarro, Pau Abrisqueta, and Andrea Kuhnl. Manuscript writing: All authors. Final approval of manuscript: All authors. Accountable for all aspects of the work: All authors.
CONFLICT OF INTEREST STATEMENT
Gloria Iacoboni: Honoraria and travel support: Novartis, Kite/Gilead, Bristol‐Myers Squibb, Abbvie, Autolus, Sandoz, Miltenyi, and AstraZeneca. Maeve O'Reilly has served on advisory boards and received honoraria from Kite/Gilead and Novartis. Mi Kwon: Consulting and lectures: Gilead and Jazz, Pfizer. Javier Delgado: Honoraria from Kite‐Gilead, Novartis, Bristol Myers Squibb, and Janssen. Claire Roddie has served on advisory boards and received honoraria from Kite/Gilead, Novartis, and BMS. Manuel Guerreiro: Consultancy: Novartis, Kite, BMS, Pierre Fabre, and MSD. Alberto Mussetti: BMS: consultancy; Takeda: Honoraria; Gilead: Research Funding; Jazz Pharmaceuticals: Consultancy. Robin Sanderson: Kite/Gilead and Novartis—speakers fees, honoraria, conference travel. Sunil Iyengar: Abbvie—Conference support; Beigene—advisory board; BMS—Conference support; Janssen—Speaker fees; Kite—advisory board; Takeda—advisory board, speaker fees, and conference support. Juan‐Manuel Sancho: Honoraria as speaker in medical education activities from Roche, Gilead‐Kite, Celgene‐BMS, Janssen, Novartis, and Incyte. Honoraria as participant in advisory boards or consulting for Roche, Gilead‐Kite, Celgene‐BMS, Janssen, Novartis, Incyte, Lilly, Beigene, and Myltenyi Biomedicine. Pere Barba: Advisory board and consultancy: Allogene, Amgen, BMS/Celgene, Kite/Gilead, Incyte, Miltenyi Biomedicine, Novartis, Nektar, Pfizer, and Pierre Fabre. Rafael Hernani: Research: Gilead. Travel support: Gilead. Honoraria: Gilead, Janssen, MSD, Celgene, and Novartis. Anna Sureda: Honoraria from Takeda, BMS, Merck, Janssen, Sanofi, Roche, Novartis, and Gilead. Alejandro Martin Garcia‐Sancho: Consultancy for Roche, BMS/Celgene, Kyowa Kirin, Novartis, Gilead/Kite, Incyte, Lilly, ADC Therapeutics America, Miltenyi, Ideogen, Abbvie, and Sobi. Honorario from Roche, BMS/Celgene, Janssen, Gilead/Kite, Takeda, Eusa Pharma, and Abbvie. Pau Abrisqueta: consulting/advisory: Roche, Genmab, Janssen, BMS, AbbVie, AstraZeneca, BeiGene; Honoraria: Roche, Genmab, Janssen, BMS, AbbVie, AstraZeneca, Gilead, and Incyte. Andrea Kuhnl has served on advisory boards and received honoraria from Kite/Gilead, Novartis, and BMS. The remaining authors declare no conflict of interest.
ETHICS STATEMENT
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
FUNDING
This research received no funding.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors thank the GETH and GELTAMO collaborative Spanish groups, together with the UK National CAR‐T Clinical Panel, for their support during the study development. Also, Angel Cedillo for his help with the database of this project.
Contributor Information
Gloria Iacoboni, Email: giacoboni@vhio.net.
Pau Abrisqueta, Email: pabrisqueta@vhio.net.
DATA AVAILABILITY STATEMENT
Data are available on request from the authors.
REFERENCES
- 1. Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B‐cell lymphoma: results from the international SCHOLAR‐1 study. Blood. 2017;130(16):1800‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Locke FL, Ghobadi A, Jacobson CA, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schuster SJ, Tam CS, Borchmann P, et al. Long‐term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B‐cell lymphomas (JULIET): a multicentre, open‐label, single‐arm, phase 2 study. Lancet Oncol. 2021;22(10):1403‐1415. [DOI] [PubMed] [Google Scholar]
- 4. Jacobson CA, Locke FL, Ma L, et al. Real‐world evidence of axicabtagene ciloleucel for the treatment of large B cell lymphoma in the United States. Transplant Cell Ther. 2022;28(9):581.e1‐581.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasquini MC, Hu ZH, Curran K, et al. Real‐world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non‐Hodgkin lymphoma. Blood Adv. 2020;4(21):5414‐5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iacoboni G, Villacampa G, Martinez‐Cibrian N, et al. Real‐world evidence of tisagenlecleucel for the treatment of relapsed or refractory large B‐cell lymphoma. Cancer Med. 2021;10(10):3214‐3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwon M, Iacoboni G, Reguera JL, et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of aggressive B‐cell lymphoma. Haematologica. 2023;108(1). 10.3324/haematol.2022.280805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bethge WA, Martus P, Schmitt M, et al. GLA/DRST real‐world outcome analysis of CAR T‐cell therapies for large B‐cell lymphoma in Germany. Blood. 2022;140(4):349‐358. [DOI] [PubMed] [Google Scholar]
- 9. Kuhnl A, Roddie C, Kirkwood AA, et al. A national service for delivering CD19 CAR‐Tin large B‐cell lymphoma—the UK real‐world experience. Br J Haematol. 2022;198(3):492‐502. [DOI] [PubMed] [Google Scholar]
- 10. Bachy E, Le Gouill S, Di Blasi R, et al. A real‐world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat Med. 2022;28(10):2145‐2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neelapu SS, Jacobson CA, Ghobadi A, et al. 5‐Year follow‐up supports curative potential of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1). Blood. 2023;141(19):2307‐2315. 10.1182/blood.2022018893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Blasi R, Le Gouill S, Bachy E, et al. Outcomes of patients with aggressive B‐cell lymphoma after failure of anti‐CD19 CAR T‐cell therapy: a DESCAR‐T analysis. Blood. 2022;140:2584‐2593. [DOI] [PubMed] [Google Scholar]
- 13. Alarcon Tomas A, Fein JA, Fried S, et al. Outcomes of first therapy after CD19‐CAR‐T treatment failure in large B‐cell lymphoma. Leukemia. 2023;37(1):154‐163. 10.1038/s41375-022-01739-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zurko J, Ramdial J, Shadman M, et al. Allogeneic transplant following CAR T‐cell therapy for large B‐cell lymphoma. Haematologica. 2023;108(1). 10.3324/haematol.2022.281242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bezerra ED, Iqbal M, Munoz J, et al. Barriers to enrollment in clinical trials of patients with aggressive B‐Cell NHL that progressed after CAR T‐cell therapy. Blood Adv. 2023;7(8):1572‐1576. 10.1182/bloodadvances.2022007868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salles G, Duell J, González Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B‐cell lymphoma (L‐MIND): a multicentre, prospective, single‐arm, phase 2 study. Lancet Oncol. 2020;21(7):978‐988. [DOI] [PubMed] [Google Scholar]
- 17. Caimi PF, Ai W, Alderuccio JP, et al. Loncastuximab tesirine in relapsed or refractory diffuse large B‐cell lymphoma (LOTIS‐2): a multicentre, open‐label, single‐arm, phase 2 trial. Lancet Oncol. 2021;22(6):790‐800. [DOI] [PubMed] [Google Scholar]
- 18. Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B‐cell lymphoma. J Clin Oncol. 2020;38(2):155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thieblemont C, Phillips T, Ghesquieres H, et al. Epcoritamab, a novel, subcutaneous CD3 × CD20 bispecific T‐cell‐engaging antibody, in relapsed or refractory large B‐cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. 2022;41(12):JCO2201725. 10.1200/JCO.22.01725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dickinson MJ, Carlo‐Stella C, Morschhauser F, et al. Glofitamab for relapsed or refractory diffuse large B‐cell lymphoma. N Engl J Med. 2022;387(24):2220‐2231. [DOI] [PubMed] [Google Scholar]
- 21. Zurko JC, Nizamuddin I, Epperla N, et al. Peri‐CAR‐T practice patterns and survival predictors for all CAR‐T patients and post‐CAR‐T failure in aggressive B‐NHL. Blood Adv. 2023;7(12):2657‐2669. 10.1182/bloodadvances.2022008240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel as second‐line therapy for large B‐cell lymphoma. N Engl J Med. 2022;386(7):640‐654. [DOI] [PubMed] [Google Scholar]
- 23. Kamdar M, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second‐line treatment in patients with relapsed or refractory large B‐cell lymphoma (TRANSFORM): results from an interim analysis of an open‐label, randomised, phase 3 trial. Lancet. 2022;399(10343):2294‐2308. [DOI] [PubMed] [Google Scholar]
- 24. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non‐Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059‐3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625‐638. [DOI] [PubMed] [Google Scholar]
- 26. Buuren S, Groothuis‐Oudshoorn K. Mice: multivariate imputation by chained equations in R. J Stat Softw. 2011;45(3):1‐67. [Google Scholar]
- 27. Chow VA, Gopal AK, Maloney DG, et al. Outcomes of patients with large B‐cell lymphomas and progressive disease following CD19‐specific CAR T‐cell therapy. Am J Hematol. 2019;94(8):E209‐E213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spiegel JY, Dahiya S, Jain MD, et al. Outcomes of patients with large B‐cell lymphoma progressing after axicabtagene ciloleucel therapy. Blood. 2021;137(13):1832‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sehn LH, Hertzberg M, Opat S, et al. Polatuzumab vedotin plus bendamustine and rituximab in relapsed/refractory DLBCL: survival update and new extension cohort data. Blood Adv. 2022;6(2):533‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gouni S, Rosenthal AC, Crombie JL, et al. A multicenter retrospective study of polatuzumab vedotin in patients with large B‐cell lymphoma after CAR T‐cell therapy. Blood Adv. 2022;6(9):2757‐2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hutchings M, Mous R, Clausen MR, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B‐cell non‐Hodgkin lymphoma: an open‐label, phase 1/2 study. Lancet. 2021;398(10306):1157‐1169. [DOI] [PubMed] [Google Scholar]
- 32. Hutchings M, Carlo‐Stella C, Morschhauser F, et al. Relapse is uncommon in patients with large B‐cell lymphoma who are in complete remission at the end of fixed‐course glofitamab treatment. In: 2022 ASH Annual Meeting and Exposition. Abstract 441, December 11, 2022.
- 33. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kasakovski D, Xu L, Li Y. T cell senescence and CAR‐T cell exhaustion in hematological malignancies. J Hematol Oncol. 2018;11(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chong EA, Alanio C, Svoboda J, et al. Pembrolizumab for B‐cell lymphomas relapsing after or refractory to CD19‐directed CAR T‐cell therapy. Blood. 2022;139(7):1026‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jäger U, Worel N, McGuirk J, et al. Safety and efficacy of tisagenlecleucel (tisa‐cel) plus pembrolizumab (pembro) in patients (pts) with relapsed/refractory diffuse large B‐cell lymphoma (r/r DLBCL): updated analysis of the phase 1b PORTIA study. J Clin Oncol. 2021;39(15_suppl):e19537. [Google Scholar]
- 37. Hirayama AV, Gauthier J, Hay KA, et al. Efficacy and toxicity of JCAR014 in combination with durvalumab for the treatment of patients with relapsed/refractory aggressive B‐cell non‐Hodgkin lymphoma. Blood. 2018;132:1680. [Google Scholar]
- 38. Major A, Yu J, Shukla N, et al. Efficacy of checkpoint inhibition after CAR‐T failure in aggressive B‐cell lymphomas: outcomes from 15 U.S. institutions. Blood Adv. 2023;7(6):4528‐4538. 10.1182/bloodadvances.2023010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Otáhal P, Průková D, Král V, et al. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. Oncoimmunology. 2016;5(4):e1115940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuramitsu S, Ohno M, Ohka F, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer Gene Ther. 2015;22(10):487‐495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
Data are available on request from the authors.