

Abstract
Proteoforms are specific molecular forms of protein products arising from a single gene that possess different structures and different functions. Therefore, a single gene can produce a large repertoire of proteoforms by means of allelic variations (mutations, indels, SNPs), alternative splicing and other pre-translational mechanisms, post-translational modifications (PTMs), conformational dynamics, and functioning. Resulting proteoforms that have different sizes, alternative splicing patterns, sets of post-translational modifications, protein–protein interactions, and protein–ligand interactions, might dramatically increase the functionality of the encoded protein. Herein, we have interrogated the tumor suppressor PTEN for its proteoforms and find that this protein exists in multiple forms with distinct functions and sub-cellular localizations. Furthermore, the levels of each PTEN proteoform in a given cell may affect its biological function. Indeed, the paradigm of the continuum model of tumor suppression by PTEN can be better explained by the presence of a continuum of PTEN proteoforms, diversity, and levels of which are associated with pathological outcomes than simply by the different roles of mutations in the PTEN gene. Consequently, understanding the mechanisms underlying the dysregulation of PTEN proteoforms by several genomic and non-genomic mechanisms in cancer and other diseases is imperative. We have identified different PTEN proteoforms, which control various aspects of cellular function and grouped them into three categories of intrinsic, function-induced, and inducible proteoforms. A special emphasis is given to the inducible PTEN proteoforms that are produced due to alternative translational initiation. The novel finding that PTEN forms dimers with biological implications supports the notion that PTEN proteoform–proteoform interactions may play hitherto unknown roles in cellular homeostasis and in pathogenic settings, including cancer. These PTEN proteoforms with unique properties and functionalities offer potential novel therapeutic opportunities in the treatment of various cancers and other diseases.
Keywords: PTEN, Proteoforms, Alternative translational initiation, Post-translational modifications
Introduction
The PTEN gene was independently discovered by two groups in 1997 while investigating chromosome 10q23, a chromosomal location that frequently showed loss of heterozygosity in advanced-stage cancers [1, 2]. The gene was named MMAC (mutated in multiple advanced cancers) or PTEN (phosphatase and tension homolog deleted on chromosome 10) [1, 2]. The protein product of the PTEN gene shared sequence homology with the protein tyrosine phosphatase (PTP) superfamily and a cytoskeletal protein tensin. Consequently, PTEN was characterized as a dual lipid and protein phosphatase, which is a non-redundant negative regulator of the PI3K/AKT pathway. The most extensively studied tumor-suppressive function of PTEN is its lipid phosphatase activity. PTEN dephosphorylates the secondary messenger PIP3 to PIP2, thereby depleting cellular PIP3, which is critical for AKT activation [3]. As a protein phosphatase, PTEN dephosphorylates itself and several proteins, such as FAK, CREB, RAB7, and IRS1 among others, to regulate oncogenic signaling [4–9]. PTEN also has several phosphatase-independent functions, particularly in the nucleus [10]. Besides its role in cancer, PTEN signaling plays an important role in neurological diseases such as autism [11].
Germline mutations in PTEN causes PTEN Hamartoma Tumor Syndrome (PHTS), increasing the susceptibility to breast, thyroid, and endometrial cancer in carriers [12]. Mutations in PTEN are also observed in Autism Spectrum Diseases (ASD) [11]. It was pointed out that in cancers, PTEN mutations are not as frequent as mutations in other tumor suppressors such as p53 [13]. However, since deregulated PTEN levels or compromised activity are often pathogenic, several non-genomic mechanisms exert a tight control on PTEN protein levels and activity [13]. Indeed, experiments in PTEN hypomorphic mouse models that express different levels of PTEN show a tissue-specific sensitivity to tumor initiation and progression [14] (Fig. 1). For example, lymph nodes and mammary glands are sensitive to small decreases in PTEN expression; a 20% decrease in PTEN expression levels is sufficient to generate tumors. While other tissues, such as the prostate, require a more profound reduction of PTEN levels for tumor initiation to occur. These observations led to the development of the “continuum model of tumor suppression” to explain the consequences of differential reduction of PTEN levels required to trigger cancer growth in a tissue selective manner [15] (Fig. 1). Unlike the two-hit hypothesis, wherein both alleles of a tumor suppressor gene must be inactivated to cause tumor initiation, the continuum model proposes that subtle changes in the tumor suppressor dosage may cause tumors without the loss/mutation of even one allele [15]. Consequently, mechanisms that alter PTEN protein stability and function are of paramount importance to understanding tumor initiation and progression.
Fig. 1.

The continuum model of tumor suppression. The tumor-suppressive functions of PTEN can be best explained by the continuum model of tumor suppression, wherein the levels of the PTEN protein dictate disease severity and tissue selectivity rather than mutations in the PTEN gene itself. Subtle reductions in PTEN protein levels, due to post-translational modifications, transcriptional repression, promoter methylation, and sub-cellular mislocalization, have a profound impact on oncogenic signaling pathways. Further, experiments in hypomorphic Pten transgenic mouse models have revealed that certain organs, such as the mammary glands, are particularly sensitive to small changes in the total PTEN levels. This is in contrast to the prostate gland, wherein a dramatic reduction in PTEN levels is required to initiate oncogenic transformation
The functionality of the PTEN protein is modulated via various processes, such as alternate splicing, alternative translational initiation, post-translational modifications, protein–protein interactions, and protein-membrane interactions. Each of these events, individually or in combination, generates a conformationally unique form (or specific conformational ensemble) of the PTEN protein (a proteoform) with varying downstream functionalities and implications. Given the importance of PTEN expression levels in tumorigenesis and oncogenic progression, it is imperative to delineate the various PTEN proteoforms and define their functions in disease pathogenesis. Consequently, this review focuses on the generation and function of a variety of PTEN proteoforms, with a special emphasis on the recently identified PTEN translational isoforms.
Classification of PTEN proteoforms
The term proteoforms refers to all the different molecular forms that a protein, arising from a single gene, can adopt [16]. Traditionally, proteoforms are defined as alternative forms of a protein that can be assigned a different primary structure and are typically produced due to alternative splicing, alternate promoter usage, alternate translational initiation, mutations, coding SNPs (Single Nucleotide Polymorphisms), and post-translational modifications (PTMs). One should keep in mind that the original idea of a proteoform, where functionally different proteins can be generated from a single gene, was crucial for moving from the classical “one gene-one protein sequence-one unique structure-one specific function” concept to the more realistic “one gene-many sequences-many structures-many functions” model. In its original meaning, the proteoform represented a group of related protein molecules arising from all combinatorial sources of variation giving rise to products arising from a single gene. Although the classical view considered only means affecting protein primary structure (genetic variation, alternatively spliced RNA, alternative transcripts, and post-translational modifications) [16], we believe that this concept should be further extended to include other mechanisms that may affect protein structure and function (such as conformational dynamics and changes induced in a protein molecule during its function). Therefore, in this review, we extend the classical definition of proteoforms to include protein variants produced due to the conformational changes, protein–protein interactions (PPIs), protein–membrane interactions, and protein–ligand interactions [17]. This extension is particularly relevant for proteins that have a tendency to misfold, that form coordination complexes with metals, that undergo conformational changes upon ligand binding, and proteins that oligomerize to execute their function. In fact, many proteins, including PTEN, undergo several conformational changes that dictate their function. These conformational changes may be induced by binding of other protein, ligand binding, interaction with membrane, or caused by the post-translational modifications. For example, Johnston et al., have clearly explained that the PTEN enzyme exists in two conformations, such as a Tense (T) conformation and a relaxed (R) conformation. The R conformation of the enzyme is bound to PIP2, while the T conformation is not. In fact, it is PIP2 (ligand) binding that allows for the transition to the R conformation. Within the R conformation, the PTEN enzyme is active and acts as a lipid phosphatase, whereas the T conformation is largely inactive. Since the T and R conformations have different functional potential (enzyme activity) and different complex structures (bound/unbound to PIP2), they may be classified as two different proteoforms of PTEN (at least in our view). EGFRs (Epidermal Growth Factor Receptors) are another example of proteins that undergo conformational changes upon ligand binding, resulting in conformational changes needed for protein–protein interaction (dimerization) [18].
Proteoforms may also dictate structural, functional, and tissue specificity, wherein, a given proteoform has a unique function within a given cell-type. Multiple modifications may occur on the protein simultaneously, thereby expanding the repertoire of each of these possible proteoforms for a given protein. Proteins can adopt several different proteoforms that are generally classified under three categories: intrinsic, function-induced, and inducible proteoforms [17]. Intrinsic or conformational proteoforms are generated due to conformational changes in a protein and may potentially impact protein function. Structural disorder in a protein usually contributes to the generation of an ensemble of intrinsic isoforms. Inducible proteoforms of a protein are produced due to specific biological processes such as alternative splicing, mutations, post-translational modifications etc. Even changes in pH, ion concentration, or redox states may generate novel proteoforms. Function-induced proteoforms are generated as a result of a protein interacting with various components of the cell in its natural environment, resulting in a physiologically and functionally relevant cellular event. This includes complexes of a given protein with other proteins, small molecules, or specialized sub-cellular components such as the cell membrane. It is important to note that a protein may exhibit a combination of the different categories of proteoforms at any given time. Alternatively, an ensemble of proteoforms can be found in a given cell, whereby they can either function in a concerted manner or have distinct biochemical and physiological roles. Conformational changes induced in the PTEN protein due to C-terminal tail (C-tail) phosphorylation represent a good example of a combination of intrinsic and inducible proteoforms for PTEN, as discussed below.
Structurally, PTEN consists of four domains. The N-terminal phosphatidylinositol 4,5-bisphosphate (PIP2)-binding module (PBM) and the C2 domain (C2D) help PTEN to bind to the cell membrane. The phosphatase domain (PD) consists of a dual specificity lipid and protein phosphatase catalytic motif. The C-tail of PTEN regulates its membrane association, stability, and enzymatic activity via phosphorylation/dephosphorylation. The C-tail region also encompasses a PDZ-binding domain, which allows PTEN to form complexes with a myriad of PDZ domain-containing proteins [13]. Different modifications occur along the various PTEN domains to generate various proteoforms as discussed below.
Intrinsic/conformational proteoforms
Structural and bioinformatics studies on PTEN have revealed that part of the PBM and the entire C-tail region of PTEN are intrinsically disordered and therefore not amenable to crystallization [19]. The relative lack of higher-order structure in the PBM and C-tail region may allow the PTEN protein to adopt several different conformations representing the intrinsic/conformational proteoforms. The most extensively studied conformational change in PTEN is induced by phosphorylation [20, 21]. Phosphorylation of a serine–threonine cluster (Ser380, Thr382, Thr383, Ser385) in the PTEN C-tail causes the protein to undergo an intra-molecular association, wherein, the disordered C-tail binds to and occludes regions of the PD and C2D. This intra-molecular association renders PTEN catalytically inactive and consequently, the PTEN C-tail is referred to as an auto-inhibitory switch for PTEN [20, 21].
Function-induced proteoforms
Function-induced proteoforms of PTEN are generated due to its interaction with other proteins, including itself, in the cellular environment. As mentioned above, the PTEN C-tail is intrinsically disordered and does not possess a stable higher-order structure. However, the PDZ-binding domain of PTEN (residues 401–403) encompass a Molecular Recognition Feature (MoRF) that adopts a secondary structure when complexed with a protein partner [19]. Consequently, crystal structures for the PTEN–PDZ-binding domain in complexes with the PDZ domains of MAST2 and PAR3 show the presence of a β-strand within the disordered PTEN C-tail [22] (Fig. 2). It is interesting to note, however, that the PTEN–PDZ domain adopts different secondary structures to bind to MAST2 and PAR3, thereby validating the concept of functional selectivity of various PTEN proteoforms. This may likely be true for several other proteins known to engage in PPIs with the PTEN C-tail [19, 23]. Further, each PTEN proteoform (constitutive or inducible) could potentially allow for an interaction with a different protein partner resulting in a highly specific well-defined downstream cellular event [24].
Fig. 2.

Protein complexes of PTEN generate new PTEN proteoforms. a The co-crystal structure of the PTEN–PDZ-binding domain with the PDZ-binding domain of MAST2 demonstrates that the disordered PTEN–PDZ-binding domain adopts a secondary structure primarily comprising a β-strand, a turn, and a bend. b In contrast, the PTEN–PDZ-binding domain adopts a slightly different secondary structure, comprising a β-strand and a bend, in complex with Par3
The PTEN protein is known to form homodimers. As compared to PTEN monomers, these PTEN dimeric complexes have higher lipid phosphatase activity at the membrane [25]. Multiple PTEN domains are involved in the dimerization process. The PBM–Phosphatase domain and the C2 domain–C-tail regions have been reported to oligomerize, forming higher-order complexes [25]. Phosphorylation and ubiquitination modifications affect PTEN dimerization, as discussed below [25, 26]. Further, disease-associated PTEN mutants that are catalytically inactive (i.e., PTEN G129E, PTEN C124S) heterodimerize with wild-type PTEN and inhibit its catalytic activity via a dominant-negative mechanism [25]. Thus, proteoform–proteoform interaction generates new proteoforms and provides another layer of complexity that may be exploited for novel biological functions.
Inducible proteoforms
A vast majority of the PTEN proteoforms are inducible and are discussed below with special emphasis on the proteoforms generated due to alternative translational initiation.
Alternative splicing
PTEN has several naturally occurring splice variants that give rise to different proteoforms with varying functions. Alternative splicing frequently results in truncated forms of the PTEN protein. Two splice variants, PTEN Δ and PTEN B are generated as a result of the inclusion of the intronic sequence after exons 8 and 5, respectively [27]. Agrawal et al. subsequently identified eight more splice variants for PTEN that retained parts of intron 3 (variants 3a, 3b, 3c), parts of intron 5 (variants 5a, 5b, 5c), excluded a portion of exon 5 (variant DelE5), or all of exon 6 (variant DelE6) [28]. These splice variants showed differential expression in breast cancer and Cowden Syndrome patients. Splice variants 5b and 5c, behaved in a manner contrary to full-length PTEN, wherein, they induced cyclin D1 promoter activity. Consequently, these oncogenic variants were found to be over-expressed in disease [28]. The differential expression of these splice variants in disease states opens up avenues for the use of these variants as molecular diagnostics.
Post-translational modifications (PTMs)
PTEN undergoes several post-translational modifications, generating a myriad of proteoforms with differently regulated catalytic activity, stability, sub-cellular localization, and protein–protein interactions (Fig. 3). A detailed description of the various PTMs and their functional relevance has been reviewed elsewhere [13, 29, 30]. However, it is important to note here that several PTEN PTMs act as priming events for further modifications, which exponentially increases the number of possible PTEN proteoforms. For instance, methylation at Lys313 residue enhances PTEN C-tail phosphorylation, whereas PTEN phosphorylation at specific sites prevents ubiquitination of this protein [31, 32]. PTMs also allow for a cross-talk between the various categories of PTEN proteoforms. For instance, phosphorylation of a serine–threonine cluster in the PTEN C-tail (inducible proteoform) results in a conformational change in the protein (intrinsic proteoform) rendering it enzymatically inactive. Further, PTEN C-tail phosphorylation and K27-linked ubiquitination (inducible proteoforms) inhibit PTEN dimerization (function-induced proteoforms) [25, 26]. Several PTMs impact the sub-cellular localization of PTEN, particularly to the nucleus, and allow PTEN to regulate the cell cycle, transcription, DNA replication, and repair processes [10, 13]. Taken together, PTMs have a profound effect on PTEN function and together with other categories of proteoforms expand the functional repertoire of PTEN.
Fig. 3.

Post-translational modifications of PTEN generates a set of PTEN proteoforms. PTEN undergoes several post-translational modifications that control its catalytic activity, stability, sub-cellular localization, and ability to engage in protein–protein interactions. (Ub ubiquitination, P phosphorylation, O oxidation, N nitrosylation, Rib ribosylation, Su sumoylation, Me methylation, Ac acetylation). Each of these modified PTEN proteins is considered a novel proteoform
Mutations
Increasingly, it has been observed that loss of PTEN and PTEN mutations are not synonymous. Marsh et al. observed that patients with mutations in the PTEN catalytic site had a more severe phenotype than patients with truncating mutations that destabilized PTEN causing lower levels of this protein [33]. This hypothesis was further validated by the findings of Papa et al., who showed that PTEN catalytic mutants can bind to and inactivate wild-type PTEN in a dominant-negative manner. In vivo experiments demonstrated that mice that were homozygous for the PTEN catalytic site mutation (i.e., Pten C124S/C124S or Pten G129E/G129E) died in utero, indicating the role of PTEN lipid phosphatase activity in embryogenesis. On the other hand, mice heterozygous for Pten C124S/+ and Pten G129E/+had malignant growth and activated AKT signaling as compared to the normal PTEN +/−heterozygous mice. These findings firmly established that expression of the mutant PTEN protein has a more severe phenotype and worst outcome as compared to the loss of expression of PTEN protein due to the loss of a Pten allele [25]. Another mutation, PTEN A126G, occurring in the catalytic core of PTEN converts it from a 3′-phosphatase to a 5′-phosphatase [34]. In this case, the point mutation creates a PTEN proteoform that functions as a novel enzyme and therefore illustrates the mutation-driven gain of new catalytic function mechanism (Fig. 4).
Fig. 4.

Mutations in the PTEN protein generate novel proteoforms with distinct functions. Different PTEN mutations have a varying impact on its function and the resulting phenotype. Point mutations in PTEN, which are outside of the catalytic core, usually have slightly reduced phosphatase activity and/or stability resulting in a mild activation of downstream oncogenic pathways compared to wild-type PTEN. In contrast, mutations in the PTEN catalytic core region result in the production of proteins that are oncogenic (i.e., PTEN C124S or PTEN G129E) or result in the production of PTEN protein with altered enzyme activity (PTEN A126G). Consequently, these point mutations have a worse phenotype compared to PTEN loss perpetuating the concept that PTEN mutations and loss are not synonymous. Truncating mutations in PTEN usually cause a decrease in stability of the PTEN protein, resulting in lower total PTEN levels (indicated in a lighter shade of green). An exception to this is the PTEN C-tail truncated mutant which behaves like an oncogene. Thus, each type of mutation in PTEN gives rise to a functionally and perhaps structurally distinct proteoform
Several truncation mutations also occur in PTEN, particularly in its C2 domain, resulting in the generation of a series of truncated proteoforms. One such example is a truncation mutation that leads to the generation of a PTEN proteoform that lacks the entire C-tail region. The C-tail-less PTEN protein exhibits a phosphatase-independent gain-of-function phenotype. The truncated PTEN protein establishes an IGF-1 autocrine signaling loop, which results in the aberrant activation of the AKT and NFκB signaling pathways, leading to the excessive growth of astrocytes in cell culture models [35]. In mouse models, heterozygous PTEN C-tail deletion causes genomic instability resulting in the generation of tumors in multiple organ systems [36]. These results demonstrate how an enzymatically active proteoform of PTEN could acquire oncogenic properties (Fig. 4).
Johnston et al. have used Differential Scanning Fluorimetry (DSF) to determine the thermostability of PTEN and its associated mutants [37]. Data from these experiments reveal that PTEN is inherently conformationally unstable with a T m value of 40.3 °C, which is only 3° above the normal human body temperature. The PTEN C124S mutant is catalytically inactive but has a T m value comparable to that of wild-type PTEN. In contrast, PTEN N82T and PTEN F337S mutants have compromised catalytic activity and decreased conformational stability. Several other mutations in PTEN, such as the autism-associated PTEN Y176C and PTEN G157G, cause a milder loss-of-function phenotype [37]. Curiously, these observations mirror the results of the thermodynamic studies of another important tumor suppressor, p53 protein, the core (DNA-binding) domain of which was shown to be marginally stable (with the T m of 42 °C) and easily destabilizable by many cancer-related mutations [38, 39]. Thus, depending upon the type of mutation(s) the PTEN proteoforms acquire distinct physicochemical properties that likely translate into their various pathophysiological roles in different diseases (Fig. 4). Given the impact of mutations on PTEN proteoforms, detailed analysis of mutations and their functional implications is warranted.
Alternate translation initiation
Introduction
Three groups independently identified and validated the existence of the PTEN alternate translational isoforms [40–42]. These translational isoforms are produced from the same mRNA as canonical PTEN and are generated due to non-AUG translational initiation. Consequently, four PTEN translational variants (PTEN-L, PTEN-M, PTEN-N, and PTEN-O) have been defined, each having a longer N-terminal extension compared to the canonical PTEN protein (Fig. 5) [41, 43]. The PTEN-M proteoform is the most predominant of the alternate translational proteoforms; however, PTEN-L remains the most studied isoform [41].
Fig. 5.
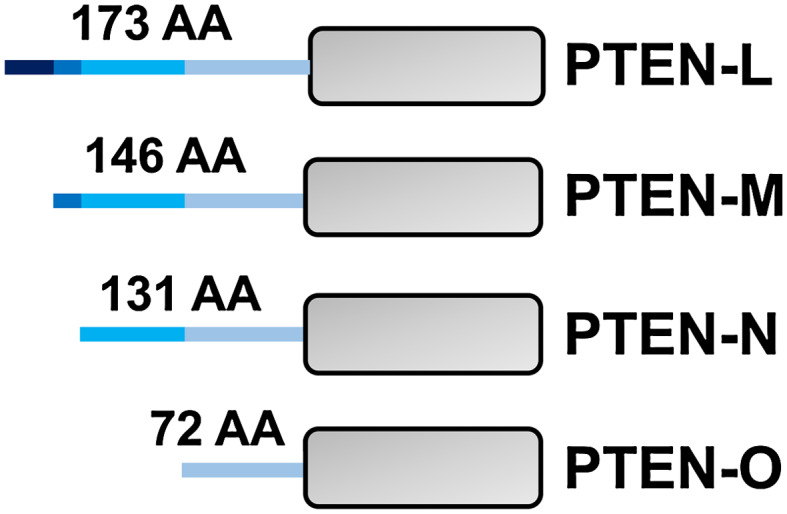
Translational variants of PTEN. PTEN has four reported translational variants, PTEN-L, -M, -N, and -O that are produced due to alternative translational initiation. Translation begins from an upstream non-AUG codon resulting in N-terminal extensions for each of the variants. It is likely that each of these proteoforms may behave differently in terms of their function and sub-cellular routing, trafficking, and localization
PTEN-L is translated from an upstream CUG codon, which is in frame with the canonical AUG start codon. Liang et al. proposed that CUG-mediated translational initiation occurs in an eIF2A-dependent mechanism and requires the presence of a palindromic sequence centered on the CUG codon [40]. Hopkins et al., however, demonstrated that a Kozak sequence is responsible for the alternate translation initiation from the CUG codon. In order to confirm the existence of PTEN-L in vivo, Liang et al. created a mouse with Pten FLAG allele, wherein a FLAG tag was inserted at the C-terminus of the Pten gene. Proteins were isolated from tissues from Pten FLAG heterozygous mice, and two bands were observed upon immunoblotting with the FLAG antibodies corresponding to the canonical PTEN and the PTEN-L, respectively [40], confirming the expression of both PTEN isoforms from the same mRNA. Ribosomal profiling, protein sequencing, and in vivo studies in mice have confirmed the existence of several PTEN translational variants. Research is now underway to define the structure, physicochemical properties, and biological relevance of these newly identified PTEN proteoforms.
Structure and stability
DSF-based experiments revealed that the Tm value for the canonical PTEN is 40.3 °C whereas that of PTEN-L is 46.7 °C in a phosphate-free buffer. PTEN-L has a T m which is 10 °C higher than the human body temperature and 6° higher than canonical PTEN, indicating that this proteoform has a higher conformational stability, which may be required for its extracellular functions, given the ability of PTEN-L to be secreted. DSF data suggested that the N-173 region of PTEN-L forms a three-dimensional structure that is closely associated with one of the domains of PTEN, in particular with the PIP2-binding module (PBM) [37]. However, bioinformatics analysis, CD, and FTIR spectroscopy data indicate that the N-173 region is primarily disordered [44–46]. Additional research efforts are required to firmly establish the structural features of the N-terminal extensions of the PTEN translational variants.
Membrane-binding and catalytic activity
Johnston et al. developed a continuous assay to measure PTEN lipid phosphatase activity and demonstrated that the catalytic activity for human PTEN is greater than previously estimated [47]. While PTEN-L has a comparable lipid phosphatase activity to the canonical PTEN, PTEN-L is 5 times more effective in binding to its substrate, PIP3 (phosphatidylinositol (3,4,5) trisphosphate), than canonical PTEN [47]. This finding indicates that PTEN conformation may dictate the binding affinity of PIP3. This notion is supported by the fact that phosphatidylinositol (4,5) bisphosphate (PIP2) allosterically activates PTEN enzymatic activity by binding to the PTEN PBM region, and mutations at the Lys13 residue in the PTEN PBM abrogate this allosteric activation [47, 48]. In contrast, the catalytic activity of PTEN-L is unaffected by PIP2 levels, indicating that PTEN-L is a constitutively active phosphatase and presumably exists in a different conformation. It is plausible that the N-terminal tail of PTEN-L associates with the PBM in a fashion mimicking that of PIP2 to maintain PTEN-L in an active state [47].
Based on PIP2 binding, a novel catalytic model for PTEN function has been put forward. Herein, PTEN exists in either of the two forms: a low activity (Tense; T) or a high activity (Relaxed; R) conformation. The T-state of PTEN is retained until PIP2 is bound, following which PTEN adopts the R conformation. PTEN-L appears to be locked in the R conformation, such that additional PIP2 binding does not alter its catalytic activity [47]. Thus, PIP2 binding reveals the two distinct proteoforms of PTEN. Likewise, using hydrogen/deuterium exchange mass spectrometry (HDX-MS), Masson et al. showed the presence of a membrane-binding helix (MBH) in PTEN-L [44]. The MBH alters the catalytic mechanism of PTEN-L to a scooting mode from a hopping mode, which is characteristic of wild-type PTEN, again demonstrating that membrane association of PTEN-L provides conditions to generate a new PTEN proteoform.
Sub-cellular localization
As mentioned above, HDX-MS-based studies have shown that PTEN-L localizes to the cell membrane via its MBH [44]. Since the other PTEN translational proteoforms, PTEN-M, PTEN-N, and PTEN-O, possess the MBH, it is very likely that they also localize to the cell membrane. Studies by Liang et al. demonstrated that PTEN-L localizes to the mitochondria, specifically the inner leaflet, where it regulates mitochondrial energetics [40]. However, neither PTEN nor PTEN-L possess a canonical mitochondrial localization sequence. It is plausible that a transporter(s) may be involved in the mitochondrial translocation of PTEN and its various proteoforms. Bioinformatics analysis using NoD, NLStradamus, and NucPred predictors reveal that the N-terminal region of PTEN-L and PTEN-M contains a putative nuclear and nucleolar localization sequence (Fig. 6) [49–52]. Given the recent identification of PTEN in the nucleolar compartment of the cell, it will be interesting to investigate whether PTEN-L and PTEN-M have any unique functions within the nucleolus [53]. This could uncover hitherto unknown functions of PTEN-L and the novel proteoform PTEN-M. Further, exogenous PTEN-L can be internalized into cells via a polyarginine stretch in the N-173 region [42]. The polyarginine stretch is present within the PTEN-M proteoform as well, raising the possibility that PTEN-M may also enter into cells. However, this function of PTEN-M remains to be determined. While PTEN-L is supposedly a secreted protein, there are conflicting reports on the secretory properties of PTEN-L [41, 42]. Therefore, secretory and extracellular functions of PTEN-L proteoform remain controversial.
Fig. 6.

PTEN-L and PTEN-M have a putative nuclear and nucleolar localization sequence, indicating their distinct sub-cellular functions. The sequence indicates the positions of the nuclear and nucleolar localization sequence as predicted by NLStradamus, NucPred, and the NoDsoftwares, respectively
Function and clinical relevance
In the mitochondria, PTEN-L increases the activity of COX (cytochrome oxidase c) in phosphatase-dependent manner. TALEN-mediated abrogation of PTEN-L resulted in the disruption of mitochondrial structure and function in HeLa cells [40] (Fig. 7). Both PTEN-L and PTEN-M reduce phospho-AKT levels, thereby inhibiting PI3K/AKT signaling [41, 42]. Exogenous PTEN-L reduces the tumor burden in xenograft and genetically engineered mouse models [42, 54, 55]. PTEN-L, once firmly established as a secretory PTEN proteoform, has tremendous potential for use as a biologic to treat various malignancies (Fig. 7).
Fig. 7.

Functional relevance of PTEN and its proteoforms. PTEN and its translational variants give rise to various proteoforms that localize to various sub-cellular compartments to regulate cell signaling. PTEN exerts its effects on the cell cycle, ribosome biogenesis, DNA damage, and repair processes in the nucleus and nucleolus. PTEN-L and -M have putative nuclear and nucleolar localization sequences and their unique roles in these compartments remain to be determined. PTEN and PTEN-L also localize to the mitochondria where they regulate apoptosis and mitochondrial energetics, respectively. Both PTEN and PTEN-L associate with the membrane and PTEN-L has an additional membrane-binding helix that facilitates this association. The other PTEN translational variants, PTEN-M, -N, and -O also possess the membrane-binding helix and whether they truly localize to the plasma membrane is unknown. PTEN-L has also been found in extracellular tissues and in circulation in the blood stream. PTEN-L, by virtue of a cell-penetrating polyarginine peptide, re-enters the cells to inhibit PI3K signaling and has been shown to be therapeutically effective in mouse models. Whether, PTEN-L is truly secretory remains controversial. However, the biochemical process underlying its secretion represents an active area of research
Consistent with the critical role of PTEN-L in dampening PI3K/AKT signaling, PTEN-L levels were found to be altered in cancers. Increased PTEN-L levels were found in the tumor microenvironment in breast cancer samples [42]. Conversely, reduced levels of PTEN-L were observed in renal-cell carcinoma samples [54].
Discussion and future directions
It appears that the relatively low number (in comparison with other tumor suppressors, e.g., p53) of disease-related mutations in PTEN severely undermines the critical role of this protein as a tumor suppressor. However, extensive studies using various Pten transgenic mouse models have demonstrated that subtle changes in PTEN protein levels can lead to the development of malignancies [14]. Unlike the two-hit model of tumor suppression, PTEN follows the continuum model wherein subtle changes in the PTEN protein levels are sufficient to cause a disease phenotype without loss or mutation of even one allele. Therefore, a systematic study of PTEN protein modifications and the proteoforms they generate is of utmost importance.
PTEN has several proteoforms that may be broadly classified as intrinsic, function-induced, and inducible. Each of these proteoforms represents a variant of PTEN with a unique functionality. This provides avenues for the use of PTEN proteoforms as biomarkers to guide therapy. For instance, PTEN, when phosphorylated at Y240, reduces the sensitivity of the cell to DNA damage and is also a predictor for the EGFR inhibitor resistance in glioblastoma [56]. HDAC inhibitors induce PTEN membrane translocation through PTEN acetylation at K163, resulting in enhanced tumor-suppressive function indicating that HDAC inhibitors may be more efficacious in tumors with functional PTEN [57]. Furthermore, phosphorylation of the PTEN C-tail inhibits catalytic activity of this protein and is frequently elevated in cancers, macular degenerative disease, myocardial infarction, and pulmonary hypertension [58–64]. While these tumor/disease samples may show a high degree of immunoreactivity with a PTEN antibody, they are in fact similar to PTEN-deficient samples, and the patients may benefit from the use of PI3K/AKT pathway inhibitors. The oncogenic BCR–ABL fusion protein induces casein kinase 2 (CK2)-mediated PTEN C-tail phosphorylation, resulting in PTEN inactivation in Chronic Myeloid Leukemia (CML). Consequently, Morotti et al. have suggested the use of CK2 inhibitors to reactivate PTEN function, which can result in the induction of apoptosis, particularly in the case of imatinib-resistant CML [65]. Studies on PTEN dimerization patterns have revealed that PTEN catalytic core mutants are oncogenic and bind to and inhibit the function of the wild-type PTEN. Additionally, data from transgenic mouse models have shown that mice harboring lesions in the PTEN catalytic core are more vulnerable to malignant growth than animals with heterozygous loss of Pten gene. These observations suggest that PTEN mutations and loss are not synonymous and must therefore be managed differently in a clinical setting with a more aggressive therapeutic regimen for patients with PTEN catalytic site mutations. Further, Liang et al. have demonstrated that PTEN-L and canonical PTEN form a complex within the mitochondria [40]. Similarly, it is possible that the various PTEN translational variants homodimerize and heterodimerize with distinct permutations and combinations, thereby exponentially expanding the repertoire of functional PTEN complexes. PTEN truncation mutations, resulting in the production of a tail-less PTEN proteoform, render the protein oncogenic due to the establishment on an autocrine IGF-1 signaling loop [35]. Consequently, treatment for patients with such PTEN truncation mutations should be tailored to dampen IGF-1 signaling pathways. Differential expression of PTEN proteoforms produced due to alternative splicing, particularly splice variants 5b and 5c, are oncogenic and are found to be over-expressed in disease states [28]. Furthermore, the discovery of PTEN-L, a putative secretory PTEN proteoform, instructs us to use PTEN proteoforms as a biologic, particularly in cancer therapy. The N-173 N-tail extension of PTEN-L, which has a cell-penetrating property, has the potential to be used as a carrier for other therapeutic molecules.
The emergence of several relatively recent techniques has made it easier for the identification and characterization of PTEN proteoforms. Refinement in top-down proteomics (TDP) approaches allows for analysis of intact proteins and discovery of novel proteoforms. Further development in TDP techniques may allow for the detection of diagnostic proteoforms in body fluids [66]. Ribosomal profiling has been used to identify PTEN translational variants [67]. In the future, the CRISPR–Cas9 technology may be employed to study the role of a given PTEN proteoform by selectively eliminating its expression from cells. Taken together, recent discovery of various PTEN proteoforms, including their property to dimerize and multimerize indicates that we have a lot to learn about biological and pathophysiological functions of PTEN, which till date had remained obscure.
Abbreviations
- PTEN
Phosphatase and tension homolog deleted on chromosome 10
- MMAC
Mutated in multiple advanced cancers
- PTP
Protein tyrosine phosphatases
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-Kinase
- AKT
V-Akt murine thymoma viral oncogene
- PIP3
Phosphatidylinositol (3,4,5)-trisphosphate
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- FAK
Focal adhesion kinase
- CREB
cAMP responsive element-binding protein
- RAB7
Ras-associated protein RAB7
- IRS1
Insulin receptor substrate 1
- PHTS
PTEN hamartoma tumor syndrome
- ASD
Autism spectrum disorder
- SNP
Single nucleotide polymorphisms
- PPI
Protein–protein interactions
- C-tail
Carboxy terminal tail
- PBM
PIP2-Binding module
- C2D
C2 domain
- PDZ
Post-synaptic density protein (PSD95), Drosophila disc-large tumor suppressor (Dlg1), and Zonula occludens-1 protein (ZO-1)
- MoRF
Molecular recognition features
- MAST2
Microtubule-associated serine/threonine kinase 2
- PAR3
Partitioning defective 3
- PTM
Post-translational modification
- NFκB
Nuclear factor kappa B
- DSF
Differential scanning fluorimetry
- CD
Circular dichroism
- FTIR
Fourier transform infrared spectroscopy
- MBH
Membrane-binding helix
- HDX-MS
Hydrogen/deuterium exchange mass spectrometry
- COX
Cytochrome oxidase c
- HDAC
Histone deacetylase
- BCR–ABL
Breakpoint cluster region–abelson fusion protein
- CK2
Casein Kinase II
- CML
Chronic myelogenous leukemia
- TDP
Top-down proteomics
- CRISPR
Clustered regularly interspaced short palindromic repeats
References
- 1.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 2.Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 3.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G(1) progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96(5):2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK. Functional analysis of the protein phosphatase activity of PTEN. Biochem J. 2012;444:457–464. doi: 10.1042/BJ20120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.You DW, Xin JP, Volk A, Wei W, Schmidt R, Scurti G, Nand S, Breuer EK, Kuo PC, Breslin P, Kini AR, Nishimura MI, Zeleznik-Le NJ, Zhang JW. FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-Null T-ALL cells. Cell Rep. 2015;10(12):2055–2068. doi: 10.1016/j.celrep.2015.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tamura M, Gu JG, Danen EHJ, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274(29):20693–20703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 7.Gu TT, Zhang Z, Wang JL, Guo JY, Shen WH, Yin YX. CREB Is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011;71(8):2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinde SR, Maddika S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat Commun. 2016 doi: 10.1038/ncomms10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi YJ, Wang JR, Chandarlapaty S, Cross J, Thompson C, Rosen N, Jiang XJ. PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol. 2014;21(6):522–527. doi: 10.1038/nsmb.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 11.McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation Study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3(3):137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 12.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18(2):400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Worby CA, Dixon JE. PTEN. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 14.Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res. 2011;71(3):629–633. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476(7359):163–169. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith LM, Kelleher NL, Proteomics CTD. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uversky VN. p53 proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016 doi: 10.3390/ijms17111874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgess AW, Cho H-S, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541–552. doi: 10.1016/S1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 19.Malaney P, Pathak RR, Xue B, Uversky VN, Dave V. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep. 2013;3:2035. doi: 10.1038/srep02035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282(32):23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 21.Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA. 2009;106(2):480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terrien E, Chaffotte A, Lafage M, Khan Z, Prehaud C, Cordier F, Simenel C, Delepierre M, Buc H, Lafon M, Wolff N. Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci Signal. 2012;5(237):ra58. doi: 10.1126/scisignal.2002941. [DOI] [PubMed] [Google Scholar]
- 23.Malaney P. Significance of PTEN phosphorylation and its nuclear function in lung cancer. Tampa: University of South Florida; 2016. [Google Scholar]
- 24.Tsai C-J, Ma B, Nussinov R. Protein–protein interaction networks: how can a hub protein bind so many different partners? Trends Biochem Sci. 2009;34(12):594–600. doi: 10.1016/j.tibs.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, Knoblauch N, Guarnerio J, Ito K, Turka LA, Beck AH, Pinton P, Bronson RT, Wei W, Pandolfi PP. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandolfi PP. Tumor suppressor phosphatases in tumorigenesis. Paper presented at the The PI3K-mTOR-PTEN network in health and disease. New York: Cold Spring Harbor Laboratory; 2016. [Google Scholar]
- 27.Sharrard RM, Maitland NJ. Alternative splicing of the human PTEN/MMAC1/TEP1 gene. Biochim Biophys Acta. 2000;1494(3):282–285. doi: 10.1016/S0167-4781(00)00210-4. [DOI] [PubMed] [Google Scholar]
- 28.Agrawal S, Eng C. Differential expression of novel naturally occurring splice variants of PTEN and their functional consequences in Cowden syndrome and sporadic breast cancer. Hum Mol Genet. 2006;15(5):777–787. doi: 10.1093/hmg/ddi492. [DOI] [PubMed] [Google Scholar]
- 29.Singh G, Chan AM. Post-translational modifications of PTEN and their potential therapeutic implications. Curr Cancer Drug Targets. 2011;11(5):536–547. doi: 10.2174/156800911795655930. [DOI] [PubMed] [Google Scholar]
- 30.Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, Pathak RR, Joerger AC. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev. 2014;114(13):6844–6879. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakakido M, Deng ZZ, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R. Dysregulation of AKT pathway by SMYD2-mediated lysine methylation on PTEN. Neoplasia. 2015;17(4):367–373. doi: 10.1016/j.neo.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20(14):5010–5018. doi: 10.1128/MCB.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PLM, Zheng ZM, Liaw D, Caron S, Duboue B, Lin AY, Richardson AL, Bonnetblanc JM, Bressieux JM, Cabarrot-Moreau A, Chompret A, Demange L, Eeles RA, Yahanda AM, Fearon ER, Fricker JP, Gorlin RJ, Hodgson SV, Huson S, Lacombe D, LePrat F, Odent S, Toulouse C, Olopade OI, Sobol H, Tishler S, Woods CG, Robinson BG, Weber HC, Parsons R, Peacocke M, Longy M, Eng C. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7(3):507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 34.Costa HA, Leitner MG, Sos ML, Mavrantoni A, Rychkova A, Johnson JR, Newton BW, Yee MC, De La Vega FM, Ford JM, Krogan NJ, Shokat KM, Oliver D, Halaszovich CR, Bustamante CD. Discovery and functional characterization of a neomorphic PTEN mutation. Proc Natl Acad Sci USA. 2015;112(45):13976–13981. doi: 10.1073/pnas.1422504112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez S, Genis L, Torres-Aleman I. A phosphatase-independent gain-of-function mutation in PTEN triggers aberrant cell growth in astrocytes through an autocrine IGF-1 loop. Oncogene. 2014;33(32):4114–4122. doi: 10.1038/onc.2013.376. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Huang CX, He JX, Lamb KL, Kang X, Gu TT, Shen WH, Yin YX. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep. 2014;6(5):844–854. doi: 10.1016/j.celrep.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston SB, Raines RT. Conformational stability and catalytic activity of PTEN variants linked to cancers and autism spectrum disorders. BioChemistry. 2015;54(7):1576–1582. doi: 10.1021/acs.biochem.5b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor MR, Lane DP, Fersht AR. Thermodynamic stability of wild-type and mutant p53 core domain. Proc Natl Acad Sci USA. 1997;94(26):14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 40.Liang H, He SM, Yang JY, Jia XY, Wang P, Chen X, Zhang Z, Zou XJ, McNutt MA, Shen WH, Yin YX. PTEN alpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014;19(5):836–848. doi: 10.1016/j.cmet.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tzani I, Ivanov IP, Andreev DE, Dmitriev RI, Dean KA, Baranov PV, Atkins JF, Loughran G. Systematic analysis of the PTEN 5′ leader identifies a major AUU initiated proteoform. Open Biol. 2016 doi: 10.1098/rsob.150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, Klein J, Pegno S, Sulis ML, Goldsteini H, Amendolara B, Lei L, Maurer M, Bruce J, Canoll P, Hibshoosh H, Parsons R. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science. 2013;341(6144):399–402. doi: 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulido R, Baker SJ, Barata JT, Carracedo A, Cid VJ, Chin-Sang ID, Dave V, den Hertog J, Devreotes P, Eickholt BJ, Eng C, Furnari FB, Georgescu MM, Gericke A, Hopkins B, Jiang X, Lee SR, Losche M, Malaney P, Matias-Guiu X, Molina M, Pandolfi PP, Parsons R, Pinton P, Rivas C, Rocha RM, Rodriguez MS, Ross AH, Serrano M, Stambolic V, Stiles B, Suzuki A, Tan SS, Tonks NK, Trotman LC, Wolff N, Woscholski R, Wu H, Leslie NR. A unified nomenclature and amino acid numbering for human PTEN. Sci Signal. 2014;7(332):e15. doi: 10.1126/scisignal.2005560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masson GR, Perisic O, Burke JE, Williams RL. The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem J. 2016;473:135–144. doi: 10.1042/BJ20150931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malaney P, Uversky VN, Dave V. The PTEN long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol Biosyst. 2013;9(11):2877–2888. doi: 10.1039/c3mb70267g. [DOI] [PubMed] [Google Scholar]
- 46.Bryant AM Structure and lipid binding preferences of the alternatively translated region of PTEN-long. Biophys J 110(3):420a. doi:10.1016/j.bpj.2015.11.2272
- 47.Johnston SB, Raines RT. Catalysis by the tumor-suppressor enzymes PTEN and PTEN-L. PLoS ONE. 2015 doi: 10.1371/journal.pone.0116898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell RB, Liu FH, Ross AH. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2003;278(36):33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen Ba AN, Pogoutse A, Provart N, Moses AM. NLStradamus: a simple hidden Markov model for nuclear localization signal prediction. BMC Bioinform. 2009;10:202. doi: 10.1186/1471-2105-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brameier M, Krings A, MacCallum RM. NucPred-predicting nuclear localization of proteins. Bioinformatics. 2007;23(9):1159–1160. doi: 10.1093/bioinformatics/btm066. [DOI] [PubMed] [Google Scholar]
- 51.Scott MS, Troshin PV, Barton GJ. NoD: a Nucleolar localization sequence detector for eukaryotic and viral proteins. BMC Bioinform. 2011;12:317. doi: 10.1186/1471-2105-12-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scott MS, Boisvert FM, McDowall MD, Lamond AI, Barton GJ. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010;38(21):7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li P, Wang D, Li H, Yu Z, Chen X, Fang J. Identification of nucleolus-localized PTEN and its function in regulating ribosome biogenesis. Mol Biol Rep. 2014;41(10):6383–6390. doi: 10.1007/s11033-014-3518-6. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Zhang P, Lin C, Yu Q, Wu J, Wang L, Cui Y, Wang K, Gao Z, Li H. Relevance and therapeutic possibility of PTEN-long in renal cell carcinoma. PLoS ONE. 2015;10(2):e114250. doi: 10.1371/journal.pone.0114250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hopkins BD. PTEN-LONG, a translational variant of the canonical tumor suppressor PTEN. New York: Columbia University; 2013. [Google Scholar]
- 56.Fenton TR, Nathanson D, de Albuquerque CP, Kuga D, Iwanami A, Dang J, Yang HJ, Tanaka K, Oba-Shinjo SM, Uno M, Inda MD, Wykosky J, Bachoo RM, James CD, DePinho RA, Vandenberg SR, Zhou HL, Marie SKN, Mischel PS, Cavenee WK, Furnari FB. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc Natl Acad Sci USA. 2012;109(35):14164–14169. doi: 10.1073/pnas.1211962109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meng Z, Jia LF, Gan YH. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene. 2016;35(18):2333–2344. doi: 10.1038/onc.2015.293. [DOI] [PubMed] [Google Scholar]
- 58.Kechagioglou P, Papi RM, Provatopoulou X, Kalogera E, Papadimitriou E, Grigoropoulos P, Nonni A, Zografos G, Kyriakidis DA, Gounaris A. Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res. 2014;34(3):1387–1400. [PubMed] [Google Scholar]
- 59.Nakahata S, Ichikawa T, Maneesaay P, Saito Y, Nagai K, Tamura T, Manachai N, Yamakawa N, Hamasaki M, Kitabayashi I, Arai Y, Kanai Y, Taki T, Abe T, Kiyonari H, Shimoda K, Ohshima K, Horii A, Shima H, Taniwaki M, Yamaguchi R, Morishita K. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat Commun. 2014;5:3393. doi: 10.1038/ncomms4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ, Lu NH. Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: a novel mechanism of PTEN inactivation. Clin Res Hepatol Gastroentrol. 2013;37(1):72–79. doi: 10.1016/j.clinre.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 61.Roy D, Dittmer DP. Phosphatase and tensin homolog on chromosome 10 Is phosphorylated in primary effusion Lymphoma and Kaposi’s Sarcoma. Am J Pathol. 2011;179(4):2108–2119. doi: 10.1016/j.ajpath.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee EJ, Kim N, Kang KH, Kim JW. Phosphorylation/inactivation of PTEN by Akt-independent PI3K signaling in retinal pigment epithelium. Biochem Biophys Res Commun. 2011;414(2):384–389. doi: 10.1016/j.bbrc.2011.09.083. [DOI] [PubMed] [Google Scholar]
- 63.Hua F, Ha TZ, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li CF. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol. 2007;178(11):7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- 64.Horita H, Furgeson SB, Ostriker A, Olszewski KA, Sullivan T, Villegas LR, Levine M, Parr JE, Cool CD, Nemenoff RA, Weiser-Evans MCM. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J Am Heart Assoc. 2013 doi: 10.1161/JAHA.113.000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morotti A, Panuzzo C, Crivellaro S, Carrà G, Fava C, Guerrasio A, Pandolfi PP, Saglio G. BCR-ABL inactivates cytosolic PTEN through casein kinase II mediated tail phosphorylation. Cell Cycle. 2015;14(7):973–979. doi: 10.1080/15384101.2015.1006970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toby TK, Fornelli L, Kelleher NL. Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem. 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michel AM, Ahern AM, Donohue CA, Baranov PV. GWIPS-viz as a tool for exploring ribosome profiling evidence supporting the synthesis of alternative proteoforms. Proteomics. 2015;15(14):2410–2416. doi: 10.1002/pmic.201400603. [DOI] [PMC free article] [PubMed] [Google Scholar]