

Abstract
Melatonin is an ancient antioxidant. After its initial development in bacteria, it has been retained throughout evolution such that it may be or may have been present in every species that have existed. Even though it has been maintained throughout evolution during the diversification of species, melatonin’s chemical structure has never changed; thus, the melatonin present in currently living humans is identical to that present in cyanobacteria that have existed on Earth for billions of years. Melatonin in the systemic circulation of mammals quickly disappears from the blood presumably due to its uptake by cells, particularly when they are under high oxidative stress conditions. The measurement of the subcellular distribution of melatonin has shown that the concentration of this indole in the mitochondria greatly exceeds that in the blood. Melatonin presumably enters mitochondria through oligopeptide transporters, PEPT1, and PEPT2. Thus, melatonin is specifically targeted to the mitochondria where it seems to function as an apex antioxidant. In addition to being taken up from the circulation, melatonin may be produced in the mitochondria as well. During evolution, mitochondria likely originated when melatonin-forming bacteria were engulfed as food by ancestral prokaryotes. Over time, engulfed bacteria evolved into mitochondria; this is known as the endosymbiotic theory of the origin of mitochondria. When they did so, the mitochondria retained the ability to synthesize melatonin. Thus, melatonin is not only taken up by mitochondria but these organelles, in addition to many other functions, also probably produce melatonin as well. Melatonin’s high concentrations and multiple actions as an antioxidant provide potent antioxidant protection to these organelles which are exposed to abundant free radicals.
Keywords: Free radical-related diseases, SIRT3, Melatonin transporters, Reactive oxygen species, Mitochondrial transition pore, Cytochrome c, Apoptosis, Inner mitochondrial membrane
Introduction
An estimated 2.5 × 109 years ago, molecular oxygen (O2) began to rise in the Earth’s atmosphere due to its persistent release from photosynthetic bacteria that had evolved an estimated half-billion years earlier [1]. The rise in atmospheric O2 was a highly selective pressure for the evolution of organisms to use O2 as the basis of their metabolism. O2-based metabolism proved to be an enormous metabolic advance for aerobic species given that the total combustion of glucose yields 36 molecules of ATP per molecule of glucose. An estimated 95% of O2 used by mammalian cells is reduced by the addition of four electrons by cytochrome c to produce two molecules of water.
There is, however, a significant downside to oxidative metabolism. The transfer of electrons between the complexes of the respiratory chain is not flawless with some electrons escaping where they chemically reduce adjacent O2 molecules [2]. This inappropriate metabolism of O2 generates free radicals and other reactive oxygen species (ROS) which are often highly toxic to molecules in the vicinity of where they are produced. Free radicals are derivatives of O2 that contain an odd number of electrons in their valence orbital; they include the superoxide anion radical (O·−2), hydroxyl radical (·OH), hydroperoxyl radical (HOO·), peroxyl radical (ROO·), and alkoxyl radical (RO·), among others [3]. Some ROS are not free radicals because they possess an even number of electrons and include hydrogen peroxide (H2O2), hypochlorous acid, etc.
O·−2 is a consequence of the univalent reduction of O2 (Fig. 1). Radicals are typically unstable and sometimes highly reactive. While O·−2 per se is not highly toxic to neighboring molecules, its most damaging actions stem from its conversion to secondary highly toxic agents, especially the ·OH and the peroxynitrite anion (ONOO−) [4, 5]. In an aqueous environment, O·−2 is quickly dismutated to H2O2. Hence, the formation of O·−2 is invariably accompanied by the production of H2O2. H2O2, a non-radical ROS, is rather stable and only sluggishly interacts with a number of organic molecules. A major aspect of H2O2 that makes it destructive is its high lipophilicity; this allows H2O2 to readily cross lipid-rich membranes thereby spreading the potential damage inflicted by free radicals. The other danger of H2O2 results when it reacts with iron, copper (and other transition metals) or with certain hemoproteins to yield the ·OH [6, 7]. These reactions are termed the Haber–Weiss reactions or, perhaps more appropriately, the O·−2 driven Fenton reaction. The ·OH is devastatingly reactive and immediately reacts with any molecule in the immediate vicinity of where it is produced; these reactions occur at diffusion limited rates. As a result, the ·OH is extremely short-lived and its “reaction cage” is very small so the damage it inflicts is site-specific. It is estimated that, of the total free radical/ROS molecular damage that occurs in organisms, the majority may be a consequence of the ·OH [6, 8]. ·OH also interacts with proteins, carbohydrates, nucleic acids, and lipids to produce ROO· as intermediates. ROO·, although less reactive than the ·OH, have a relatively long half-life and, therefore, they damage molecules at some distance from their site of production. The most thoroughly studied reaction of ROO· involves the peroxidation of polyunsaturated fatty acids (PUFA). The destruction of PUFA in cell membranes is a major factor that leads to the functional deterioration of cells and their eventual death. ROO· can also oxidize carbohydrates, proteins and some sulfhydryl components of hemoproteins.
Fig. 1.

A small percentage of oxygen inhaled/utilized by aerobic organisms generates oxygen-based derivatives, often called reactive oxygen species (ROS), that can damage critical molecules within cells. Some of the derivatives are free radicals [with an unpaired electron represented by the dot (·)] and others are not, e.g., hydrogen peroxide. The superoxide anion radical is quickly metabolized by superoxide dismutase (SOD) to hydrogen peroxide which can be removed from the intracellular environment by either catalase (CAT) or glutathione peroxidase (GPx). Oxidized glutathione (GSSG) is converted back to reduced glutathione (GSH) by glutathione reductase (GRd). The most destructive derivatives of oxygen are the hydroxyl radical and peroxynitrite. The hydroxyl radical is formed from hydrogen peroxide during the Fenton reaction and peroxynitrite is generated when the superoxide anion couples with nitric oxide. A large percentage of the ROS formed within cells is produced in mitochondria as a consequence of the leakage of electrons (e−) from the electron transport chain
While O2 is considered poisonous as noted above, the predominate theory to explain its toxicity is a consequence of its chemical reduction to O·−2. The most obvious source of O·−2 in vivo in aerobic cells is generally considered to be the mitochondrial electron transport chain (ETC) (Fig. 2). In addition to the presence of the ETC in the mitochondria of all mammalian cells (except erythrocytes since they lack mitochondria), it is also present in the membranes of many bacteria; in plant chloroplasts and some other less studied sites [9, 10].
Fig. 2.
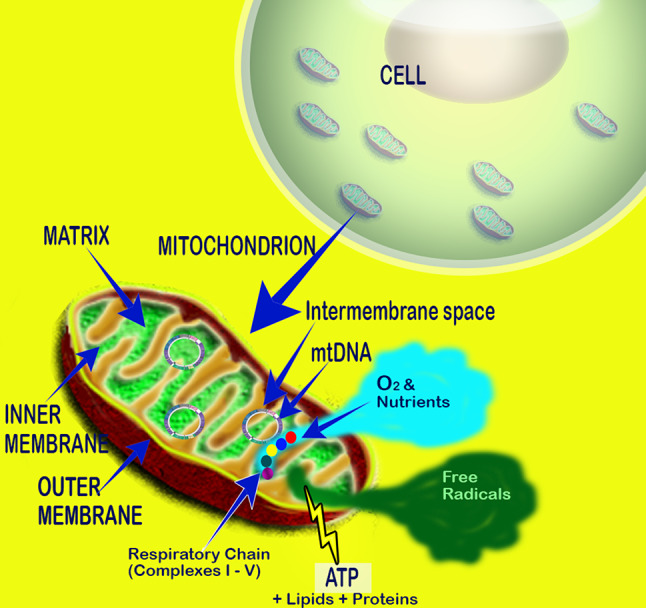
The mitochondrial respiratory chain utilizes oxygen to generate energy in the form of ATP. Free radicals and reactive oxygen species are formed when electrons that are passed between successive complexes are fumbled and chemically reduce adjacent oxygen molecules. The toxic derivatives of oxygen, since the majority are formed in mitochondria, especially damage mitochondrial DNA, proteins and lipids. Because mitochondria are a primary source of toxic derivatives of oxygen, an antioxidant positioned in mitochondria would be especially important in reducing oxidative damage
While the mitochondrial ETC is efficient in shunting electrons between successive components (which constitute the complexes), some electrons are fumbled and reduce nearby oxygen molecules to O·−2. The quantity of O·−2 produced is related to the O2 tension; thus O·−2 production increases as the concentration of O2 rises. Under physiological O2 levels and when the ETC is functioning optimally, an estimated 1–3% of the O2 is converted to O·−2. In the event of damage to the components of the ETC, they function suboptimally, so electron leakage increases as does ROS formation. This occurs in aged individuals, during toxin exposure, etc. Other sources of ROS include enzymes, e.g., xanthine oxidase, auto-oxidation reactions and haem proteins [11].
Evolution of melatonin’s multiple functions
Melatonin predictably evolved an estimated 3.0–2.5 billion years ago, probably in photosynthetic bacteria [12, 13], where it was specifically designed to neutralize the toxic O2 derivatives that were produced during photosynthesis [14]. Once melatonin (N-acetyl-5-methoxytryptamine) appeared, for the next 3 billion years evolution never tinkered with the chemical structure of this agent such that the melatonin in cyanobacteria [15, 16] is structurally identical to melatonin that exists in present-day mammals, including the human [17, 18]. While melatonin’s structure has remained stable, its functions have become highly diversified. Thus, its original antioxidant function has been retained and supplemented with a variety of other actions during various stages of evolution (Fig. 3).
Fig. 3.

Melatonin is believed to exist in most, possibly all, animal and plant species. It predictably evolved 3.0–2.5 billion years ago in photosynthetic cyanobacteria as an antioxidant; this function has been retained to the present day including in humans. Other functions of melatonin, many more than are shown in this figure, appeared at later stages of evolution.
Reprinted with permission from Manchester et al. [13]
The first action of melatonin that was identified after its discovery in 1958 [19] was its ability to regulate the reproductive capability of photosensitive mammals [20, 21]. Thus, the duration of nocturnally elevated melatonin levels, which changed seasonally, were shown to drive the waxing and waning of reproductive competence in temperate and arctic species that are seasonal breeders [22, 23]. It soon became apparent that this action of melatonin was not its sole, and likely, not its most important action. Since then, a baffling array of functions have been assigned to this molecule, e.g., oncostatic [24, 25], anti-inflammatory [26–30], circadian rhythm modulation [31, 32], sleep promotion [33, 34], anti-venom [35, 36], body weight regulation [37, 38], anti-diabetic [39, 40], anti-fibrotic [41, 42], and others. The action of melatonin that has the longest history, however, is likely its ability to maintain redox homeostasis.
Melatonin as a free radical scavenger and as an antioxidant
Melatonin is uncommonly effective in reducing oxidative stress because of the number of means it has as a direct free radical scavenger and indirect antioxidant. Thus, melatonin (a) functions in this capacity in both the aqueous and lipid portions of the cell [43], (b) as a result, it protects lipids [44, 45], proteins [46, 47], and DNA [48, 49] from oxidative damage, (c) it is more highly concentrated in the regions of the cells where many of the free radicals are formed, e.g., mitochondria [50, 51], (d) it may be synthesized in the mitochondria [52] and at this site its synthesis may be inducible [53], (e) not only melatonin but a number of its metabolites also function as radical scavengers [54–56], (f) melatonin binds transition metals which reduces the formation of the most aggressive ROS, i.e., ·OH [57, 58], (g) melatonin stimulates the activity of a number of antioxidative enzymes [59–61], and (h) it promotes the synthesis of another important antioxidant, glutathione [62]. Finally, SIRT3—a class III histone deacetylase, which is primarily located in the mitochondrial matrix, has critical functions in protecting these organelles from oxidative stress [63]. While the role of melatonin in impacting mitochondrial SIRT3 is not mechanistically well defined, data indicate that SIRT3 may mediate at least some of the antioxidative actions of melatonin [64–66]. Considering these diverse functions, it is not always possible to determine the relative importance of each of these processes in a given highly oxidizing environment.
The detoxification of ROS/RNS is achieved by melatonin and a number of its metabolic kin in what is later referred to as the antioxidant cascade [67, 68]. Hence, the derivatives of melatonin that are formed when it directly neutralizes a free radical, often by electron donation [69], are equally as effective, and sometimes more so, than melatonin itself in reducing oxidative stress [70]. There are a number of comprehensive reviews that summarize the details by which melatonin functions in the reduction of oxidative damage. Rather than re-iterating these multiple actions here, the reader is directed to the associated publications [13, 15, 61, 71–78].
Immunocytochemical evidence for melatonin as a mitochondria-targeted antioxidant
That melatonin acts at the level of the mitochondria to prevent ROS toxicity [79, 80] was documented within a decade after the indole was discovered to be a direct potent free radical scavenger [67, 81, 82] and indirect antioxidant [58–60, 62, 83]. Using cyanide, an ETC complex IV inhibitor, Yamamoto and Yang [84] showed that this drug’s ability to cause seizures and kill mice was reversed by melatonin; the implication of these findings was that melatonin entered mitochondria and interfered with the negative actions of cyanide. Similarly, the actions of neurotoxins including 6-hydroxydopamine (6-OHDA) [85], kainic acid [86] and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [87]-mediated dysfunction of mitochondrial Complex I are also overcome by melatonin. During the same time frame, melatonin was reported to augment the activities of respiratory chain Complexes I and IV and to reduce mitochondrial damage resulting from the treatment of rats with ruthenium red, a molecule that inhibits the mitochondrial Ca2+ uniporter leading to oxidative stress and mitochondrial uncoupling [88, 89]. These actions were not duplicated when vitamins C or E were used as replacements for melatonin. Martin and coworkers [90] also verified that melatonin stimulated oxidative phosphorylation and promoted a rise in ATP production in neuronal and hepatic cell mitochondria. Finally, hepatocyte respiratory physiology of aging mice was restored by melatonin, particularly at the levels of Complexes I and IV [91] and it limited ischemia/reperfusion-mediated mitochondrial dysfunction in rat liver [92]. Finally, the group led by Jou [93, 94] found that melatonin curtailed laser irradiation-induced ROS formation in astrocyte mitochondria and protected mitochondrial DNA from mutations/deletions and the cells from apoptosis.
The first comprehensive investigation designed to visualize melatonin’s ability to both quench ROS formed in mitochondria and to elucidate the series of molecular events that culminate in ROS-mediated cellular death was carried out by Jou et al. [95]. Using a combination of time lapse conventional and confocal microscopy with the aid of fluorescent probes, we monitored the actions of melatonin at the mitochondrial level. Initially, melatonin (100 µM) was found to highly effectively resist 10 mM H2O2-induced mitochondrial swelling and other cellular changes related to apoptosis of astrocytes as seen using phase contrast microscopy. Moreover, confocal microscopy imaging of the cells after they were treated with fluorescent probes to identify the health of mitochondria (Mito G) and of the nuclei (propidium iodide) documented that H2O2-mediated loss of functional mitochondria and nuclear impulsion was prevented by melatonin.
To determine whether the mitochondrial protection associated with melatonin treatment of H2O2-exposed astrocytes related to the ability of the indole to neutralize free radicals in these subcellular organelles, ROS levels in mitochondria were evaluated using dichlorofluorescein (DCF) and dichlororhodamine (D-123) [95]. The findings showed that after H2O2 treatment, there was a massive rise in free radical fluorescence in mitochondria within 10 min. Concurrent melatonin treatment prevented the rapid increase in mitochondrial ROS concentrations and maintained them at the levels seen in untreated astrocytes (Fig. 4). To achieve this marked inhibition of ROS fluorescence, it is likely that melatonin penetrated to the mitochondrial matrix where it either scavenged the ROS as they were formed or it prevented their formation in the mitochondria.
Fig. 4.
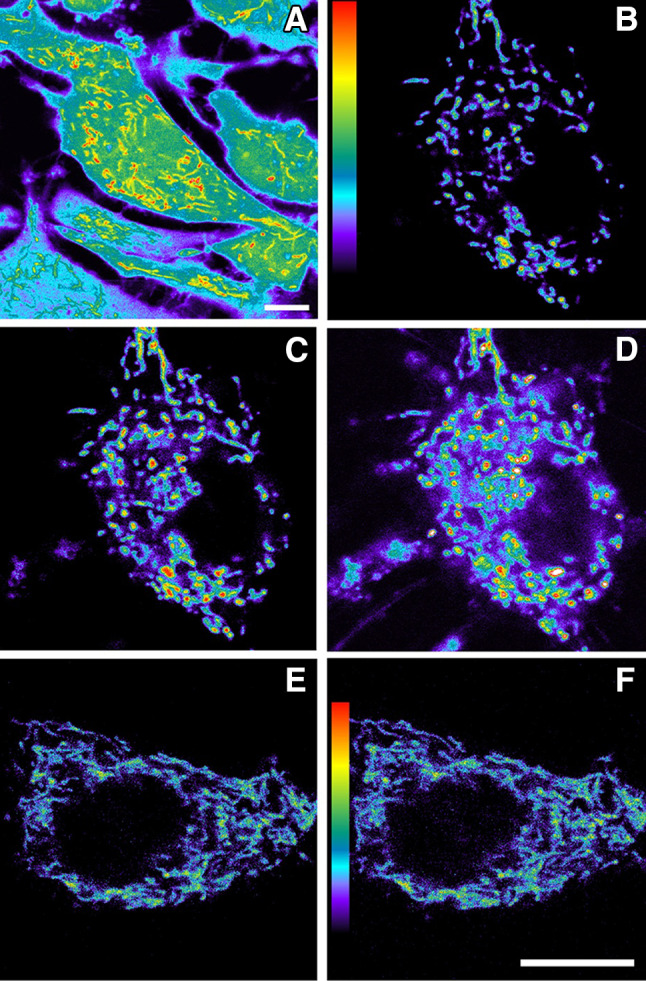
Fluorescence imaging of reactive oxygen species generation in cultured astrocytes, especially in the mitochondria, and the inhibition of ROS by melatonin. a An enhanced pseudocolor image which documents the higher ROS levels in mitochondria (yellow to red) than in other subcellular compartments. b–d Rapid increase in mitochondrial ROS formation in astrocytes exposed to an oxidant, H2O2, as visualized using dihydrorhodamine 123; a before exposure to H2O2; b at 5 min and c at 10 min following the addition of H2O2. In addition to being much brighter, the mitochondria in the H2O2-exposed cells are swollen. e, f When melatonin was added to the culture medium simultaneously with H2O2, ROS levels in the mitochondria did not increase at either 5 or 10 min, consistent with the ability of melatonin to enter the mitochondria and neutralize the ROS.
Reprinted with permission Jou et al. [95]
The reduction in ROS due to melatonin was further documented using FACS (fluorescence detected by flow cytometry) coupled with the use of ROS sensitive probes [95]. In this case, ROS formation was promoted by the exposure of astrocytes to either H2O2 or to short chain oxidants, i.e., tert-butyl hydroperoxide (t-BuOOH) or cumene hydroperoxide (Cu-OOH). Again, using these technologies, melatonin dose-dependently inhibited ROS formation (Fig. 5). Melatonin not only restricted ROS formation in oxidant-treated cells, but also lowered the concentration of ROS in otherwise untreated resting cells. When an equivalent concentration of vitamin E was used as an antioxidant as a replacement for melatonin, it proved far less effective than the indole in reversing the toxic actions of oxidant treatment.
Fig. 5.
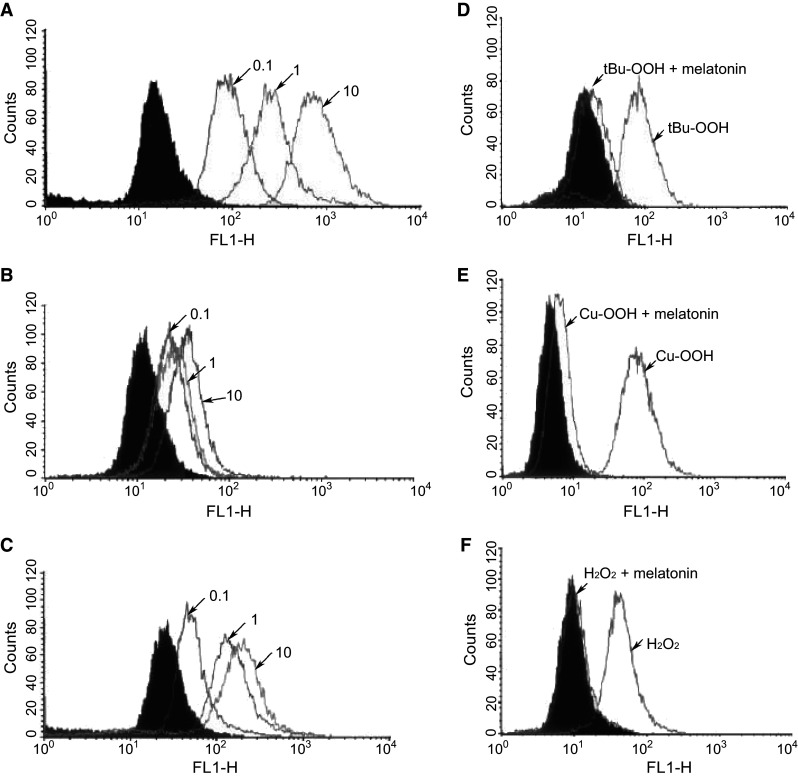
Fluorescence detected by flow cytometry (FACS) analysis of ROS generation in astrocytes after exposure to one of several oxidants: H2O2, tert-butyl hydroperoxide (t-BuOOH) or cumene hydroperoxide (Cu-OOH). a Illustrates the dose-dependent increase in mitochondrial ROS induced by H2O2 (0.1, 1.0 or 10 mM). b The addition of melatonin (100 µM) with H2O2 greatly diminished ROS fluorescence. c When 100 µM vitamin E was exchanged for melatonin, it was much less effective than melatonin in reducing mitochondrial ROS. d, e Melatonin also significantly lowered ROS generation in cells exposed to either t-BuOOH or Cu-OOH, respectively. f Melatonin inhibition of ROS-mediated by H2O2 exposure.
Reprinted with permission from Jou et al. [95]
As a continuation of these studies, we [95] examined the dysregulation of intracellular calcium as an early indication of opening of the mitochondrial permeability transition pore (MPTP) in cells exposed to an oxidant. Under control conditions, astrocyte mitochondrial Ca2+ concentrations were low but increased rapidly after H2O2 treatment with opening of the MPTP. The events resulting from mitochondrial Ca2+ overload were prevented by melatonin. Likewise, mitochondrial membrane potential depolarization induced by H2O2 was suppressed by melatonin leading to a reduction in the escape of lethal cytochrome c (Fig. 6) and caspase-mediated apoptosis.
Fig. 6.
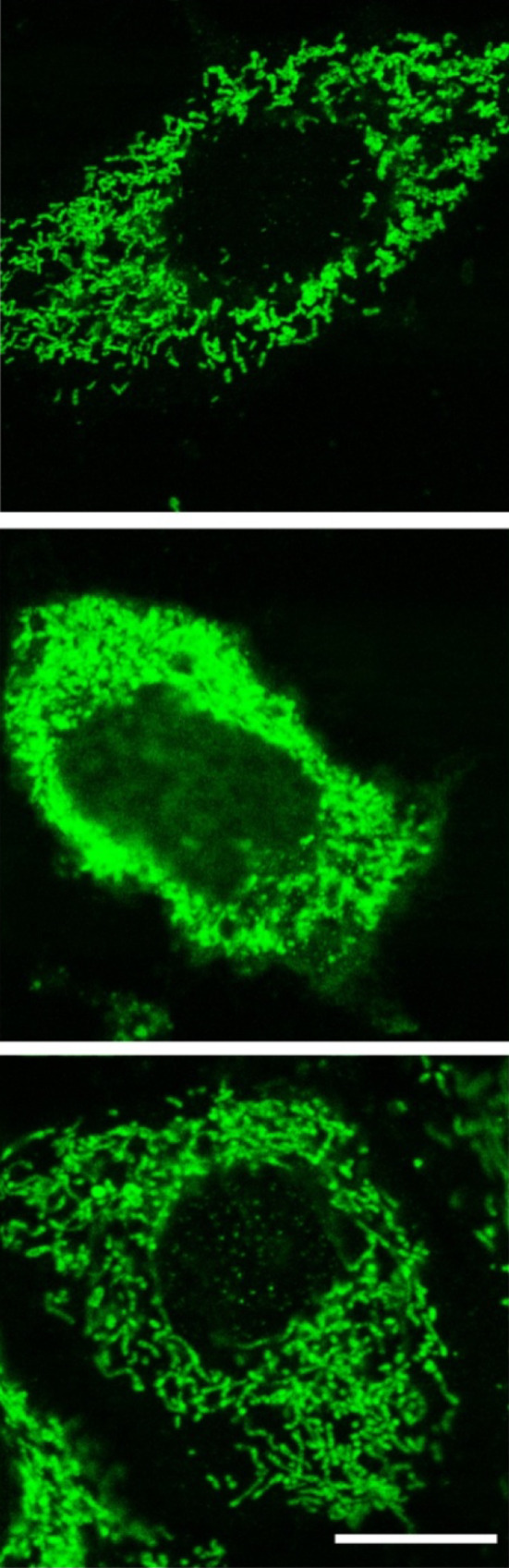
The induction of cellular apoptosis after exposure to an oxidant involves the release of cytochrome c from damaged mitochondria with the subsequent activation of caspases leading to programmed cell death. This figure illustrates the localizations of cytochrome c in the mitochondria of astrocytes not exposed to an oxidant (top); middle after 90 min exposure to H2O2; bottom after exposure to H2O2 plus melatonin. Clearly, H2O2 treatment caused a massive release of cytochrome c into the cytosol and much less into the nucleus with melatonin almost totally preventing this escape. The release of cytochrome c occurred simultaneously with retraction of cell processes, shrinkage of the cells and an irregular plasma membrane. Cytochrome c was detected using immunocytochemistry and laser scanning confocal microscopy. Bar 10 µM.
Reprinted with permission from Jou et al. [95]
This series of investigations illustrate that by intervening at the early phases of the mitochondrial-mediated apoptotic process, melatonin prevents all the downstream events associated with the loss of cells resulting from massive free radical damage due to oxidant exposure. The findings also are consistent with melatonin’s ability to quickly gain access to the mitochondrial matrix where it readily soaks up free radicals.
In the studies summarized and reported by others [88, 96], melatonin also was superior to other antioxidants in preventing oxidative damage normally meted out by toxic ROS. The large scale deletion of 4577 bp from mitochondrial DNA is known as the common deletion (CD); this deletion eliminates roughly one-third of the mitochondrial base pairs and severely damages the efficiency of the respiratory chain. This defect greatly augments mitochondrial ROS generation in cells which leads to an elevated rate of apoptosis. When the CD occurs in humans it is responsible for respiratory chain defect-associated diseases; treatments for these conditions have limited efficacy [97].
Using appropriate fluorescent probes, Jou and colleagues [98] and Peng et al. [99] were able to visualize the increased mitochondrial ROS production in single cells suffering with the CD. Considering the high efficacy of melatonin as an ROS scavenger, we [100] tested how effective the indole would be in protecting cells with CD-augmented mitochondrial oxidative stress and apoptosis and especially from secondary oxidative stress (that which occurs when such already-damaged cells are exposed to H2O2). In these situations, melatonin lowered basal as well as secondary oxidative stress. Moreover, melatonin prevented mitochondrial ROS-mediated depolarization of the mitochondrial membrane and counteracted opening of the MPTP. Melatonin also reduced cardiolipin depletion and halted apoptosis. Finally, melatonin suppressed mitochondrial Ca2+ dysregulation with this protection exceeding that provided by either vitamin E or synthetic mitochondria-targeted co-enzyme Q, i.e., Mito Q. Based on these observations, Jou and coworkers [100] feel melatonin may prove effective as a therapy in clinical situations that involve mitochondrial malfunction.
A follow-up report revealed that melatonin reduced apoptosis of astrocytes in which mitochondrial Ca2+ dysregulation was suppressed. Also under some conditions, melatonin effectively attenuates MPTP opening and apoptosis in the cells where its antioxidative actions are prevented, indicating that melatonin may directly target the MPTP [101].
Melatonin in multiple organs and routes of release
Based on the discovery of melatonin in pineal tissue [19] and the fact that surgical removal of this organ (or its sympathetic denervation or decentralization) reduces circulating levels to near zero [102, 103] and eliminates some of its circadian and circannual functions [20, 104–106], for many years the pineal was considered the exclusive source of melatonin in vertebrates. In all mammalian species, the pineal synthesizes and releases melatonin in a circadian fashion with the highest circulating levels at night and much lower values during the day. The details of the circadian control of melatonin synthesis in the pineal gland have been well defined [18].
While it was initially thought that the primary secretory route of pineal melatonin is into the rich vascular network in the gland, we believe that the major pathway of secretion is in fact directly into the cerebrospinal fluid (CSF) [107]. This is consistent with the very large amplitude nocturnal rise in CSF melatonin relative to the much lower nighttime increase in the blood and also with the much more precise rise and fall of melatonin in the fluid present in the third ventricle of the brain [108]. Thus, the circadian regulation of the central biological clock, i.e., the suprachiasmatic nuclei (SCN), is reliant on the CSF melatonin rhythm rather than the cycle of melatonin in the blood [107, 109]. The blood melatonin rhythm, however, along with the autonomic nervous system is presumably left to cue circadian clock genes that exist in peripheral tissues [110].
We also surmise that another important function of the large nightly increases in CSF melatonin is for its antioxidant protection of the brain [107], which has a high metabolic rate and a very high utilization of O2 which puts the mitochondria of neurons and glial in excessive oxidative jeopardy [111]. The consequences of the high use of O2 by the brain, because of the generation of partially reduced derivatives, is seen in many neurodegenerative disorders all of which have a prominent oxidative component [112–114]. Besides melatonin of pineal origin providing neuronal/glial mitochondrial protection from oxidative stress, possibly all cells in the central nervous system generate melatonin for their restricted use in resisting the toxicity of ROS/RNS.
While the derivation of CSF and blood melatonin rhythms are undoubtedly a function of melatonin released from the pineal gland, it is now obvious that melatonin synthesis is not unique to this organ. The first structure, after the pineal, in which melatonin was found to be produced was another neural structure, the retinas [115], where, like the pineal gland, its synthesis is rhythmic. Subsequently, melatonin production has been uncovered in many non-neural tissues.
In some peripheral organs, local melatonin synthesis is inferred because surgical removal of the pineal gland, which depletes circulating melatonin values to barely measureable values, does not decrease values in peripheral cells. For example, in hepatocytes not only does pinealectomy not diminish intracellular concentrations of the indole, but melatonin levels actually rise in some subcellular organelles [50], a presumed compensatory response to the reduction of circulating melatonin. The rise in tissue levels of melatonin after pinealectomy, implies that melatonin synthesis is inducible in animals [53] as it is in plants [13]. Even in the presence of an intact pineal gland, melatonin concentrations in mitochondria and cell membranes of hepatocytes greatly exceed those in the blood [50]. Interestingly, bile, which is produced by hepatocytes, has exceptionally high concentrations of melatonin [116] potentially also originating from the mitochondria of liver-associated cells. The uncommonly high levels of melatonin in bile are predictably for the purpose of protecting the epithelium of the biliary tree from oxidative damage inflicted by highly toxic biliary constituents [117]. The atypically elevated levels of melatonin in the bile may also be augmented due to its recirculation in the enterohepatic circulation. Finally, melatonin produced in the gut microbiome or consumed in the diet is taken up by the capillary bed of the hepatic venous system which may then transfer it to the hepatocyte from where it may be shunted into the bile. Melatonin, which was discovered in land plants in 1995 [118, 119], is in much higher concentrations in plant products that are common in the human and animal diet. Melatonin in plants serves a similar function as in animals, i.e., as an antioxidant [120] and it also functions in growth promotion, not unlike an auxin [121].
The widespread production of melatonin in peripheral organs should not be unexpected considering the evolutionary origin of mitochondria. We recently proposed that the ability of all eukaryotic cells to produce melatonin [12] stems from the likely bacterial origin of mitochondria (and chloroplasts of plants) [122]. According to the endosymbiotic theory, mitochondria and chloroplasts developed from bacteria that were engulfed by ancestral prokaryotic organisms (Fig. 7). Since the devoured bacteria already produced melatonin [15], we speculate that this function was preserved by both the evolving mitochondria and chloroplasts [12]. Since the former exists in most cells of eukaryotes, essentially every cell may have the capability of forming its own melatonin, not for distribution throughout the organism, but use locally in protection against an oxidative challenge (Fig. 8). In cells other than the pinealocytes, the melatonin synthetic pathway in mitochondria and chloroplasts may be functioning at a low level or may be dormant under conditions of minimal oxidative stress, but it is upregulated under circumstances where a compensatory rise in melatonin production would be required to resist an augmented production of ROS/RNS. Such a compensatory stimulation of melatonin formation has already been reported in plants subjected to either abiotic [123] or biotic stress [124] and is consistent with the findings of Venegas and colleagues [50] who reported loss of pineal melatonin is accompanied by a rise in cell membrane melatonin concentrations in cerebrocortical cells and in hepatocytes. Many stresses are known to promote free radical production in cells, e.g., toxic drugs (doxorubicin), chemical toxins (paraquat), heavy metals, ionizing radiation, ultraviolet radiation, hypoxia (stroke/heart attack), hyperoxia, drought, excessive cold or heat, environmental air pollutants (soot/smog), bacterial invasion, excessive exertion, inflammation, allergens, etc. Each of these stresses presumably induces a compensatory rise in intracellular melatonin synthesis in the affected cell for its own protection, i.e., as a firewall against oxidative stress. This is clearly one means among several features by which melatonin differs from other free radical scavengers, i.e., it may be produced at the local level where maximal free radical generation occurs and its synthesis in these cells can be upregulated.
Fig. 7.

The endosymbiotic theory of the origin of mitochondria and chloroplasts. Mitochondria arose from engulfed bacteria that were initially taken in and digested for their nutrients. During evolution, the ingested bacteria developed a symbiotic relationship with the host cell and evolved into mitochondria. Likewise, photosynthetic bacteria were also taken in as food but eventually evolved to form chloroplasts. Since, the ingested bacteria (which formed both mitochondria and chloroplasts) produced melatonin, we proposed this function was retained such that in current day animals and plants, both mitochondria and chloroplasts retain the ability to produce melatonin. Emerging evidence supports this assumption.
Reprinted with permission from Manchester et al. [13]
Fig. 8.

The targeting of melatonin to the mitochondria; evidence suggests that melatonin enters the mitochondria through specific transporters, PETP 1/2 (oligopeptide transporters). The actions of melatonin in mitochondria are multiple. These actions, particularly including its ability to reduce oxidative damage to critical mitochondrial molecules, preserve the function of these organelles and benefit diseases in which mitochondrial malfunction is a feature. Melatonin increases the efficiency of the electron transport chain (I, II, III and IV) and improves ATP production (ATP synthase). Reactive oxygen species (ROS) produced when electrons leak from the ETC are directly scavenged by melatonin and its metabolite [N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK)]. ROS are also metabolized by mitochondria superoxide dismutase (SOD2) and scavenged by glutathione (GSH) and SIRT3. Melatonin also modulates uncoupling protein (UCP2) to maintain an optimal inner mitochondrial membrane potential and prevents opening of the mitochondrial permeability transition pore (MPTP). This limits the escape of cytochrome c when the mitochondrion is damaged by ROS. Recent evidence suggests that melatonin, in addition to quickly entering the mitochondria, may also be synthesized in this organelle (5HT → Mel) where it is also metabolized to AFMK. Not shown in this figure are other actions of melatonin that prevent mitochondrial damage and cell death. These include a reduction in oxidative damage to lipids, proteins and DNA, upregulation of antiapoptotic proteins and downregulation of proapoptotic proteins and a suppression of the activities of caspases which execute the apoptosis pathway
Evidence for the uptake of melatonin and its synthesis in mitochondria
The phagocytosis of melatonin-forming bacteria by primitive eukaryotes (the endosymbiotic theory, Fig. 7) theoretically explains the origin of both mitochondria and chloroplasts that exist in present-day unicellular and multicellular organisms. When these engulfed bacteria evolved into these cellular organelles, they retained their melatonin-forming ability and horizontally transferred this machinery to the eukaryotic cell [15]. This inherited capability has persisted throughout evolution.
The ability of mitochondria and chloroplasts to produce melatonin is currently under intensive investigation. With regard to the production of melatonin by mitochondria, it is known that these organelles contain much higher melatonin concentrations than exist in the circulation [50, 125]. This could mean that mitochondria are capable of concentrating melatonin from the blood against a gradient; however, since pinealectomy rather than depleting mitochondrial melatonin levels actually causes them to rise [50] argues against this possibility. An alternative explanation is that the mitochondria produce their own melatonin.
In the pineal gland, where melatonin is abundantly produced on a nightly basis, the immunocytochemical localization of the AANAT at the ultrastructural level seems to be restricted to the mitochondria [126, 127]. Assuming the validity of this finding, it suggests that the melatonin’s synthetic pathway in pinealocytes is at least partially located in mitochondria. If this is the case for pinealocytes, it may extend to other cells as well.
This indirect evidence is supported by a recent observation related to melatonin synthesis in the mammalian oocyte. These critically important cells have been shown to synthesize melatonin to ensure that they are adequately protected from oxidative damage during their maturation, at ovulation and during their fallopian tube transfer and implantation [128–130]. Based on the data of He and coworkers [52], the mitochondria of oocytes are the major site of melatonin production in these cells. When oocyte mitochondria were cultured in a medium containing the melatonin precursor, serotonin, the melatonin concentration in the medium quickly increased and within 15 min, the values were about 12-fold higher than levels in medium from mitochondria not supplemented with serotonin. Similarly, AANAT (alkylamine-N-acetyl transferase) estimated from immunocytochemical images, was shown to exist in both mitochondria and cytosol of mouse oocytes. The production of melatonin in oocyte mitochondria has implications for the more general implications that mitochondria of other cells also produce this indole. Hopefully, these observations will prompt other investigators to examine melatonin synthesis in mitochondria of somatic cells. For chloroplasts, the data relative to their capacity to produce melatonin are more complete than the data for the synthesis of this indole in mammalian mitochondria [131–133].
While the production of melatonin in mitochondria seems likely and evidence of this has emerged [134, 135], the synthesis of melatonin in the cytosol is not precluded. Certainly, in reference to the pineal gland it has always been assumed that melatonin synthesis is primarily confined to the cytosol [18]. However, an analysis of the kinetics of AANAT as well as substrate availability, i.e., acetyl CoA, we predict that mitochondria more efficiently convert serotonin to melatonin than does the cytosol [136]. This is also consistent with the much higher concentrations of melatonin in this organelle than in the cytosol or in the blood, even after pinealectomy which lowers circulating melatonin values to near zero [50, 137].
The high efficacy of melatonin in limiting mitochondrial damage in diseases/disorders that have a high oxidative component [138–141] would generally require elevated levels of this antioxidant, either as a result of its local synthesis or rapid uptake. Even before melatonin was discovered to be a free radical scavenger, we noted that an intensive mitochondrial oxidation-producing situation, i.e., forced swimming, caused the rapid disappearance of melatonin from the pineal and the blood [142–144]. These studies did not prove that melatonin was rapidly extracted from the blood to be concentrated in the mitochondria of cells experiencing high oxidative stress. However, the very high utilization of O2 in the mitochondria of the stressed cells to produce ATP is known to be associated with elevated O·−2 generation and oxidative damage [145–147]. Thus, the increased concentration of melatonin at these sites would provide the necessary reductive equivalents to combat the extensive oxidative damage that would normally occur under such conditions.
Melatonin uptake into cells and eventually into mitochondria was long assumed to be related to its high lipid solubility which would allow it to rapidly diffuse through lipid-rich plasma and mitochondrial membranes [148, 149]. While this explanation may suffice following the exogenous administration of pharmacological amounts of melatonin where blood levels greatly exceed the normally high concentrations in the mitochondria [50], this would not be adequate to explain melatonin uptake by mitochondria that have levels significantly higher than physiological blood concentrations. The implication is that there may be an active uptake mechanism to concentrate melatonin in mitochondria. We recently suggested that melatonin enters cells through the glucose transporters (GLUT1 and GLUT4) [150]. According to this report, melatonin uptake was unexpectedly slow (one-to-several hours) and would seemingly not be consistent with the rapid disappearance of melatonin from the blood of highly stressed animals (seconds to minutes) [144]. The mechanism identified by Hevia and colleagues [150] does not provide information or the uptake of melatonin into mitochondria although the GLUT1 and GLUT4 transporters are located in the mitochondrial membrane.
In an attempt to resolve the issue as to whether there is a means, other than simple diffusion, by which melatonin may enter the mitochondria, Huo and coworkers [151] considered the possibility that either the human oligopeptide transporters (PEPT) 1/2 or the organic anion transporter (OAT) 3 aided this process. The authors used two human cancer cell lines, PC3 and U118, to perform their study. While the OAT3 transporter was not found to be involved, docking analysis of melatonin with PEPT 1/2 which are located in the mitochondrial membrane, showed that melatonin readily embedded into the active site of the transporters. PEPT 1/2 facilitated the transfer of melatonin into the mitochondria which correlated with the intraorganellar concentration of the indole (Fig. 8). The authors concluded that the oligopeptide transporters PEPT 1/2 play a crucial role in determining the high levels of melatonin in mitochondria. As such, melatonin is in a pivotal position to neutralize radicals that are generated by the less-than-perfectly functioning ETC.
Melatonin receptors/binding sites have been described in the plasma membrane, in the cytosol and in the nucleus of cells [26, 152–154]. Additionally, there is a single report claiming that a melatonin receptor also exists in the mitochondrial membrane. This report, published by Wang et al. [155], used cells from an animal model of Huntington disease. If this receptor does exist in the mitochondrial membrane, it could help to explain the high efficiency of melatonin in protecting this organelle from oxidative damage, e.g., by mediating the upregulation of mitochondrial antioxidative enzymes. The presence of this receptor, which has yet to be confirmed, would seemingly have no impact in determining the intramitochondrial concentration of melatonin, but could be related to the indirect antioxidative processes of melatonin in mitochondria, e.g., stimulating antioxidant enzyme activities, enhancing SIRT3 activity, etc.
Diseases where mitochondrial dysfunction occurs and where melatonin is beneficial
Sepsis, severe sepsis, and septic shock are serious medical conditions that are associated with extensive oxidative destruction of key molecules within cells. When several essential organs are damaged to the extent that they functionally fail, the condition is referred to as septic shock and is accompanied by multiple organ failure with a high degree of mortality being the result. A major predicted causative factor for multiple organ failure/septic shock is mitochondrial dysfunction resulting from damage inflicted by locally produced toxic reactive oxygen and nitrogen species [156–158]. In both humans [157, 159] and animals [160, 161] severe mitochondrial malfunction is central in this condition.
In 2001, a report was published by Gitto and colleagues [162] in which melatonin was used to treat sepsis in humans. In that report, we observed that giving septic human neonates intravenously administered melatonin both reduced the degree of oxidative damage (reduced levels of lipid peroxidation products in blood) and significantly limited the rate of mortality in these newborns. Subsequently, there have been numerous similar studies published using experimental animals with all the data pointing to the ability of melatonin to overcome many of the negative molecular consequences of sepsis including preventing the death of animals [163–165], as was shown by Gitto et al. [162] in humans. Since the mitochondria are a central organelle for the site of oxidative damage resulting from the bacterial toxins associated with septic infections, it would be expected that antioxidant supplementation may be beneficial in resisting molecular damage in this critical illness in animal and human sepsis [166, 167].
Perhaps related to the very high degree of damage that occurs or due to the limited ability of classic antioxidants to gain access to mitochondria, conventional free radical scavengers provide little protection, even when they are given in very high doses, against septic infections [168–171]. To overcome this deficiency, a new strategy has been to design antioxidants that target the mitochondria and concentrate in these organelles thereby increasing their effectiveness as a treatment. This therapy would require that the antioxidant be (a) delivered especially to the mitochondria, (b) be located at the proper site in the mitochondria to best scavenge newly formed radicals, and (c) to stimulate other processes that reduce local free radical levels. Examples of this latter function would be the augmentation of the activity of antioxidative enzymes and the reduction of the number of free radicals formed, i.e., radical avoidance [172]. Finally, (d) the optimal antioxidant should also have significant anti-inflammatory actions to quell the cytokine storm that is elicited during a septic event since many cytokines promote free radical generation [153, 173, 174].
The most common means to enhance the cellular and organellar uptake of conventional antioxidants has been to couple them with a lipophilic cation which allows them to more easily permeate lipid bilayers and concentrate in mitochondria. These so-called mitochondria-targeted antioxidants concentrate in the mitochondria up to 500-fold. The triphenylphosphonium cation is most frequently conjugated to an antioxidant to improve its uptake (Fig. 9) [175, 176]. The best known mitochondria-targeted antioxidants are MitoQ (based on the antioxidant co-enzyme Q10 [177]) and MitoE (based on α-tocopherol [178]).
Fig. 9.

The structure of synthetically produced, mitochondria-targeted antioxidants, i.e., MitoE and MitoQ. When vitamin E or co-enzyme Q10 is coupled to the triphosphonium cation, they more readily accumulate in the cytosol and in the mitochondria due to their increased lipid solubility. With regard to mitochondria, these synthetically produced antioxidants accumulate in concentrations of 200-–500-fold greater than unconjugated vitamin E and co-enzyme Q10. Despite this high concentration, when compared under in vivo experimental conditions, melatonin at equimolar concentrations was as good as or better than the fabricated antioxidants in protecting against cellular oxidative stress. For this and other reasons, we consider melatonin to be a mitochondria-targeted antioxidant. IMM inner mitochondrial membrane, OMM outer mitochondrial membrane
Using a sepsis-mediated organ failure model, Lowes et al. [51] compared the ability of melatonin to that of equimolar concentrations of either MitoQ and MitoE in resisting oxidative damage and the inflammatory response of rats given two bacterial endotoxins, lipopolysaccharide and peptidoglycan. Each of the antioxidants was given as a bolus injection followed by a 5-h infusion period. While each of the antioxidants had beneficial actions, melatonin was superior in terms of reducing plasma levels of lipid peroxidation products and hepatic protein damage, in restoring organ dysfunction (estimated using plasma aspartate transaminase and urinary creatinine levels) and in improving mitochondrial oxidative phosphorylation. Of the agents tested, Lowes et al. [51] concluded that melatonin would be the most effective treatment to counteract endotoxin toxicity, i.e., in humans. The findings also speak to the fact that melatonin readily accumulates in the mitochondria, i.e., it is likely a mitochondria-targeted antioxidant. Certainly, the very extensive work of the group of Acuna-Castroviejo [179] is consistent with the high efficacy of melatonin in reducing the consequences of sepsis at the mitochondrial level.
Considering the obviously high production of ROS in mitochondria, it seems obvious that a free radical scavenger specifically designed to localize in mitochondria would be highly beneficial in reducing the total oxidative burden that cells sustain. Free radical scavengers and enzymatic antioxidants are essential in maintaining redox homeostasis so as to reduce excessive damage to critical molecules.
While the idea of designing mitochondria-targeted antioxidants has been around for at least two decades [168, 170, 171, 180] and since melatonin is highly effective in reducing oxidative stress at the mitochondrial level [88, 91, 92, 163, 166, 181–184], we recently proposed that in fact melatonin is a mitochondria-targeted antioxidant [185]. The rationale for this classification was further elaborated in two subsequent publications [77, 167].
An example of a neurodegenerative condition that is clearly linked to aberrant mitochondrial structure and function is multiple sclerosis (MS) [186]; in fact, this progressive disease is believed to be primarily a mitochondria-related condition [187]. In an experimental model of this disease, Kashani and colleagues [188] reported that the use of melatonin as a treatment of MS-like pathology in mice was followed by an obvious improvement of mitochondrial function and a reduced disease progression. More recently, we [189] reported on an MS patient who was treated with 5–300 mg melatonin daily for 4 years. Melatonin treatment was initiated after the patient had failed to respond to glucocorticoid medications and was judged to be at the Expanded Disability Status Scale (EDSS) 8.0 of the disease (patient restricted to bed or wheelchair) [189]. During the use of melatonin daily for 4 years, the patient showed steady improvement and was diagnosed as being at EDSS 6.0 (walks with cane, crutch or brace up to 100 m without resting). While the symptoms of this disease spontaneously wax and wane in the short term, we feel that melatonin definitely reduced the severity of this condition because of the degree of improvement and the duration of the beneficial effects (4 years). Given that MS is generally considered a condition in which mitochondrial physiology is compromised, coupled with the known action of melatonin at the mitochondrial level, the findings suggest a more comprehensive evaluation of melatonin in MS patients should be considered.
A physiological deficiency of a number of progressive neurodegenerative conditions also involves mitochondrial malfunction as a primary or secondary disturbance in neurons or glia, e.g., Alzheimer, Parkinson, and Huntington disease [190–192]. In addition to a genetic predisposition, the onset of these conditions typically occurs when endogenous melatonin levels have deteriorated to chronically low values. Moreover, melatonin treatment of models of these diseases in experimental animals have often shown benefits in terms of prevention or slowed progression of both the neuropathology and behavior. Based on the outcomes reported, melatonin supplementation would likely be more useful in the prevention/slowing of these diseases rather than as a treatment of these conditions after they are at an advanced stage. Finally, a patient suffering with Duchenne muscular dystrophy, a condition that has a major mitochondrial dysfunction component, also responded to melatonin treatment with a substantial slowing of the disease [193].
The findings summarized above represent only a small percentage of the studies that have described an improvement of mitochondrial function when they are exposed to melatonin. Collectively, the data from both experimental and clinical publications, which are numerous, attest to melatonin having specific beneficial effects at the mitochondrial level.
Concluding remarks
Melatonin seems to meet the criteria as a mitochondria-targeted antioxidant. Abundant immunocytochemical evidence supports the conclusion that melatonin has ready access to the intermembrane space and matrix of mitochondria (Fig. 8). Certainly, this ubiquitously acting antioxidant, particularly in situations where mitochondrial ROS production is exaggerated, has the capability of reducing intramitochondrial free radical levels, visualized using appropriate fluorescent probes, as well as minimizing the potential molecular damage that is a normal consequence of enhanced ROS generation. Moreover, melatonin’s ability to achieve these healthful actions may be aided by its local production in the mitochondria.
We have surmised that mitochondria originated in early prokaryotes when these cells engulfed melatonin-producing bacteria as a source of nutrients. Eventually, the engulfed bacteria established a symbiotic relationship with these cells and evolved into mitochondria; when they did so they retained the melatonin-producing ability that their precursors possessed.
The likelihood of mitochondria being a source of melatonin is supported by other data as well. Thus, mitochondria have melatonin levels that greatly exceed those in the blood and, additionally, surgical removal of the pineal gland which lowers circulating melatonin to near zero and deprives cells of exogenously produced melatonin, does not cause a concomitant drop in mitochondrial melatonin concentrations. In fact, the loss of exogenously available melatonin may cause a compensatory rise in melatonin production. Based on kinetic considerations and substrate availability, melatonin production is calculated to more likely take place in the mitochondria rather than in the cytosol. Finally, incubation of isolated mitochondria with serotonin, a necessary precursor of melatonin, is followed by a large rise in melatonin concentrations in this organelle.
The number of reports proving that melatonin reduces molecular damage under severely enhanced oxidative experimental and clinical situations has accumulated for two decades and are now numerous. A plethora of toxins and disease models which cause or are accompanied by greatly exaggerated free radical generation have been used to challenge the ability of melatonin to prevent or forestall the massive damage that would normally occur. In these situations, melatonin has never failed to be protective.
Given the numerous findings that document the highly significant protective actions of melatonin in very high oxidative stress conditions, this endogenously produced indoleamine has proven itself as an essential and worthy “firewall” against toxic free radicals. Considering its high efficacy along with its mitochondria targeting, melatonin should be more extensively exploited for its mitochondrial protective actions and its antioxidant potential, particularly at the clinical level.
Acknowledgements
This work was supported in part by Grants CMRPD1C0511-3 (from the Chang Gung Memorial Hospital, Taiwan), MOST 105-2320-B-182-011 and MOST 104-2320-B-182-008 (to MJJ).
References
- 1.Izon G, Al Zerkle, Williford KH, Farquhar J, Paulton SW, Claire MW. Biological regulation of atmospheric chemistry en route to planetary oxygenation. Proc Natl Acad Sci USA. 2017;114:e2571–e2579. doi: 10.1073/pnas.1618798114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halliwell B. Free radicals, proteins, DNA: oxidative damage versus redox regulation. Biochem Soc Trans. 1996;24:1023–1027. doi: 10.1042/bst0241023. [DOI] [PubMed] [Google Scholar]
- 3.Fridovich I. Oxygen: how do we stand it? Med Princ Pract. 2013;22:131–137. doi: 10.1159/000339212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 5.Borg DC. Oxygen free radicals and tissue injury. In: Tarr M, Samson F, editors. Oxygen free radicals in tissue damage. Cambridge: Birkhauser; 1993. pp. 12–53. [Google Scholar]
- 6.Goldstein S, Meyerstein D, Czapski G. The Fenton reagents. Free Radic Biol Med. 1993;15:435–445. doi: 10.1016/0891-5849(93)90043-T. [DOI] [PubMed] [Google Scholar]
- 7.Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause or consequence? Lancet. 1994;344:721–734. doi: 10.1016/S0140-6736(94)92211-X. [DOI] [PubMed] [Google Scholar]
- 8.Kehrer JP. Free radicals as mediators of tissue injury. Crit Rev Toxicol. 1993;23:21–48. doi: 10.3109/10408449309104073. [DOI] [PubMed] [Google Scholar]
- 9.Felix JA, Lundgren DG. Electron transport system associated with membranes of Bacillus cereus during vegetative growth and sporulation. J Bacteriol. 1973;115:552–559. doi: 10.1128/jb.115.2.552-559.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu LN. Distribution and dynamics of electron transport complexes in cyanobacterial thylakoid membranes. Biochim Biophys Acta. 2016;1857:256–265. doi: 10.1016/j.bbabio.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreno SN, Docampo R. Reduction of the metallochromic indicators murexide and tetramethylmurexide to their free radical metabolites by cytoplasmic enzymes and reducing agents. Chem Biol Interact. 1986;57:17–25. doi: 10.1016/0009-2797(86)90045-1. [DOI] [PubMed] [Google Scholar]
- 12.Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ. Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes. J Pineal Res. 2013;54:127–138. doi: 10.1111/jpi.12026. [DOI] [PubMed] [Google Scholar]
- 13.Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, Vriend J, Tan DX, Reiter RJ. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res. 2015;59:403–419. doi: 10.1111/jpi.12267. [DOI] [PubMed] [Google Scholar]
- 14.Noctor G, Veljovic-Jovanovic S, Foyer CH. Peroxide processing in photosynthesis: antioxidant coupling and redox signaling. Philos Trans R Soc Lond B Biol Sci. 2000;355:1465–1475. doi: 10.1098/rstb.2000.0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manchester LC, Poeggeler B, Alvarez FL, Ogden GB, Reiter RJ. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: implications for an ancient antioxidant system. Cell Mol Biol Res. 1995;41:391–395. [PubMed] [Google Scholar]
- 16.Byeon Y, Lee K, Park Y, Park S, Back K. Molecular cloning and functional analysis of serotonin-N-acetyltransferase from the cyanobacterium Synechocystis sp. PCC 6803. J Pineal Res. 2013;55:371–376. doi: 10.1111/jpi.12080. [DOI] [PubMed] [Google Scholar]
- 17.Champney TH, Holtorf AP, Steger RW, Reiter RJ. Concurrent determination of enzymatic activities and substrate concentrations in the melatonin synthetic pathway within the same rat pineal gland. J Neurosci Res. 1984;11:59–66. doi: 10.1002/jnr.490110107. [DOI] [PubMed] [Google Scholar]
- 18.Stehle JH, Saade A, Rawashdeh O, Ackermann K, Jilg A, Sebesteny T, Maronde E. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res. 2011;51:17–43. doi: 10.1111/j.1600-079X.2011.00856.x. [DOI] [PubMed] [Google Scholar]
- 19.Lerner AB, Case JD, Takahashi Y, Lee TH, Mori W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J Am Chem Soc. 1958;80:2587. doi: 10.1021/ja01543a060. [DOI] [Google Scholar]
- 20.Hoffman RA, Reiter RJ. Pineal gland: influence of gonads on male hamsters. Science. 1965;148:1609–1611. doi: 10.1126/science.148.3677.1609. [DOI] [PubMed] [Google Scholar]
- 21.Reiter RJ, Fraschini F. Endocrine aspects of the mammalian pineal gland: a review. Neuroendocrinology. 1969;5:219–255. doi: 10.1159/000121862. [DOI] [PubMed] [Google Scholar]
- 22.Reiter RJ. Evidence for refractoriness of the pituitary-gonadal axis to the pineal gland in golden hamsters and its possible implications for annual reproductive rhythms. Anat Rec. 1972;173:365–371. doi: 10.1002/ar.1091730311. [DOI] [PubMed] [Google Scholar]
- 23.Dardente H, Lomet D, Robert V, Decourt C, Beltrano M, Pellicer-Rubio MT. Seasonal breeding in mammals: from basic science to applications and back. Theriogenology. 2016;86(324):332. doi: 10.1016/j.theriogenology.2016.04.045. [DOI] [PubMed] [Google Scholar]
- 24.Hill SM, Belancio VP, Dauchy RT, Xiang S, Brimer S, Mao L, Hauch A, Lundberg PW, Summers W, Yuan L, Frasch T, Blask DE. Melatonin: an inhibitor of breast cancer. Endocr Relat Cancer. 2015;22:R183–R204. doi: 10.1530/ERC-15-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiter RJ, Rosales-Corral SA, Tan DX, Acuna-Castroviejo D, Qin L, Yang SF, Xu K. Melatonin, a full service anti-cancer agent: inhibition of initiation, progression and metastasis. Int J Mol Sci. 2017;18:e843. doi: 10.3390/ijms18040843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ. A review of the multiple actions of melatonin on the immune system. Endocrine. 2005;27:189–200. doi: 10.1385/ENDO:27:2:189. [DOI] [PubMed] [Google Scholar]
- 27.Carrillo-Vico A, Reiter RJ, Lardone PJ, Herrera JL. The modulatory role of melatonin on immune responsiveness. Curr Opin Investig Drugs. 2006;7:423–431. [PubMed] [Google Scholar]
- 28.Mauriz JL, Collado PS, Veneraso C, Reiter RJ, Gonzalez-Gallego J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: recent insights and new perspectives. J Pineal Res. 2013;54:1–14. doi: 10.1111/j.1600-079X.2012.01014.x. [DOI] [PubMed] [Google Scholar]
- 29.Hosseinzadeh A, Kamrava SK, Joghataei MT, Darabi R, Shakeri-Zadeh A, Shahriari M, Reiter RJ, Ghaznavi H, Mehrzadi S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J Pineal Res. 2016;61:411–425. doi: 10.1111/jpi.12362. [DOI] [PubMed] [Google Scholar]
- 30.Cardinali DP, Hardeland R. Inflammaging, metabolic syndrome and melatonin: a call for treatment studies. Neuroendocrinology. 2017;104:382–397. doi: 10.1159/000446543. [DOI] [PubMed] [Google Scholar]
- 31.Wetterberg L. Melatonin in humans physiological and clinical studies. J Neural Transm. 1978;13:289–310. [PubMed] [Google Scholar]
- 32.Wurtman RJ, Liebermann HR. Melatonin secretion as a mediator of circadian variations in sleep and sleepiness. J Pineal Res. 1985;2:301–303. doi: 10.1111/j.1600-079X.1985.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 33.Kanji S, Mera A, Hutton B, Burry L, Rosenberg E, MacDonald E, Luks V. Pharmacological interventions to improve sleep in hospitalized adults: a systematic review. BMJ Open. 2016;6:e12108. doi: 10.1136/bmjopen-2016-012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie Z, Chen F, Li WA, Geng X, Li C, Meng X, Feng Y, Liu W, Yu F. A review of sleep disorders and melatonin. Neurol Res. 2017;1:1–7. doi: 10.1080/01616412.2017.1315864. [DOI] [PubMed] [Google Scholar]
- 35.Abdel Moneim AE, Ortiz F, Leonardo-Mendonca RC, Vergano-Villodres R, Guerrero-Martinez JA, Lopez LC, Acuna-Castroviejo D, Escames G. Protective effects of melatonin against oxidative damage induced by Egyptian cobra (Naja haje) crude venom in rats. Acta Trop. 2015;143:58–65. doi: 10.1016/j.actatropica.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Sharma RD, Katkar GD, Sundaram MS, Paul M, Naveen Kumar SK, Swethakumar B, Hemshekhar M, Girish KS, Kemparaju K. Oxidative stress induced methemoglobinemia is the silent killer during snakebite: a novel and strategic neutralization by melatonin. J Pineal Res. 2015;59:240–254. doi: 10.1111/jpi.12256. [DOI] [PubMed] [Google Scholar]
- 37.Xu F, Wang J, Hong F, Wang S, Jin X, Xue T, Jia L, Zhai Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J Pineal Res. 2017;62:e12399. doi: 10.1111/jpi.12399. [DOI] [PubMed] [Google Scholar]
- 38.Puchalski SS, Green JN, Rasmussen DD. Melatonin effect on rat body weight regulation in response to high-fat diet at middle age. Endocrine. 2003;21:163–167. doi: 10.1385/ENDO:21:2:163. [DOI] [PubMed] [Google Scholar]
- 39.Peschke E, Bahr I, Muhlbauer E. Experimental and clinical aspects of melatonin and clock genes in diabetes. J Pineal Res. 2015;59:1–23. doi: 10.1111/jpi.12240. [DOI] [PubMed] [Google Scholar]
- 40.Gao L, Zhao YC, Liang Y, Lin XH, Tan YJ, Wu DD, Li XZ, Ye BZ, Keng FQ, Sheng JZ, Huang HF. The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J Pineal Res. 2016;61:340–352. doi: 10.1111/jpi.12351. [DOI] [PubMed] [Google Scholar]
- 41.Hu W, Ma Z, Jiang S, Fan C, Deng C, Yan X, Di S, Lv J, Reiter RJ, Yang Y. Melatonin: the dawning of a treatment for fibrosis? J Pineal Res. 2016;60:121–131. doi: 10.1111/jpi.12302. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Fernandez B, Sanchez DI, Crespo I, San Miguel B, Alvarez M, Tunon MJ, Gonzalez-Gallego J. Inhibition of the Sphk1/S1P signaling pathway by melatonin in mice with liver fibrosis and human hepatic stellate cells. BioFactors. 2017;43:272–282. doi: 10.1002/biof.1342. [DOI] [PubMed] [Google Scholar]
- 43.Reiter RJ, Tan DX, Rosales-Corral SA, Manchester LC. The universal nature, unequal distribution and antioxidant functions of melatonin. Mini Rev Med Chem. 2013;13:373–384. doi: 10.2174/1389557511313030006. [DOI] [PubMed] [Google Scholar]
- 44.Garcia JJ, Lopez-Pingarron L, Almeida-Sauza P, Tres A, Escudero P, Garcia-Gil FA, Tan DX, Reiter RJ, Ramirez JM, Bernal-Perez M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review. J Pineal Res. 2014;58:225–237. doi: 10.1111/jpi.12128. [DOI] [PubMed] [Google Scholar]
- 45.Deng MS, Xu Q, Liu YE, Jiang CH, Zhou H, Gu L. Effects of melatonin on liver function and lipid peroxidation in a rat model of hepatic ischemia/reperfusion injury. Exp Ther Med. 2016;11:1955–1960. doi: 10.3892/etm.2016.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mollaoglu H, Topal T, Ozler M, Uysal B, Reiter RJ, Korkmaz A, Oter S. Antioxidant effects of melatonin in rats during chronic exposure to hyperbaric oxygen. J Pineal Res. 2007;42:50–54. doi: 10.1111/j.1600-079X.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 47.Waseem M, Tabassum H, Parnez S. Neuroprotective effects of melatonin as evidenced by abrogation of oxaliplatin induced behavioral alterations, mitochondrial dysfunction and neurotoxicity in rat brain. Mitochondrion. 2016;30:168–176. doi: 10.1016/j.mito.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Reiter RJ, Tan DX, Kim SJ, Qi W. Melatonin as a pharmacological agent against oxidative damage to lipids and DNA. Proc West Pharmacol Soc. 1998;41:229–236. [PubMed] [Google Scholar]
- 49.Chua S, Lee FY, Chiang HJ, Chen KH, Lu HI, Chen YT, Yang CC, Lin KC, Chen YL, Kao GS, Chen CH, Chang HW, Yip HK. The cardioprotective effect of melatonin and exendin-4 in a rat model of cardiorenal syndrome. J Pineal Res. 2016;61:438–456. doi: 10.1111/jpi.12357. [DOI] [PubMed] [Google Scholar]
- 50.Venegas C, Garcia JA, Escames G, Ortiz F, Lopez A, Doerrier C, Garcia-Corso L, Lopez LC, Reiter RJ, Acuna-Castroviejo D. Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res. 2012;52:217–227. doi: 10.1111/j.1600-079X.2011.00931.x. [DOI] [PubMed] [Google Scholar]
- 51.Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Brit J Anaesth. 2013;110:472–480. doi: 10.1093/bja/aes577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He C, Wang J, Zhang Z, Yang M, Li Y, Tian X, Ma T, Tao J, Zhu K, Song Y, Ji P, Liu G. Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte’s quality under in vitro conditions. Int J Mol Sci. 2016;17:939–955. doi: 10.3390/ijms17060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan DX, Manchester LC, Esteban-Zubero E, Zhou Z, Reiter RJ. Melatonin as a potent and inducible endogenous antioxidant: synthesis and metabolism. Molecules. 2015;20:18886–18906. doi: 10.3390/molecules201018886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan DX, Reiter RJ, Manchester LC, Yan MT, El-Sawi M, Sainz RM, Mayo JC, Kohen R, Allegra M, Hardeland R. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem. 2002;2:181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- 55.Galano A, Tan DX, Reiter RJ. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J Pineal Res. 2013;54:245–257. doi: 10.1111/jpi.12010. [DOI] [PubMed] [Google Scholar]
- 56.Janjetovic Z, Jarrett SC, Lee EF, Duprey C, Reiter RJ, Slominski AT. Melatonin and its metabolites protect human melanocytes against UVB-induced damage: involvement of NRF2-mediated pathways. Sci Rep. 2017;7:1274. doi: 10.1038/s41598-017-01305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Limson J, Nyokong T, Daya S. The interaction of melatonin and its precursors with aluminum, cadmium, copper, iron, lead, and zinc: an adsorptive voltammetric study. J Pineal Res. 1998;24:15–21. doi: 10.1111/j.1600-079X.1998.tb00361.x. [DOI] [PubMed] [Google Scholar]
- 58.Galano A, Medina ME, Tan DX, Reiter RJ. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: a physicochemical analysis. J Pineal Res. 2015;58:107–116. doi: 10.1111/jpi.12196. [DOI] [PubMed] [Google Scholar]
- 59.Barlow-Walden LR, Reiter RJ, Abe M, Pablos M, Menendez-Pelaez A, Chen LD, Poeggeler B. Melatonin stimulates glutathione peroxidase. Neurochem Int. 1995;26:497–502. doi: 10.1016/0197-0186(94)00154-M. [DOI] [PubMed] [Google Scholar]
- 60.Reiter RJ, Tan DX, Osuna C, Gitto E. Actions of melatonin in the reduction of oxidative stress. J Biomed Sci. 2000;7:444–458. doi: 10.1007/BF02253360. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez C, Mayo JC, Sainz RM, Antolin I, Herrera F, Martin V, Reiter RJ. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079X.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 62.Urata Y, Honma S, Goto S, Todoroki S, Iida T, Cho S, Honma K, Kendo T. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic Biol Med. 1999;27:838–847. doi: 10.1016/S0891-5849(99)00131-8. [DOI] [PubMed] [Google Scholar]
- 63.Mayo JC, Sainz RM, Gonzalez-Menendez P, Cepas V, Tan DX, Reiter RJ. Melatonin and sirtuins: a “not-so unexpected” relationship. J Pineal Res. 2017;62:e12391. doi: 10.1111/jpi.12391. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y, Qing W, Sun M, Lv L, Guo D, Jiang Y. Melatonin protects hepatocytes against bile acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radic Res. 2015;49:1275–1284. doi: 10.3109/10715762.2015.1067806. [DOI] [PubMed] [Google Scholar]
- 65.Pi H, Xu S, Reiter RJ, Guo P, Zhang L, Yi Y, Tian L, Zhang R, Cao Z, He M, Lu Y, Duan W, Yu Z, Zhou Z. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11:1037–1051. doi: 10.1080/15548627.2015.1052208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhai M, Li B, Duan W, Jing L, Zhang B, Zhang M, Yu L, Liu Z, Yu B, Ren K, Gao E, Yang Y, Liang H, Jin Z, Yu S. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63:e12419. doi: 10.1111/jpi.12419. [DOI] [PubMed] [Google Scholar]
- 67.Tan DX, Chen LD, Poeggeler B, Manchester LC, Reiter RJ. Melatonin: a potent, endogenous hydroxyl radical scavenger. Endocr J. 1993;1:57–63. [Google Scholar]
- 68.Poeggeler B, Saarela S, Reiter RJ, Tan DX, Chen LD, Manchester LC, Barlow-Walden LR. Melatonin—highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole assessed in vitro. Ann NY Acad Sci. 1994;738:419–420. doi: 10.1111/j.1749-6632.1994.tb21831.x. [DOI] [PubMed] [Google Scholar]
- 69.Galano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res. 2011;51:1–16. doi: 10.1111/j.1600-079X.2011.00916.x. [DOI] [PubMed] [Google Scholar]
- 70.Reiter RJ, Tan DX, Galano A. Melatonin reduces lipid peroxidation and membrane viscosity. Front Physiol. 2014;5:377. doi: 10.3389/fphys.2014.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan DX, Hardeland R, Manchester LC, Poeggler B, Lopez-Burillo S, Mayo JC, Sainz RM, Reiter RJ. Mechanistic and comparative studies of melatonin and classic antioxidants in terms of their interaction with the ABTS cation radical. J Pineal Res. 2003;34:349–359. doi: 10.1034/j.1600-079x.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 72.Galano A. On the direct scavenging activity of melatonin towards hydroxyl and a series of peroxyl radicals. Phys Chem Chem Phys. 2011;13:7178–7188. doi: 10.1039/c0cp02801k. [DOI] [PubMed] [Google Scholar]
- 73.Hardeland R. Melatonin and the theories of aging: a critical appraisal of melatonin’s role in antiaging mechanisms. J Pineal Res. 2013;55:325–356. doi: 10.1111/jpi.12090. [DOI] [PubMed] [Google Scholar]
- 74.Alvarez-Diduk R, Galano A, Tan DX, Reiter RJ. N-acetylserotonin and 6-hydroxymelatonin against oxidative stress: implications for the overall protection exerted by melatonin. J Phys Chem B. 2015;119:8535–8543. doi: 10.1021/acs.jpcb.5b04920. [DOI] [PubMed] [Google Scholar]
- 75.Shin IS, Shin NR, Park JW, Jeon JM, Kwon OK, Kim JS, Lee IC, Kim JC, Oh SR, Ahn KS. Melatonin attenuates neutrophil inflammation and mucus secretion in cigarette smoke-induced chronic obstructive pulmonary disease via the suppression of ErK-Sp1 signaling. J Pineal Res. 2015;58:50–60. doi: 10.1111/jpi.12192. [DOI] [PubMed] [Google Scholar]
- 76.Galano A, Castaneda-Arriga R, Perez-Gonzales A, Tan DX, Reiter RJ. Phenolic melatonin related compounds: their role as chemical protectors against oxidative stress. Molecules. 2016;21:1442. doi: 10.3390/molecules21111442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. 2016;61:253–278. doi: 10.1111/jpi.12360. [DOI] [PubMed] [Google Scholar]
- 78.Galano A, Tan DX, Reiter RJ (2017) Melatonin and related compounds: chemical insights into their protective effects against oxidative stress. Curr Org Chem 21 (in press)
- 79.Acuna-Castroviejo D, Martin M, Macias M, Escames G, Leon J, Khaldy H, Reiter RJ. Melatonin, mitochondria and cellular bioenergetics. J Pineal Res. 2001;30:65–74. doi: 10.1034/j.1600-079X.2001.300201.x. [DOI] [PubMed] [Google Scholar]
- 80.Okatani Y, Wakatsaki A, Reiter RJ. Melatonin and mitochondrial respiration. In: Pandi-Perumal SR, Cardinali DP, editors. Melatonin: biological basis of its function in health and disease. Georgetown: Lander Bioscience; 2004. pp. 11–24. [Google Scholar]
- 81.Bromme HJ, Morke W, Peschke D, Ebelt H. Scavenging effect of melatonin on hydroxyl radicals generated by alloxan. J Pineal Res. 2000;29:201–208. doi: 10.1034/j.1600-0633.2002.290402.x. [DOI] [PubMed] [Google Scholar]
- 82.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 83.Pablos MI, Agopito MT, Gutierrez R, Recio JM, Reiter RJ, Barlow-Walden L, Acuna-Castroviejo D, Menendez-Pelaez A. Melatonin stimulates the activity of the detoxifying enzyme glutathione peroxidase in several tissues of chickens. J Pineal Res. 1995;19:111–115. doi: 10.1111/j.1600-079X.1995.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto H, Yang HW. Preventive effect of melatonin against cyanide-induced seizures and lipid peroxidation in mice. Neurosci Lett. 1996;207:89–92. doi: 10.1016/0304-3940(96)12493-9. [DOI] [PubMed] [Google Scholar]
- 85.Dabbeni-Sala F, Di Santo S, Franceschini D, Skaper SD, Giusti P. Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J. 2001;15:167–170. doi: 10.1096/fj.00-0129com. [DOI] [PubMed] [Google Scholar]
- 86.Dabbeni S, Floreani M, Franceschini D, Skaper SD, Giusti P. Kainic acid induces selective mitochondrial oxidative phosphorylation enzyme dysfunction in cerebellar granule neurons: protective effects of melatonin and GSH ethyl ester. FASEB J. 2001;15:1786–1788. doi: 10.1096/fj.00-0427fje. [DOI] [PubMed] [Google Scholar]
- 87.Absi E, Ayala A, Machado A, Parrado J. Protective effect of melatonin against the 1-methyl-4-phenylpyridinium-induced inhibition of complex I of the mitochondrial respiratory chain. J Pineal Res. 2000;29:40–47. doi: 10.1034/j.1600-079X.2000.290106.x. [DOI] [PubMed] [Google Scholar]
- 88.Martin M, Macias M, Escames G, Leon J, Acuna-Castroviejo D. Melatonin but not vitamins C or E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000;14:1677–1679. doi: 10.1096/fj.99-0865fje. [DOI] [PubMed] [Google Scholar]
- 89.Martin M, Macias M, Escames G, Reiter RJ, Agapito MT, Ortiz GG, Acuna-Castroviejo D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J Pineal Res. 2000;28:242–248. doi: 10.1034/j.1600-079X.2000.280407.x. [DOI] [PubMed] [Google Scholar]
- 90.Martin M, Macias M, Leon J, Escames G, Khaldy Acuna-Castroviejo D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int J Biochem Cell Biol. 2002;34:348–357. doi: 10.1016/S1357-2725(01)00138-8. [DOI] [PubMed] [Google Scholar]
- 91.Okatani Y, Wakatsuki A, Reiter RJ, Enzan H, Miyahara Y. Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur J Pharmacol. 2003;469:145–152. doi: 10.1016/S0014-2999(03)01643-1. [DOI] [PubMed] [Google Scholar]
- 92.Okatani Y, Wakatsuki A, Reiter RJ, Miyahara Y. Acutely administered melatonin restores hepatic mitochondrial physiology in old mice. Int J Biochem Cell Biol. 2003;35:367–375. doi: 10.1016/S1357-2725(02)00260-1. [DOI] [PubMed] [Google Scholar]
- 93.Jou MJ, Jou SB, Chen HM, Lin CH, Peng TI. Critical role of mitochondrial reactive oxygen species formation in visible laser irradiation-induced apoptosis in rat brain astrocytes (RBA-1) J Biomed Sci. 2002;9:507–516. doi: 10.1007/BF02254977. [DOI] [PubMed] [Google Scholar]
- 94.Peng TI, Wei YH, Wu HY, Jou MJ. Mitochondrial calcium and ROS mediated apoptosis plays a potential pathogenic role in diseases associated with mitochondrial DNA 4977 bp deletion. Biophys J. 2003;84:205a. doi: 10.1016/S0006-3495(03)74843-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jou MJ, Peng TI, Reiter RJ, Jou SB, Wu HY, Wen ST. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J Pineal Res. 2004;37:55–70. doi: 10.1111/j.1600-079X.2004.00140.x. [DOI] [PubMed] [Google Scholar]
- 96.Gitto E, Tan DX, Reiter RJ, Karbownik M, Manchester LC, Cuzzocrea S, Fulia F, Barberi I. Individual and synergistic antioxidative actions of melatonin: studies with vitamin E, vitamin C, glutathione and desferrioxamine (desferoxamine) in liver homogenates. J Pharm Pharmacol. 2001;53:1393–1401. doi: 10.1211/0022357011777747. [DOI] [PubMed] [Google Scholar]
- 97.Dimauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 98.Jou MJ, Peng TI, Wu HY, Wei YH. Enhanced generation of mitochondrial reactive oxygen species in cybrids containing 4977-bp mitochondrial DNA deletion. Ann NY Acad Sci. 2005;1042:221–228. doi: 10.1196/annals.1338.024. [DOI] [PubMed] [Google Scholar]
- 99.Peng TI, Yu PR, Chen JY, Wang HL, Wu HY, Wei YH, Jou MJ. Visualizing common deletion of mitochondrial DNA-augmented mitochondrial reactive oxygen species generation and apoptosis upon oxidative stress. Biochem Biophys Acta. 2006;1762:241–255. doi: 10.1016/j.bbadis.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 100.Jou MJ, Peng TI, Yu PZ, Jou SB, Reiter RJ, Chen JY, Wu HY, Chen CC, Hsu LF. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J Pineal Res. 2007;43:389–403. doi: 10.1111/j.1600-079X.2007.00490.x. [DOI] [PubMed] [Google Scholar]
- 101.Jou MJ, Peng TI, Hsu LF, Jou SB, Reiter RJ, Yang CM, Chiao CC, Lin YF, Chen CC. Visualization of melatonin’s multiple mitochondrial levels of protection against mitochondrial Ca2+-mediated permeability transition and beyond in rat brain astrocytes. J Pineal Res. 2010;48:20–38. doi: 10.1111/j.1600-079X.2009.00721.x. [DOI] [PubMed] [Google Scholar]
- 102.Ozaki Y, Lynch HJ. Presence of melatonin in plasma and urine of pinealectomized rats. Endocrinology. 1976;99:641–644. doi: 10.1210/endo-99-2-641. [DOI] [PubMed] [Google Scholar]
- 103.Mori Y, Okamura H. Effects of timed melatonin infusion on prolactin secretion in pineal denervated goats. J Pineal Res. 1986;3:77–86. doi: 10.1111/j.1600-079X.1986.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 104.Hoffman RA, Reiter RJ. Influence of compensatory mechanisms and the pineal gland on dark-induced gonadal atrophy in male hamsters. Nature. 1965;207:658–659. doi: 10.1038/207658a0. [DOI] [PubMed] [Google Scholar]
- 105.Reiter RJ, Hester RJ. Interrelationships of the pineal gland, the superior cervical ganglia and the photoperiod in the regulation of the endocrine systems of hamsters. Endocrinology. 1966;79:1168–1170. doi: 10.1210/endo-79-6-1168. [DOI] [PubMed] [Google Scholar]
- 106.Quay MB. Temporal mitotic patterns around a brain lesion: cellular and regional asynchronism and an effect of pinealectomy. Chronobiologia. 1974;1:237–258. [PubMed] [Google Scholar]
- 107.Reiter RJ, Tan DX, Kim SK, Cruz MH. Delivery of pineal melatonin to the brain and SCN: role of canaliculi, cerebrospinal fluid, tanycytes and Virchow–Robin perivascular spaces. Brain Struct Funct. 2014;219:1873–1887. doi: 10.1007/s00429-014-0719-7. [DOI] [PubMed] [Google Scholar]
- 108.Tricoire H, Locatelli A, Chemineau P, Malpaux B. Melatonin enters the cerebrospinal fluid through the pineal recess. Endocrinology. 2002;143:84–90. doi: 10.1210/endo.143.1.8585. [DOI] [PubMed] [Google Scholar]
- 109.Vriend J, Reiter RJ. Melatonin feedback on clock genes: a theory involving the proteasome. J Pineal Res. 2015;58:1–11. doi: 10.1111/jpi.12189. [DOI] [PubMed] [Google Scholar]
- 110.Matsumura R, Node K, Akaski M. Estimation methods for human circadian phase by use of peripheral tissues. Hypertens Res. 2016;39:623–627. doi: 10.1038/hr.2016.68. [DOI] [PubMed] [Google Scholar]
- 111.Muller MJ, Geisler C. From the past to future: from energy expenditure to energy intake to energy expenditure. Eur J Clin Nutr. 2017;71:358–364. doi: 10.1038/ejcn.2016.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosales-Corral SA, Acuna-Castroviejo D, Coto-Montes A, Boga JA, Manchester LC, Fuentes-Broto L, Korkmaz A, Tan DX, Reiter RJ. Alzheimer’s disease: pathological mechanisms and the beneficial role of melatonin. J Pineal Res. 2012;52:167–202. doi: 10.1111/j.1600-079X.2011.00937.x. [DOI] [PubMed] [Google Scholar]
- 113.Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X, Deng C, Di S, Xin Z, Wu G, Yang Y, Reiter RJ, Liang G. Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J Pineal Res. 2016;60:253–262. doi: 10.1111/jpi.12300. [DOI] [PubMed] [Google Scholar]
- 114.Salim S. Oxidative stress and the central nervous system. J Pharmacol Exp Ther. 2017;380:201–205. doi: 10.1124/jpet.116.237503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cardinali DP, Rosner JM. Retinal localization of the hydroxyindole-O-methyl transferase (HIOMT) in the rat. Endocrinology. 1971;89:301–303. doi: 10.1210/endo-89-1-301. [DOI] [PubMed] [Google Scholar]
- 116.Tan DX, Manchester LC, Reiter RJ, Qi W, Hanes MA, Farley NJ. High levels of melatonin in the bile of mammals. Life Sci. 1999;65:2523–2529. doi: 10.1016/S0024-3205(99)00519-6. [DOI] [PubMed] [Google Scholar]
- 117.Reiter RJ, Rosales-Corral SA, Manchester LC, Liu X, Tan DX. Melatonin in the biliary tract and liver: health implications. Curr Pharm Res. 2014;20:4788–4801. doi: 10.2174/1381612819666131119105826. [DOI] [PubMed] [Google Scholar]
- 118.Dubbels R, Reiter RJ, Klenke E, Goebel A, Schnakenberg E, Ehlers C, Schiwara HW, Schloot W. Melatonin in edible plants identified by radioimmunoassay and by high performance liquid chromatography-mass spectrometry. J Pineal Res. 1995;18:28–31. doi: 10.1111/j.1600-079X.1995.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 119.Hattori A, Migitaka H, Iigo M, Itoh M, Yamamoto K, Ohtani-Kaneko R, Hara M, Suzuki T, Reiter RJ. Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors n vertebrates. Biochem Mol Bio Int. 1995;35:627–634. [PubMed] [Google Scholar]
- 120.Reiter RJ, Tan DX, Zhou Z, Cruz MH, Fuentes-Broto L, Galano A. Phytomelatonin: assisting plants to survive and thrive. Molecules. 2015;20:7396–7437. doi: 10.3390/molecules20047396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arnao MB, Ruiz-Hernandez J. Function of melatonin in plants: a review. J Pineal Res. 2015;59:133–150. doi: 10.1111/jpi.12253. [DOI] [PubMed] [Google Scholar]
- 122.Archibald JM. Endosymbiosis and eukaryotic cell evolution. Curr Biol. 2015;25:R911–R921. doi: 10.1016/j.cub.2015.07.055. [DOI] [PubMed] [Google Scholar]
- 123.Bajwa VS, Shukla MR, Sherif SM, Murch SJ, Saxena PK. Role of melatonin in alleviating cold stress in Arabidopsis thaliana . J Pineal Res. 2014;56:238–245. doi: 10.1111/jpi.12115. [DOI] [PubMed] [Google Scholar]
- 124.Shi H, Qian Y, Tan DX, Reiter RJ. Melatonin reduces the transcripts of CBF-DREB1S and their involvement in both abiotic and biotic stresses in Arabidopsis. J Pineal Res. 2015;59:334–342. doi: 10.1111/jpi.12262. [DOI] [PubMed] [Google Scholar]
- 125.Acuna-Castroviejo D, Escames G, Venegas C, Diaz-Casado ME, Lima-Cabello E, Lopez LC, Rosales-Corral SA, Tan DX, Reiter RJ. Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. Cell Mol Life Sci. 2014;71:2997–3025. doi: 10.1007/s00018-014-1579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kerenyi NA, Sotonyi P, Somogyi E. Localizing acetylserotonin transferase by electron microscopy. Histochemistry. 1975;46:77–80. doi: 10.1007/BF02463562. [DOI] [Google Scholar]
- 127.Kerenyi NA, Balogh I, Somogyi E, Sotonyi P. Cytochemical investigation of acetylserotonin-transferase activity in the pineal gland. Cell Mol Biol Incl Cyto Enzymol. 1979;25:259–262. [PubMed] [Google Scholar]
- 128.Tamura H, Takasaki A, Ichias M, Taniquchi K, Maekawa R, Asada H, Taketani T, Matsuoka A, Yamagata Y, Mori M, Ishikawa H, Reiter RJ. Oxidative stress impairs oocyte quality and melatonin protects oocytes from free radical damage and improves fertilization rate. J Pineal Res. 2008;44:222–226. doi: 10.1111/j.1600-079X.2007.00516.x. [DOI] [PubMed] [Google Scholar]
- 129.Sakaguchi K, Itoh MT, Takahashi N, Tarumi W, Ishizuka B. The rat oocyte synthesizes melatonin. Reprod Fertil Rev. 2013;25:674–682. doi: 10.1071/RD12091. [DOI] [PubMed] [Google Scholar]
- 130.Coelho LA, Peres R, Amaral FG, Reiter RJ, Cipolla-Neto J. Daily differential expression of melatonin-related genes and clock genes in rat cumulus-oocyte complex: changes after pinealectomy. J Pineal Res. 2015;58:490–499. doi: 10.1111/jpi.12234. [DOI] [PubMed] [Google Scholar]
- 131.Back K, Tan DX, Reiter RJ. Melatonin biosynthesis in plants: multiple pathways catalyze tryptophan to melatonin in the cytoplasm or chloroplasts. J Pineal Res. 2016;61:426–437. doi: 10.1111/jpi.12364. [DOI] [PubMed] [Google Scholar]
- 132.Zheng X, Tan DX, Allan AC, Zuo B, Zhao Y, Reiter RJ, Wang L, Wang Z, Guo Y, Zhou J, Shan D, Li Q, Han Z, Kong J. Chloroplastic biosynthesis of melatonin and its involvement in protection of plants from salt stress. Sci Rep. 2017;7:41236. doi: 10.1038/srep41236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Byeon Y, Lee HY, Lee K, Back K. Cellular localization and kinetics of the rice melatonin biosynthetic enzymes SNAT and ASMT. J Pineal Res. 2014;56:107–114. doi: 10.1111/jpi.12103. [DOI] [PubMed] [Google Scholar]
- 134.Tan DX, Hardeland R, Manchester LC, Korkmaz A, Ma S, Rosales-Corral S, Reiter RJ. Functional roles of melatonin in plants and perspectives in nutritional and agricultural sciences. J Exp Bot. 2013;63:577–597. doi: 10.1093/jxb/err256. [DOI] [PubMed] [Google Scholar]
- 135.Wang L, Feng L, Zheng X, Guo Y, Zhou F, Shan D, Liu X, Kong J. Plant mitochondria synthesize melatonin and enhance the tolerance of plants to drought stress. J Pineal Res. 2017;69:e12429. doi: 10.1111/jpi.12429. [DOI] [PubMed] [Google Scholar]
- 136.Tan DX, Hardeland R, Back K, Manchester LC, Alatorre-Jimenez MA, Reiter RJ. A hypothesis concerning the predominant melatonin synthetic pathway: inversion of the terminal enzymatic steps. J Pineal Res. 2016;61:27–40. doi: 10.1111/jpi.12336. [DOI] [PubMed] [Google Scholar]
- 137.Acuna-Castroviejo D, Lopez LC, Escames G, Lopez A, Garcia JJ. Melatonin-mitochondria interplay in health and disease. Curr Top Med Chem. 2011;11:221–240. doi: 10.2174/156802611794863517. [DOI] [PubMed] [Google Scholar]
- 138.Yu L, Liang H, Lu Z, Zhao G, Zhai M, Yang Y, Yang J, Yi D, Chen W, Wang X, Duan W, Jin Z, Yu S. Membrane receptor-dependent Notch1/Hes1 activation by melatonin protects against myocardial ischemia-reperfusion injury: in vivo and in vitro studies. J Pineal Res. 2015;59:420–433. doi: 10.1111/jpi.12272. [DOI] [PubMed] [Google Scholar]
- 139.Stefanova NA, Maksimova KY, Kiseleva E, Rudnitskaya EA, Muraleva NA, Kolosova NG. Melatonin attenuates impairments of structural hippocampal neuroplasticity in OXYS rats during active progression of Alzheimer’s disease-like pathology. J Pineal Res. 2015;59:163–177. doi: 10.1111/jpi.12248. [DOI] [PubMed] [Google Scholar]
- 140.Chuang JI, Pan IL, Hsieh CY, Huang CY, Chen PC, Shin JW. Melatonin prevents the Drp 1-dependent mitochondrial fission and oxidative insult in the cortical neurons after MPP treatment. J Pineal Res. 2016;61:230–240. doi: 10.1111/jpi.12343. [DOI] [PubMed] [Google Scholar]
- 141.Prieto-Dominguez N, Ordonez R, Fernandez A, Mendez-Blanco C, Baulies A, Garcia-Ruiz L, Fernandez-Checa JC, Mauriz JL, Gonzalez-Gallego J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J Pineal Res. 2016;61:396–407. doi: 10.1111/jpi.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ueck M, Troiani ME, Reiter RJ. Transient reduction in pineal melatonin levels but not N-acetyltransferase activity in rats to swim for 15 minutes at night. Neuroendocrinol Lett. 1988;10:81–90. [Google Scholar]
- 143.Wu W, Reiter RJ, Troiani ME, Vaughan GM. Elevated daytime rat pineal and serum melatonin levels induced by isoproterenol are depressed by swimming. Life Sci. 1987;41:1473–1479. doi: 10.1016/0024-3205(87)90712-0. [DOI] [PubMed] [Google Scholar]
- 144.Troiani ME, Reiter RJ, Vaughan MK, Oaknin S, Vaughan GM. Swimming depresses nighttime melatonin content without changing N-acetyltransferase activity in the rat pineal gland. Neuroendocrinology. 1987;47:55–60. doi: 10.1159/000124891. [DOI] [PubMed] [Google Scholar]
- 145.Sewerynek E, Melchiorri D, Chen LD, Reiter RJ. Melatonin reduces both basal and bacterial lipopolysaccharide-induced lipid peroxidation. Free Radic Biol Med. 1995;19:903–909. doi: 10.1016/0891-5849(95)00101-3. [DOI] [PubMed] [Google Scholar]
- 146.Sewerynek E, Ortiz GG, Reiter RJ, Pablos MI, Melchiorri D, Daniels WMV. Lipopolysaccharide-induced DNA damage is greatly reduced in rats treated with the pineal hormone melatonin. Mol Cell Endocrinol. 1996;17:183–188. doi: 10.1016/0303-7207(95)03742-X. [DOI] [PubMed] [Google Scholar]
- 147.Andrabi SA, et al. Direct inhibition of the mitochondrial transition pore: a possible mechanism for antiapoptotic effects of melatonin. FASEB J. 2008;18:869–887. doi: 10.1096/fj.03-1031fje. [DOI] [PubMed] [Google Scholar]
- 148.Le Bars D, Thivolle P, Vitte RA, Bojkowski C, Chazot G, Arendt J, Frackowiak RS, Claustrat B. PET and plasma pharmacokinetic studies after bolus intravenous administration of [11C]melatonin in humans. Int J Radiat Appl Instrum B. 1991;18:357–362. doi: 10.1016/0883-2897(91)90132-5. [DOI] [PubMed] [Google Scholar]
- 149.Costa EJ, Shida CS, Baggi MH, Ito AS, Laney-Fruend MT. How melatonin interacts with lipid bilayers: a study by fluorescence and ESR spectroscopies. FEBS Lett. 1997;416:103–106. doi: 10.1016/S0014-5793(97)01178-2. [DOI] [PubMed] [Google Scholar]
- 150.Hevia D, Gonzalez-Menendez P, Quiros-Gonzalez L, Miar A, Rodriguez-Garcia A, Tan DX, Reiter RJ, Mayo JC, Sainz RM. Melatonin uptake through glucose transporters: a new target for melatonin inhibition of cancer. J Pineal Res. 2015;58:234–250. doi: 10.1111/jpi.12210. [DOI] [PubMed] [Google Scholar]
- 151.Huo X, Wang C, Yu Z, Peng Y, Wang S, Feng S, Zhang S, Tian X, Sun C, Liu K, Deng S, Ma X. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: an implication of the therapeutic potential. J Pineal Res. 2017;62:e12390. doi: 10.1111/jpi.12390. [DOI] [PubMed] [Google Scholar]
- 152.Pozo D, Reiter RJ, Calvo JR, Guerrero JM. Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J Cell Biochem. 1997;64:430–442. doi: 10.1002/(SICI)1097-4644(19970601)65:3<430::AID-JCB12>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 153.Dubocovich ML, Rivera-Bermudez MA, Gerdin MJ, Masana MI. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front Biosci. 2003;8:d1093–d1108. doi: 10.2741/1089. [DOI] [PubMed] [Google Scholar]
- 154.Boutin JA. Quinone reductase 2 as a promising of target melatonin therapeutic actions. Expert Opin Ther Targets. 2016;20:303–317. doi: 10.1517/14728222.2016.1091882. [DOI] [PubMed] [Google Scholar]
- 155.Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H. The melatonin MT1 receptor axis modulates mutant Huntington-mediated toxicity. J Neurosci. 2011;31:14496–14507. doi: 10.1523/JNEUROSCI.3059-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep. 2003;5:365–371. doi: 10.1007/s11908-003-0015-9. [DOI] [PubMed] [Google Scholar]
- 157.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4:729–741. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 158.Volt H, Garcia JA, Doerrier C, Diaz-Casado ME, Guerra-Librero A, Lopez LC, Escames G, Tresguerres JA, Acuna-Castroviejo D. Same molecule but different expression: aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J Pineal Res. 2016;60:193–205. doi: 10.1111/jpi.12303. [DOI] [PubMed] [Google Scholar]
- 159.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski P, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 160.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286:R491–R497. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 161.Gellerich FN, Trumbeckaite S, Hertel K, Zierz S, Müller-Werdan V, Werdan K, Redl H, Schlag G. Impaired energy metabolism in hearts of septic baboons: diminished activities of Complex I and Complex II of the mitochondrial respiratory chain. Shock. 1999;11:336–341. doi: 10.1097/00024382-199905000-00006. [DOI] [PubMed] [Google Scholar]
- 162.Gitto E, Karbownik M, Reiter RJ, Tan DX, Cuzzocrea S, Chiurazzi P, Cordaro S, Corona G, Trimarchi G, Barberi I. Effects of melatonin treatment in septic newborns. Pediatr Res. 2001;50:756–760. doi: 10.1203/00006450-200112000-00021. [DOI] [PubMed] [Google Scholar]
- 163.Escames G, Acuna-Castroviejo D, Lopez LC, Maldonado MD, Sanchez-Hidalgo M, Lun J, Reiter RJ. The pharmacological utility of melatonin in the treatment of septic shock. J Pharm Pharmacol. 2006;58:1153–1165. doi: 10.1211/jpp.58.9.0001. [DOI] [PubMed] [Google Scholar]
- 164.Reiter RJ, Carneiro RC, Oh CS. Melatonin in relation to cellular antioxidative defense mechanisms. Horm Metab Res. 1997;29:363–372. doi: 10.1055/s-2007-979057. [DOI] [PubMed] [Google Scholar]
- 165.Wu CC, Chiao CW, Hsiao G, Chen A, Yen MH. Melatonin prevents endotoxin-induced circulatory failure. J Pineal Res. 2001;30:147–156. doi: 10.1034/j.1600-079X.2001.300303.x. [DOI] [PubMed] [Google Scholar]
- 166.Leon J, Acuna-Castroviejo D, Escames G, Tan DX, Reiter RJ. Melatonin mitigates mitochondrial malfunction. J Pineal Res. 2005;38:1–9. doi: 10.1111/j.1600-079X.2004.00181.x. [DOI] [PubMed] [Google Scholar]
- 167.Tan DX, Manchester LC, Qin L, Reiter RJ. Melatonin: a mitochondrial targeting molecule involving mitochondrial functioning and dynamics. Int J Mol Sci. 2016;17:2124. doi: 10.3390/ijms17122124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RAJ, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 169.Echtay KS, Murphy MP, Smith RAJ, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:47129–47135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- 170.Jauslin ML, Meier T, Smith RAJ, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 171.Ramis MR, Esteban S, Miralles A, Tan DX, Reiter RJ. Protective effects of mitochondria and mitochondria-targeted antioxidants against oxidative stress. Curr Med Chem. 2015;22:2690–2711. doi: 10.2174/0929867322666150619104143. [DOI] [PubMed] [Google Scholar]
- 172.Hardeland R. Antioxidative protection by melatonin: multiplicity of mechanisms from radical neutralization to radical avoidance. Endocrine. 2005;27:119–130. doi: 10.1385/ENDO:27:2:119. [DOI] [PubMed] [Google Scholar]
- 173.Muxel SM, Laranjeira-Silva MF, Carvalho-Sousa CE, Floeter-Winter LM, Markus RP. The ReIA/cRel nuclear factor-κB (NF-κB) dimer, crucial for inflammation resolution, mediates the transcription of the key enzyme in melatonin synthesis in RAW 264.7 macrophages. J Pineal Res. 2016;60:394–404. doi: 10.1111/jpi.12321. [DOI] [PubMed] [Google Scholar]
- 174.Lin C, Chao H, Li Z, Xu X, Liu Y, Hou L, Liu N, Ji J. Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J Pineal Res. 2016;61:177–186. doi: 10.1111/jpi.12337. [DOI] [PubMed] [Google Scholar]
- 175.Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1095–R1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochem Biophys Acta. 2014;1842:1282–1294. doi: 10.1016/j.bbadis.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Smith RAJ, Porteous AM, Coulter CV, Murphy MP. Selective targeting of an antioxidant in mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 179.Doerrier C, Garcia JA, Volt H, Diaz-Casado ME, Luna-Sanchez M, Fernandez-Gil B, Escames G, Lopez LC, Acuna-Castroviejo D. Permeabilized myocardial fibers as model to detect mitochondrial dysfunction during sepsis and melatonin effects without disruption of mitochondrial network. Mitochondrion. 2016;27:56–63. doi: 10.1016/j.mito.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 180.Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J. 2015;29:4766–4771. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 181.Reiter RJ, Paredes SD, Korkmaz A, Jou MJ, Tan DX. Melatonin combats molecular terrorism at the mitochondrial level. Interdisc Toxicol. 2008;1:101–113. doi: 10.2478/v10102-010-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cippolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ. Melatonin, energy metabolism, and obesity. J Pineal Res. 2014;56:371–381. doi: 10.1111/jpi.12137. [DOI] [PubMed] [Google Scholar]
- 183.Sharma S, Singh H, Ahmad N, Mishra P, Tiwari A. The role of melatonin in diabetes: therapeutic implications. Arch Endocrinol Metab. 2015;59:391–399. doi: 10.1590/2359-3997000000098. [DOI] [PubMed] [Google Scholar]
- 184.Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang H, Tang J, Li H, Feng M, Deng P, Guo P, Tian L, Xie J, He M, Lu Y, Zhang M, Zhang Y, Wang W, Reiter RJ, Yu Z, Zhou Z. Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J Pineal Res. 2016;60:291–302. doi: 10.1111/jpi.12310. [DOI] [PubMed] [Google Scholar]
- 185.Reiter RJ, Tan DX, Galano A. Melatonin: exceeding expectations. Physiology (Bethesda) 2014;29:325–333. doi: 10.1152/physiol.00011.2014. [DOI] [PubMed] [Google Scholar]
- 186.Sadeghian M, Mastrolia V, Rezaei Haddad A, Mosley A, Mullali G, Schiza D, Sajic M, Hargreaves I, Heales S, Duchen MR, Smith KJ. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci Rep. 2016;6:33249. doi: 10.1038/srep33249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mao P, Reddy PH. Is multiple sclerosis: a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66–79. doi: 10.1016/j.bbadis.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kashani IR, Rajabi Z, Akbari M, Hassanzadeh G, Mohseni A, Eramsadati MK, Rafiee K, Beyer C, Kipp M, Zendedel A. Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res. 2014;232:2835–2846. doi: 10.1007/s00221-014-3946-5. [DOI] [PubMed] [Google Scholar]
- 189.Lopez-Gonzalez A, Alvarez-Sanchez N, Lardone PJ, Cruz-Chamorro L, Martinez-Lopez A, Guerrero JM, Reiter RJ, Carrillo-Vico A. Melatonin improves primary progressive multiple sclerosis: a case report. J Pineal Res. 2015;58:173–177. doi: 10.1111/jpi.12203. [DOI] [PubMed] [Google Scholar]
- 190.Swerdlow RH, Koppel S, Weidling I, Hayley C, Ji Y, Wilkins HM. Mitochondria, cybrids, aging and Alzheimer’s disease. Prog Mol Biol Transl Sci. 2017;146:259–302. doi: 10.1016/bs.pmbts.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Giannoccaro MP, La Morgia C, Rizzo G, Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov Disord. 2017;32:346–363. doi: 10.1002/mds.26966. [DOI] [PubMed] [Google Scholar]
- 192.Liot G, Valette J, Pepin J, Flament J, Braulliet E. Energy defects in Huntington’s disease: why “in vivo” evidence matters. Biochem Biophys Res Commun. 2017;483:1084–1095. doi: 10.1016/j.bbrc.2016.09.065. [DOI] [PubMed] [Google Scholar]
- 193.Chahbouni M, Escames G, Venegas C, Sevilla B, Garcia JA, Lopez LC, Munoz-Hoyos A, Molina-Carballo A, Acuna-Castroviejo D. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J Pineal Res. 2010;48:282–289. doi: 10.1111/j.1600-079X.2010.00752.x. [DOI] [PubMed] [Google Scholar]