

Abstract
The combinatorial expression of Hox genes is an evolutionarily ancient program underlying body axis patterning in all Bilateria. In the head, the neural crest (NC)––a vertebrate innovation that contributes to evolutionarily novel skeletal and neural features––develops as a structure free of Hox-gene expression. The activation of Hoxa2 in the Hox-free facial NC (FNC) leads to severe craniofacial and brain defects. Here, we show that this condition unveils the requirement of three Six genes, Six1, Six2, and Six4, for brain development and morphogenesis of the maxillo-mandibular and nasofrontal skeleton. Inactivation of each of these Six genes in FNC generates diverse brain defects, ranging from plexus agenesis to mild or severe holoprosencephaly, and entails facial hypoplasia or truncation of the craniofacial skeleton. The triple silencing of these genes reveals their complementary role in face and brain morphogenesis. Furthermore, we show that the perturbation of the intrinsic genetic FNC program, by either Hoxa2 expression or Six gene inactivation, affects Bmp signaling through the downregulation of Bmp antagonists in the FNC cells. When upregulated in the FNC, Bmp antagonists suppress the adverse skeletal and cerebral effects of Hoxa2 expression. These results demonstrate that the combinatorial expression of Six1, Six2, and Six4 is required for the molecular programs governing craniofacial and cerebral development. These genes are crucial for the signaling system of FNC origin, which regulates normal growth and patterning of the cephalic neuroepithelium. Our results strongly suggest that several congenital craniofacial and cerebral malformations could be attributed to Six genes’ misregulation.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1477-z) contains supplementary material, which is available to authorized users.
Keywords: Neural crest, Hoxa2, Signaling, Head skeleton, Holoprosencephaly, Electroporation, RNAi
Introduction
The face and the brain are phylogenetic innovations, which have evolved in the chordates phylum from the origin of vertebrate evolution. They develop from the early neurula stage by a series of concatenated events, which rely on definite morphogenetic movements, and cell migrations, proliferations, and differentiations. The precise arrangement of cell interaction and fate is coordinated by signaling pathways, where the morphogenetic action of signaling molecules are reciprocally linked to the activation of transcription factors. During evolution, the emergence of the face and brain has coincided with the specification of a pluripotent cell population in the vertebrate embryo, the neural crest (NC) [1, 2]. The NC develops from the lateral borders of the neural epithelium, and builds up the vertebrate head by providing its skeletal, dermal, and connective tissues [3, 4]. In humans, the association of craniofacial and neural defects in some congenital malformations points to the importance of reciprocal interactions between the cephalic NC and the anterior cephalic neuroepithelium. However, the molecular basis of the joint malformations of the face and the brain is still poorly understood.
Recent embryological studies have provided further evidence for the importance of the NC in cephalic ontogeny. The removal of the cephalic NC before its cells begin to migrate in the avian embryo results in the absence of a craniofacial skeleton and severe malformations of the pre-otic brain [5, 6], including anencephaly and absence of the telencephalon. The absence of the most rostral part of the NC, which is destined to yield the facial skeleton [also referred as the facial NC (FNC); Fig. 1a; Fig. S1], results in lower levels of Fgf8 production in both secondary brain organizers—the anterior neural ridge (ANR) and the isthmus—known to play a critical role in fore- and midbrain neurogenesis [7–9]. Moreover, the treatment of embryos from which the FNC has been removed with exogenous Fgf8 has been shown to rescue the phenotype of the concerned embryos [5, 6]. We have then shown that, at critical stages of neurogenesis, the cephalic NC is responsible for the regulation of Fgf8 production through the secretion of Bmp antagonists that counteract signals produced by the prechordal plate, the neural epithelium, and the superficial ectoderm [10]. Fgf8 production is itself regulated by Bmps [10, 11].
Fig. 1.

Hoxa2 expression in the FNC alters gene expression in the developing head. a Experimental design for FNC transfection and implantation into a stage-matched naive embryo at 5ss. a′ Bilateral electroporation of NC at cephalic level with Fluorescein Dextran in a 5ss-embryo. The FNC, which extends from the mide-diencephalon down to r2, is delineated with dotted line; once transfected, the FNC is subjected for transplantation into a naive untransfected embryo. a″ At 7ss, fluorescent FNC cells that have been engrafted have started to migrate (b–e) Head morphology 24 h after eletroporation and surgery. b, c External appearance and d, e internal morphology of cephalic vesicles (dotted lines) in control (b, d) and Hoxa2-transfected (c, e) embryos, showing that Hoxa2 expression in the FNC alters face and brain development. f, g HNK1-Mab labeling of migrating FNC cells; despite morphological changes, the forced expression of Hoxa2 in the FNC does not alter FNC cell migration. h–y Analysis of the expression of Fgf8 (h–j), Bmp4 (k–m), Shh (n–p), Wnt8b (q–s, arrowheads; dotted lines indicate the boundary between the diencephalon posteriorly and the telencephalon anteriorly), Noggin (t–v) and Six2 (w–y) in control (h, k, n, q, t, w), Hoxa2-transfected (i, l, o, r, u, x), and Six2-silenced (j, m, p, s, v, y) embryos. While Six2 expression is inhibited in the FNC-derived mesenchyme, its expression in the pharyngeal endoderm persists after Hoxa2 activation or Six2 silencing in FNC cells. Changes in gene expression are shown with arrowheads (z) RT-PCR analysis of Hoxa2, Noggin, Dan, and Six2 expression in NF-derived cells grown in vitro for 24 h, taken from r4 to r8 NC, FNC, and Hoxa2-transfected and Six2-silenced FNC. Di diencephalon, Mes mesencephalon, Rh rhombencephalon, Tel telencephalon
We have also demonstrated that the effects of NC ablation can be mimicked by the overexpression of either Hoxa2 alone or a combination of Hoxa3 and Hoxb4 in FNC cells, which do not normally express Hox genes. No facial structures develop when these genes are overexpressed. Moreover, the cephalic vesicles collapse and eventually degenerate, resulting in a severe, anencephalic condition [12]. These observations indicate that the molecular mechanisms governing craniofacial development differ from those controlling the ontogeny of the posterior hindbrain and trunk, in which Hox genes are active. It has been previously shown that, in the pharyngeal region, where a combinatorial expression of Hox gene regulates morphogenesis, Hoxa2 interferes with a transcription factor of the Six family: Hoxa2 directly targets the expression of Six2 and represses its activity in the NC-derived mesenchyme populating the hyoid arch [13]. This raises questions about how Hoxa2 interferes with the morphogenetic program required for skeletogenesis in the Hox-free domain of the head.
We have investigated the mechanisms involved in these processes, by triggering the ectopic expression of Hoxa2 in the FNC. We used the chick embryo as a model, which offers the unique advantage of allowing a precisely controlled time-, space-, and tissue-specific transgenesis. We show here that the forced expression of Hoxa2 in FNC cells, while suppressing their skeletogenic potential, decreases the expression of genes of the Six family (Six1, Six2, and Six4) in a variable manner. The Six1, Six2, and Six4 had similar patterns of activity in premigratory and migrating FNC cells, but their selective silencing had diverse effects, resulting in an overall reduction or partial truncation of the facial skeleton. Under these conditions, dramatic perturbations of cerebral development were observed ranging from the loss of choroid plexus and mild septal defects to alobar holoprosencephaly. By contrast, the simultaneous silencing of all three genes reproduces the adverse effects of Hoxa2 expression, indicating that they cooperate in the control of head skeletogenesis and brain morphogenesis.
We also show that one major effect of Hoxa2 expression or Six gene silencing is an upregulation of Bmp signaling in the head. Under these conditions, the overexpression of a Bmp antagonist in the FNC circumvents the deleterious effects of Hoxa2 expression, thus rescuing face and brain development.
Materials and methods
Microsurgery
Experiments were carried out on chick embryos at the 5- to 6-somite stage (5–6 ss, i.e. 30 h of incubation at 38 °C). Chick embryos were prepared for in ovo surgery as previously described [14]. The fate map of the cephalic neural primordium [15] was used as a reference for determining the presumptive level of the encephalic structures extending from the mid-diencephalon down to r8. According to these fate maps, the surgical and molecular manipulations were performed on the FNC, which extends from the mid-diencephalon down to rhombomere2 (r2) included (Fig. 1a).
Double-stranded RNA synthesis and electrophoration with exogenous nucleic acid
dsRNA were synthesized from cDNAs encoding the targeted genes [16]. In contrast to studies carried out in mammals, in which dsRNA yield unspecific blockade of protein synthesis, no such effects were ever detected in chicken embryos. ([17] and references therein). dsRNA-driven gene silencing results in highly specific gene inactivation.
According to these procedures, dsRNA molecules were synthesized from the cDNA encoding Six1, Six2, and Six4. For control series, solutions of non-annealed sense plus anti-sense RNA strands corresponding to the sequences of the targeted genes were used at the same concentration for in ovo electroporation, according to the same paradigm. In parallel, we also did some control experiments using dsRNA designed against Hoxa2 for electroporation of FNC cells. As these cells are normally devoid of Hoxa2 expression, the outcome of these experiments was similar to the control series, thus confirming the specificity of the dsRNA-driven gene silencing strategy.
Depending on the experimental series, the dsRNA molecules were used at different concentrations: functional assays on brain development were yielded with solutions of 600 ng/μl dsRNA but resulted in a high proportion of dead embryos beyond E5. The shortcoming of these series precluded the analysis of the consequences of Six gene silencing on craniofacial development. So, for examination of E8 skeletogenic development, silencing experiments were carried out with solutions of 300 ng/μl dsRNA. Gain-of-function experiments involving Hoxa2 retroviral construct [12] and Six2 gene expression vector [18] were performed with solutions of 1 μg/μl of constructs. Replication competent retroviral constructs used in this study are designed to allow for the overexpression of the insert but not produce viral particles. The ectopic activity of the gene of interest is therefore restricted to the transfected cells (i.e. FNC cells) and their progenies.
In ovo electroporations were performed in 5–6ss chick embryos. Briefly, exogenous nucleic acids in 0.01 % Fast Green (Sigma) were injected into the cephalic neural groove. Together with exogenous nucleic acid sequences, Fluorescein Dextran was coelectroporated in order to visualize and track the FNC-derived cells. Bilateral electroporation was achieved by placing two anodes on the vitelline membrane flanking the head of the embryo and one cathode against the anterior region of the embryo [12] The dsRNA molecules, retroviral or plasmidic constructs were delivered to the FNC cells by a series of five 27-V pulses (T830 BTX; Genetronics, San Diego, CA, USA). The bilaterally transfected chick neural folds, extending from the diencephalon down to r2, were then surgically excised and homotopically implanted into a stage-matched untransfected host.
Embryo processing
For whole-mount preparations, the control and experimental embryos were harvested in sterile PBS then fixed in 4 % formaldehyde. After extensive washes in PBS, embryos were dehydrated in graduate methanol and permeabilized by cold-shock in 100 % methanol at −20 °C for 45 min. After rehydratation in PBS, embryos were subjected to hybridization for gene expression analysis or immunocytochemsitry. Embryos subjected to histological analysis were fixed in 60 % ethanol, 10 % formaldehyde, 10 % acetic acid, dehydrated, and embedded in paraffin. Six-μm-thick sections were collected on Superfost-Plus slides, and processed for either hybridization or immunocytochemistry.
Whole-mount LTR staining and immunocytochemistry
In control and experimental series, NC cell migration and cell proliferation were visualized by whole-mount immunocytochemistry with monoclonal antibodies (Mab) against HNK1 (CD57; Santa Cruz Biotechnology) and phosphorylated histone3 (PhH3; Sigma), respectively, in E2.5 embryos [12]. Cell death was detected by whole-mount Lysotracker Red (LTR; Invitrogen) staining, as previously described [19]. Quantification of cell proliferation and death was then performed on vibratome sections.
Cartilage staining
Chondrogenic structures from E8 embryos were visualized after fixation in 80 % ethanol, 20 % acetic acid, and 0.015 % Alcian Blue 8GX for 16 h, extensive washing in 100 % ethanol, and clearing in 1 % KOH and 20 % glycerol.
In situ hybridization
Whole-mount in situ hybridization [20] was performed with the probes for Fgf8 [21], Shh [22], Bmp4 [23], Noggin [24], and Wnt8b [25] transcripts. When necessary, double hybridizations with FITC-tagged riboprobes were performed by tyramide signal amplification (Perkin-Elmer). Six gene riboprobes were generated with the following specific primer pairs for cDNA amplification by PCR and cloning with the pCR®-TOPO system (Invitrogen): Six1 (For.5′- CGAACCCCGGATCCCATTGCG -3′;Rev.5′- GCAGCACCGCCGCGTTAAGA -3′), Six2 (For.5′- GCGTCAATGAACGGGAATAA -3′; Rev.5′- AGCGGTTTAAGAGCCCAGAT -3′), and Six4 (For.5′- CCACATCCGCTCTCCAGCTCG -3′; Rev.5′- TGATGTCAGTGATGGACACCCCG -3′). GAPDH expression was used as internal reference in all PCR experiments. SyBR green assays were normalized to GAPDH activity using the following primer pair: For.5′- TCCAGGAGCGTGACCCCAGC -3′; Rev.5′- TGCCAGGCAGTTGGTGGTGC -3′. RNA probes were synthesized with the Riboprobe Combination System kit (Promega) and labeled with UTP-digoxygenin (Boehringer). After whole-mount hybridization, some embryos were dehydrated in ethanol, permeabilized in toluene, and embedded in paraffin for sectioning.
Microscope image acquisition
Images of whole-mount embryos were acquired on a LEICA MZ-FLIII stereomicroscope equipped with a X-Cite series 120Q, as a source of UV light, and a Qimaging cooled micropublisher camera driven by the Qcapture software.
RT-PCR analysis of gene expression in vitro
The molecular profile of the electroporated neural folds was determined by RT-PCR. The Hoxa2-treated, Six2-silenced FNC and r4–r8 NC were microdissected and cultured in vitro for 24 h in DMEM supplemented with 10 % FCS. NC cells were then harvested for cDNA preparation (SuperScript™ III; Invitrogen) and RT-PCR analysis for Hoxa2, Six2, Noggin, Dan, Sox9, Runx2, and GAPDH. RT-PCR primers for specific target genes were designed in Primer-BLAST tools: Hoxa2 For.5′- TTTTTCTCCGCGGGGGCTGC -3′; Rev.5′- GGCTGGGGATGGTCTGCTCG -3′. Noggin For.5′- GGCTGGGGATGGTCTGCTCG -3′; Rev.5′- TATAGGACCGGGCAGAAGGT -3′; Dan For.5′- AGGAGAACATGCCCGCAGAG -3′; Rev.5′- ATCCTGCAGACAGCCCTTGG -3′; Sox9 For.5′- TCTCCGTTTTCTCCTCCCCT -3′; Rev.5′ –CTTGAGGTCGGGTGTTCTCC -3′.
Quantitative RT-PCR
The isolation of RNA was performed using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. After isolation, RNA was treated with DNase I (Invitrogen) to digest contaminating DNA. 1 μg of RNA sample was then used for reverse transcription and synthesis of cDNA using Superscript III system (Invitrogen). Quantitative real-time PCR was performed on an AB7300 (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems). The level of target gene expression was compared to GAPDH gene activity and measured by comparative Pfaffl method [26]. The statistic analysis was performed using ANOVA/Turkey tests.
Results
Hoxa2 effects on gene expression in the developing head
We investigated the molecular changes resulting from Hoxa2 activation in FNC cells, by bilaterally electroporating these cells with an RCAS construct driving Hoxa2 expression before they began to migrate, in chick embryos at the 5–6 somite stage (ss). Hoxa2 activity was restricted to FNC cells, by dissecting out the transfected neural folds and transplanting them into a naive recipient chick at the same stage (Fig. 1a). In parallel, control experiments were carried out with an empty RCAS construct, according to the same procedure. Morphological differences were observed between experimental embryos and controls 24 h after surgery (E2.5; n = 8). Transgenic embryos had much smaller heads, with a different morphology, including conspicuous atrophy of the pre-otic encephalic vesicles (Fig. 1b–e). However, NCC emigration from the FNC was similar in control and Hoxa2-transfected embryos: NC-derived mesenchymal cells massively colonized the 1st branchial arch (BA1) and the nasofrontal bud (Fig. 1f, g).
We hypothesized that the morphological defects in Hoxa2-transgenic embryos might result from the disturbance of signaling pathways, such as those involving Fgf8, Bmp4, Shh, and Wnt8b. Fgf8 transcript levels in the ANR, BAs, and isthmus were significantly lower than normal [21] after Hoxa2 activation in the FNC (Fig. 1h, i; n = 5), this situation being reminiscent of that resulting from the removal of the FNC [5, 6]. Under this condition, Bmp4 expression has been shown to counteract Fgf8 signaling at this stage [11]. However, in Hoxa2-transgenic embryos, the amount of Bmp4 transcripts was unaffected in the maxillo-mandibular, retro-ocular, nasofrontal, and BA ectoderm (Fig. 1k, l; n = 5/5). The absence of the FNC has been shown to result in a ventralization of the encephalic neuroepithelium, as demonstrated by laterally extended Shh expression [6]. Ectopic Hoxa2 activation in FNC cells resulted in a similar lateral expansion of Shh expression (Fig. 1n, o; arrowheads; n = 6/6), together a decrease in the size of the prosencephalic and mesencephalic alar plates. At that stage, the domain of Wnt8b expression straddles the dorsal part of the anterior diencephalon and posterior telencephalon (Fig. 1q, white arrowheads), from which the choroid plexi normally develop in the lateral and third ventricles (Fig. 1q, dotted lines); [25, 27]. It is also expressed at the dorsal rhombencephalic midline. In Hoxa2 transgenic embryos, Wnt8b was much less strongly expressed in the prosencephalon (Fig. 1r; white arrowheads) and not expressed at all in the rhombencephalon (Fig. 1r; black arrowhead; n = 6). Thus, Hoxa2 expression in the FNC-derived mesenchyme had a deleterious effect on the dorso-ventral molecular patterning of the developing encephalic vesicles. Overall, Hoxa2 activation in FNC cells resulted in atrophy of the fore- and midbrain, the downregulation of Fgf8 in the ANR, isthmus and BA ectoderm, the downregulation of Wnt8b, and a lateral expansion of Shh expression.
Effect of Hoxa2 on Bmp antagonists
We previously showed that the amounts of Bmp antagonists (Gremlin and Noggin) produced by FNC cells indirectly regulate Fgf8 expression in the ANR. Surgical ablation of the FNC results in brain defects due to an increase in Bmp signaling in response to the loss of Bmp inhibitor production by the NC cells [10]. In normal development, Noggin is expressed in the migrating FNC cells responsible for populating the facial processes and BAs [28] (Fig. 1t; arrowheads). When Hoxa2 expression was ectopically triggered in FNC cells, these cells displayed very low levels of Noggin expression in the maxillo-mandibular process (Fig. 1u; n = 5).
We analyzed the molecular profile of control and Hoxa2-transfected FNC embryos by PCR: the cephalic NC was surgically explanted after either sham or Hoxa2-RCAS transfection and grown in vitro for 24 h. PCR analyses demonstrated a loss of Noggin expression in Hoxa2-transgenic FNC (Fig. 1z). A similar effect on the expression of Dan, encoding another Bmp-antagonist produced by FNC cells [29, 30], was also found (Fig. 1z).
Effect of Six2 silencing on gene expression in the developing head
Six2 has been shown to be a direct downstream target of Hoxa2 in BA2, in which it is downregulated by the product of Hoxa2 at specific sites [13]. Six2 expression is observed in the developing head from 5ss onward, and is detected in migrating FNC cells as early as 10ss (Fig. S2a, b; arrowheads). At 25ss, Six2 expression in FNC cells was detected in the vicinity of the trigeminal, facial and vestibulo-acoustic ganglionic anlagen, in the maxillo-mandibular processes and, to a lesser extent, in the naso-frontal bud (Fig. 1w; Fig. S2c–e). From this stage onwards, Six2 expression became increasingly strong in the NC-derived mesenchyme and endodermal pouches (Fig. S2f, g).
In experimental embryos, ectopic Hoxa2 activity prevented the expression of Six2 in FNC-derived cells, but did not affect the expression of Six2 in the pharyngeal endoderm (Fig. 1x; n = 6). Similarly, PCR analysis of Hoxa2-expressing NC cells grown in vitro confirmed the downregulation of Six2 following transfection with Hoxa2 (Fig. 1z).
We investigated the intrinsic role of Six2 in head development by silencing the Six2 gene in the FNC. We used the same method to transfect the FNC cells of 5–6ss embryos bilaterally with a dsRNA targeting Six2, and then transferring these cells into naive embryos (Fig. 1a). At E2.5, Six2-depleted embryos displayed severe facial hypoplasia and atrophy of the pre-otic brain. As described above, gene expression analyses in experimental embryos were performed by in situ hybridization and RT-PCR on explanted FNC cells. Both approaches confirmed the inhibition of Six2 in FNC in vivo and in vitro (Fig. 1y, z). By contrast, the endodermal expression of Six2 was unaffected after surgery.
Influence of Six2 silencing on gene expression in the developing head
We analyzed the pattern of expression of the Fgf8, Bmp4, Wnt8b, and Shh genes in Six2-depleted embryos, at E2.5. The loss of Six2 function in FNC cells modified Fgf8 expression in a similar manner to Hoxa2 transfection: Fgf8 activity was inhibited in the ANR, the isthmus, and the BA1 ectoderm (Fig. 1j; n = 5). Bmp4 transcript accumulation was maintained in the nasofrontal and maxillary regions (Fig. 1m). Following the silencing of Six2, a considerable expansion was observed in the domain of Shh expression in the pre-otic neuroepithelium, as in the Hoxa2-transfected embryos (Fig. 1p, arrowheads; n = 6), and an upregulation of Shh expression was observed in the maxillo-mandibular commissural ectoderm (asterisk). Similarly, along the prosencephalic and rhombencephalic midline, Six2 silencing narrowed the region of Wnt8b expression (Fig. 1s; white arrowheads).
We then investigated the consequences of Six2 silencing for the expression of genes encoding Bmp-antagonists: Six2 inactivation led to the downregulation of both Noggin (Fig. 1v, z) and Dan (Fig. 1z).
Further head and brain development in Six2-silenced embryos
As the ectopic activation of Hoxa2 in FNC severely compromised face and brain development [12], we looked for cerebral and skeletal defects after Six2 depletion in FNC cells.
At E5, the cephalic vesicles of isolated brains from Six2-dsRNA-treated embryos exhibited major defects. The telencephalic vesicles were smaller than in controls: the dorsolateral development of the pallium, the thalamus, and the optic tectum were abnormal (Fig. 2a–d; n = 5). Six2 silencing in the FNC also had detrimental effects on the development of the prosencephalic and rhombencephalic choroid plexi, which were totally disorganized (Fig. 2d, asterisk). More specifically, at prosencephalic level, the inhibition of Six2 activity in FNC cells affected the development of the pallium and septum pellucidum, which were atrophied compared to the control (Fig. 2e, f). Similarly, the thalamus appeared underdeveloped (Fig. 2e, f).
Fig. 2.
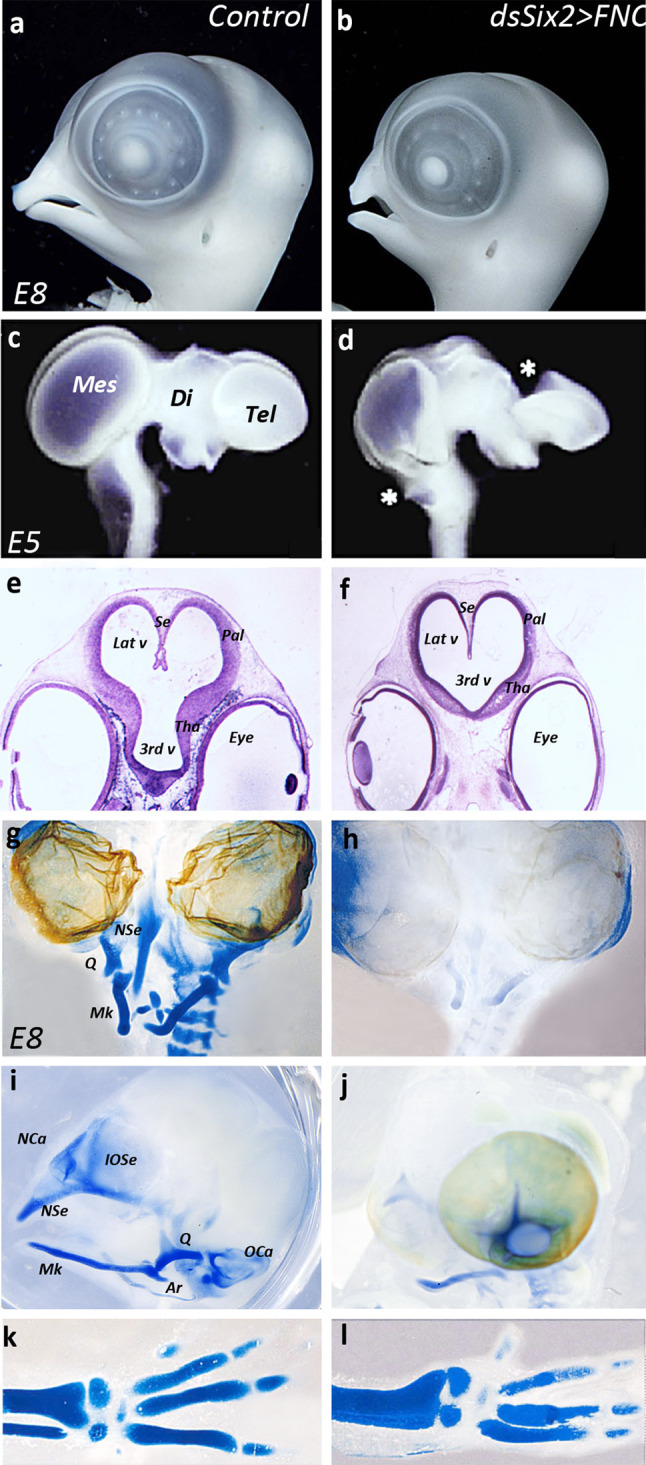
Six2-silencing in the FNC hampers head and brain development. a, b Morphology and c, d whole-mount brain preparation in control (a, c) and experimental (b, d) embryos. In Six2-silenced embryos, the nasofrontal and maxillo-mandiblar development is atrophied (b), and brain morphology is altered along the dorsal midline (d): choroid plexi are missing (stars). Cresyl-violet staining of paraffin sections (e, f) on E5 control (e) and experimental (f) embryos showing the defective development of prosencephalic structures in experimental series: the pallial and thalamic neuroepithelium are thinner than in control (f). In addition, the inter-hemispheric septum fails to form normally. g–j Whole-mount staining of cartilage in the head skeleton of E8 (g, i) control and (h, j) Six2-silenced embryos. On frontal views (g, h), much of the nasal and mandibular cartilages are absent in experimental embryos (h). On profile views (i, j), the skeletal defects resulting from the silencing of Six2 consist in the absence of the interorbitary and nasal septa, nasal and otic capsules, quadrate and articular (j); on control embryo, eyeballs have been removed to evidence skeletal structures at the midline. By contrast, the development of the appendicular skeleton is similar in the control and Six2-silenced series, as shown by the normal development of the radius, ulna, and digits (k, l). Ar articular, Di diencephalon, IOSe interorbital septum, Lat v lateral ventricle, Mes mesencephalon, Mk Meckel’s cartilage, NCa nasal capsule, NSe nasal septum, OCa otic capsule, Pal pallium, Q quadrate, Se septum, Tel telencephalon, Tha thalamus, 3rd v third ventricule
Six2 loss-of-function in the FNC led to severe defects of head skeletal development at E8: the nasal septum and capsule were almost completely absent and the mandible was reduced in its distal-most part (Fig. 2g–j). Moreover, the articular, the quadrate, the interorbital septum, the sclera, and the otic capsule were totally missing (Fig. 2i, j). In contrast, the appendicular skeleton of Six2-depleted embryos developed normally (Fig. 2k, l).
Rescue of the Hoxa2 phenotype by Six2
As Six2 silencing reproduced the molecular and morphological changes induced by Hoxa2 translocation, we tried to rescue the dysmorphologies generated by Hoxa2 by cotransfection of Hoxa2 and Six2. This experiment was performed by simultaneously transfecting FNC cells with Hoxa2 and Six2 retroviral constructs, in the same experimental design as for the preceding experiments. The embryos were observed at 24ss: the combined expression of Six2 and Hoxa2 led to normal head development (Fig. 3a–i; n = 5). Similarly, the gene expression pattern of Fgf8, Wnt8b, Shh, and Noggin tended to normalize when Six2 was cotransfected with Hoxa2 in FNC cells (Fig. S3; compare with Fig. 1i, o, r). These observations were confirmed by the analysis of the skeletal phenotype resulting from these experimental series at E8: while Hoxa2 totally hampered the skeletal differentiation, cotransfection of Hoxa2 and Six2 in FNC cells fully restored the development of nasofrontal and maxillo-mandibular structures (Fig. 3j–l; n = 3/3). The elimination of the deleterious effects of Hoxa2 expression by the upregulation of Six2 revealed that Six2 encodes a major transcription factor controlling the early steps of head development.
Fig. 3.

Molecular interactions between Hoxa2, Six2, Six1 and Six4. a–c Hoxa2 expression in control, Hoxa2, and Hoxa2+Six2 transgenic embryos: whole-mount in situ hybridizations show the expansion of Hoxa2 transcript accumulation at the expense of facial territories in embryos engrafted with Hoxa2-transfected FNC (b, c). d–f Six2 transcript accumulation evidenced by fluorescent hybridization (red) in the same embryos; embryos are delineated with dotted lines. The forced expression of Hoxa2 in FNC cells inhibits the activity of Six2 (e), which can be restored when Hoxa2 and Six2 are cotransfected in FNC cells (f). g–i Superimposed expression of Hoxa2 and Six2: Six2 overexpression in the FNC can rescue the Hoxa2 phenotype at E2.5 (i). j–l Whole-mount skeletal preparation of control, Hoxa2, and Hoxa2+Six2 transgenic embryos. Alcian blue staining shows the complete set of skeletal elements in controls at E8 (j). Hoxa2 expression in FNC totally hampers skeletal differentiation at this stage (k), but cotransfection of Hoxa2+Six2 fully restores the development of maxillo-facial and mandibular skeleton (l). m–o Six1 and p-r Six4 expression in (m, p) control, (n, q) Hoxa2-transfected, (o, r) dsSix2-transfected embryos. Hoxa2 ectopic expression strongly affects the activity of Six1 and Six4 in FNC cells (n, q), while the silencing of Six2 has a limited effect (o, r). s qRT-PCR analysis of Six gene activity on explanted FNC cells following Hoxa2 transfection. Control FNC cells express high level of Six1, Six2, and Six4. After induction of Hoxa2, the activity of all of the three genes is strongly affected. ***P < 0.001. Error bars SEM from three independent experiments, which have been performed in triplicate
Involvement of Six1 and Six4 in head morphogenesis
As the cerebral and skeletal defects resulting from Six2 silencing in the FNC did not amount to a full phenocopy of those resulting from Hoxa2 activation, we searched for other genes from the same family that might serve as downstream targets of Hoxa2 in the developing head. We investigated whether the transfection of FNC cells with Hoxa2 affected the expression of Six1 and Six4, both of which are activated in FNC-derived mesenchyme. During normal development, Six1 and Six4 display similar patterns of expression at 24ss: transcripts are present in the maxillo-mandibular region, more caudal BAs, and in the otic vesicles (Fig. 3m, p). The ectopic activation of Hoxa2 expression greatly reduced the expression of Six1 and Six4 in the maxillo-mandibular region (Fig. 3n, q). In contrast, Six2 silencing in FNC cells slightly impacted on Six1 and Six4 expression pattern at 24ss (Fig. 3o, r).
In order to quantify the changes in Six gene expression resulting from the ectopic activation of Hoxa2 in the FNC, we performed a qRT-PCR analysis on control and Hoxa2-transfected FNC cells that were grown for 24 h in vitro before RNA preparation. The SYBR green assays were normalized on GAPDH activity. The quantification of gene expression revealed that Hoxa2 chiefly affected the activity of Six2, which was significantly reduced when compared to control series (Six2∆ = 710 %; Fig. 3s). Similarly, Six1 and Six4 expression activity shrank in Hoxa2-transfected FNC cells (Six1∆ = 78 %, Six4∆ = 110 %; Fig. 3s).
We explored the respective roles of Six1 and Six4 genes in brain development by selectively modifying the expression of these two genes through dsRNA-driven gene silencing at 5–6ss followed by functional screening of effects on the morphogenesis of cephalic vesicles at E5. Six1 silencing in FNC cells entailed profound changes in brain development with respect to the control (Fig. 4a–d). Structural defects in Six1-depleted embryos consisted in a dramatic microcephaly (Fig. 4c). The telencephalic septation failed to form, and embryos virtually exhibited an alobar holoprosencephaly together with a severe atrophy of the prosencephalic neuroepithelium (Fig. 4d; asterisk and arrowheads). In contrast, depriving FNC cells of Six4 activity resulted in a different outcome. The cephalic vesicles were smaller than normal (Fig. 4e, f). Notably, the diverticulation of the anterior primitive prosencephalon occurred: the cerebral hemispheres and the lateral ventricles formed. However, in these embryos, the inter-hemispheric septum was broader than normal and the cavum failed to develop (Fig. 4f; asterisk).
Fig. 4.

Silencing of Six gene impairs brain development. a–l Analysis of brain development in E5 (a–c) control, (d–f) Six1-, (g–i) Six4-, (j–l) Six1/Six2/Six4-silenced embryos. At this stage, in control embryos (a), the telencephalic hemispheres are well developed (b). c, d Knocking-down Six1 in FNC cells hampers the development of pallial and subpallial structures (d, arrowheads) and inhibits the septation of the anterior prosencephalon (d, asterisk). e, f Inhibition of Six4 results in mild defects, which affect the formation of the septum pellucidum (f, asterisk). The triple silencing degenerates towards a dramatic anencephaly (g), which extends from the telencephalon down to the anterior rhombencephalon (h, arrowheads). Cav cavum pellucidum, Lat v lateral ventricle, Pal pallium, Se septum pellucidum, Tha thalamus, 3rd v third ventricle
In order to further analyze the requirement of Six gene activity in FNC cells for brain development, we decided to silence the three genes, Six1, Six2, and Six4, simultaneously in FNC cells at 5–6ss. At E5, the triple silencing generated a dramatic anencephalic condition (Fig. 4g). Embryos exhibited a profound brain defect that encompassed the tel-, di-, and mesencephalon (Fig. 4h; arrowheads). Such a phenotype was reminiscent of that resulting from either FNC ablation or Hoxa2 activity in FNC [12], hence pointing to the requirement of Six gene activity in the Hox-negative NC for the proper development of the pre-otic brain.
Later on, we also examined the impact of Six1 and Six4 loss-of-function on craniofacial development at E8. Embryos subjected to Six1 downregulation displayed no loss of skeletal elements with respect to the control (Fig. 5a–c). However, Six1 silencing strongly affected facial cartilage size, leading to a global homothetic decrease in the size of the head skeleton (Fig. 5d–f). This phenotype was different from that resulting from the silencing of Six4. At E8, embryos subjected to Six4 silencing displayed a striking skeletal phenotype (Fig. 5g–i). All but the proximal and distal tips of the nasofrontal and mandibular cartilages were missing; the remaining skeletal structures, part of the quadrate, the rostral end of Meckel’s cartilages, the nasal septum, and the entoglossum were all smaller than normal (Fig. 5h, i).
Fig. 5.

Six genes have complementary role in head skeletogenesis. a–l Skeletal preparation of E8 (a–c) control, (d–f) Six1-, (g–i) Six4-, (j–l) Six1/Six2/Six4-silenced embryos. The genes Six1 and Six4 genes, when knocked-down, generate different skeletal phenotypes. The phenotype resulting from the triple silencing of Six1/Six2/Six4 sums up the requirement of each targeted gene for skeletal development and reveals their complementary role in the skeletogenic patterning. Ar articular, Bb basibranchial, Bh basihyal, Cb ceratobranchial, Eb epibranchial, Mk Meckel’s cartilage, NCa nasal capsule, NSe nasal septum, Q quadrate
Pursuing the idea that ectopic Hoxa2 activity might inhibit the expression of Six1, Six2, and Six4, we decided to switch off the expression of all three of these genes simultaneously, by electroporating FNC cells with dsRNA molecules targeting these three genes at 5–6ss. At E8, the triple silencing of Six1, Six2, and Six4 completely prevented skeletogenic differentiation (Fig. 5j–l; n = 5/5). None of the elements of the facial skeleton developed (Fig. 5j) and whole-mount skeletal preparations were totally transparent (Fig. 5k, l), mimicking the skeletal phenotype resulting from the transfection of FNC cells with Hoxa2 [12].
Thus, the various Six genes involved in head skeletogenesis have different, complementary roles in this process, suggesting that craniofacial skeletogenesis, which normally takes place in a Hox-free domain, closely depends on the combinatorial expression of Six genes for growth and patterning.
Relationship between Six genes and Noggin expression in the FNC
As mentioned above, the adverse effects of Hoxa2 activity in the FNC were accompanied by the downregulation of Bmp antagonists. We therefore hypothesized that Hoxa2 expression might exert its negative effects on face and brain development by interfering with the regulation of Bmp signaling. We decided to counteract the effects of Hoxa2 through the forced expression of Noggin in the same system. FNC cells were cotransfected with retroviral constructs driving the expression of Hoxa2 and Noggin at 5–6ss. As previously indicated, the electroporated tissue was subsequently transplanted into a naive recipient embryo (see Fig. 1a). We first investigated whether Noggin overexpression could restore normal levels of Six2 expression. We found that Hoxa2 expression completely prevented Six2 gene activity in the developing head (Fig. 6a, b), and that the overexpression of Noggin and Hoxa2 together did not rescue Six2 activity in the FNC-derived mesenchyme (Fig. 6c). However, even in the absence of Six2 activity, the forced expression of Noggin induced a significant restoration of normal head development (Fig. 6c; n = 7/7). We investigated the cellular events accounting for these morphological changes by studying cell proliferation and death in parallel in control, Six2-depleted, Hoxa2, and Hoxa2 + Noggin transgenic embryos at 18ss, on frontal views (Fig. 6d–l). In control embryos, particularly high levels of cell proliferation were detected by PhH3 immunolabeling in the dorsal parts of the prosencephalic and optic vesicles, and in BA1 (Fig. 6d). Cell death was observed only rarely, rostrally and mostly along the anterior midline, at the level of the ANR (Fig. 6e). By contrast, both dsSix2 (n = 5/5) and Hoxa2-treated (n = 10/10) embryos displayed much lower levels of cell proliferation, particularly in the dorsolateral areas (Fig. 6f, h; green arrows). In these embryos, the territory in which cell death occurred was larger, encompassing much of the prosencephalic territory along with the lateroventral part of the diencephalon and the optic stalk (Fig. 6g, i; red arrows). In addition, the expansion of cell death along the dorsal midline was associated with the initiation of brain defects (Fig. 6g, i). When Hoxa2 expression was combined with Noggin expression, cell proliferation patterns tended to normalize (Fig. 6j, l). Additionally, cell death was reduced and occurred only in the very anterior midline (Fig. 6k, l; n = 12/12). The effects of Noggin on head development in the presence of ectopic Hoxa2 activity could therefore be attributed to both an increase in proliferation and lower levels of cell death.
Fig. 6.

Noggin supplementation in Hoxa2-treated embryos can bypass Six inhibition to restore cell proliferation and reduce cell death. a–c Comparison for Six2 expression between (a) control, (b) Hoxa2, and (c) Hoxa2+Noggin transgenic embryos at E2. Hoxa2 represses Six2 expression (b), but Noggin can restore normal facial development (c). Analysis of (d, f, h, j) cell proliferation through PhH3 immunodetection and (e, g, i, k) cell death by whole-mount LTR staining in (d, e) control, (f, g) dsSix2-treated, (h, i) Hoxa2, and (j, k) Hoxa2+Noggin-tranfected embryos. Hoxa2 activity in FNC cells early biases cell proliferation and death in the developing head towards a deleterious condition. l Quantification of cell proliferation and death in control and experimental series. Supplementation with Noggin tends to normalize the balance between cell proliferation and death. BA1 1st branchial arch, Ey eye, Pro prosencephalon, St stomodeum
Noggin can rescue the effects of Hoxa2
As Noggin activity in the migrating FNC cells was demonstrated to be critical for encephalization, through the control of Fgf8 production in the ANR, we explored the regulation of Fgf8 activity in these series. The forced expression of Hoxa2 in FNC cells resulted in much lower levels of Fgf8 transcripts in the ANR and isthmus (Fig. 7a, b). Following the cotransfection of FNC cells with both Noggin and Hoxa2, Noggin activity prevented the effects of Hoxa2 and normalized Fgf8 activity in the ANR, isthmus, and BAs (Fig. 7c; n = 6/6). We investigated the effect of this treatment on brain development. Isolated brain preparations from E4 embryos revealed normal development of the pre-otic encephalic vesicles, with morphological subdivisions into telencephalic hemispheres, thalamus, and optic tectum (Fig. 7d). In E4 embryos transfected with Hoxa2 alone, the dorsal part of the brain was missing, such that the cephalic vesicles were no longer recognizable (Fig. 7e). By contrast, transfection with both Hoxa2 and Noggin restored brain development to normal morphology (Fig. 7f).
Fig. 7.

Noggin expression can overcome the deleterious effects of Hoxa2 on brain and skeletal development. Comparisons between (a, d, g, j, m) control, (b, e, h, k, n) Hoxa2, (c, f, i, l, o) Hoxa2+Noggin transgenic embryos (a–c) in Fgf8 expression, (d–f) E4 brain morphology, (g–l) E5 Sox9 expression. Supplementation of Hoxa2-treated embryos with Noggin can restore the effect of Hoxa2 on Fgf8 activity in ANR, BAs and isthmus (b, c). Under this condition, the brain development, which is severely compromised by Hoxa2 (e), is restored and the cephalic vesicles of the fore- and midbrain develop (f). g–i At E5, whole-mount hybridization for Sox9 shows that Noggin activity can counterbalance the inhibition of Sox9 expression by Hoxa2 to control the chondrogenic commitment of FNC cells. On paraffin sections (j–l), the mesenchyme in the maxillo-mandibular processes normally expresses Sox9 (j); Hoxa2 inhibits its expression (k), but Noggin can restore the chondrogenic potential of mesenchymal cells by stimulating Sox9 activity (l). m qRT-PCR analysis performed on FNC cells subjected to normal condition, Hoxa2-, and Hoxa2+Noggin-transfection and grown in vitro for 24 h, shows that Hoxa2 represses Sox9 activity, but Noggin can overcome its effect. Error bars SEM from three independent experiments, which have been performed in triplicate (***P < 0.001). n–p Alcian blue staining of whole-mount skeletal preparation of control, Hoxa2 and Hoxa2+Noggin transgenic embryos. The skeletal development at E8 (n), which is completely abolished by Hoxa2 (o), is restored by Noggin (p). q, r Alcian blue and alizarin red staining of skeletal preparation at E10 Similarly, when Noggin is cotransfected with the three dsRNA against Six1, Six2, and Six4, its activity is sufficient to fully restore skeletogenic development (r). Md mandibular bud, Mx maxillary process, OTe optic tectum, Pal pallium, T tongue, Tha thalamus
We investigated whether Noggin expression could counteract the effects of Hoxa2 on facial skeletogenesis, by assessing the expression of Sox9, a master gene of chondrogenesis [31], in control, Hoxa2, and Hoxa2 + Noggin transgenic embryos at E5. In control embryos, Sox9 was homogeneously expressed throughout the facial processes, including the nasofrontal region, as well as in the maxillary and mandibular buds (Fig. 7g, j). But its expression was strongly downregulated in Hoxa2-transfected embryos (Fig. 7h, k; arrowheads). Noggin overexpression restored Sox9 expression in the developing face (Fig. 7i, l). These results were further confirmed by a qRT-PCR analysis performed on in vitro experiments. According to the paradigm previously evoked, FNC cells taken from control, Hoxa2-transfected, and Hoxa2 + Noggin cotransfected embryos were grown for 24 h in vitro before harvesting RNA. The quantification of Sox9 gene expression revealed that Hoxa2 reduced its activity (Sox9∆ = −67.53 %; Fig. 7m). In contrast, in Hoxa2 + Noggin-cotransfected FNC cells, Sox9 activity was restored to the level of control series (Sox9∆ = 21.93 %; Fig. 7m).
These observations suggested that Noggin might rescue skeletogenesis in Hoxa2-transfected FNC cells. Skeletogenic phenotypes were analyzed on whole-mount skeletal preparations at E8. Alcian blue staining showed a normal pattern for the facial skeleton in control embryos (Fig. 7n), whereas the facial skeleton was totally missing in Hoxa2-transgenic embryos (Fig. 7o; n = 9/9). The entire set of skeletal elements corresponding to the upper face (the nasal capsule and septum) and lower jaw (Meckel’s cartilage, the quadrate, and entoglossum) was restored by the overexpression of Noggin, which counteracted the effects of Hoxa2 in FNC cells (Fig. 7p; n = 5/5).
In order to see whether the activity of Noggin is downstream of Six genes’ activity, we tried to rescue the lack of skeletal development resulting from the triple silencing of Six1, Six2, and Six4 (Fig. 5k, l) by cotransfecting the three dsRNA with the retroviral construct driving Noggin expression. At E10, we observed the complete restoration of the skeletal nasofrontal and maxillo-mandibular structures (Fig. 7q, r). These results therefore showed that the control of head morphogenesis operated by the combination of Six genes is mediated by Noggin activity.
Discussion
In this work, we explored the molecular mechanisms by which Hoxa2 expression affects the developmental program of the FNC, preventing the development of both facial structures and pre-otic brain. Most of the adverse effects of Hoxa2 expression in the FNC on the development of the facial skeleton and brain were reproduced by the inhibition of Six2 expression in the NC-derived mesenchyme. The Six2 gene, which is a direct target of Hoxa2 in BA2 [13, 32], is strongly expressed in the Hox-free NC cells from the beginning of their migration until E5, the last time point recorded in our observations. When Hoxa2 expression is triggered in the normally Hox-free cephalic neural fold, which gives rise to the facial and cranial skeleton and connective tissue, NC cells lose their ability to express Six2. In parallel, we showed that Six2 silencing resulted in the loss of much of the NC-derived head skeleton and profound abnormalities of brain development, such as defects of the dorsal pre-otic structures, which develop from the alar plate anlage of the anterior neural plate. These defects, evident from E2 onwards, are consistent with the dorsalizing effect of the migrating FNC cells on the development of the cephalic vesicles as already demonstrated [6, 10]. In particular, Hoxa2 activation or Six2 inactivation caused the loss of Fgf8 and Wnt8b expression. The molecular change in Fgf8 expression in the ANR due to the repression of Noggin expression in the migrating FNC cells, as observed in both situations, had a profound impact on the development of the prosencephalon and mesencephalon. Similarly, changes of the pattern of Wnt8b gene expression preceded alterations of choroid plexus development.
The parts of the brain that turn out to mostly depend on the regulatory activity of the FNC are those that have evolved more recently: the telencephalon and the dorsal part of the di- and mesencephalon. We show here that the development of these structures is incompatible with the expression of genes of the Hox clusters, which pattern the rest of the body plan in all Bilateria. Furthermore, the lack of choroid plexus formation after transfection with Hoxa2 or Six2 silencing suggests that the development of these essential structures for brain homeostasis benefits from the input of the FNC via Six gene activity.
Two other Six genes in addition to Six2 were found to be essential for head development. Despite their overlapping patterns of expression in premigratory and migrating NC cells, silencing of Six1, Six2, and Six4 in FNC cells had detrimental effects that were differently distributed on the head structures. As far as brain morphogenesis is concerned, each Six gene when separately switched off through RNAi exerts a complementary role in patterning the dorsal midline of the pre-otic brain: Six2 silencing results in the loss of the choroid plexus at both prosencephalic and rhombencephalic levels, while dsSix4 perturbs the formation of the septum pellucidum, a condition reminiscent of lobar holoprosencephaly, and dsSix1 affects the diverticulation of the prosencephalon, hence leading to a flagrant alobar holoprosencephaly. So, individually, Six1, Six2, and Six4 genes cannot reciprocally compensate their effect on brain development for the loss of the others. The phenotype resulting from the triple silencing gives rise to a severe anencephaly, a worsened condition, which recapitulates all the defaults mentioned above, and phenocopies the activation of Hoxa2. Altogether, these data point to the complementary role of Six gene expression in the FNC for the proper development of the brain.
Regarding the facial skeleton, Six2 silencing selectively hampered nasofrontal skeleton, joint, and mandibular symphysis development, but had no effect on development of the medial part of Meckel’s cartilage. Six1 silencing resulted in an overall homothetic reduction of the head skeleton and Six4 silencing, specifically targeted the nasofrontal and mandibular skeleton, but had no effect on proximal and distal structures. The simultaneous silencing of all three genes completely prevented skeletal development. Our data therefore demonstrate the synergistic roles of the Six1, Six2, and Six4 genes, which cooperate to provide the scaffolding of craniofacial structures (Fig. S4). The ectopic expression of Hoxa2 thus reveals a requirement for Six genes for the spatial layout of skeletogenic NC derivatives.
Recently, the mutation of a chromatin-remodeling gene, Cecr2, has been associated with neural tube defects and facial abnormalities, the severity of which depends on the genetic background. This mutation affects much of the genes early expressed by the FNC cells, among which is Six1 [33]. These data strongly suggest that the activity of Six gene in FNC cells can be epigenetically regulated; such a mechanism could account for the congenital malformations altering face and brain development, and deserves to be further explored.
Six genes have been conserved throughout evolution and encode transcription factors that can function either as transcription activators or repressors. Six homologs (Sine oculis, Optrix, and DSix4) in embryos were initially shown to play a role in eye development, through mutations in fly [34]. The ontogenetic importance of this gene family was subsequently documented for thymic and kidney morphogenesis and for muscle development in mammals [18]. These genes were shown to be essential for the ontogeny of all sense organs [35, 36]. In Brachyrrine (Br) mutants, generated by irradiation, the transcriptional repression of Six2 is associated with frontonasal dysplasia and renal hypoplasia. In homozygous Br/Br mutants, despite the abolition of Six2 expression throughout the body, only the facial skeleton was affected [37]. Thus, the mechanisms regulating bone and cartilage development is different in the head and trunk.
In our experiments, the forced expression of Six2 in the FNC cells of embryos transfected with Hoxa2 competitively overcame the adverse effects of Hoxa2 expression, restoring the competence of FNC cells for cephalic development. These observations are consistent with the results of genetic manipulations of BA2 development: Hoxa2 knockout has been shown to expand the skeletogenic program in BA2 [38–41], leading to the development of BA1-like structures from BA2 territories in which Six2 expression was relieved of Hoxa2 activity. Furthermore, Six2 overexpression confers on BA2 cells an ability to form BA1-like structures in a spatially restricted manner. Similarly, Six2 overexpression in BA2 results in a phenocopy of Hoxa2 inactivation. This indicates that, in normal BA2 development, Hoxa2 switches off Six2 expression in rhombencephalic NC cells [13, 32]. Concomitantly, Hoxa2 represses the activation of Pitx1 transcription by Fgf8 from the ectoderm, preventing the initiation of a “pro-mandible” program and restricting the domain of activation of Sox9 and Cbfa1, two transcription factors essential for chondrogenic and membranous bone differentiation, respectively [42, 43]. Thus, Six gene expression in the developing head is essential, to drive the formation of craniofacial structures derived from the Hox-free domain of the NC, and to counteract the effect of the Hox genes involved in the patterning of pharyngeal structures.
Our experiments show that the treatment of FNC cells, by either Hoxa2 activation or silencing of its downstream Six gene targets, results in an upregulation of Bmp signaling mediated by the loss of Bmp antagonists. In both cases, the upregulation of Bmp signaling is achieved through a decrease in Noggin and Dan production by FNC cells. Bmps decrease the amount of Fgf8 produced by the secondary brain organizers (ANR and isthmus) and BA1 ectoderm. The BA1 ectoderm is an important signaling center for the development of the jaw [5, 43, 44], whereas the ANR and isthmus are critical for brain development [6, 7, 11]. Thus, Hoxa2 expression affects the development not only of the head skeleton but also of the telencephalon, thalamus, and optic tectum. The major role of anti-Bmp signaling by NC cells is further demonstrated by the abolition of the adverse effect of Hoxa2 expression on Fgf8 production by the cotransfection of FNC cells with Noggin. Under this condition, forcing the expression of Noggin bypasses the inactivation of Six-gene pathway to rescue Fgf8 activity (Fig. S5). Such a restoration of normal development results from intense proliferation in the dorsal neuroepithelium and the limitation of cell death at the anterior midline, leading eventually to the normalization of cephalic vesicle development.
Noggin overexpression in Hoxa2-transfected embryos also causes a striking restoration of head skeleton development. In these experiments, the total absence of skeleton formation due to Hoxa2 expression, mediated by the potent repression of Sox9 expression, was reversed (Fig. S5). In this case, the entire set of skeletal structures was formed, owing to the restoration of Sox9 activity.
In conclusion, the production by Hox-negative FNC cells of Bmp antagonists, under the control of Six genes, appears to be a key mechanism regulating head morphogenesis in vertebrates.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dr. Françoise Dieterlen for critical reading of the manuscript, Drs. Andréa Trentin and Jörg Kobarg for the technical help with qRT-PCR, Drs. Brickell, Monsoro-Burq, Dressler, Oliver, and Niswander for providing plasmids. This work was supported by the Centre National de la Recherche Scientifique, and grants from the Agence Nationale de la Recherche (ANR-BLAN–0153) and the Ecole des Neurosciences de Paris (Réseau Technologique de Recherche Avancée:-Stem Cells). R.C.G. held fellowships from Fondation des Treilles, Egide, Conselho Nacional de Desenvolvimento Científico e Tecnológico/Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CNPq/CAPES––Brazil).
References
- 1.Gans C, Northcutt RG. Neural crest and the origin of vertebrates: a new head. Science. 1983;20:268–274. doi: 10.1126/science.220.4594.268. [DOI] [PubMed] [Google Scholar]
- 2.Hall BK. Germ layers and the germ-layer theory revisited: primary and secondary germ layers, neural crest as a fourth germ layer, homology, and demise of the germ-layer theory. Evol Biol. 1998;30:121–186. doi: 10.1007/978-1-4899-1751-5_5. [DOI] [Google Scholar]
- 3.Le Douarin NM. The neural crest. Cambridge: Cambridge University Press; 1982. [Google Scholar]
- 4.Le Douarin NM, Kalcheim C. The neural crest. 2. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 5.Creuzet S, Schuler B, Couly G, Le Douarin NM. Reciprocal relationships between Fgf8 and neural crest cells in development. Proc Natl Acad Sci USA. 2004;101:4843–4847. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creuzet SE, Martinez S, Le Douarin NM. The cephalic neural crest exerts a critical effect on forebrain and midbrain development. Proc Natl Acad Sci USA. 2006;103:1433–1438. doi: 10.1073/pnas.0605899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- 8.Martinez S, Crossley PH, Cobos I, Rubenstein JLR, Martin GR. FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development. 1999;126:1189–1200. doi: 10.1242/dev.126.6.1189. [DOI] [PubMed] [Google Scholar]
- 9.Echevarría D, Vieira C, Gimeno L, Martínez S. Neuroepithelial secondary organizers and cell fate specification in the developing brain. Brain Res Rev. 2003;43:179–191. doi: 10.1016/j.brainresrev.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Creuzet SE. Regulation of pre-otic brain development by the cephalic neural crest. Proc Natl Acad Sci USA. 2009;106:15774–15779. doi: 10.1073/pnas.0906072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohkubo Y, Chiang C, Rubenstein JLR. Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience. 2002;111:1–17. doi: 10.1016/S0306-4522(01)00616-9. [DOI] [PubMed] [Google Scholar]
- 12.Creuzet S, Couly G, Bennaceur S, Vincent C, Le Douarin NM. Negative effect of hox gene expression on the development of the neural crest-derived facial skeleton. Development. 2002;129:4301–4313. doi: 10.1242/dev.129.18.4301. [DOI] [PubMed] [Google Scholar]
- 13.Kutejova E, Engist B, Mallo M, Kanzler B, Bobola N. Hoxa2 downregulates Six2 in the neural crest-derived mesenchyme. Development. 2005;132:469–478. doi: 10.1242/dev.01536. [DOI] [PubMed] [Google Scholar]
- 14.Le Douarin N, Dieterlen-Lièvre F, Creuzet S, Teillet M-A. Quail––chick transplantations. Methods Cell Biol. 2008;87:19–58. doi: 10.1016/S0091-679X(08)00202-1. [DOI] [PubMed] [Google Scholar]
- 15.Grapin-Botton A, Bonnin M-A, McNaughton LA, Krumlauf R, Le Douarin NM. Plasticity of transposed rhombomeres: Hox gene induction is correlated with phenotypic modifications. Development. 1995;121:2707–2721. doi: 10.1242/dev.121.9.2707. [DOI] [PubMed] [Google Scholar]
- 16.Pekarik V, Bourikas D, Miglino N, Joset P, Stoekli E. Screening for gene function in chicken embryo using RNAi and electroporation. Nat Biotech. 2003;21:93–96. doi: 10.1038/nbt770. [DOI] [PubMed] [Google Scholar]
- 17.Stoeckli ET (2003) RNAi in avian embryos. In RNAi: a guide to gene silencing, 297–312. Cold Spring Harbor Laboratory, New York. ISBN 978-087969704-4
- 18.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrovová E, Sedmera D, Míšek I, Lešník F, Luptáková L. Bendiocarbamate toxicity in the chick embryo. Folia Biol (Praha) 2009;55:61–65. [PubMed] [Google Scholar]
- 20.Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Expression of a delta homologue in prospective neurons in the chick. Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- 21.Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996;380:66–68. doi: 10.1038/380066a0. [DOI] [PubMed] [Google Scholar]
- 22.Riddle RD, Johnson RL, Laufer E, Tabin CJ. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;25:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 23.Francis PH, Richardson MK, Brickell PM, Tickle C. Bone morphogenetic proteins and a signaling pathway that controls patterning in the developing chick limb. Development. 1994;120:209–218. doi: 10.1242/dev.120.1.209. [DOI] [PubMed] [Google Scholar]
- 24.Connolly DJ, Patel K, Cooke J. Chick noggin is expressed in the organizer and neural plate during axial development, but offers no evidence of involvement in primary axis formation. Int J Dev Biol. 1997;41:389–396. [PubMed] [Google Scholar]
- 25.Garda AL, Puelles L, Rubenstein JLR. Expression patterns of Wnt8b and Wnt7b in the chicken embryonic brain suggest a correlation with forebrain patterning centers and morphogenesis. Neuroscience. 2002;113:689–698. doi: 10.1016/S0306-4522(02)00171-9. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lako M, Lindsay S, Bullen P, Wilson DI, Robson SC, Strachan T. A novel mammalian Wnt gene, WNT8B, shows brain-restricted expression in early development, with sharply delimited expression boundaries in the developing forebrain. Hum Mol Gen. 1998;7:813–822. doi: 10.1093/hmg/7.5.813. [DOI] [PubMed] [Google Scholar]
- 28.Tzahor E, Kempf H, Mootoosamy RC, Poon AC, Abzhanov A, Tabin CJ, Dietrich S, Lassar AB. Antagonists of Wnt and BMP signaling promote the formation of vertebrate head muscle. Gen Dev. 2003;17:3087–3099. doi: 10.1101/gad.1154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu DR, Economides AN, Wang X, Eimon PM, Harland RM. The Xenopus dorsalizing factor gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol Cell. 1998;1:673–683. doi: 10.1016/S1097-2765(00)80067-2. [DOI] [PubMed] [Google Scholar]
- 30.Eimon PM, Harland RM. Xenopus dan, a member of the dan gene family of BMP antagonists, is expressed in derivatives of the cranial and trunk neural crest. Mech Dev. 2001;107:187–189. doi: 10.1016/S0925-4773(01)00462-2. [DOI] [PubMed] [Google Scholar]
- 31.Wright E, Hargrave MR, Christiansen J, Cooper L, Kun J, Evans T, Gangadharan U, Greenfield A, Koopman P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat Genet. 1995;9:15–20. doi: 10.1038/ng0195-15. [DOI] [PubMed] [Google Scholar]
- 32.Kutejova E, Engist B, Self M, Oliver G, Kirikenko P, Bobola N. Six2 functions redundantly immediately downstream of Hoxa2 . Development. 2008;135:1463–1470. doi: 10.1242/dev.017624. [DOI] [PubMed] [Google Scholar]
- 33.Fairbridge NA, Nicholas A, Dawe CE, Niri FH, Kooistra MK, King-Jones K, McDermid HE. Cecr2 mutations causing exencephaly trigger misregulation of mesenchymal/ectodermal transcription factors. Birth Def Res A. 2010;88:619–625. doi: 10.1002/bdra.20695. [DOI] [PubMed] [Google Scholar]
- 34.Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. 2009;66:565–583. doi: 10.1007/s00018-008-8335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christophorou NA, Bailey AP, Hanson S, Streit A. Activation of Six1 target genes is required for sensory placode formation. Dev Biol. 2009;336:327–336. doi: 10.1016/j.ydbio.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki Y, Keiko I, Kawakami K. Development of gustatory papilla in the absence of Six1 and Six4 . J Anat. 2011;219:710–721. doi: 10.1111/j.1469-7580.2011.01435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fogelgren B, Kuroyama MC, McBratney-Owen B, Spence AA, Malahn LE, Anawati M, Cabatbat C, Alarcon VB, Marikawa Y, Lozanoff S. Misexpression of Six2 is associated with heritable frontonasal dysplasia and renal hypoplasia in 3H1 Br mice. Dev Dyn. 2008;237:1767–1779. doi: 10.1002/dvdy.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gendron-Maguire M, Mallo M, Zhang M, Gridley T. Hoxa2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell. 1993;75:1317–1331. doi: 10.1016/0092-8674(93)90619-2. [DOI] [PubMed] [Google Scholar]
- 39.Rijli F, Mark M, Lakkaraju S, Dierich A, Dolle P, Chambon P. A homeotic transformation is generated in the rostral branchial region of the head by disruption of Hoxa2, which acts as a selector gene. Cell. 1993;75:1333–1349. doi: 10.1016/0092-8674(93)90620-6. [DOI] [PubMed] [Google Scholar]
- 40.Kanzler B, Kuschert SJ, Liu YH, Mallo M. Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development. 1998;125:2587–2597. doi: 10.1242/dev.125.14.2587. [DOI] [PubMed] [Google Scholar]
- 41.Barrow JR, Capecchi MR. Compensatory defects associated with mutations in Hoxa1 restore normal palatogenesis to Hoxa2 mutants. Development. 1999;126:5011–5026. doi: 10.1242/dev.126.22.5011. [DOI] [PubMed] [Google Scholar]
- 42.Bobola N, Carapuco M, Ohnemus S, Kanzler B, Leibbrandt A, Neubuser A, Drouin J, Mallo M. Mesenchymal patterning by Hoxa2 requires blocking Fgf-dependent activation of Pitx1. Development. 2003;130:3403–3414. doi: 10.1242/dev.00554. [DOI] [PubMed] [Google Scholar]
- 43.Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Gen Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.