

Abstract
Nesfatin-1 is an anorexic nucleobindin-2 (NUCB2)-derived hypothalamic peptide. It controls feeding behavior, water intake, and glucose homeostasis. If intracerebrally administered, it induces hypertension, thus suggesting a role in central cardiovascular control. However, it is not known whether it is able to directly control heart performance. We aimed to verify the hypothesis that, as in the case of other hypothalamic satiety peptides, Nesfatin-1 acts as a peripheral cardiac modulator. By western blotting and QT-PCR, we identified the presence of both Nesfatin-1 protein and NUCB2 mRNA in rat cardiac extracts. On isolated and Langendorff-perfused rat heart preparations, we found that exogenous Nesfatin-1 depresses contractility and relaxation without affecting coronary motility. These effects did not involve Nitric oxide, but recruited the particulate guanylate cyclase (pGC) known as natriuretic peptide receptor A (NPR-A), protein kinase G (PKG) and extracellular signal-regulated kinases1/2 (ERK1/2). Co-immunoprecipitation and bioinformatic analyses supported an interaction between Nesfatin-1 and NPR-A. Lastly, we preliminarily observed, through post-conditioning experiments, that Nesfatin-1 protects against ischemia/reperfusion (I/R) injury by reducing infarct size, lactate dehydrogenase release, and postischemic contracture. This protection involves multiple prosurvival kinases such as PKCε, ERK1/2, signal transducer and activator of transcription 3, and mitochondrial KATP channels. It also ameliorates contractility recovery. Our data indicate that: (1) the heart expresses Nesfatin-1, (2) Nesfatin-1 directly affects myocardial performance, possibly involving pGC-linked NPR-A, the pGC/PKG pathway, and ERK1/2, (3) the peptide protects the heart against I/R injury. Results pave the way to include Nesfatin-1 in the neuroendocrine modulators of the cardiac function, also encouraging the clarification of its clinical potential in the presence of nutrition-dependent physio-pathologic cardiovascular diseases.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1138-7) contains supplementary material, which is available to authorized users.
Keywords: Nesfatin-1, Natriuretic peptide receptor A, Cardioprotection, pGC/PKG, Signal transduction
Introduction
The cardiovascular system and the gastrointestinal (GI) apparatus are targets of both humoral and nervous networks which control their activity in the short, medium, and long term, acting directly on heart, vessels, and GI districts, and indirectly by controlling appetite, fluid intake, and plasma osmolarity. Under physio-pathological conditions these circuits are impaired and this compromises both cardiovascular and GI performance [1]. Crossroads for many circuits is the hypothalamus which produces several neuropeptides involved in cardiovascular, GI, and alimentary homeostasis. Some of these neuromediators (i.e., neuropeptide Y, NPY) act peripherally on the GI system by directly controlling mucosal function, motility, and secretion; they also influence the cardiovascular system by eliciting direct cardiac and/or vascular effects [2, 3].
Recently, growing attention has been directed to the novel hypothalamic anorexigenic peptide, Nesfatin-1, an 82-amino-terminal fragment derived from the larger protein nucleobindin-2 (NUCB2) [4]. In the rat, the Nesfatin-1 gene and protein are expressed in the hypothalamic nuclei involved in feeding behavior, food intake, body weight, and glucose homeostasis [5]. In arcuate (ARC), paraventricular (PVN), and supraoptic nuclei (SON), Nesfatin-1 colocalizes with pro-opiomelanocortin/cocaine and amphetamine responsive transcript (POMC/CART), NPY, oxytocin, and vasopressin, suggesting the presence of multiple peptidergic correlations. NUCB2 gene expression is significantly regulated by nutritional status, suggesting a role of Nesfatin-1 in energy homeostasis [6]. In fact, Nesfatin-1 elicits anti-hyperglycemic effects, as shown by the reduced blood glucose observed after intravenous injection of the peptide in hyperglycemic db/db mice [7]. Notably, Nesfatin-1 is able to cross the blood–brain barrier in both the blood-to-brain and brain-to-blood directions [8], and this indicates that peripheral sources of the peptide may affect brain activity and vice versa. Additionally, and/or alternatively, central Nesfatin-1 can regulate peripheral functions. This is supported by the inhibition of gastric emptying observed after central administration of the peptide [6].
Nesfatin-1 effects are attributed to an unknown metabotropic G-protein-coupled receptor, although no conclusive evidence is available [9]. The peptide was found to increase intracellular calcium via a PTX-sensitive Gi/o protein [9]. At the same time, it seems to influence the ATP-dependent potassium channels Kir6.2, to elicit hyperpolarization in neuropeptide Y/Agouti-related peptide (NPY/AgRP) neurons [10]. Lastly, Nesfatin-1 interacts with melanocortin signaling [11].
Recently, in addition to its role in nutrition and energy balance, Nesfatin-1 was proposed to contribute to vascular control. In fact, central Nesfatin-1 was shown to activate the nervous circuits which are responsible for hypertension [12], an effect which is presumed to also occur via hypothalamic melanocortin-3/4 receptors [13]. However, although these findings suggest Nesfatin-1 to be involved in central cardiovascular homeostasis, it is currently unknown whether the peptide is able to directly control heart performance. To fill this gap, we investigated on the rat heart: (1) the cardiac expression of Nesfatin-1, (2) the effects of the direct exposure of isolated and Langendorff-perfused cardiac preparations to exogenous Nesfatin-1 and the mechanism of action which sustains its effects, and (3) whether the peptide is able to elicit cardioprotection against ischemia/reperfusion (I/R) injuries.
Materials and methods
Animals
Adult Wistar rats (250–300 g body weight; Harlan, Italy), fed a standard diet and water ad libitum, were used. All studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (Publication No. 85-23, revised 1996).
Perfusion technique
Rats were anesthetized by ethyl carbamate (2 g/kg body weight, i.p.). Hearts were rapidly excised, placed in ice-cold perfusion buffer, cannulated via the aorta, and perfused in the Langendorff mode at a constant flow rate of 12 ml/min and temperature of 37° C. The perfusion solution was a modified Krebs–Henseleit solution (KHs) gassed with 95 % O2 and 5 % CO2 (pH 7.4) containing (in mmol/L): NaCl 113.0; KCl 4.7; MgSO4 1.2; NaHCO3 25.0; KH2PO4 1.2; CaCl2 1.8; glucose 11; mannitol 1.1; and Na-pyruvate 5 [14].
Left ventricular pressure (LVP) was measured by means of a latex water-filled balloon inserted into the left ventricle via the left atrium [adjusted to obtain left ventricular end-diastolic pressure (LVEDP) of 5–7 mmHg] and connected to a pressure transducer (BLPR gauge; WRI, USA). The maximal values of the first derivative of LVP, [+(LVdP/dt)max, mmHg/s], which indicates the maximal rate of left ventricular contraction, the time to peak tension of isometric twitch (TTP, s), the maximal rate of left ventricular pressure decline of LVP [−(LVdP/dt)max, mmHg/s)], the half-time relaxation (HTR, s), which is the time required for intraventricular pressure to fall from the peak to 50 %, Tau (τ, s) which is the relaxation time constant, the T/−t ratio obtained by +(LVdP/dt)max/−(LVdP/dt)max, as indexes of contraction and relaxation, and LVEDP were used to assess cardiac function. Mean coronary pressure (CP, mmHg) was calculated as the average of values obtained during several cardiac cycles.
Western blotting analysis
Cardiac ventricles perfused with Nesfatin-1 (100 pmol/L) were homogenized in ice-cold RIPA buffer (Sigma-Aldrich, Milan, Italy) containing a mixture of protease inhibitors (1 mmol/L aprotinin, 20 mmol/L phenylmethylsulfonyl fluoride, and 200 mmol/L sodium orthovanadate). Cardiac tissue homogenates were then centrifuged at 200g for 10 min at 4 °C to remove tissue debris.
Amounts of 10–50 μg protein of cardiac tissue were electrophoresed through a reducing SDS/10 % (w/v) or SDS/15 % (w/v) (for detection of Nesfatin-1 protein) polyacrylamide gel and electroblotted on to a nitrocellulose membrane. After the transfer, the membranes were stained with Red Poinseau to confirm the equal loading and transfer. The membrane was blocked and incubated with the polyclonal IgG for pERK1/2, pAKT, signal transducer and activator of transcription 3 (STAT3), endothelial nitric oxide synthase (eNOS), neuronal NOS (nNOS), β-actin (Santa Cruz, DBA, Milan, Italy) and Nesfatin-1 (Phoenix Pharmaceuticals, Germany); β-actin was used as loading control. The levels of proteins and phosphoproteins were detected with horseradish peroxidase-linked secondary antibodies and the ECL® (enhanced chemiluminescence) System (GE Healthcare, Milan, Italy). Autoradiographs were scanned to obtain arbitrary densitometric units. Data were normalized against those of the corresponding β-actin. The experiments were performed in triplicate and the results calculated as mean ± SD, and expressed as protein change (%).
The same protocols were applied to verify Akt phosphorylation on skeletal muscle, liver, and heart homogenates of food-deprived rat after injection of a single dose (0.25 nmol/g bw) of Nesfatin-1 (30 min before sacrifice) [15].
Q-RT-PCR
mRNA expression level of selected genes was quantified by real-time PCR using Step One sequence detection system (Applied Biosystems, Milan, Italy). TaqMan assay (Applied Biosystems) (Rn00573037_m1, Rn00597158_m1 and Rn99999916_s1) was used to quantify mRNA NUCB2. The relative mRNA amounts were calculated by comparative cycle threshold [16] and normalized by GAPDH expression.
S-nitrosylation (SNO)
The SNO of proteins was obtained through Biotin switch assay. This protocol was performed as previously described [17]. Extracts were adjusted to 0.5 mg/mL of protein and equal amounts were blocked with 4 volumes of blocking buffer (225 mmol/L Hepes, pH 7.7, 0.9 mmol/L EDTA, 0.09 mmol/L Neocuproine, 2.5 % SDS, and 20 mmol/L MMTS) at 50 °C for 20 min with agitation. After blocking, extracts were precipitated with 2 volumes of cold (−20 °C) acetone, chilled at −20 °C for 10 min, centrifuged at 2,000g, 4 °C for 5 min, washed with acetone, dried out at room temperature, and resuspended in 0.1 mL HENS buffer (250 mmol/L Hepes, pH 7.7, 1 mmol/L EDTA, 0.1 mmol/L Neocuproine, and 1 % SDS) per mg of protein. Until this step, all operations were carried out in the dark. A 1/3 volume of biotin-HPDP 4 mmol/L in DMF and ascorbate 1 mmol/L were added and incubated for 1 h at room temperature. Proteins were acetone precipitated again and resuspended in the same volume of HENS buffer. To detect biotinylated proteins by western blot, samples from the biotin switch assay were separated on 15 % SDS–PAGE gels, transferred to PVDF membranes, blocked with non-fat dried milk, and incubated with streptavidin-peroxidase diluted 1/5,000 for 1 h. Blots were developed by enhanced chemioluminescence (ECL) and were placed in a film cassette with photograph film. Films were exposed for 30 s, developed, and fixed.
Experimental protocols
Isolated Langendorff preparation
Nesfatin-1 stimulated preparations
Preliminary experiments (data not shown) obtained by repetitive exposure of each heart to one concentration of Nesfatin-1 (100 pmol/L) revealed absence of desensitization. Thus, concentration–response curves were obtained by perfusing the cardiac preparation with KHs enriched with increasing concentrations of Nesfatin-1 (0.1 pmol/L–10 nmol/L) for 10 min.
Involvement of membrane receptors
To evaluate whether Gi/o proteins are involved in the cardiac action of Nesfatin-1, hearts were pre-incubated for 60 min with KHs enriched with pertussis toxin (PTx) (10 pmol/L) and then exposed for 10 min to Nesfatin-1 (100 pmol/L). As shown in the rat heart [18], PTx catalyses the ADP ribosylation of the a-subunit of Gi/o proteins and uncouples the interaction between Gi and inhibitory receptors of adenylate cyclase.
Based on the results obtained from dose–response curves, antagonists were used at a concentration which did not affect cardiac performance.
To analyze whether the NPR-A is involved in Nesfatin-1 mediated effects, hearts were perfused with 100 pmol/L Nesfatin-1 for 10 min and washed out with KHs. Subsequently, cardiac preparations were perfused with Anantin (100 nmol/L), a NPR-A antagonist, for 10 min, followed by perfusion with KHs containing a single concentration of Nesfatin-1 (100 pmol/L) plus Anantin (100 nmol/L) for another 10 min.
Inhibitor-stimulated preparations
To verify the pathways involved in the Nesfatin-1 mechanism, hearts, stabilized for 20 min with KHs, were perfused with 100 pmol/L of Nesfatin-1 for 10 min and then washed out with KHs. After returning to control conditions, each cardiac preparation was perfused with a specific inhibitor for 10 min; then it was perfused with KHs containing a single concentration of Nesfatin-1 (100 pmol/L) plus the inhibitor for an additional 10 min. In particular, the involvement of ERK, NOSs, protein Kinase G (PKG), and phosphodiesterase 2 (PDE2) was evaluated by perfusing the hearts with PD (100 nmol/L), L-NAME (10 μmol/L), KT5823 (100 nmol/L), and EHNA (100 nmol/L), respectively.
Ischemia/reperfusion protocols
Each heart was allowed to stabilize for 40 min; at this time, baseline parameters were recorded. After stabilization, hearts were randomly assigned to one of the treatment groups described below and then subjected to 30 min of global, no-flow ischemia followed by 120 min of reperfusion (I/R). Pacing was discontinued at the beginning of the ischaemic period and restarted after the third minute of reperfusion [19].
Cardiac function on and infarct size studies
Group 1 (I/R group, n = 6) hearts were stabilized and subjected to I/R protocol only.
Group 2 (PostC group, n = 6) hearts underwent to a post-conditioning protocol (PostC) (i.e., 5 cycles of 10-s reperfusion and ischemia at beginning of reperfusion).
Group 3 (Nesfatin-1 group, n = 6) Nesfatin-1 (100 pmol/L) was infused for 20 min at the beginning of 120-min reperfusion.
Group 4 (group, n = 6) hearts were perfused with Nesfatin-1 (100 pmol/L) plus one of the following inhibitors: epsilon-V1-2 (1 μmol/L; Nesfatin-1 + epsilon-V1-2) or 5HD (10 μmol/L; Nesfatin-1 + 5HD); perfusion with inhibitors started 5 min before ischaemia and continued during the early 7 min of reperfusion in the presence of Nesfatin-1 (100 pmol/L).
To study, whether Nesfatin-1 induces phosphorylation of Extracellular signal-regulated kinases1/2 (ERK1/2) and STAT3, after 40-min stabilization hearts underwent 30-min global ischemia followed by 7-min reperfusion (n = 4 for each group). Therefore, one group received I/R only (7-min reperfusion), one group received Nesfatin-1 in the early reperfusion (for 7 min) and one group was perfused with physiological solution only (Sham) for 77 min.
The concentration of Nesfatin-1 was chosen on the basis of a preliminary dose–response curve as the dose that induced the highest infarct size reduction (data not shown).
Cardiac performance before and after ischaemia was evaluated by analyzing LVP recovery as an index of contractile activity, and LVEDP for contracture. Contracture is defined as an increase in LVEDP of 4 mmHg above the baseline level [19].
Assessment of myocardial injury
To obtain infarct areas, hearts were rapidly removed from the perfusion apparatus at the end of reperfusion, and the left ventricle was dissected into 2- to 3-mm circumferential slices. After 20 min of incubation at 37 °C in 0.1 % solution of nitro blue tetrazolium in phosphate buffer, unstained necrotic tissue was carefully separated from stained viable tissue by an independent observer who was not aware of the nature of the intervention. The weights of the necrotic and non-necrotic tissues were then determined, and the necrotic mass was expressed as a percentage of total left ventricular mass, including septum [19, 20].
Lactate dehydrogenase
Since, in isolated rat hearts, ischemic postconditioning is known to reduce the production of lactate dehydrogenase (LDH) during reperfusion [21], the release of this enzyme during each experimental group was tested. Samples of coronary effluent were withdrawn with a catheter inserted into the right ventricle via the pulmonary artery. Samples were taken during reperfusion. LDH release was measured as described by Penna and collaborators [21]. Data are expressed as cumulative values for the entire reperfusion period.
Cyclic guanosine monophosphate (cGMP) and Nesfatin-1 measurements
For cGMP determination, frozen tissues (200–300 mg) were treated with 6 % trichloroacetic acid at 0 °C and centrifuged at 1,000g for 10 min. Supernatants were extracted three times with 3 mL diethyl ether saturated with water, and the aqueous phases were collected and stored at −80 °C [cGMP Biotrack Enzyme Immunoassay (EIA) System; GE Healthcare].
The same protocol was used for plasma and tissue Nesfatin-1 concentrations, which were measured by using commercial enzyme immunoassay kits (Nesfatin-1: ELISA Kit Protocol; Phoenix Pharmaceuticals).
Co-immunoprecipitation
To identify a physiological interaction between Nesfatin-1 and the NPR-A, a co-immunoprecipitation technique was used. Hearts were homogenized in lysis buffer as described before. The homogenates were incubated with Nesfatin-1 (100 pmol/L) for 10 min. Equal amounts of proteins were incubated overnight at 4 °C with anti-Nesfatin-1 or with anti-NPR-A (Santa Cruz) primary antibodies. Proteins were then immunoprecipitated by Protein A/G PLUS-Agarose (immunoprecipitation reagent; Santa Cruz). The immunoprecipitated proteins were washed three times with lysis buffer and subjected to a common western blotting. Membranes were probed with anti-Nesfatin-1 and anti-NPR-A primary antibodies and subsequently with peroxidase-coupled anti-IgG secondary antibodies, revealed using the ECL system. Nesfatin-1 protein was used as positive control.
Local sequence alignment
The local alignment was performed with the LALIGN software (http://www.ch.embnet.org/software/LALIGN_form.html) by aligning the loop of BNP interacting with NPR-C (aa residues 5-16; [22]) and Nesfatin-1.
Drugs and chemicals
Nesfatin-1 was purchased from Phoenix Pharmaceuticals. KT5823 was purchased from Calbiochem (VWR International, Milan, Italy). Pertussis toxin (PTx), Anantin, PD98059 (PD), N-Nitro-l-arginine methyl ester hydrochloride (L-NAME), erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA), 5-Hydroxydecanoate (5-HD), epsilon-V1-2 were purchased from Sigma-Aldrich. Nesfatin-1 and L-NAME were dissolved in water. All compounds were solubilized in DMSO, except PD which was dissolved in ethanol. Preliminary experiments showed that the presence of equivalent amounts of ethanol or DMSO in KHs solution in the absence of any drug did not modify the basal cardiac performance.
Statistics
Data are the mean ± SEM. Since each heart represents its own control, the statistical significance of differences within-group was assessed using the ANOVA test (p < 0.05). Comparison between groups was made by using a one-way analysis of variance (ANOVA) followed by the Bonferroni correction for post hoc t tests. Differences were considered to be statistically significant at p < 0.05.
Results
Cardiac Nesfatin-1 identification
To evaluate whether the heart expresses Nesfatin-1, rat ventricular extracts were exposed to Nesfatin-1 antibody. Western blotting analyses revealed that Nesfatin-1 is expressed in the ventricle (Fig. 1a, lane 1), as well as in brain and gastric extracts used as positive controls [4, 6] (Fig. 1a, lanes 2, 3). Nesfatin-1 ventricular expression was confirmed by ELISA assay (Fig. 1c) and by the identification of the mRNA of its precursor NUCB2 by TaqMan Q-RT PCR (Fig. 1b). Nesfatin-1 concentrations were evaluated by ELISA in plasma, heart, and lung extracts. Concentrations were: 12.73 ± 1 nmol/L in plasma, 3.41 ± 1.5 nmol/L in heart, and 0.67 ± 1.2 nmol/L in lung.
Fig. 1.

a Immunoblots of brain (lane 1), heart (lane 2), lung (lane 3), and stomach (lane 4) rat homogenates showing expression of Nesfatin-1 and β-actin as housekeeping protein, b real-time PCR of Nucleobindin 2 mRNA expression in heart, lung, and brain, c Nesfatin-1 ELISA assay of heart, plasma and lung. Changes were evaluated as mean ± SE of six experiments for each group. Significant difference from control values (one-way ANOVA), *p < 0.05
Nesfatin-1 effects on cardiac parameters
We aimed to evaluate whether exogenous Nesfatin-1 directly affects basal cardiac performance. By exposing the Langendorff-perfused heart to increasing concentrations (0.1 pmol/L–10 nmol/L) of Nesfatin-1, we found that the peptide induced negative inotropic and lusitropic effects, revealed by the dose-dependent decrease of all cardiac parameters, which was significant starting from 1 pmol/L (Fig. 2) [basal values: LVP 88 ± 2.8 mmHg, +(LVdP/dt)max 2,489 ± 124 mmHg/s, CP 63 ± 3 mmHg, −(LVdP/dt)max = −1,664 ± 70 mmHg/s; T/−t = −1.48 ± 1.85 mmHg/s, τ = 0.04 ± 0.01/s, HTR = 0.05 ± 0.01/s]. The peptide did not affect CP (Fig. 2). Experiments performed on electrically paced hearts revealed similar inotropic and lusitropic effects, thus confirming that changes of HR did not influence the cardiodepression elicited by Nesfatin-1 (data not shown).
Fig. 2.

Concentration–response curves of increasing concentrations (0.1 pmol/L–10 nmol/L) of Nesfatin-1 on LVP, +(LVdP/dt)max, −(LVdP/dt)max, T/−t, τ, HTR, and CP on the isolated and Langendorff-perfused rat heart. For abbreviations and basal values, see “Results”. Percentage changes were evaluated as mean ± SE of six experiments for each group. Significant difference from control values (one-way ANOVA), *p < 0.05
Mechanisms of action of Nesfatin-1
The intracellular transduction pathways involved in the Nesfatin-1-dependent cardiac effects were assessed by using specific inhibitors that, used alone, did not significantly influence the cardiac performance. It has been reported that, in PVN, Nesfatin-1 interacts with a G-protein-coupled receptor [10]. To verify the involvement of Gi/o proteins in the mechanism of action activated by Nesfatin-1, cardiac preparations were perfused with KHs containing PTx (10 pmol/L) in the presence of the peptide. We found that the toxin did not affect the Nesfatin-1-dependent negative inotropic and lusitropic influence (Fig. 3a), ruling out the involvement of Gi/o-protein-coupled receptors.
Fig. 3.
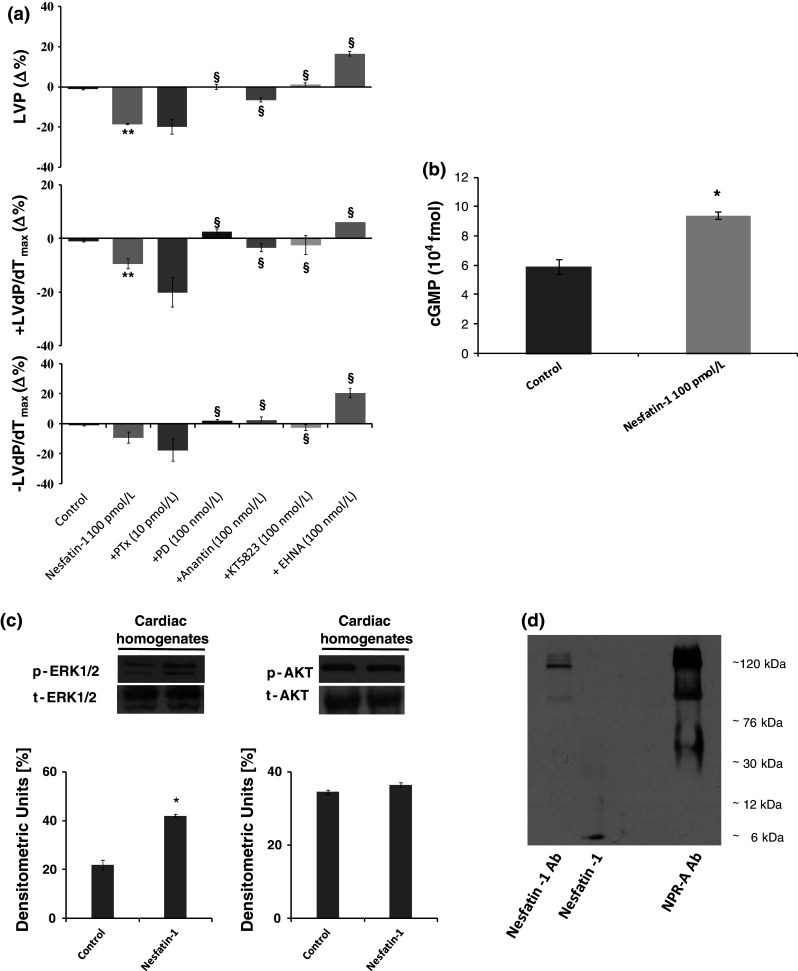
a Effects of Nesfatin-1 before and after treatment with PTx (10 pmol/L), Anantin (100 nmol/L), PD (100 nmol/L), KT5823 (100 nmol/L), and EHNA (100 nmol/L) on LVP, +(LVdP/dt)max, −(LVdP/dt)max, in the isolated and Langendorff-perfused rat heart. b cGMP concentrations in control and Nesfatin-1-treated heart extracts. c Immunoblots of pERK1/2, ERK total, pAKT, and AKT total in control and Nesfatin-1-treated hearts (cardiac homogenates). d Nesfatin-1 co-immunoprecipitation with NPR-A. Percentage changes were evaluated as mean ± SE of six experiments for each group. Significant difference (one-way ANOVA) from control values of Nesfatin-1 versus Krebs–Henseleit solution (KHs), *p < 0.05. Comparison between groups treated with Nesfatin-1 alone versus antagonist, § p < 0.05
It is well known that the mammalian heart expresses particulate guanylyl cyclase (pGC)-linked receptors, namely natriuretic peptide receptor type A (NPR-A) and natriuretic peptide receptor type B (NPR-B), whose activation by natriuretic peptides elicit a negative cardiac modulation [23]. To verify whether pGC-linked receptors contribute to the negative inotropic and lusitropic effects elicited by Nesfatin-1, NPR-A was inhibited by using the selective antagonist, Anantin (100 nmol/L). We found that Nesfatin-1-dependent cardiodepression was abolished by Anantin (Fig. 3a), thus suggesting NPR-A to be involved. This possibility was supported by the increased cGMP levels observed in cardiac extracts after exposure to Nesfatin-1 (100 pmol/L) (Fig. 3b).
To identify the intracellular mediators involved in Nesfatin-1-dependent effects, hearts were perfused with either PD (100 nmol/L), a selective ERK1/2 blocker, or KT5823, a selective PKG blocker, or EHNA, a selective PDE2 blocker. We found that all these inhibitors abolished the effects of Nesfatin-1 (Fig. 3a). By western blotting analyses of Nesfatin-1-exposed ventricular homogenates, we found that the peptide induced an increase of pERK1/2, but not of pAKT (Fig. 3c). We also observed, a decreased Akt phosphorylation in skeletal muscle, liver, and heart in food-deprived rats,, with respect to control (ad libitum fed) animals. In both skeletal muscle and liver, a single injection of Nesfatin-1 (0.25 nmol/g bw) induced a slight increase of Akt phosphorylation, which however, remained lower with respect to the control. In contrast, in the heart, exposure to Nesfatin-1 does not change phospho-Akt levels (data not shown).
Co-immunoprecipitation of membranes incubated with both anti-Nesfatin-1 and anti-NPR-A revealed a band of about 120 kDa corresponding to the molecular mass of NPR-A.
Further evidence of the possible interaction between Nesfatin-1 and NPR-A was obtained by bioinformatic analysis. We have found 33 % identity and 83 % similarity between the analyzed amino acids (Fig. 4).
Fig. 4.

Local sequence alignment of the BNP and Nesfatin-1. The loop of BNP interacting with NPR-A (aa residues 5–16) has been aligned with Nesfatin-1 sequence by LALIGN software (http://www.ch.embnet.org/software/LALIGN_form.html). Hydrophobic amino acids which can interact with Pocket I and Pocket II of NPR-A are reported in bold and italic bold, respectively
NO pathway involvement
Very recently, it was reported that Nesfatin-1 affects peripheral blood vessels contractile reactivity by inhibiting the NO donor-induced smooth muscle relaxation via an impaired cGMP production [23]. To verify whether NO is involved in Nesfatin-1 negative inotropism and lusitropism, Langendorff-perfused hearts were exposed to L-NAME (a non-selective NOSs inhibitor) (Fig. 5a). The results revealed that L-NAME did not influence Nesfatin-1 cardiac effects. The NO independence of the Nesfatin-1 cardiodepression was confirmed by the unchanged eNOS and nNOS phosphorylation and protein SNO after exposure to Nesfatin-1 (Fig. 5c).
Fig. 5.

a Effects of Nesfatin-1 before and after treatment with L-NAME (10 μmol/L) on LVP on the isolated and Langendorff-perfused rat heart preparation, b immunoblots of peNOS, eNOS, pnNOS, and nNOS in control and Nesfatin-1 treated-hearts (cardiac homogenates), c biotin switch assay of S-nitrosylated proteins in the membrane fraction of homogenates from control and Nesfatin-1-treated hearts. Percentage changes were evaluated as mean ± SE of five experiments for each group. Significant difference from control values and from control values of Nesfatin-1 alone versus Krebs–Henseleit solution (KHs) (one-way ANOVA), *p < 0.05. Comparison between groups of Nesfatin-1 alone versus L-NAME, § p < 0.05
Improvement of post-ischemic cardiac function by Nesfatin-1
The possibility that Nesfatin-1 elicits cardioprotection was investigated by comparing the effects induced by PostC maneuvers with those elicited by the peptide administered after I/R. Both systolic and diastolic functions were analyzed. Systolic function is represented by the level of inotropic activity (i.e., LVP recovery). Hearts of the I/R group presented a limited LVP recovery; in fact, at the end of reperfusion, LVP was 16.5 ± 3.8 mmHg instead of baseline value, 87.75 ± 13.9 mmHg. At the end of PostC protocols, LVP was 80 ± 5 mmHg instead of baseline value, 82 ± 5 mmHg. Nesfatin-1 markedly improved LVP recovery during reperfusion, being LVP at the end of reperfusion 72 ± 10 mmHg (baseline value 87 ± 7 mmHg) (Fig. 6b). The Nesfatin-1-dependent improvement of post-ischaemic LVP was abolished when the hearts were co-treated with Nesfatin-1 plus, either the PKCε inhibitor (epsilon-V1-2) or the mitoKATP channels blocker (5HD) (Fig. 7).
Fig. 6.
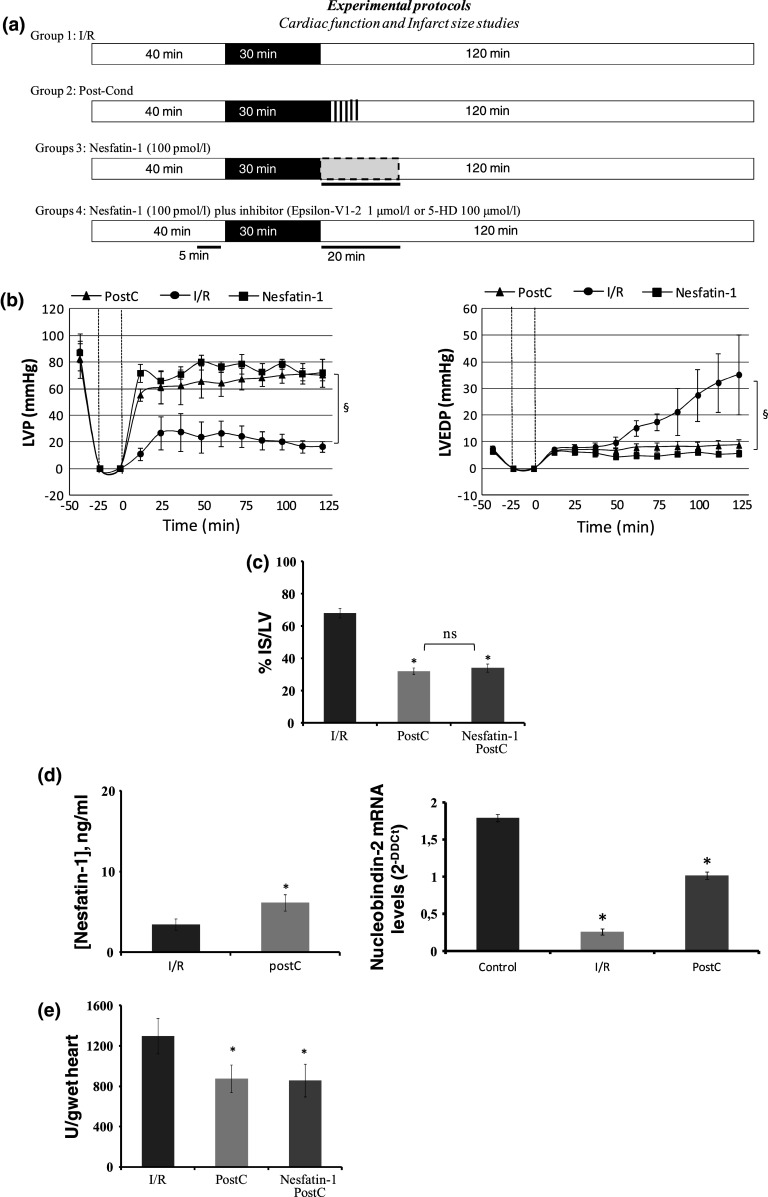
a Protocol groups. b LVP and LVEDP variations. Data are expressed as changes of LVP and LVEDP values (mmHg) from the stabilization to the end of the 120 min of reperfusion with respect to the baseline values for each group. Vertical lines indicate ischemic administration. Comparison between groups, § p < 0.05. c Infarct size. The amount of necrotic tissue measured after 30-min global ischaemia and 120-min reperfusion is expressed as percentage of left ventricle (IS/LV %); *p < 0.05 with respect to I/R. Significant differences from control values of PostC or Nesfatin-1 PostC alone versus I/R (one-way ANOVA), *p < 0.05. d Nesfatin-1 ELISA measurement and real-time PCR of NUCB-2 of I/R and PostC hearts. Significant differences (one-way ANOVA), *p < 0.05. e Effects of Nesfatin-1 on LDH release. Values are expressed as mean ± SE of absolute data (U/wet wt, units per g of wet heart). Significant difference from control values (one-way ANOVA), *p < 0.05; changes were evaluated as mean ± SE of eight experiments for each group
Fig. 7.

Cardiac performance. a LVP variations. Data are expressed as changes of LVP values (mmHg) at the end of 120-min reperfusion with respect to baseline values for each group. Significant differences from control values, *p < 0.05 with respect to I/R, for Nesfatin-1-Post (100 pmol/L), Nesfatin-1-Post (100 pmol/L) + epsilon-V1-2 (1 μmol/L), and Nesfatin-1-PostC (100 pmol/L) + 5HD (100 μmol/L). b LVEDP variations. Data are expressed as changes of LVEDP variations (mmHg) at the end of 120-min reperfusion with respect to baseline values for each group. Significant differences from control values, *p < 0.05 with respect to I/R, for Nesfatin-1-Post, Nesfatin-1-Post + epsilon-V1-2, and Nesfatin-1-Post + 5HD. c Infarct size. The amount of necrotic tissue measured after 30-min global ischaemia and 120-min reperfusion is expressed as percentage of the left ventricle (IS/LV %); *p < 0.05 with respect to I/R and each antagonist group
Diastolic function is represented by the level of contracture (i.e., LVEDP 4 mmHg or more above baseline level) [18]. I/R markedly increased LVEDP (from 7.5 ± 1.1 mmHg in the baseline to 35 ± 15 mmHg at the end of reperfusion). LVEDP was not significantly modified by PostC, being 8.8 ± 2.1 mmHg at the end of the experimental protocol. During reperfusion, Nesfatin-1 abolished contracture development; in fact, LVEDP at the end of reperfusion was 5.7 ± 1 mmHg (Fig. 6b). The Nesfatin-1-dependent reduction of post-ischaemic contracture was abolished by co-treatment with Nesfatin-1 plus, either the PKCε inhibitor (epsilon-V1-2) or the mitoKATP channels blocker (5HD) (Fig. 7).
Total infarct size was expressed as a percentage of left ventricular (LV) mass. LV mass was similar in all groups (LV weight was 935 ± 18 mg; range 559–1,105 mg). Infarct size was 68 ± 3 % in I/R, 32 ± 2 % in PostC and 34 ± 2.5 % in the heart perfused with Nesfatin-1 (Fig. 6c). This protection disappears in the presence of either PKCε inhibitor (epsilon-V1-2) or the mitoKATP channels blocker (5HD) (Fig. 7). LDH release in the I/R group during reperfusion was 1,297 ± 175 U/g (units per g of wet heart), while it was significantly reduced after both PostC (874 ± 137 U/g) and reperfusion with Nesfatin-1 (859 ± 162 U/g) (Fig. 6e).
Lastly, with respect to control hearts, in I/R hearts a significant decrease of Nesfatin-1 protein and NUCB-2 mRNA levels was observed by ELISA and Q-RT-PCR analyses, respectively. In contrast, hearts reperfused with Nesfatin-1 showed an increase in NUCB-2 (Fig. 6d).
Improvement of the cardioprotective pathway
The involvement of the protective kinases ERK1/2 and STAT3 in the Nesfatin-1-activated mechanism of action was evaluated by western blotting. Representative bands and densitometric analysis of the scanned blots are reported in Fig. 8. Data are normalized with respect to the control group. We found that Nesfatin-1 significantly increased the phosphorylation levels of both ERK1/2 and STAT3 (Fig. 8).
Fig. 8.

Representative western blots and relative densitometric analysis of STAT3 and ERK1/2 phosphorylation in Sham, I/R, PostC, and Nesfatin-1-treated hearts. Percentage changes were evaluated as mean ± SE of four experiments for each group. Significant difference (one-way ANOVA) from control values, *p < 0.05
Discussion
This study provided to the best of our knowledge the first evidence that, under basal conditions, the rat heart expresses Nesfatin-1 protein and the mRNA of its precursor, NUCB2. We also found that direct exposure to exogenous Nesfatin-1 induces dose-dependent negative inotropism and lusitropism, which involve pGC-NPR-A, the cGMP/PKG pathway, and ERK1/2. Of importance, in a biomedical perspective, Nesfatin-1 elicits cardioprotection against I/R injury, acting as a post-conditioning agent.
Nesfatin-1 as a cardioinhibitory peptide
We observed, on the isolated and Langendorff-perfused rat heart, that exogenous Nesfatin-1 affects contractility and relaxation in a dose-dependent manner. As shown by the reduced LVP and +(LVdt/dp)max, it induced a negative inotropism starting at 0.1 pM, reaching a maximum at 1 nM. Contrarily, the peptide elicited a biphasic modulation of lusitropism, positive at lower concentrations and negative at higher concentrations. These data indicated that, as in the case of other satiety molecules (i.e., Glucagon-like peptide 2 [25]), peripheral Nesfatin-1 is able to modify the performance of the unstimulated mammalian heart. Notably, the cardiac effects are evident from picomolar concentrations of the peptide. These doses are in the same range of concentrations which inhibit water and food intake in the rat [26]. They are also lower than those detected in the plasma of ad libitum-fed rat in which we found that peptide circulates at a concentration of 12.73 ± 1.5 nmol/L. The high cardiac sensitivity observed in the present study is not surprising, since other neuropeptides have been found to affect myocardial performance at concentrations which are lower than those found in the blood (i.e., HCNP [18]). It should be underlined that, as we observed by immunoblotting and Q-RT-PCR, the unstimulated rat heart constitutively expresses both the precursor NUCB2 and the protein Nesfatin-1. This expression is comparable to that detected in the brain and the stomach, which are known to be Nesfatin-1-producing organs [6, 27]. Accordingly, the cardiac tissue appears to be a source for Nesfatin-1, which may act in a paracrine/autocrine manner on the heart itself, in addition and/or as an alternative to the centrally released peptide. Studies are needed to establish whether, and to what, extent the picomolar amounts of exogenous Nesfatin-1 administered to the perfused heart encounter the intracardiac peptide, thus eliciting a joined effect on contractility and relaxation.
Recently, a hypertensive role has been suggested for Nesfatin-1. It was found that the peptide counteracts the NO-dependent vasodilation of peripheral arteries [24]. In addition, central Nesfatin-1 activates the nervous circuits which are responsible for hypertension [12], an effect which is presumed to also occur via hypothalamic melanocortin-3/4 receptors [13]. It is known that, in vivo under basal conditions, the increase in vascular resistance is counterbalanced by cardioinhibition [28]. This contributes on the one hand to preserve normal blood pressure values and, on the other hand, to prevent the cardiac compensatory mechanisms which are activated by hypertension (i.e., ventricular hypertrophy) [29]. Other cardioactive peptides, such as catestatin, vasostatin, and GLP-2, induce negative inotropism. This cardiodepression is accompanied by a non-significant coronary constriction which, however, does not influence the myocardial activity of the peptides. The vasoconstrictory effects suggested for Nesfatin-1 appear in contrast with the cardiodepression observed in our study. However, tissue-specific effects (peripheral arteries vs. heart), different methods for Nesfatin-1 administration (central vs. direct in vitro perfusion) and the possible involvement of other intermediates (i.e., indirect melanocortin-dependent sympathetic activation), must be taken into account. Notably, the direct negative inotropism and lusitropism induced by Nesfatin-1, together with the indirect hypertensive activity [24], recall the classic α-adrenergic effects. In fact, it is known that activation of α-adrenergic receptors subtype reduces the left ventricular pressure and induces peripheral vasoconstriction [30].
Signalling pathways for Nesfatin-1
In the absence of knowledge about a specific receptor for Nesfatin-1, and on the basis of the data obtained on PVN, it has been suggested that Nesfatin-1 elicits its effects by recruiting Gi/o proteins [10]. This possibility is not confirmed by our observations, since Gi/o protein inhibition by PTx did not change Nesfatin-1-induced negative inotropism and lusitropism.
It has been reported that NO is an important negative modulator of cardiac performance. In the mammalian heart, the NOS-NO-cGMP-PKG system mediates specific intracardiac signaling involved in the beat-to-beat control of the contractile performance [31]. In rat ventricular myocytes, the NOS-produced NO, by targeting soluble GC, and thus PKG, negatively affects contractility by reducing ICa and L and by phosphorylating troponin I [31]. The functional consequence of this concerted cascade is inhibition of contractility. Interestingly, in our experiments, we found that Nesfatin-1-induced negative inotropic and lusitropic effects are not influenced by the inhibition of the NOS-NO system by L-NIO. This NO independence was confirmed by eNOS phosphorylation and protein SNO analyses. In both cases, exposure to Nesfatin-1 did not change the basal levels of p-eNOS and protein SNO. However, ELISA assay showed that Nesfatin-1-dependent negative inotropism and lusitropism are accompanied by increased cGMP levels. Alternative to the NO-dependent route, intracellular cGMP may be generated by natriuretic peptides binding to their pGC receptors (NPR-A and NPR-B). Based on these premises, we analyzed whether Nesfatin-1-dependent cardiodepression involves the NP-activated pGC. We found that pGC-NPR-A inhibition by Anantin abolished the effects induced by Nesfatin-1 on both contractility and relaxation. In the rat heart, NPR-A was found to be predominant in ventricular myocytes, with respect to other cardiac cells [32]. This may contribute to explaining the prevalent myocardial effects elicited by Nesfatin-1 and the lack of coronary responsiveness to the peptide. The possibility of a Nesfatin-1/NPR-A interaction was supported by co-immunoprecipitation studies which revealed a band of about 120 kDa corresponding to the molecular mass of NPR-A. Further evidence of the possible interaction between Nesfatin-1 and NPR-A was obtained by bioinformatic analysis. We have selected the loop region (aa 5–16) of the natriuretic peptide B (involved in NPR-A binding) and we have performed a local alignment between these 12 amino acids and the Nesfatin-1 sequence. In particular, we have found 33 % identity and 83 % similarity (see Fig. 4), and this strongly suggests that Nesfatin-1 could interact with NPR-A, since it shares a very high similarity with its physiological ligand, BNP. Interestingly, this portion of the Nesfatin-1 sequence contains hydrophobic amino acids that could interact with both pocket I and II of NPR-A [22].
Downstream, cGMP controls the activity of cGMP-dependent PKG and phosphodiesterases (PDEs). In particular, cGMP activates PDE2, and this decreases intracellular cAMP concentrations [33]. In our study, pre-treatment with the specific PDE2 inhibitor, EHNA, and the PKG blocker, KT5823, abolished the Nesfatin-1-dependent cardiac effects, confirming the involvement of these targets in the signalling activated by the peptide. Of note, Nesfatin-1 cardiomodulation is also mediated by increased ERK1/2 phosphorylation. In the heart, ERK1/2 mediates short- and long-term responses elicited by many cardioactive substances [34, 35]. It is also involved in infarct reduction achieved with ischaemic post-conditioning [36].
Nesfatin-1-dependent cardioprotection
It is known that many cardiodepressive peptides (i.e., the chromogranin-A-derived vasostatin and catestatin; [37, 38]) protect the heart from I/R damage. Accordingly, we investigated whether, in addition to its negative influence on basal cardiac performance, Nesfatin-1 is able to elicit cardioprotection. For this purpose, we used a pharmacological post-conditioning protocol [39]. We observed that I/R is accompanied by a reduced expression of NUCB-2, which returns to normal after the post-conditioning maneuver. In addition, Nesfatin-1, given in the early reperfusion, induced a significant protective effect against myocardial I/R injury, similar to post-conditioning. This effect is revealed by the significant reduction of the infarct size, and the marked improvement of post-ischemic contractile function expressed as a decrease of contracture development. Physiological analyses of the mechanism of action which sustains Nesfatin-1-dependent cardioprotection revealed the involvement of mito-KATP channels, and of pro-survival kinases, namely PKCε. In particular, PKCε may act on mitochondria to affect cellular survival reducing both necrosis and apoptosis [40, 41]. At the same time, mitoKATP channels activation may facilitate PKCε re-activation via reactive oxygen species (ROS) signaling. This PKCε-dependent circuit of activation and re-activation takes part in the so-called “memory-associated protection” [40, 41]. Of note, this protective signaling was found to also be involved in the cardioprotection induced by NPR-A activation [42].
By biomolecular studies, we observed that STAT3 and ERK1/2 take part to the Nesfatin-1-mediated post-conditioning. It is known that STAT is an important membrane-to-nucleus signaling for many stress responses, which include I/R, oxidative stress, and hypoxia [43–45]. It is also a crucial member of protective cascades (i.e., SAFE) that, when activated, induce survival signals in the infracted myocardium [46, 47]. Since cardioprotection by Nesfatin-1 also involves ERK1/2, a known component of the RISK pathway [48], we suggest that the peptide acts in pharmacological post-conditioning by activating both RISK and SAFE cascades.
Our data are particularly relevant since alterations in post-infarction left ventricular function are major determinants of mortality and prognosis [40, 49]. After an acute myocardial infarction, reperfusion is necessary for saving the viable myocardium. However, this is not without risk, as the reperfusion itself may result in myocyte death. Accordingly, in the search for novel pharmacological approaches that target and attenuate reperfusion-induced cell death, to improve the classic reperfusion strategy aimed at limiting myocardial infarction, our results on Nesfatin-1-induced cardioprotection deserve further attention.
In conclusion, by using physio-pharmacological and molecular approaches, we demonstrated that the anorexic hypothalamic peptide, Nesfatin-1, elicits a direct influence on the heart by depressing contractility and relaxation via a cGMP-linked pathway, and by inducing cardioprotection against I/R injuries. The finding that the cardiac tissue itself expresses both Nesfatin-1 and the precursor NUCB-2 allows the inclusion of the peptide in the growing list of cardiac hormones. Notably, we suggest an interaction with pGC/NPR-A, thus opening a novel perspective for research. In fact, any progress on the therapeutic application of Nesfatin-1 is limited by the absence of conclusive evidence concerning a specific Nesfatin-1 receptor. Taken together, our data are of relevance since they enrich the knowledge on the multilevel circuits that, by involving several signaling peptides, orchestrate hypothalamic, GI, alimentary, and cardiovascular functions. They also open the way for clarifying the clinical potential of Nesfatin-1 in nutrition-dependent cardiovascular diseases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This research was supported by grants from Ministero dell’Università e Ricerca Scientifica e Tecnologica (ex 60 % T.A. and M.C.C.), “Progetto Giovani Ricercatori 2010.” (E.F.) and National Institute of Cardiovascular Research (INRC).
Conflict of interest
None.
Abbreviations
- 5-HD
5-Hydroxydecanoate
- ARC
Arcuate nuclei
- CP
Coronary pressure
- eNOS
Endothelial nitric oxide synthase
- ERK1/2
Extracellular signal-regulated kinases1/2
- I/R
Ischemia/reperfusion injury
- −(LVdP/dt)max
Maximal rate of left ventricular pressure decline of LVP
- +(LVdP/dt)max
Maximal values of the first derivative of LVP
- LVP
Left ventricular pressure
- NO
Nitric oxide
- NPR-A
Natriuretic peptide receptor A
- NPY/AgRP
Neuropeptide Y/agouti-related peptide
- NUCB2
Nucleobindin-2
- pGC
Particulate guanylate cyclase
- POMC/CART
Pro-opiomelanocortin/cocaine and amphetamine responsive transcript
- PostC
Post-conditioning protocol
- PVN
Paraventricular nuclei
- SNO
S-nitrosylation
- SON
Supraoptic nuclei
- STAT3
Signal transducer and activator of transcription 3
Footnotes
T. Angelone and E. Filice contributed equally to the work.
References
- 1.Angelone T, Quintieri AM, Amodio N, Cerra MC. Endocrine orchestration of cardiovascular, gastrointestinal and hypothalamic control. Curr Med Chem. 2011;18(32):4976–4986. doi: 10.2174/092986711797535236. [DOI] [PubMed] [Google Scholar]
- 2.Vona-Davis LC, McFadden DW. NPY family of hormones: clinical relevance and potential use in gastrointestinal disease. Curr Top Med Chem. 2007;7(17):1710–1720. doi: 10.2174/156802607782340966. [DOI] [PubMed] [Google Scholar]
- 3.Pedrazzini T, Seydoux J, Künstner P, Aubert JF, Grouzmann E, Beermann F, Brunner HR. Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY Y1 receptor. Nat Med. 1998;4(6):722–726. doi: 10.1038/nm0698-722. [DOI] [PubMed] [Google Scholar]
- 4.Oh-I S, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, Eguchi H, Yamamoto M, Imaki T, Hashimoto K, Tsuchiya T, Monden T, Horiguchi K, Yamada M, Mori M. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature. 2006;443(7112):709–712. doi: 10.1038/nature05162. [DOI] [PubMed] [Google Scholar]
- 5.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol. 2005;493(1):63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 6.Stengel A, Goebel M, Yakubov I, Wang L, Witcher D, Coskun T, Taché Y, Sachs G, Lambrecht NW. Identification and characterization of nesfatin-1 immunoreactivity in endocrine cell types of the rat gastric oxyntic mucosa. Endocrinology. 2009;150(1):232–238. doi: 10.1210/en.2008-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Y, Zhang J, Tang Y, Bi F, Liu JN. The novel function of nesfatin-1: anti-hyperglycemia. Biochem Biophys Res Commun. 2010;391(1):1039–1042. doi: 10.1016/j.bbrc.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 8.Price TO, Samson WK, Niehoff ML, Banks WA. Permeability of the blood-brain barrier to a novel satiety molecule nesfatin-1. Peptides. 2007;28(12):2372–2381. doi: 10.1016/j.peptides.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Brailoiu GC, Dun SL, Brailoiu E, Inan S, Yang J, Chang JK, Dun NJ. Nesfatin-1: distribution and interaction with a G protein-coupled receptor in the rat brain. Endocrinology. 2007;148(10):5088–5094. doi: 10.1210/en.2007-0701. [DOI] [PubMed] [Google Scholar]
- 10.Pałasz A, Krzystanek M, Worthington J, Czajkowska B, Kostro K, Wiaderkiewicz R, Bajor G (2012) Nesfatin-1, a unique regulatory neuropeptide of the brain. Neuropeptides 10.1016/j.npep.2011.12.002 [DOI] [PubMed]
- 11.Maejima Y, Sedbazar U, Suyama S, Kohno D, Onaka T, Takano E, Yoshida N, Koike M, Uchiyama Y, Fujiwara K, Yashiro T, Horvath TL, Dietrich MO, Tanaka S, Dezaki K, Oh-I S, Hashimoto K, Shimizu H, Nakata M, Mori M, Yada T. Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. Cell Metab. 2009;10(5):355–365. doi: 10.1016/j.cmet.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Mimee A, Smith PM, Ferguson AV. Nesfatin-1 influences the excitability of neurons in the nucleus of the solitary tract and regulates cardiovascular function. Am J Physiol Regul Integr Comp Physiol. 2012 doi: 10.1152/ajpregu.00266.2011. [DOI] [PubMed] [Google Scholar]
- 13.Yosten GL, Samson WK. Nesfatin-1 exerts cardiovascular actions in brain: possible interaction with the central melanocortin system. Am J Physiol Regul Integr Comp Physiol. 2009;297(2):R330–R336. doi: 10.1152/ajpregu.90867.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerra MC, De Iuri L, Angelone T, Corti A, Tota B. Recombinant N-terminal fragments of chromogranin-A modulate cardiac function of the Langendorff-perfused rat heart. Basic Res Cardiol. 2006;101(1):43–52. doi: 10.1007/s00395-005-0547-2. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu H, Oh-I S, Hashimoto K, Nakata M, Yamamoto S, Yoshida N, Eguchi H, Kato I, Inoue K, Satoh T, Okada S, Yamada M, Yada T, Mori M. Peripheral administration of nesfatin-1 reduces food intake in mice: the leptin-independent mechanism. Endocrinology. 2009;150(2):662–671. doi: 10.1210/en.2008-0598. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Cerra MC, Angelone T, Parisella ML, Pellegrino D, Tota B. Nitrite modulates contractility of teleost (Anguilla anguilla and Chionodraco hamatus, i.e. the Antarctic hemoglobinless icefish) and frog (Rana esculenta) hearts. Biochim Biophys Acta. 2009;1787(7):849–855. doi: 10.1016/j.bbabio.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Angelone T, Goumon Y, Cerra MC, Metz-Boutigue MH, Aunis D, Tota B. The emerging cardioinhibitory role of the hippocampal cholinergic neurostimulating peptide. J Pharmacol Exp Ther. 2006;318(1):336–344. doi: 10.1124/jpet.106.102103. [DOI] [PubMed] [Google Scholar]
- 19.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34(1):33–43. doi: 10.1016/S0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 20.Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P. Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res. 2007;75(1):168–177. doi: 10.1016/j.cardiores.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006;101(2):180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- 22.Parat M, McNicoll N, Wilkes B, Fournier A, De Léan A. Role of extracellular domain dimerization in agonist-induced activation of natriuretic peptide receptor A. Mol Pharmacol. 2008;73(2):431–440. doi: 10.1124/mol.107.039982. [DOI] [PubMed] [Google Scholar]
- 23.Springer J, Azer J, Hua R, Robbins C, Adamczyk A, McBoyle S, Bissell MB, Rose RA (2012) The natriuretic peptides BNP and CNP increase heart rate and electrical conduction by stimulating ionic currents in the sinoatrial node and atrial myocardium following activation of guanylyl cyclase-linked natriuretic peptide receptors. J Mol Cell Cardiol 10.1016/j.yjmcc.2012.01.018 [DOI] [PubMed]
- 24.Yamawaki H, Takahashi M, Mukohda M, Morita T, Okada M, Hara Y. A novel adipocytokine, nesfatin-1 modulates peripheral arterial contractility and blood pressure in rats. Biochem Biophys Res Commun. 2012;418(4):676–681. doi: 10.1016/j.bbrc.2012.01.076. [DOI] [PubMed] [Google Scholar]
- 25.Angelone T, Filice E, Quintieri AM, Imbrogno S, Amodio N, Pasqua T, Pellegrino D, Mulè F, Cerra MC (2010) Receptor identification and physiological characterisation of glucagon-like peptide-2 in the rat heart. Nutr Metab Cardiovasc Dis 10.1016/j.numecd.2010.07.014 [DOI] [PubMed]
- 26.Yosten GL, Redlinger L, Samson WK. Evidence for a role of endogenous nesfatin-1 in the control of water drinking. J Neuroendocrinol. 2012 doi: 10.1111/j.1365-2826.2012.02304.x. [DOI] [PubMed] [Google Scholar]
- 27.Mohan H, Unniappan S. Ontogenic pattern of nucleobindin-2/nesfatin-1 expression in the gastroenteropancreatic tissues and serum of Sprague Dawley rats. Regul Pept. 2012;175(1–3):61–69. doi: 10.1016/j.regpep.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Opie LH (2004) Heart physiology. From cell to circulation. 4th edn. Lippincott, Philadelphia
- 29.Sperelakis N, Kurachi Y, Terzic A, Cohen MV. Heart physiology and pathophysiology. 4. San Diego: Academic; 2001. [Google Scholar]
- 30.Piascik MT, Perez DM. Alpha1-adrenergic receptors: new insights and directions. J Pharmacol Exp Ther. 2001;298(2):403–410. [PubMed] [Google Scholar]
- 31.Abi-Gerges N, Fischmeister R, Méry PF. G protein-mediated inhibitory effect of a nitric oxide donor on the L-type Ca2+ current in rat ventricular myocytes. J Physiol. 2001;531(Pt 1):117–130. doi: 10.1111/j.1469-7793.2001.0117j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin X, Hänze J, Heese F, Sodmann R, Rudolf E. Lang gene expression of natriuretic peptide receptors in myocardial cells. Circ Res. 1995;77:750–758. doi: 10.1161/01.RES.77.4.750. [DOI] [PubMed] [Google Scholar]
- 33.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27(1):47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 34.Dube′ PE, Forse CL, Bahrami J, Brubaker PL. The essential role of insulin-like growth factor-1 in the intestinal tropic effects of glucagon-like peptide-2 in mice. Gastroenterology. 2006;131:589–605. doi: 10.1053/j.gastro.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 35.Clerk A, Sugden PH. Signaling through the extracellular signal regulated kinase 1/2 cascade in cardiac myocytes. Biochem Cell Biol. 2004;82(6):603–609. doi: 10.1139/o04-110. [DOI] [PubMed] [Google Scholar]
- 36.Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol. 2005;289(4):H1618–H1626. doi: 10.1152/ajpheart.00055.2005. [DOI] [PubMed] [Google Scholar]
- 37.Cappello S, Angelone T, Tota B, Pagliaro P, Penna C, Rastaldo R, Corti A, Losano G, Cerra MC. Human recombinant chromogranin A-derived vasostatin-1 mimics preconditioning via an adenosine/nitric oxide signaling mechanism. Am J Physiol Heart Circ Physiol. 2007;293(1):H719–H727. doi: 10.1152/ajpheart.01352.2006. [DOI] [PubMed] [Google Scholar]
- 38.Penna C, Alloatti G, Gallo MP, Cerra MC, Levi R, Tullio F, Bassino E, Dolgetta S, Mahata SK, Tota B, Pagliaro P. Catestatin improves post-ischemic left ventricular function and decreases ischemia/reperfusion injury in heart. Cell Mol Neurobiol. 2010;30(8):1171–1179. doi: 10.1007/s10571-010-9598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penna C, Abbadessa G, Mancardi D, Tullio F, Piccione F, Spaccamiglio A, Racca S, Pagliaro P. Synergistic effects against post-ischemic cardiac dysfunction by sub-chronic nandrolone pretreatment and postconditioning: role of beta2-adrenoceptor. J Physiol Pharmacol. 2008;59(4):645–659. [PubMed] [Google Scholar]
- 40.Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C. Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal. 2011;14(5):833–850. doi: 10.1089/ars.2010.3245. [DOI] [PubMed] [Google Scholar]
- 41.Hausenloy DJ, Lecour S, Yellon DM. Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal. 2011;14(5):893–907. doi: 10.1089/ars.2010.3360. [DOI] [PubMed] [Google Scholar]
- 42.Nishikimi T, Maeda N, Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovasc Res. 2006;9(2):318–328. doi: 10.1016/j.cardiores.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 43.Bolli R, Dawn B, Xuan YT. Role of the JAK-STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc Med. 2003;13:72–79. doi: 10.1016/S1050-1738(02)00230-X. [DOI] [PubMed] [Google Scholar]
- 44.Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, Sack MN, Jonassen AK, Mjøs OD, Opie LH, Lecour S. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol. 2008;103:444–453. doi: 10.1007/s00395-008-0728-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mascareno E, El-Shafei M, Maulik N, Sato M, Guo Y, Das DK, Siddiqui MA. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001;104:325–329. doi: 10.1161/01.CIR.104.3.325. [DOI] [PubMed] [Google Scholar]
- 46.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–785. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA. 2001;98:9050–9055. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 49.Penna C, Abbadessa G, Mancardi D, Spaccamiglio A, Racca S, Pagliaro P. Nandrolone-pretreatment enhances cardiac beta(2)-adrenoceptor expression and reverses heart contractile down-regulation in the post-stress period of acute-stressed rats. J Steroid Biochem Mol Biol. 2007;107(1–2):106–113. doi: 10.1016/j.jsbmb.2007.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.