

Abstract
A series of novel thio-derivatives of d-glucosamine has been synthesized using double inversion procedures at the C3 atom. New compounds were applied as ligands for the diethylzinc addition to benzaldehyde and the products of the addition were obtained with a low to good enantiomeric ratio. The direction and the level of the asymmetric induction were highly dependent on the type of protecting groups on the nitrogen and sulfur atoms.
Keywords: benzaldehyde, enantioselectivity, d-glucosamine, diethylzinc, thiol
1. Introduction
The asymmetric addition of dialkylzinc compounds to aldehydes is an important method for the synthesis of optically active secondary alcohols. Due to the low reactivity of dialkylzinc reagents toward the carbonyl group, the addition requires the presence of a catalyst, usually an amino alcohol, which increases the rate of reaction and often controls the stereochemical outcome of the alcohol product. Since the first efficient chiral catalyst, (-)-3-exo (dimethylamino)isoborneol (DAIB), was introduced by Noyori [1], syntheses and applications of several efficient chiral catalysts have been reported [2,3].
Derivatives of common, simple carbohydrates are not frequently reported among the many different forms of chiral ligands that are accessible. The benefit of sugar derivatives is their modular synthesis, which allows for easy ligand structure modification throughout synthesis, resulting in different ligands possessing the same chiral precursor. We previously reported the synthesis of hydroxy sulfonamides 2 derived from d-glucosamine 1, which were used as ligands in the titanium-promoted additions of diethyl- [4,5], and alkynylzincs [6] to aldehydes (Figure 1).
Figure 1.

Ligands for the enantioselective additions of organozinc compounds.
It has been observed that β-amino thiol can be viable alternative to β-amino alcohol as a catalyst for asymmetric organozinc addition because of certain crucial features, including the diminished tendency of metal thiolates to reduce the Lewis acidity of the metal in comparison to metal alcoholates, higher affinity of thiols to zinc and increased polarizability of sulfur in comparison to the oxygen atom [7].
Several amino thiol ligands were reported, and one of the most efficient was the l-valine-derived amino thiols 3 developed by Tseng and Yang [8]. We have recently proved the efficacy of 3a in the highly enantioselective alkenylation of aldehydes [9].
Herein, we present our studies on the synthesis of d-glucosamine derivatives bearing thiol functionality at the C3 position (sugar nomenclature) and their application for the diethylzinc addition to aldehydes.
The enantioselective addition of organozinc compounds is usually catalyzed by two types of ligands, namely β-hydroxy amines, as described by Noyori, and β-hydroxy sulfonamides or diols in the presence of tetraisopropoxytitanium, according to the mechanism described by Gau [10] and Walsh [11]. We decided to use our experience with the titanium-promoted addition of organozinc compounds in the presence of β-hydroxy sulfonamides 2 and investigate the respective thio-analogs. Additionally, as we expected the incompatibility of the hard Ti(OiPr)4 with the soft sulfur atom, we planned a synthesis of the thio-analogs of the classical β-hydroxy amines earlier described by Davis and co-workers [12].
2. Results
At first, we choose the known [4] amino alcohol 4 as a most versatile starting material for the synthesis of all the ligands. The synthesis of methyl 4,6-O-benzylidene-2,3-dideoxy-3-thio-2-p-toluenesulfonamido-α-d-glucopyranoside 10 proceeded easily. The reaction at step (v) required some optimization, as the reaction in DMF gave a major product 9 contaminated with some side products; however, the same reaction performed in acetonitrile furnished 9 as a single compound (Scheme 1).
Scheme 1.

The synthesis of methyl 4,6-O-benzylidene-2,3-dideoxy-3-thio-2-p-toluenesulfonamido-α-d-glucopyranoside 10. Reaction conditions: (i) p-TsCl, Na2CO3, 1,4-dioxane:H2O (1:1), 4 h, 92%; (ii) MsCl, Et3N, DCM. 0 °C to r.t., 3 h, 74%; (iii) NaOAc, 2-methoxyethanol:H2O (10:1), 110 °C, 46 h, 95%; (iv) MsCl, Et3N, DCM. 0 °C to r.t., 3 h, 90%; (v) KSAc, MeCN, 75 °C, 1.5 h, 89%; and (vi) LiAlH4, THF, 0 °C, 4 h, 41%.
Successful synthesis of the sulfonamide 10 prompted us to apply the same method to the morpholine ligand 16, belonging to the second series of ligands. The synthesis proceeded reasonably well; however, we encountered some problems. In particular, the inversion of C3 hydroxyl (step iii) was very slow and the product 13 was obtained with a moderate yield (Scheme 2).
Scheme 2.

Synthesis of methyl 4,6-O-benzylidene-2,3-dideoxy-2-(4-morpholinyl)-3-thio-α-d-glucopyranoside 16. Reaction conditions: (i) bis(2-bromoetyl) ether, MeCN, K2CO3, 80 °C, 20 h, 47%; (ii) MsCl, pyridine, r.t., 4 h, 73%; (iii) NaOAc, 2-methoxyethanol, 115 °C, 72 h, 46%; (iv) MsCl, pyridine, 4 h, r.t. 68%; (v) KSAc, DMF, 24 h, 100 °C, 70%; and (vi) LiAlH4, THF, 0 °C to r.t., 76%.
Next, we applied this approach for the synthesis of a piperidine derivative, namely methyl 4,6-O-benzylidene-2,3-dideoxy-2-(1-piperidynyl)-3-thio-α-d-glucopyranoside. Unfortunately, even after the long optimization of the conditions, we could not achieve the inversion of C3 hydroxyl from the equatorial to axial position with a yield higher than 20% and the prolonged reaction times did not increase the conversion over 50% with a simultaneous decomposition of the product. Therefore, we abandoned this path for the synthesis.
We reached the conclusion that the presence of cyclic amine at C2 could be responsible for the problems with the inversion of the configuration at C3. Therefore, we decided to synthesize an analog of the amino alcohol 4, the methyl 2-amino-4,6-O-benzylidene-2-deoxy-α-d-allopyranoside 20.
We decided to try a not very popular but often highly efficient method: Lattrell–Dax epimerization [13], which has been successfully used in carbohydrate chemistry [14].
We started from the carbamate 17, an easily available precursor of amino alcohol 4 [4]. The synthesis of triflate 18 was straightforward and furnished the expected product with a high yield. The subsequent Lattrell–Dax epimerization proceeded smoothly, providing 19, which was converted to the expected allopyranoside 20 (Scheme 3).
Scheme 3.

Synthesis of methyl 2-amino-4,6-O-benzylidene-2-deoxy-α-d-allopyranoside 20. Reaction conditions: (i) Tf2O, pyridine, DCM, −20 °C, 2 h, 93%; (ii) NaNO2, DMF, r.t., 6 days, 67%; and (iii) 4N KOH, EtOH:2-methoxyethanol (2:1), 80 °C, 20 h, 52%.
The formation of the pyrrolidine 21 and piperidine 22 derivatives was achieved using the respective dibromides; however, the mesylation of these compounds at C3 was only possible using pyridine and not triethylamine as a base. We then tried to convert both mesylates 23 and 24 into thioacetate. Much to our disappointment, treating the mesylate derivatives with potassium thioacetate failed to produce any product (Scheme 4).
Scheme 4.

Attempted synthesis of pyrrolidinyl and piperidinyl-3-thio derivatives. Reaction conditions: (i) 21: C4H8Br2, K2CO3, MeCN, reflux, 24 h, 88%; 22 C5H10Br2, K2CO3, MeCN, reflux, 40 h, 70%; (ii) MsCl, DCM, r.t., 4 h, 23 52%, 24 60%; and (iii) KSAc, DMF or MeCN or DMPU, reflux, 0%.
The problems we encountered prompted us to cease our efforts toward the synthesis of pyrrolidinyl and piperidinyl derivatives and to expand the pool of C2 sulfonamides.
First, the amino alcohol 20 was treated with an excess of methanesulfonyl chloride in the presence of pyridine. The resulting dimethanesulfonyl derivative 25 was subjected to the nucleophilic substitution reaction with potassium thioacetate in DMPU, which appeared to be the solvent of choice for this transformation. However, the expected product was accompanied by a by-product, presumably resulting from the elimination of the mesylate, and the mixture was inseparable at this stage. Fortunately, the reduction of thioacetate 26 furnished thiosulfonamide 27 in a good yield and the by-product could be removed at this final stage (Scheme 5).
Scheme 5.

Synthesis of methyl 4,6-O-benzylidene-2,3-dideoxy-2-methanesulfonamido-3-thio-α-d-glucopyranoside 27. Reaction conditions: (i) MsCl, pyridine, 0 °C, 24 h 75%; (ii) KSAc, DMPU, 100 °C, 6 h, 60%; and (iii) LiAlH4, 0 °C to r.t., 4 h, 65%.
The same strategy could be applied for the synthesis of trifluoromethylsulfonamide derivative 31. The double protection of amino alcohol 20 could be achieved using an excess of triflic anhydride (3 equiv.) in DCM and pyridine (10 equiv.), resulting in a mixture of mono- and di-triflate products, approximately 40% to 60%. Fortunately, it was possible to separate these two compounds and the triflation of the remaining hydroxyl derivatives worked without any problems under similar conditions (Scheme 6).
Scheme 6.

Synthesis of methyl 2,3-dideoxy-4,6-O-benzylidene-3-thio-2-trifloromethanesulfonamido-α-d-glucopyranoside 31. Reaction conditions: (i) Tf2O, pyridine, DCM, −20 °C to 0 °C, 2 h, 51% (29), 34% (28); (ii) Tf2O, pyridine, DCM −20 °C, 2 h, 61%; (iii) KSAc, MeCN, 5 to 20 °C, 40%; and (iv) NaOMe, MeOH, r.t., 18 h, 76%.
The inversion procedure (KSAc, DMPU) developed for mesylate 25 did not work for the triflate 28. The expected product 30 was obtained with a very low yield (15%), probably due to competitive elimination. The optimization of this reaction showed that acetonitrile is the solvent of choice for this transformation and 30 was obtained with 40% yield; however, the elimination was still a major problem. In the final conversion to free thiol 31 using standard approach, the reduction of thioacetate with LiAlH4 was unsuccessful and it could not be obtained by this method. We decided to hydrolyze the acetate under the Zemplen conditions [15]. The expected ligand 31 was obtained in a 76% yield.
Having to hand all the available ligands, we began studies on the enantioselective additions of diethylzinc to benzaldehyde. We started with the p-toluenesulfonamide ligand 10 using the titanium tetraisopropoxide-based method. Not very surprisingly, the enantioselectivity in the toluene was very poor (e.r. 57:43, S/R), and it was even lower when using methylene chloride as a solvent (e.r. 54.5:45.5). Supposedly, the required titanium–sulfur complex was not formed as expected; however, the ligands exhibited very good catalytic properties—the chemical yields of 1-phenylpropanol were high at –95% and 85%, respectively (Table 1, entries 1 and 2). Then, we tried the morpholine ligand 16 in the absence of Ti(OiPr)4 and we obtained the expected product with a very good chemical yield of 83%, but again with a surprisingly low enantioselectivity, where the enantiomeric ratio was only 55:45, R/S, much lower than that for the respective amino alcohol reported by Davis and co-workers (Table 1, entry 3) [12]. It became obvious that the 3-thio-glucosamine ligands did not follow the expected modes of action; therefore, we decided to check the inductive properties of the sulfonamide ligands in the absence of Ti(OiPr)4 Lewis acid. First, we tried the ligand 10 and noticed a substantial improvement of the enantiodiscrimination to 74.5:25.5, again with a good yield and the S enantiomer prevailing (Table 1, entry 4). The bulky trifluoromethanesulfonamide ligand 31 was less efficient and the observed e.r. was only 64:36 (Table 1, entry 5). Unexpectedly, the highest enantiomer ratio 80.5:19.5 was obtained for the methanesulfonamide derivative 27 (Table 1, entry 6).
Table 1.
Results of the diethylzinc additions to benzaldehyde.
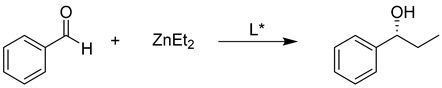
| ||||||
|---|---|---|---|---|---|---|
| Entry a | ZnEt2 (equiv.) | L* (equiv.) | Solvent | Time [h] | Yield [%] | e.r. (Config.) |
| 1 b | 3 | 10 (0.1) | DCM | 18 | 85 | 54.5:45.5 (S) |
| 2 b | 3 | 10 (0.1) | Toluene | 18 | 95 | 57:43 (S) |
| 3 | 2 | 16 (0.1) | Toluene | 20 | 83 | 55:45 (R) |
| 4 | 2 | 10 (0.1) | Toluene | 20 | 87 | 74.5:25.5 (S) |
| 5 | 2 | 31 (0.1) | Toluene | 24 | 82 | 64:36 (S) |
| 6 | 2 | 27 (0.1) | Toluene | 24 | 86 | 80.5:19.5 (S) |
| 7 | 2 | 15 (0.1) | Toluene | 20 | 35 | 53:47 (R) |
| 8 | 2 | 9 (0.1) | Toluene | 20 | 61 | 71.5:28.5 (S) |
| 9 | 2 | 26 (0.1) | Toluene | 20 | 73 | 54.5:45.5 (R) |
| 10 | 2 | 30 (0.1) | Toluene | 20 | 70 | 51.5:48.5 (R) |
a All reactions performed at r.t.; b In the presence of Ti(OiPr)4 (1.4 equiv.).
The literature reports indicate that thioacetate derivative may also be an effective chiral ligand [16,17].
Therefore, we decided to check four intermediate compounds, 9, 15, 26, and 30, as ligands. They appeared to be much less efficient ligands than the respective thiols; the results are presented in Table 1, entries 7–10.
3. Discussion
The data presented in the introduction, indicating that amino thiols can be equally good, and often better, ligands in the addition of organozinc reagents to carbonyl compounds, are not confirmed in the case presented here of 3-thio-glucosamine derivatives.
The low asymmetric induction of the reaction catalyzed by the sulfonamides in the presence of Ti(OiPr)4 was not very surprising in the context of hard and soft acids and bases theory (Table 1, entries 1 and 2). However, the very low e.r. in the reaction catalyzed by the ligand 16 was unexpected (Table 1, entry 3). The earlier-reported hydroxy analogue [12] was quite efficient and we expected at least comparable enantioselectivity. Supposedly, the geometry of the transition state can be adversely influenced by the length of the zinc–sulfur bond, 2.2–2.4 Å vs. 1.9–2.2 Å for the zinc–oxygen bond. We performed MM2 energy minimalization in order to analyze the geometry of the most probable transition state based on the Noyori’s model (Figure 2).
Figure 2.

Transition state models: (A)—Davis’ ligand (ref. [12]); and (B)—ligand 16 (this work).
The left image shows the model for the addition of Davis’ ligand [12], which exhibited good enantioselectivity (e.r. 82.5:17.5). As indicated in Noyori’s model, the ethyl group attached to the same zinc atom as the benzaldehyde efficiently hinders the rotation of the aldehyde, securing high enantiodiscrimination. A quite different situation is observed in the case of the thiol ligand 16. That ethyl group is directed backwards relative to the aldehyde, which creates much less steric hindrance and does not prevent rotation of the aldehyde molecule. As a result, an attack on both faces of the carbonyl group is almost equally probable, what results in poor selectivity.
The reaction proceeded quite well for the sulfonamide ligands in the absence of the Lewis acid. The best results were obtained for the least sterically demanding mesyl derivative 27 and not for the highly hindered trifluorosulfonamide 31 (Table 1, entry 6 and 5), which, on the basis of our previous studies and the very high selectivity observed in the presence of the 3-hydroxy analog of 31, should be the most efficient ligand.
In the case of sulfonamides, the main factor influencing enantiodiscrimination is probably the interaction of the anomeric methoxyl group with the methyl- and trifluoromethylsulfonamide groups in 27 and 31, respectively (Figure 3A,B). The bigger CF3 substituent has to be placed farther from the anomeric OCH3 (Figure 3A) and that allows for the easier rotation of the aldehyde, leading to diminished enantioselectivity. The smaller methanesulfonyl group locates itself in close proximity of the anomeric methoxy (Figure 3B) and hinders the rotation of the carbonyl group, securing higher enantiodiscrimination.
Figure 3.

Transition state models: (A)—31; and (B)—27.
Also, the results for the acetate derivatives tested suggest that various coordination modes are possible for the ligand–diethylzinc system, thus highly altering the transition state geometry and, as a consequence, the observed asymmetric induction.
In conclusion, we have synthesized a series of novel 3-thio-derivatives of d-glucosamine. New compounds were applied as ligands for the diethylzinc addition to benzaldehyde and the products of the addition were obtained with a low to good enantiomeric ratio.
4. Material and Methods
Benzaldehyde was distilled under reduced pressure and stored under argon. Diethylzinc solution (1.1 M in toluene) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All the reactions were performed under an argon atmosphere in oven-dried glassware using the Schlenk technique when necessary. The 1H and 13C NMR spectra were recorded in CDCl3 using a Bruker AVANCE 300 MHz spectrometer (Bruker, Billerica, MA, USA). All the chemical shifts are quoted in parts per million relatives to tetramethylsilane (δ, 0.00 ppm) as the internal standard. The 1H-NMR splitting patterns are abbreviated as follows: singlet (s), doublet (d), triplet (t), quartet (q), dd (doublet of doublets), and m (multiplets). The high-resolution mass spectra were recorded on a Quattro LC Micromass, LCT Micromass TOF HiRes, and Shimadzu LCMS-9030 apparatus. Flash column chromatography was performed on silica gel (Kieselgel-60, Merck, 230–400 mesh). TLC analyses were performed on 250 μm Silica Gel 60 F254 plates (Merck, Rahway, NJ, USA) and visualized by the quenching of the UV fluorescence at 254 nm or by staining with molybdic acid–cerium (II) sulphate solution. High-performance liquid chromatography was conducted on a Knauer chromatograph (Berlin, Germany) equipped with a diode array detector using a Diacel Chiralcel OD-H column (Osaka, Japan). Compounds 4, 5 and 17 were synthesized according to published procedures [4].
The Supplementary Material (Figures S1–S27) contains the 1H and 13C NMR spectra as PDF copies.
4.1. General Procedure for Diethylzinc Addition to Benzaldehyde
In the Schlenk tube equipped with a stirring element, a ligand was placed (0.1 equiv.), and the tube was flushed three times with argon. Then, the solvent was added (1.25 mL). After 10 min, 1.1 M ZnEt2 in toluene (2 equiv., 0.945 mL) was added. After 30 min of stirring at room temperature, freshly distilled benzaldehyde (0.5 mmol, 51 μL) was added, and then the reaction was stirred at the indicated time until TLC showed the disappearance of the aldehyde. The reaction was diluted with diethyl ether and quenched with 1 M HCl. The organic layer was separated, and the aqueous layer was extracted twice with ether. The combined organic layers were then concentrated and purified by column chromatography (hexane-EtOAc 7:3) to give 1-phenyl-1-propanol as a colorless oil. The 1H-NMR (300 MHz, CDCl3) δ 7.43–7.23 (m, 5H, C6H5), 4.62 (t, J 6.6 Hz, 1H, PhCH(OH)Et), 1.90–1.70 (m, 2H, CH2), and 0.95 (t, J 7.4 Hz, 3H, CH3).
4.2. General Procedure for Diethylzinc Addition to Benzaldehyde in the Presence of Ti(OiPr4)
In the Schlenk tube equipped with a stirring element, a ligand was placed (0.1 equiv.), and the tube was flushed three times with argon. Then, methylene chloride was added (3 mL), followed by Ti(OiPr)4 (1.4 equiv., 0.24 mL). After 1.5 h, the reaction mixture was cooled to 0 °C and 1.1 M ZnEt2 (3 equiv., 1.35 mL) was added. After 45 min, an aldehyde (0.5 mmol) was added and stirred at 0 °C for an additional 30 min, then allowed to warm up to room temperature. The reaction was stirred at this temperature until TLC showed the disappearance of the aldehyde. The reaction was diluted with diethyl ether and quenched with 1 M HCl. The organic layer was separated, and the aqueous layer was extracted twice with ether. The combined organic layers were then concentrated and purified by column chromatography (hexane-EtOAc 7:3) to give 1-phenyl-1-propanol as a colorless oil. The 1H-NMR (300 MHz, CDCl3) δ 7.43–7.23 (m, 5H, C6H5), 4.62 (t, J 6.6 Hz, 1H, PhCH(OH)Et), 1.90–1.70 (m, 2H, CH2), and 0.95 (t, J 7.4 Hz, 3H, CH3).
4.3. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-p-toluenesulfonamide-α-d-glucopyranoside (6)
To the stirred solution of 5 (3.4 g, 7.9 mmol) in DCM (60 mL) at 0 °C, triethylamine (1.6 g, 15.8 mmol) and MsCl (1.22 mL, 15.8 mmol) were added. The reaction was left at 0 °C for 30 min, then stirred at room temperature for 3 h. The volatiles were removed under a vacuum. The organic phases were washed with water and brine and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 3:2) to yield methanesulfonamide 6 as a white solid (2.9 g, 74%). The 1H-NMR (300 MHz, CDCl3) δ 7.84 (d, J 8.2 Hz, 2H), 7.51–7.30 (m, 7H), 5.52 (s, 1H), 5.21 (d, J 9.8 Hz, 1H), 4.82–4.63 (m, 2H), 4.31 (dd, J 9.8, 4.2 Hz, 1H), 3.91–3.79 (m, 1H), 3.80–3.63 (m, 2H), 3.62–3.50 (m, 1H), 3.39 (s, 3H), 2.93 (s, 3H), and 2.45 (s, 3H). The 13C-NMR (75 MHz, CDCl3) δ 144.0, 137.1, 136.5, 129.9, 129.3, 128.4, 127.4, 126.0, 101.8, 99.7, 79.1, 68.7, 62.7, 56.3, 55.9, 52.6, 38.9, and 21.6. HRMS (ESI-TOF) calcd. for C22H27NO9S2Na [M + Na]+: 536.1023; found 536.1013.
4.4. Methyl 4,6-O-Benzylidene-2-deoxy-2-p-toluenesulfonamide-α-d-allopyranoside (7)
To the stirred solution of a substrate, 6 (2.6 g, 5 mmol) in 2-methoxyethanol/H2O (10:1, 83 mL), sodium acetate anhydrous (4 g, 48 mmol) was added. The reaction was stirred at 110 °C for 46 h. Then, the reaction was cooled to room temperature, the solvent was removed under a vacuum, and the crude was diluted with DCM. The organic phase was washed with water and brine, and dried over anhydrous MgSO4. The yellow residue was purified by column chromatography (hexane-EtOAc 3:2) to give 7 as a white solid (2.1 g, 95%). The 1H-NMR (300 MHz, CDCl3) δ 7.81–7.61 (m, 2H), 7.40–7.16 (m, 5H), 7.05–6.93 (m, 2H), 5.71 (d, J 9.4 Hz, 1H), 5.48 (s, 1H), 4.68 (d, J 1.2, 0.6 Hz, 1H), 4.27 (dd, J 10.1, 4.7 Hz, 1H), 4.08 (ddd, J 5.7, 2.9, 1.2 Hz, 1H), 4.02 (dd, J 9.8, 4.1 Hz, 1H), 3.94–3.83 (m, 2H), 3.76 (t, J 9.9 Hz, 1H), 3.47 (s, 3H), 2.83 (d, J 5.8 Hz, 1H), and 2.29 (s, 3H). The 13C-NMR (75 MHz, CDCl3) δ 143.1, 137.5, 137.2, 129.3, 128.9, 128.1, 127.4, 126.2, 101.9, 101.7, 73.7, 70.3, 69.1, 59.6, 55.8, 53.6, and 21.6. HRMS (ESI-TOF) calcd. for C21H25NO7SNa [M + Na]+: 458.1249; found 458.1249.
4.5. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-p-toluenesulfonamido-α-d-allopyranoside (8)
To the stirred solution of 7 (2.1 g, 4.9 mmol) in DCM (60 mL) at 0 °C, triethylamine (1.38 mL, 9.9 mmol) and mesyl chloride (0.7 mL, 9.9 mmol) were added. The reaction was left at 0 °C for 30 min, then stirred at room temperature for 3 h. The solvent was removed under a vacuum, and the organic phase was washed with water and brine and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 3:2) to yield methanesulfonamide 8 as a white solid (2.3 g, 90%). The 1H-NMR (300 MHz, CDCl3) δ 7.75 (d, J 8.3 Hz, 2H), 7.45–7.25 (m, 5H), 7.16–6.98 (m, 2H), 5.58 (d, J 8.8 Hz, 1H), 5.51 (s, 1H), 4.83 (t, J 1.2 Hz, 1H), 4.77 (dd, J 2.7, 1.3 Hz, 1H), 4.33 (dd, J 9.4, 3.2 Hz, 1H), 4.04–3.90 (m, 3H), 3.87–3.72 (m, 1H), 3.49 (s, 3H), 3.15 (s, J 4.9 Hz, 3H), and 2.36 (s, 3H). The 13C-NMR (75 MHz, CDCl3) δ 143.5, 136.8, 136.7, 129.4, 129.05, 128.0, 127.4, 126.1, 101.7, 99.3, 75.9, 72.8, 68.9, 58.9, 56.0, 51.6, 37.9, and 21.5. HRMS (ESI-TOF) calcd. for C22H27NO9S2Na [M + Na]+: 536.1025; found 536.1038.
4.6. Methyl 4,6-O-Benzylidene-2,3-dideoxy-3-thioacetyl-2-p-toluenesulfonamido-α-d-glucopyranoside (9)
Mesylate 8 (200 mg, 0.38 mmol) was stirred in acetonitrile (8 mL) under argon, and then potassium thioacetate (222 mg, 1.94 mmol) was added. The reaction was stirred at 75 °C for 1.5 h. Then, the solvent was removed under a vacuum, and the crude was diluted with dichloromethane. The organic phases were washed with water and brine, and dried over anhydrous MgSO4. The brown residue was purified by column chromatography (hexane-EtOAc 3:2) to give thioacetate 9 as a yellow solid (171 mg, 89%). The 1H-NMR (300 MHz, CDCl3) δ 7.81–7.70 (m, 2H), 7.42–7.23 (m, 5H), 7.18–7.00 (m, 2H), 5.92 (d, J 9.2 Hz, 1H), 5.52 (s, 1H), 4.59 (q, J 0.8 Hz, 1H), 4.26 (dd, J 10.0, 4.2 Hz, 1H), 4.06–3.89 (m, 3H), 3.–3.63 (m, 2H), 3.48 (s, 3H), 2.35 (s, 3H), and 2.32 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 191.7, 143.0, 138.0, 136.9, 129.4, 129.0, 128.1, 127.3, 126.2, 101.8, 101.6, 75.2, 69.0, 59.5, 56.1, 53.2, 46.0, 30.4, and 21.6. HRMS (ESI-TOF) calcd. for C23H27NO7S2[M + Na]+: 516.1127; found 516.1138.
4.7. Methyl 4,6-O-Benzylidene-2,3-dideoxy-3-thio-2-p-toluenesulfonamido-α-d-glucopyranoside (10)
Lithium aluminum hydride (202 mg, 5.32 mmol) was stirred in dry THF (12 mL) at 0 °C, and then thioacetate substrate 9 (376 mg, 0.76 mmol) in dry THF (12 mL) was added. The reaction was stirred at 0 °C for 30 min, then continued stirring at room temperature for 4 h. After completion of the reaction, the reaction was worked up according to the Fieser workup: diluted with diethyl ether, followed by water, 15% aq. NaOH solution, water again, then dried with anhydrous MgSO4. The organic solvent was evaporated under reduced pressure, and the crude was purified by column chromatography (hexane-EtOAc 7:3) to yield 10 as a white solid (145 mg, 41%). The 1H-NMR (300 MHz, CDCl3) δ 7.79–7.54 (m, 2H), 7.44–7.24 (m, 5H), 7.08–6.95 (m, 2H), 5.83 (d, J 9.5 Hz, 1H), 5.52 (s, 1H), 4.75 (q, J 0.9 Hz, 1H), 4.27 (dd, J 10.2, 4.9 Hz, 1H), 4.13 (dd, J 9.9, 4.2 Hz, 1H), and 4.04–3.87 (m, 2H), 3.79 (t, J 10.1 Hz), 3.45 (s, 3H), 3.36 (ddd, J 9.9, 2.4, 0.9 Hz, 1H), 2.34 (s, 3H), 2.13 (d, J 9.8 Hz, 1H). The 13C-NMR (75 MHz, CDCl3) δ 143.0, 137.4, 136.9, 129.2, 128.9, 128.0, 127.3, 126.2, 102.9, 101.8, 72.7, 69.0, 59.7, 55.8, 55.4, 41.8, and 21.5. HRMS (ESI-TOF) calcd. for C21H25NO6S2Na [M + Na]+: 474.1021; found 474.1017.
4.8. Methyl 4,6-O-Benzylidene-2-deoxy-2-(4-morpholinyl)-α-d-glucopyranoside (11)
To the stirred solution of amino alcohol 4 (1.83 g, 6.624 mmol) solution in acetonitrile, potassium carbonate (1.83 g, 13.24 mmol) and bis(2-bromoetyl)-diethyl ether (1.24 mL, 9.936 mmol) were added. The reaction stirred for 20 h under reflux. The solvent was evaporated under reduced pressure, and the crude was diluted in DCM. The organic phase was washed with water and brine, and dried over anhydrous MgSO4. Purification by flash column chromatography (hexane-EtOAc 1:5) gave 11 as a white solid (1.1 g, 47%). The 1H NMR (300 MHz, CDCl3) δ 7.59–7.46 (m, 2H), 7.44–7.32 (m, 3H), 5.59 (s, 1H), 4.87 (d, J 3.1 Hz, 1H), 4.32–4.26 (m, 1H), 4.20 (dd, J 10.5, 8.7 Hz, 1H), 3.90–3.77 (m, 2H), 3.70–3.63 (m, 4H), 3.59 (t, J 8.9 Hz, 1H), 3.42 (s, 3H), 3.13 (d, J 0.9 Hz, 1H), 2.86 (dt, J 6.0, 3.1 Hz, 4H), and 2.72 (dd, J 10.6, 3.2 Hz, 1H).
4.9. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-(4-morpholinyl)-α-d-glucopyranoside (12)
The substrate 11 (1000 mg, 2.84 mmol) was stirred in dry dichloromethane (10 mL) and pyridine (10 mL) for 5 min at room temperature, and then mesyl chloride (977.68 mg, 8.53 mmol) was added dropwise. The reaction was stirred under argon for 4 h, then quenched with methanol. The solvent was removed under reduced pressure and diluted with dichloromethane. The organic phases were washed with water and brine and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:2) to give mesylate 12 as a white solid (900 mg, 73%). The 1H NMR (300 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.42–7.37 (m, 3H), 5.57 (s, 1H), 5.17 (dd, J 10.8, 9.0 Hz, 1H), 4.90 (d, J 3.2 Hz, 1H), 4.32 (dd, J 10.1, 4.6 Hz, 1H), 3.91 (td, J 9.8, 4.5 Hz, 1H), 3.83–3.73 (m, 2H), 3.73–3.64 (m, 4H), 3.43 (s, 3H), 2.98 (s, 3H), 2.96 (d, J 3.2 Hz, 1H), and 2.91 (m, 4H). The 13C-NMR (75 MHz, CDCl3) δ 136.7, 129.4, 128.4, 126.1, 101.9, 100.4, 81.0, 76.8, 69.1, 67.8, 67.1, 62.4, 54.8, 50.5, and 39.2. HRMS (ESI-TOF) calcd. for C19H27NO8SNa [M + Na]+: 452.1355; found 452.1364.
4.10. Methyl 4,6-O-Benzylidene-2-deoxy-2-(4-morpholinyl)-α-d-allopyranoside (13)
The substrate 12 (1.15 g, 2.67 mmol) was suspended in 2-methoxyethanol/H2O (10:1, 36 mL) and reacted with anhydrous sodium acetate (1.7 g, 20 mmol) at 115–125 °C for 72–120 h. The solvent was removed under reduced pressure, the residue was diluted with DCM, and the organic phases were washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (4% MeOH-DCM) to give hydroxyl 13 as a white solid (440 mg, 46%). The 1H NMR (300 MHz, CDCl3) δ 7.50–7.44 (m, 2H) 7.42–7.36 (m, 3H), 5.51 (s, 1H), 4.55 (d, J 1.9 Hz, 1H), 4.40–4.28 (m, 2H), 4.26–4.11 (m, 2H), 3.79–3.64 (m, 6H), 3.43 (s, 3H), 3.09–2.94 (m, 3H), and 2.87–2.78 (m, 2H). The 13C-NMR (75 MHz, CDCl3) δ 137.7, 129.1, 128.4, 126.2, 102.6, 101.5, 78.7, 69.8, 68.8, 67.6, 64.9, 60.0, 55.3, and 52.9. HRMS (ESI-TOF) calcd. for C18H26NO6 [M + H]+: 352.1760; found 352.1749.
4.11. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-(4-morpholinyl)-α-d-allopyranoside (14)
The substrate 13 (600 mg, 1.7 mmol) was stirred in dry DCM (6 mL) and pyridine (6 mL) for 5 min at room temperature, and then mesyl chloride (586 mg, 5.12 mmol) was added dropwise. The reaction was stirred under argon for 4 h, then quenched with MeOH. The solvent was removed under reduced pressure and diluted with DCM. The organic phases were washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:2) to give mesylate 14 as a white solid (500 mg, 68%). The 1H-NMR (300 MHz, CDCl3) δ 7.51–7.43 (m, 2H), 7.43–7.35 (m, 3H), 5.52 (s, 1H), 5.05 (dd, J 2.5, 1.2 Hz, 1H), 4.72 (p, J 0.6 Hz, 1H), 4.42–4.31 (m, 2H), 4.16–4.06 (m, 1H), 3.76–3.67 (m, 5H) 3.41 (s, 3H), 3.18 (ddd, J 4.6, 2.5, 0.8 Hz, 1H) 3.11 (s, 3H), 3.11–3.04 (m, 2H), and 2.79 (m, 2H). The 13C-NMR (75 MHz, CDCl3) δ 137.4, 129.2, 128.4, 126.1, 102.7, 98.6, 77.6, 75.9, 69.5, 67.4, 63.3, 59.1, 55.5, 52.9, and 38.7. HRMS (ESI-TOF) calcd. for C19H27NO8SNa [M + Na]+: 452.1355; found 452.1369.
4.12. Methyl 4,6-O-Benzylidene-2,3-dideoxy-2-(4-morpholinyl)-3-thioacetyl-α-d-glucopyranoside (15)
Mesylate 14 (311 mg, 0.72 mmol) was diluted in anhydrous DMF (20 mL) under argon, and then potassium thioacetate (331 mg, 2.89 mmol) was added. The reaction was stirred at 100 °C for 24 h. Then, the solvent was removed under a vacuum, and the crude was diluted with EtOAc. The organic phases were washed with water, saturated aq. NH4Cl solution, and brine, and dried over anhydrous MgSO4. The brown residue was purified by column chromatography (hexane-EtOAc 7:3) to give thioacetate 15 as a white solid (210 mg, 70%). The 1H-NMR (300 MHz, CDCl3) δ 7.43–7.34 (m, 5H), 5.46 (s, 1H), 4.54 (dt, J 0.9, 0.5 Hz, 1H), 4.43 (td, J 9.9, 5.5 Hz, 1H), 4.31 (d, J 1.2 Hz, 1H), 4.28 (t, J 5.0 Hz, 1H), 3.93 (dd, J 9.6, 3.8 Hz, 1H), 3.81–3.70 (m, 4H), 3.72–3.60 (m, 1H), 3.39 (s, 3H), 3.10–2.99 (m, 2H), 2.98 (dd, J 1.7 Hz, 1H), 2.88 (m, 2H), and 2.40 (s, 3H). The 13C-NMR (75 MHz, CDCl3) δ 193.2, 137.6, 129.2, 128.4, 126.2, 102.6, 100.6, 79.9, 69.5, 67.5, 64.7, 58.8, 55.4, 53.5, 43.7, and 30.4. HRMS (ESI-TOF) calcd. for C20H27NO6SNa [M + Na]+: 432.1457; found 432.1472.
4.13. Methyl 4,6-O-Benzylidene-2,3-dideoxy-2-(4-morpholinyl)-3-thio-α-d-glucopyranoside (16)
Lithium aluminum hydride (130 mg, 3.42 mmol) was stirred in dry THF (7 mL) at 0 °C, and then thioacetate 16 (175 mg, 0.42 mmol) in dry THF (7 mL) was added. The reaction was stirred at 0 °C for 30 min, then continued at room temperature for 2.5 h. After completion of the reaction, the reaction was quenched according to the Fieser workup: diluted with diethyl ether, followed by water, 15% aq. NaOH solution (0.5 mL), water again, then dried over anhydrous MgSO4. The organic solvent was evaporated under reduced pressure, and the crude was purified by column chromatography (hexane-Et2O 1:1) to obtain ligand 16 as a white solid (120 mg, 76%). The 1H NMR (300 MHz, CDCl3) δ 7.53–7.43 (m, 2H), 7.42–7.35 (m, 3H), 5.51 (s, 1H), 4.69 (d, J 1.4 Hz, 1H), 4.43–4.23 (m, 3H), 3.81–3.61 (m, 5H), 3.58 (ddd, J 8.5, 2.7, 1.5 Hz, 1H), 3.39 (s, 3H), 3.15 (dd, J 4.3, 2.7 Hz, 1H), 3.10–2.95 (m, 2H), 2.95–2.72 (m, 2H), and 2.24 (d, J 8.5 Hz, 1H). The 13C NMR (75 MHz, CDCl3) δ 137.7, 129.1, 128.4, 126.2, 102.7, 102.3, 77.8, 69.7, 67.7, 66.6, 59.8, 55.3, 52.8, and 39.6. HRMS (ESI-TOF) calcd. for C18H25NO5SNa [M + Na]+: 390.1351; found 390.1367.
4.14. Methyl 4,6-O-Benzylidene-2-deoxy-2-methoxycarbonylamido-3-O-trifluoromethanesulfonyl-α-d-glucopyranoside (18)
The substrate 17 (4 g, 11.7 mmol) was dissolved in dry dichloromethane (117 mL) and pyridine (9.4 mL, 117 mmol) was added at −20 °C. The reaction was stirred for 10 min before triflic anhydride (7.4 g, 4.36 mL, 35.3 mmol) was added dropwise under argon and left at that temperature for 2 h. The reaction was then quenched with saturated aq. NaHCO3, and the organic phase were separated. The organic phase was washed three times with 3% aq. CuSO4 solution, then with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:2) to provide the triflate 18 a white–yellowish solid (5.1 g, 94% yield). The 1H-NMR (300 MHz, CDCl3) δ 7.50–7.43 (m, 2H), 7.39–7.33 (m, 3H), 5.59 (s, 1H), 5.15 (d, J 10.4 Hz, 1H), 5.03–4.90 (m, 1H), 4.78 (d, J 3.7 Hz, 1H), 4.37–4.20 (m, 2H), 3.88–3.77 (m, 3H), 3.72 (s, 3H), and 3.41 (s, 3H). The 13C-NMR (75 MHz, CDCl3) δ 156.4, 136.4, 129.2, 128.3, 125.9, 101.4, 99.7, 84.4, 78.5, 68.6, 63.0, 55.7, 53.8, and 52.8. HRMS (ESI-TOF) calcd. for C19H27NO5Na [M + Na]+: 372.17814; found 372.17850.
4.15. Methyl 4,6-O-Benzylidene-2-deoxy-2-methoxycarbonylamido-α-d-allopyranoside (19)
The triflate 18 (5.2 g, 11.03 mmol) was dissolved in dry DMF (55 mL), and sodium nitrite (5.2 g, 77.2 mmol) was added. The reaction was stirred at room temperature under argon for six days before the solvent was evaporated under a vacuum and the residue was extracted with ethyl acetate, and the organic phase was washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:2) to provide the hydroxyl 19 as a white solid (3 g, 67%). The 1H NMR (300 MHz, CDCl3) δ 7.54–7.44 (m, 2H), 7.44–7.31 (m, 3H), 5.61 (s, 1H), 5.52 (d, J 9.6 Hz, 1H), 4.76 (d, J 4.0 Hz, 1H), 4.38 (dd J 10.4 Hz, 1H) 4.25–4.18 (m, 1H), 4.18–4.07 (m, 1H), 3.99 (dd, J 9.4, 3.9 Hz, 1H), 3.79 (t, J 10.3 Hz, 1H), 3.71 (s, 3H), 3.64 (dd, J 9.4, 2.8 Hz, 1H), 3.45 (s, 3H), and 2.62 (d, J 6.5 Hz, 1H). The 13C NMR (75 MHz, CDCl3) δ 156.5, 137.1, 129.3, 128.4, 126.3, 102.0, 99.5, 78.6, 69.2, 68.4, 57.5, 56.3, 52.4, and 51.3. HRMS (ESI-TOF) calcd. for C16H21NO7Na [M + Na]+: 362.1202; found 362.12198.
4.16. Methyl 4,6-O-Benzylidene-2-deoxy-2-amino-α-d-allopyranoside (20)
The substrate 19 (800 mg, 2.37 mmol) was dissolved in a solution of 4 N KOH in ethanol:2-methoxyethanol (2:1, 0.15 M, 15.7 mL) and the mixture was refluxed overnight. The dark brown residue was neutralized with a few amounts of 1 N HCl and extracted with chloroform three times. The organic phase was separated and washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (EtOAc-MeOH 5:2) to afford the amino alcohol 20 as a white solid (332 mg, 50%). The 1H NMR (300 MHz, CDCl3) δ 7.54–7.47 (m, 2H), 7.40–7.30 (m, 3H), 5.57 (s, 1H), 4.64 (dd, J 3.8, 0.7 Hz, 1H), 4.36 (dd, J 10.2, 5.1 Hz, 1H), 4.15–3.99 (m, 2H), 3.75 (t, J 10.3 Hz, 1H), 3.52 (dd, J 9.7, 2.7 Hz, 1H), 3.44 (s, 3H), 2.94 (t, J 3.8, 3.1 Hz, 1H), and 2.06 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 137.3, 129.2, 128.3, 126.3, 102.1, 102.0, 79.4, 70.7, 69.4, 57.4, 56.2, and 52.5. HRMS (ESI-TOF) calcd. for C14H20NO5 [M + H]+: 282.13360; found 282.13348.
4.17. Methyl 4,6-O-Benzylidene-2-deoxy-2-(1-pyrrolidinyl)-α-d-allopyranoside (21)
To the stirred solution of amino alcohol substrate 20 (150 mg, 0.54 mmol) in acetonitrile (5 mL), potassium carbonate (149 mg, 1.08 mmol), and 1,4-dibromobutane (350 mg, 1.62 mmol) were added. The reaction was stirred for 24 h under reflux. The solvent was evaporated under reduced pressure, and the crude was diluted in chloroform. The organic phase was washed with water and brine, and dried over anhydrous MgSO4. Purification by flash column chromatography (EtOAc-MeOH 5:2) gave 21 as a white solid (160 mg, 88%). The 1H NMR (300 MHz, CDCl3) δ 7.57–7.47 (m, 2H), 7.44–7.26 (m, 3H), 5.59 (s, 1H), 4.79 (d, J 3.5, 0.8 Hz, 1H), 4.37 (dd, J 10.3, 5.3 Hz, 1H), 4.31 (t, J 2.5 Hz, 1H), 4.21 (td, J 9.8, 5.1 Hz, 1H), 3.76 (t, J 10.2 Hz, 1H), 3.54 (dd, J 9.7, 2.7 Hz, 1H), 3.45 (s, 3H), 2.82–2.68 (m, 2H), 2.66–2.52 (m, 2H), 2.37–2.28 (t, J 3.5, 2.8 Hz, 1H), and 1.95–1.82 (m, 4H). The 13C NMR (75 MHz, CDCl3) δ 137.3, 129.0, 128.2, 126.4, 102.1, 99.9, 79.6, 69.3, 67.1, 66.6, 57.8, 55.7, 51.5, and 23.0. HRMS (ESI-TOF) calcd. for C18H25NO5SNa [M + Na]+: 358.16249; found 358.16291.
4.18. Methyl 4,6-O-Benzylidene-2-deoxy-2-(1-piperidinyl)-α-d-allopyranoside (22)
To the stirred solution of 20 (400 mg, 1.44 mmol) in acetonitrile (18 mL), potassium carbonate (397 mg, 4.32 mmol) and 1,5-dibromobutane (0.58 mL, 993 mg, 4.32 mmol) were added. The reaction was stirred for 40 h under reflux. The solvent was evaporated under reduced pressure, and the crude was diluted in chloroform. The organic phase was washed with water and brine, and dried over anhydrous MgSO4. Purification by flash column chromatography (EtOAc-MeOH 5:2) gave 22 as a white solid (355 mg, 70%). The 1H NMR (300 MHz, CDCl3) δ 7.58–7.46 (m, 2H), 7.40–7.30 (m, 3H), 5.58 (s, 1H), 4.90 (d, J 3.4, 1H), 4.43 (t, J 2.7, 2.6 Hz, 1H), 4.36 (dd, J 10.1, 5.2 Hz, 1H), 4.20 (td, J 10.0, 5.1 Hz, 1H), 3.75 (t, J 10.2 Hz, 1H), 3.49 (dd, J 9.7, 2.7 Hz, 1H), 3.43 (s, 3H), 2.76–2.61 (m, 2H), 2.55–2.40 (m, 2H), 2.39 (t, J 3.5, 2.7 Hz, 1H), 1.70–1.60 (m, 4H), and 1.48 (m, J 6.1 Hz, 2H). The 13C NMR (75 MHz, CDCl3) δ 135.4, 129.1, 128.3, 126.4, 102.2, 99.1, 80.0, 69.4, 66.1, 65.1, 57.8, 55.5, 51.1, 28.5, and 25.8. HRMS (ESI-TOF) calcd. for C19H27NO5Na [M + Na]+: 372.17814; found 372.1770.
4.19. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-(1-pyrrolidinyl)-α-d-allopyranoside (23)
The pyrrolidine substrate 21 (80 mg, 0.23 mmol) was stirred in 1:1 dry DCM:pyridine (1.5 mL) for 5 min at 0 °C, and then mesyl chloride (81 mg, 0.05 mL, 0.71 mmol) was added dropwise. The reaction was stirred under argon for 4 h at room temperature, then quenched with methanol. The solvent was removed under reduced pressure and diluted with dichloromethane. The organic phases were washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:4) to give product 23 as a white solid (50 mg, 52%). The 1H NMR (300 MHz, CDCl3) δ 7.53–7.45 (m, 2H), 7.36 (dd, J 5.1, 1.8 Hz, 3H), 5.60 (s, 1H), 5.37 (t, J 2.8 Hz, 1H), 4.78 (d, J 3.7 Hz, 1H), 4.37 (dd, J 10.3, 5.2 Hz, 1H), 4.21 (td, J 10.0, 5.2 Hz, 1H), 3.79–3.68 (m, 2H), 3.44 (s, 3H), 3.05 (s, 3H), 2.79 (m, 2H), 2.58 (m, 2H), 2.46 (t, J 3.3 Hz, 1H), and 1.84 (ddd, J 6.7, 4.6, 2.1 Hz, 4H). The 13C NMR (75 MHz, CDCl3) δ 136.9, 129.2, 128.4, 126.1, 101.8, 99.2, 75.8, 69.2, 65.7, 58.1, 55.8, 51.9, 39.5, and 23.0. HRMS (ESI-TOF) calcd. for C19H27NO7SNa [M + Na]+: 436.14004; found 436.13993.
4.20. Methyl 4,6-O-Benzylidene-2-deoxy-3-O-methanesulfonyl-2-(1-piperidinyl)-α-d-allopyranoside (24)
The substrate 22 (100 mg, 0.28 mmol) was stirred in dry DCM:pyridine (1:1, 3 mL) for 5 min at room temperature, and then mesyl chloride (98 mg, 0.85 mmol) was added dropwise. The reaction was stirred under argon for 4 h at room temperature, then quenched with methanol. The solvent was removed under reduced pressure and diluted with DCM. The organic phases were washed with water and brine and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:3) to give the product 24 as a white solid (72 mg, 60%). The 1H NMR (300 MHz, CDCl3) δ 7.56–7.43 (m, 2H), 7.42–7.32 (m, 3H), 5.58 (s, 1H), 5.46 (t, J 2.8 Hz, 1H), 4.85 (d, J 3.0 Hz, 1H), 4.36 (dd, J 10.3, 5.2 Hz, 1H), 4.20 (td, J 10.0, 5.2 Hz, 1H), 3.73 (t, J 10.3 Hz, 1H),3.66 (dd, J 9.7, 2.7 Hz, 1H), 3.42 (s, 3H), 3.09 (s, 3H), 2.77 (m, 2H), 2.54 (m, 3H), 1.70–1.55 (m, 4H), and 1.51–1.41 (m, 2H). The 13C NMR (75 MHz, CDCl3) δ 136.5, 128.7, 127.9, 125.7, 101.3, 98.5, 76.8, 74.3, 68.8, 65.0, 57.8, 55.0, 51.3 39.1, 25.6, 25.1, and 23.9. HRMS (ESI-TOF) calcd. for C20H29NO7SNa [M + Na]+: 450.1556; found 450.1558.
4.21. Methyl 4,6-O-Benzylidene-2-deoxy-2-methanesulfonamido-3-O-methanesulfonyl-α-d-allopyranoside (25)
To the stirred solution of amino alcohol 20 (300 mg, 1.06 mmol) in dry pyridine (5 mL) at 0 °C, mesyl chloride (0.24 mL, 3.19 mmol) was added dropwise. The reaction was left stirring for 24 h, before the solvent was evaporated under a vacuum, and was then diluted with dichloromethane. The organic phase was washed with saturated NaHCO3, water, and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:1) to afford the product 25 as a white solid (350 mg, 75%). The 1H NMR (300 MHz, CDCl3) δ 7.52–7.31 (m, 5H), 5.59 (s, 1H), 5.20 (t, J 3.2 Hz, 1H), 5.13 (d, J 9.7 Hz, 1H), 4.75 (d, J 4.4 Hz, 1H), 4.39 (dd, J 10.6, 5.2 Hz, 1H), 4.13 (td, J 10.1, 5.2 Hz, 1H), 4.00 (dt, J 9.7, 4.0 Hz, 1H) 3.82–3.67 (m, 2H), 3.45 (s, 3H), 3.08 (s, 3H), and 2.97 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 136.7, 129.5, 128.5, 126.0, 102.0, 98.9, 77.5, 76.1, 69.1, 57.6, 56.5, 51.9, 43.6, and 39.3. HRMS (ESI-TOF) calcd. for C16H23NO9S2Na [M + Na]+: 460.07064; found 460.07192.
4.22. Methyl 4,6-O-Benzylidene-2,3-dideoxy-2-methanesulfonamido-3-thioacetyl-α-d-glucopyranoside (26)
The mesylate substrate 25 (350 mg, 0.822 mmol) was dissolved in DMPU (8 mL) and treated with potassium thioacetate (469 mg, 4.11 mmol) at 100 °C for 6 h. The suspension was cooled and diluted with 1:1 EtOAc-Et2O (100 mL) and washed with saturated NaHCO3 solution (3 × 40 mL). The organic phase was washed with water and brine, and dried over anhydrous MgSO4. The crude was purified by column chromatography (hexane-EtOAc 1:1) to afford the inseparable mixture of the thioacetyl product 26 and the elimination adduct (180 mg, 60% product). The pure thioacetyl compound 26 was obtained by following this method: to the stirred a solution of thiol 27 (10 mg, 0.02 mmol) in dichloromethane (1 mL), triethylamine (0.01 mL, 0.06 mmol) and acetic anhydride (0.01 mL, 0.09 mmol) were added dropwise at 0 °C and stirred for 30 min. The solvent was then evaporated, and the crude was directly purified by column chromatography (hexane-EtOAc 1:1) to afford pure thioacetyl 26 (15 mg, 75%) as a white solid. The 1H NMR (300 MHz, CDCl3) δ 7.52–7.43 (m, 2H), 7.43–7.30 (m, 3H), 5.50 (s, 1H), 4.90 (d, J 10.1 Hz, 1H), 4.82 (d, J 3.5 Hz, 1H), 4.28 (dd, J 10.3, 4.7 Hz, 1H), 3.95 (t, J 11.4 Hz, 1H), 3.93–3.84 (m, 1H), 3.71 (t, J 10.3 Hz, 1H), 3.65 (ddd, J 11.7, 10.1, 3.5 Hz, 1H), 3.54 (dd, J 11.2, 9.0 Hz, 1H), 3.47 (s, 3H), 2.99 (s, 3H), and 2.37 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 195.0, 137.0, 129.2, 128.4, 126.3, 102.0, 99.7, 78.0, 69.0, 64.5, 57.5, 55.7, 45.3, 42.4, and 31.0. HRMS (ESI-TOF) calcd. for C17H23NO7S2Na [M + Na]+: 440.08082; found 440.08197.
4.23. Methyl 4,6-O-Benzylidene-2,3-dideoxy-2-methanesulfonamido-3-thio-α-d-glucopyranoside (27)
Lithium aluminum hydride (76 mg, 2.01 mmol) was stirred in dry THF (5 mL) at 0 °C, and then the mixture of thioacetate substrate 26 and the elimination by-product (220 mg, 0.287 mmol) in dry THF (5 mL) was added. The reaction was stirred at 0 °C for 30 min, then continued at room temperature for 4 h. After completion of the reaction, the reaction was worked up according to Fieser’s workup: diluted with diethyl ether, followed by water, 15% aq. NaOH solution, water again, then dried over anhydrous MgSO4. The organic solvent was evaporated under reduced pressure, and the crude was purified by column chromatography (hexane: EtOAc 7:3, then with 3:2), yielding 27 as a white solid (70 mg, 65%). The 1H NMR (300 MHz, CDCl3) δ 7.49 (qd, J 4.7, 1.7 Hz, 2H), 7.42–7.33 (m, 3H), 5.55 (s, 1H), 4.86 (d, J 10.3 Hz, 1H), 4.79 (d, J 3.5 Hz, 1H), 4.27 (dd, J 9.3, 3.8 Hz, 1H), 3.82–3.66 (m, 2H), 3.55 (ddd, J 11.1, 10.3, 3.5 Hz, 1H), 3.44 (s, 3H), 3.42–3.27 (m, 2H), 3.13 (s, 3H), and 2.20 (d, J 3.4 Hz, 1H). The 13C NMR (75 MHz, CDCl3) δ 136.9, 129.3, 128.4, 126.2, 102.0, 99.3, 82.2, 68.9, 63.9, 58.9, 55.7, 42.4, and 41.5. HRMS (ESI-TOF) calcd. for C17H23NO7S2Na [M + Na]+: 398.07025; found 398.0703.
4.24. Methyl 4,6-O-Benzylidene-2-deoxy-2-trifluoromethanesulfonamido-α-d-allopyranoside (28) and Methyl 4,6-O-Benzylidene-2-deoxy-2-trifluoromethanesulfonamido-3-O-trifluoromethanesulfonyl-α-d-allopyranoside (29)
To the stirred solution of amino alcohol 20 (300 mg, 1 mmol) in dichloromethane (10 mL) at −20 °C, pyridine (0.9 mL, 10 mmol) was added. The reaction was left stirring for 10 min before triflic anhydride (0.53 mL, 3.1 mmol) was added dropwise under argon. After 2 h of reaction, the saturated aqueous solution of NaHCO3 was added, the aqueous phase was washed with dichloromethane, and the organic phase was washed with water and brine, and dried over anhydrous MgSO4. The crude was purified using column chromatography (hexane-EtOAc 7:3) to afford triflamide 28 (50 mg, 34%), and triflate 29 (200 mg, 51%) as white–yellowish solids. The 1H NMR (300 MHz, CDCl3) δ 7.52–7.43 (m, 2H), 7.42–7.32 (m, 3H), 5.60 (s, 1H), 4.76 (d, J 4.3 Hz, 1H), 4.38 (dd, J 10.3, 4.9 Hz, 1H), 4.28 (t, J 3.2 Hz, 1H), 4.13 (dd, J 10.1, 4.8 Hz, 1H), 3.82–3.71 (m, 2H), 3.62 (dd, J 9.7, 2.8 Hz, 1H), and 3.47 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 136.8, 129.5, 128.5, 126.3, 102.0, 99.3, 78.0, 69.0, 68.7, 57.2, 56.6, and 54.5. The 19F NMR (282 MHz, CDCl3) δ −78.05. HRMS (ESI-TOF) calcd. for C15H18F3NO7S [M−H]−: 412.06833; found 412.66700.
To the stirred solution of triflamide 28 (50 mg, 0.12 mmol) in dichloromethane (2.5 mL) at −20 °C, pyridine (0.1 mL, 1.2 mmol) was added. The reaction was left stirring for 10 min before triflic anhydride (0.02 mL, 0.24 mmol) was added dropwise under argon. After 2 h of reaction, the saturated aqueous solution of NaHCO3 was added, and the aqueous phase was washed with dichloromethane. The organic phase was washed with water and brine, and dried over anhydrous MgSO4. The crude was purified using column chromatography (hexane-EtOAc 7:3) to afford triflate 29 (40 mg, 61%) as a yellow solid. The 1H NMR (300 MHz, CDCl3) δ 7.52–7.44 (m, 2H), 7.44–7.32 (m, 3H), 5.59 (s, 1H), 5.37 (t, J 2.9 Hz, 1H), 4.78 (d, J 4.3 Hz, 1H), 4.38 (dd, J 10.5, 5.2 Hz, 1H), 4.15 (dd, J 9.9, 5.2 Hz, 1H), 4.01 (t, J 3.9 Hz, 1H), 3.85–3.69 (m, 2H), and 3.49 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 135.70, 129.07, 127.91, 125.86, 102.06, 97.07, 81.62, 74.51, 68.27, 57.22, 56.04, and 52.76. The 19F NMR (282 MHz, CDCl3) δ −73.75 and −77.80. HRMS (ESI-TOF) calcd. for C16H17F6NO9S2 [M−H]−: 544.01762; found 544.01646.
4.25. Methyl 4,6-O-Benzylidene-2,3-dideoxy-3-thioacetyl-2-trifluoromethanesulfonamido-α-d-glucopyranoside (30)
To the stirred substrate 29 (92 mg, 0.16 mmol) in dry acetonitrile (3 mL), potassium thioacetate (96 mg, 0.84 mmol) was added under argon. The reaction was stirred at 5 °C to 20 °C for 3 h before the solvent was evaporated and diluted with ethyl acetate. The organic phase was then washed with water and brine, and dried over anhydrous MgSO4. The crude was then purified with column chromatography (hexane-EtOAc 8:2) to afford 30 (30 mg, 40%) as a white solid. The 1H NMR (300 MHz, CDCl3) δ 7.53–7.38 (m, 2H), 7.37 (ddd, J 4.6, 3.2, 2.6 Hz, 3H), 5.51 (s, 1H), 4.79 (d, J 3.4 Hz, 1H), 4.30 (dd, J 10.3, 4.8 Hz, 1H), 4.02 (t, J 11.4 Hz, 1H), 3.91 (td, J 9.7, 4.7 Hz, 1H), 3.79–3.65 (m, 2H), 3.62–3.45 (m, 1H), 3.50 (s, 3H), and 2.39 (s, 3H). The 13C NMR (75 MHz, CDCl3) δ 196.9, 136.8, 129.3, 128.4, 126.3, 102.1, 99.0, 68.9, 64.7, 59.3, 55.9, 44.7, 30.9, 29.8, and 27.0. The 19F NMR (282 MHz, CDCl3) δ −77.55. HRMS (ESI-TOF) calcd. for C17H20F3NO7S2 [M−H]−: 470.05605; found 470.05588.
4.26. Methyl 4,6-O-Benzylidene-2,3-dideoxy-2-trifluoromethanesulfonamido-3-thio-α-d-glucopyranoside (31)
To the stirred substrate 30 (10 mg, 0.02 mmol) in dry methanol (2 mL), sodium methoxide in methanol (0.06 mmol, 0.15 M) was added under argon. The reaction was stirred at room temperature overnight before the solvent was evaporated and diluted with dichloromethane and aqueous solution of NH4Cl. The water phase was acidified with 1 M HCl and then extracted with dichloromethane. The organic phase was then washed with water and brine, and dried over anhydrous MgSO4. The crude was then purified with column chromatography (hexane-EtOAc 8:2) to afford the thiol 31 (7 mg, 76%) as a white solid. The 1H NMR (300 MHz, CDCl3) δ 7.56–7.44 (m, 2H), 7.44–7.32 (m, 3H), 5.57 (s, 1H), 4.76 (d, J 3.5 Hz, 1H), 4.29 (dd, J 9.8, 4.3 Hz, 1H), 3.79 (dd, J 8.7, 4.5 Hz, 1H), 3.71 (t, J 9.9 Hz, 1H), 3.67–3.61 (m, 1H), 3.47 (s, 3H), 3.57–3.35 (m, 1H), 3.35 (dd, J 7.5, 3.3 Hz, 1H), and 2.12 (d, J 4.2 Hz, 1H). The 13C NMR (75 MHz, CDCl3) δ 136.8, 129.4, 128.5, 126.2, 102.1, 98.4, 82.0, 68.8, 64.2, 59.7, 55.8, 41.18, and 29.86. The 19F NMR (282 MHz, CDCl3) δ −77.22. HRMS (ESI-TOF) calcd. for C15H18F3NO6S2 [M−H]−: 428.04549; found 428.04488.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25105542/s1.
Author Contributions
T.B. conceived and designed the experiments; Y.Z.H. performed the experiments, analyzed the data, and wrote the experimental part; T.B. and Y.Z.H. discussed the results and contributed to writing the paper. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data are available from the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
The financial support from the University of Warsaw (5011000301) is gratefully acknowledged.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Kitamura M., Suga S., Kawai K., Noyori R. Catalytic asymmetric induction. highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986;108:6071–6072. doi: 10.1021/ja00279a083. [DOI] [PubMed] [Google Scholar]
- 2.Bauer T. Enantioselective dialkylzinc-mediated alkynylation, arylation and alkenylation of carbonyl groups. Coord. Chem. Rev. 2015;299:83–150. doi: 10.1016/j.ccr.2015.03.025. [DOI] [Google Scholar]
- 3.Bauer T. Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. Elsevier; Amsterdam, The Netherlands: 2024. Organozinc. [DOI] [Google Scholar]
- 4.Bauer T., Tarasiuk J., Paśniczek K. Highly enantioselective diethylzinc addition to aldehydes catalyzed by d-glucosamine derivatives. Tetrahedron Asymmetry. 2002;13:77–82. doi: 10.1016/S0957-4166(02)00053-8. [DOI] [Google Scholar]
- 5.Bauer T., Gajewiak J. α-Hydroxy carboxylic acids as ligands for enantioselective diethylzinc additions to aromatic and aliphatic aldehydes. Tetrahedron. 2004;60:9163–9170. doi: 10.1016/j.tet.2004.07.060. [DOI] [Google Scholar]
- 6.Bauer T., Smoliński S. Enantioselective addition of diethylzinc to aldehydes catalyzed by d-glucosamine derivatives: Highly pronounced effect of trifluoromethylsulfonamide. Appl. Catal. A. 2010;375:247–251. doi: 10.1016/j.apcata.2010.01.009. [DOI] [Google Scholar]
- 7.Kang J., Lee J.W., Kim J.I. Enantioselective addition of diethylzinc to α-branched aldehydes. Chem. Commun. 1994;17:2009–2010. doi: 10.1039/c39940002009. [DOI] [Google Scholar]
- 8.Tseng S., Yang T. The application of chiral amino thiols as catalysts in the enantioselective addition of diethylzinc to aldehydes. Tetrahedron Asymmetry. 2004;15:3375–3380. doi: 10.1002/chin.200512035. [DOI] [Google Scholar]
- 9.Maliszewski B., Bauer T. Enantioselective alkenylation of aldehydes with protected propargylic alcohols in the presence of a crown ether as an additive. Adv. Synth. Catal. 2019;361:3689–3693. doi: 10.1002/adsc.201900363. [DOI] [Google Scholar]
- 10.Wu K.-H., Gau H.-M. Mechanism of Asymmetric Dialkylzinc Addition to Aldehydes Catalyzed by Titanium(IV) Complexes of N-Sulfonylated β-Amino Alcohols. Organometallics. 2004;23:580–588. doi: 10.1021/om034298o. [DOI] [Google Scholar]
- 11.Balsells J.T., Davis J., Carroll P., Walsh P.J. Insight into the Mechanism of the Asymmetric Addition of Alkyl Groups to Aldehydes Catalyzed by Titanium−BINOLate Species. J. Am. Chem. Soc. 2002;124:10336–10348. doi: 10.1021/ja0171658. [DOI] [PubMed] [Google Scholar]
- 12.Emmerson D.P.G., Villard R., Mugnaini C., Batsanov A., Howard J.A.K., Hems W.P., Tooze R.P., Davis B.G. Precise structure-activity relationships in asymmetric catalysis using carbohydrate scaffolds to allow ready fine-tuning: Dialkylzinc-aldehyde additions. Org. Biomol. Chem. 2003;21:3826–3838. doi: 10.1039/b309715n. [DOI] [PubMed] [Google Scholar]
- 13.Albert R., Dax K., Link R.W., Stuetz A.E. Carbohydrate triflates: Reaction with nitrite, leading directly to epi-hydroxy compounds. Carbohydr. Res. 1983;118:C5–C6. doi: 10.1016/0008-6215(83)88062-8. [DOI] [Google Scholar]
- 14.Dong H., Pei Z., Ramström O. Stereospecific Ester Activation in Nitrite-Mediated Carbohydrate Epimerization. J. Org. Chem. 2006;71:3306–3309. doi: 10.1021/jo052662i. [DOI] [PubMed] [Google Scholar]
- 15.Zemplén G., Kuntz A. Studien über Amygdalin, IV: Synthese des natürlichen l-Amygdalins. Berichte Dtsch. Chem. Ges. (A B Ser.) 1924;57:1357–1359. doi: 10.1002/cber.19240570825. [DOI] [Google Scholar]
- 16.Jin M.J., Ahn S.J., Lee K.S. New chiral catalysts for the highly enantioselective addition of diethylzinc to aldehydes. Tetrahedron Lett. 1996;37:8767–8770. doi: 10.1016/S0040-4039(96)02027-8. [DOI] [Google Scholar]
- 17.Jin M.-J., Sarkar S.M., Lee D.-H., Qiu H. Highly Enantioselective Aryl Transfer to Aldehydes: A Remarkable Effect of Sulfur Substitution in Amino Thioacetate Ligands. Org. Lett. 2008;10:1235–1237. doi: 10.1021/ol8001249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available from the authors.