

Abstract
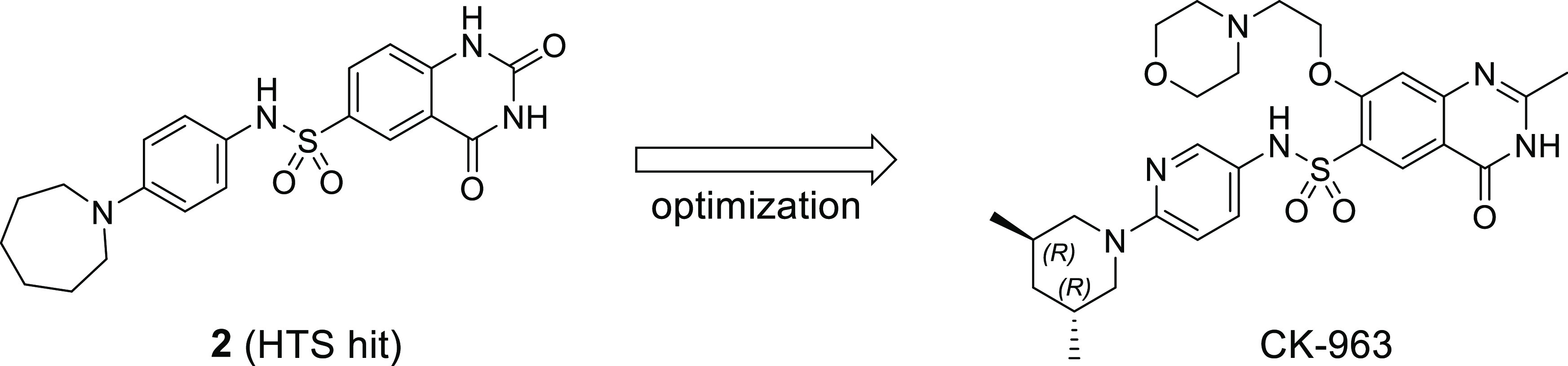
Novel cardiac troponin activators were identified using a high throughput cardiac myofibril ATPase assay and confirmed using a series of biochemical and biophysical assays. HTS hit 2 increased rat cardiomyocyte fractional shortening without increasing intracellular calcium concentrations, and the biological target of 1 and 2 was determined to be the cardiac thin filament. Subsequent optimization to increase solubility and remove PDE-3 inhibition led to the discovery of CK-963 and enabled pharmacological evaluation of cardiac troponin activation without the competing effects of PDE-3 inhibition. Rat echocardiography studies using CK-963 demonstrated concentration-dependent increases in cardiac fractional shortening up to 95%. Isothermal calorimetry studies confirmed a direct interaction between CK-963 and a cardiac troponin chimera with a dissociation constant of 11.5 ± 3.2 μM. These results provide evidence that direct activation of cardiac troponin without the confounding effects of PDE-3 inhibition may provide benefit for patients with cardiovascular conditions where contractility is reduced.
Introduction
Heart failure (HF) is a heterogeneous condition that is defined by the inability of the heart to pump enough blood through the body to enable normal physiological function.1,2 HF affects over 64 million people worldwide, and approximately half will die within five years of initial hospitalization.3 The prognosis for patients remains poor despite recent advances in medical treatment, and the total HF cost is expected to reach $160 billion dollars in the United States by 2030.4,5 New drugs with improved efficacy and safety profiles are clearly needed to support this increasingly large patient population.
Calcitrope6 drugs such as dobutamine, milrinone, and digoxin are among the numerous options for the treatment of HF.7 These compounds show beneficial improvements in cardiac contractility through the activation of secondary messenger pathways that increase cardiomyocyte calcium concentrations, but this mechanism also makes the heart less efficient by increasing myocardial oxygen consumption and activating calcium-dependent signaling cascades.8 Subsequent detrimental changes in myocardial energetics lead to negative clinical outcomes with long-term calcitrope drug use as exemplified by the malignant arrythmias and systemic hypotension that have been observed with the phosphodiesterase-3 (PDE-3) inhibitor milrinone.9,10
Heart Failure with reduced ejection fraction (HFrEF) is a form of HF that affects over 23 million people and occurs when decreased cardiac contractility reduces left ventricular ejection fraction to less than 40% with concomitant progressive left ventricular dilatation and adverse cardiac remodeling.11 Direct activation of the cardiac sarcomere using myotropes,6 compounds that directly interact with the molecular motor or scaffolding of the heart, to increase cardiac contractility without increasing intracellular calcium concentrations is a recent approach for the treatment of HFrEF.12–14 Omecamtiv mecarbil (OM, Figure 1) is the first direct cardiac sarcomere activator that allosterically binds to cardiac myosin and improves cardiac muscle contractility by increasing the rate of phosphate release and the proportion of myosin heads forming force-generating interactions with actin (otherwise known as the duty ratio)15,16 In a phase 3 trial in patients with HFrEF, OM reduced the number of HF events, with patients having the lowest ejection fractions showing the largest response.17 OM did not show an increase in the frequency of cardiac ischemic and ventricular arrhythmia events.
Figure 1.

Structure of OM, a cardiac myosin activator.
The ability of cardiac myosin activator OM to improve clinical outcomes for HF patients prompted our research team to explore the activation of other targets within the cardiac sarcomere to find differentiated and complementary approaches to the treatment of HF. There have been numerous reports of compounds known as calcium sensitizers that sensitize cardiac muscle to calcium and increase force production without increasing intracellular calcium concentrations (Figure 2).14 Most reported calcium sensitizers also have biological activity against other targets like PDE-3 that are known to modulate cardiac function and could obfuscate the pharmacological effect of a selective calcium sensitizer. Levosimendan is a calcium sensitizer that has been approved in many countries for the treatment of acutely decompensated congestive HF.18 Levosimendan binds to cTnC (Kd = 0.1–0.7 mM) but also has numerous other biological activities including single digit nanomolar PDE-3 inhibition that have been proposed to account for the observed clinical effect and could explain an adverse event profile that is similar to other PDE-3 inhibitors.19−24 MCI-154 sensitizes cardiac muscle to calcium but also inhibits PDE-3 at similar potencies.25 EMD 57033 shows similar calcium sensitization and PDE-3 inhibitory activity with increased myofilament ATPase activity in systems devoid of troponin/tropomyosin.26,27 CGP 48506 was the first reported compound to sensitize cardiac muscle to calcium without PDE-3 inhibitory activity, but no in vivo pharmacological studies have been reported with this compound.28,29
Figure 2.
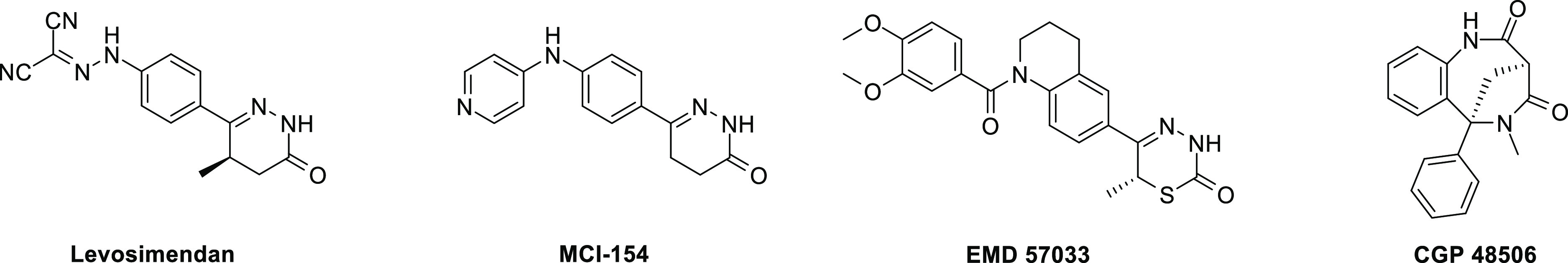
Example of previously reported calcium sensitizers.
Results and Discussion
A research program was initiated with the goal of finding compounds that directly sensitize cardiac troponin to calcium without inhibition of PDE-3 to enable pharmacological investigation. A high throughput screen of an internal compound collection with a pyruvate kinase and lactate dehydrogenase reporter system that measures the rate of adenosine triphosphate (ATP) hydrolysis in cardiac myofibrils was performed to discover new cardiac sarcomere modulators.30−35 Sulfonamides 1 and 2 were identified as structurally related screening hits with cardiac myofibril biochemical AC40 values of 5.5 and 1.1 μM respectively (AC40 is defined as the concentration of compound that increases the myofibril ATP hydrolysis rate by 40% at calcium concentrations that produce 25% of the maximum calcium-dependent activation in a control experiment.) Compound 2 was inactive against fast skeletal myofibrils and smooth muscle myosin but showed similar activity in the cardiac and slow skeletal myofibril assays (Figure 3). The lack of selectivity between cardiac and slow skeletal myofibrils suggests that 2 may primarily interact with either cardiac myosin or the cardiac thin filament component troponin C (TnC) since both sarcomere constituents are shared between cardiac and slow skeletal muscle.36,37
Figure 3.

(a) HTS hits 1 and 2. (b) Dose–response relationship for the ATP hydrolysis rate of various muscle types treated with 2.
Cardiomyocyte experiments were performed to determine if 2 met the key objective of activating cardiac myofibrils without increasing calcium flux. Adult male Sprague–Dawley rat cardiomyocytes were stimulated at 1 Hz at 37 °C, and quiescent myocytes with well-defined striations were selected for contractility assessment. Subsequent treatment with 2 (10 μM) significantly increased myocyte fractional shortening compared to untreated cells over a single contraction cycle without changing intracellular calcium concentrations (Figure 4). The myocyte relaxation velocity was 55.4 ± 18.2 μm/s (23% greater than basal) and the relaxation time to baseline (T50) was 0.308 ± 0.059 s (30% greater than basal). These results demonstrate the translation of cardiac myofibril ATPase biochemical activity to an increase in cardiomyocyte contractility without changing intracellular calcium levels.
Figure 4.

(a) Cardiomyocyte contractility profile at 1 Hz pacing and treatment with sulfonamide 2 (10 μM) vs vehicle. (b) Effect of 2 (10 μM) on cardiomyocyte calcium concentration compared to vehicle as determined by a Fura dye.
The biological target of 1 and 2 was investigated using a sarcomere component swap experiment. Reconstituted sarcomeres14 composed of the four possible combinations of cardiac myosin S1, fast skeletal myosin S1, cardiac thin filament, and fast skeletal thin filament were treated with 1 and 2 at a single concentration (40 μM). Only the reconstituted sarcomeres that contain the cardiac thin filament showed activation when treated with the cardiac myofibril activators (Figure 5), implying that the target of these compounds is within the cardiac thin filament and the compounds were not direct cardiac myosin activators.
Figure 5.
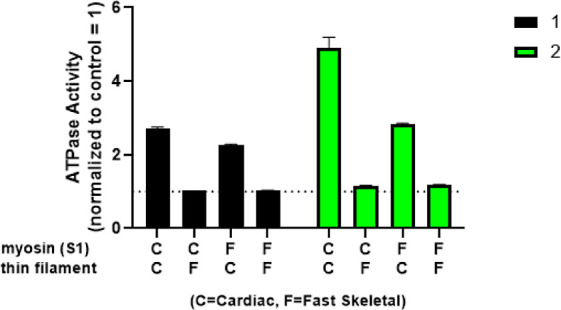
Determination of sarcomere component activity using the component swap experiment for 1 (n = 2, black bar) and 2 (n = 1, green bar).
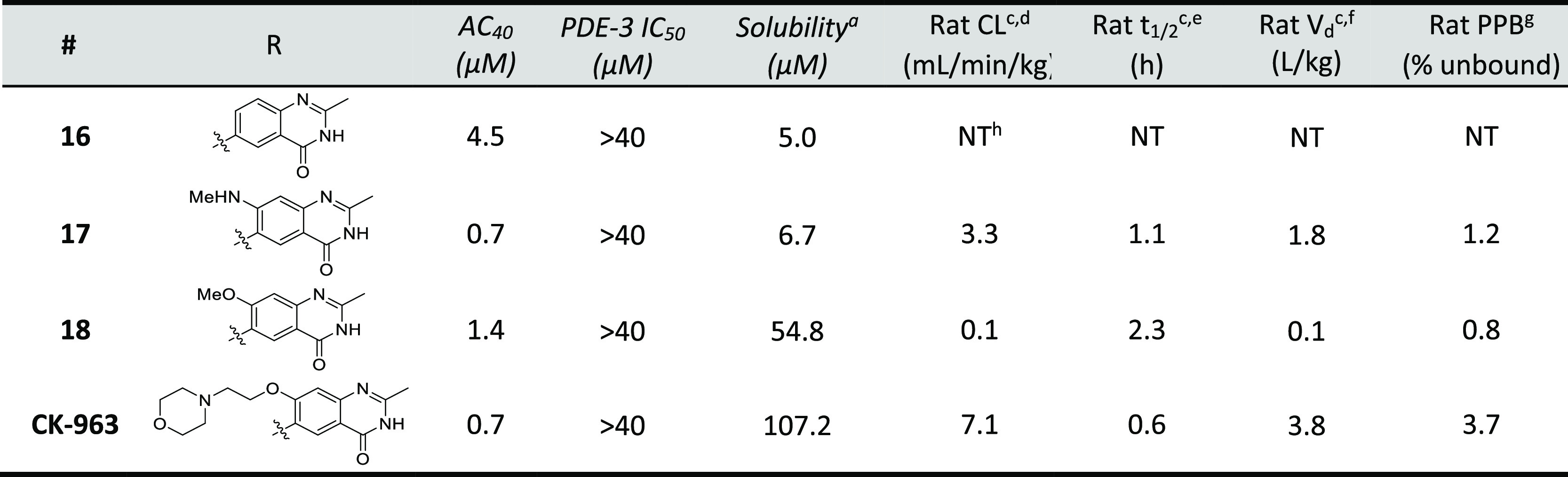
After confirmation that the target was a component of the cardiac thin filament, a series of structurally related analogs of sulfonamide 2 were synthesized to identify key binding interactions (Table 1). Hybridization of 1 and 2 provided biarylsulfonamide 3 with significantly improved cardiac myofibril biochemical potency (AC40 = 0.1 μM) compared to the HTS hits, but this compound also possessed PDE-3 inhibitory activity (IC50 = 4.6 μM). Incorporation of a methyl group at the 1- position (5) did not change biochemical potency or PDE-3 inhibition, but removal of the sulfonamide hydrogen bond donor by either methylation (4) or replacement with oxygen or carbon atoms (data not shown) led to a significant loss in biochemical potency. The solubility of 5 in the PDE-3 buffer solution was 1.2 μM, indicating potentially greater PDE-3 inhibition than is reflected by the IC50 measurement of 4.6 μM.
Table 1. HTS Hits and Early Structure-Activity Relationship.

Cardiac myofibril biochemical potency. AC40 represents the concentration needed to increase the ATPase rate by 40%.
Not tested.
A summary of properties for 2 and 3 is shown in Table 2. These compounds were potent in both cardiac and slow skeletal myofibrils but did not activate fast skeletal myofibrils or smooth muscle myosin. As stated above, cardiac and slow skeletal muscle have identical myosin and TnC, and the similar level of biochemical activity observed in cardiac and slow skeletal muscle for 2 and 3 along with the results of the reconstituted sarcomere assay suggest the possibility of a molecular interaction that involves TnC. Both compounds possess properties that were expected to make pharmacological assessment challenging, specifically PDE-3 IC50 < 5 μM and low single digit μM aqueous solubility, but were still considered to be reasonable starting points for optimization.
Table 2. Properties of 2 and 3.

| property | 2 | 3 |
|---|---|---|
| cardiac myofibril AC40, μM | 1.1 | <0.1 |
| slow skeletal myofibril AC40, μM | 1.7 | <0.1 |
| fast skeletal myofibril AC40, μM | >39.2 | >39.2 |
| smooth muscle myosin IC50, μM | >39.2 | >39.2 |
| PDE-3 IC50, μM | 2.8 | 4.6 |
| solubility, μM | 2.3 | 2.4 |
| cLogPa | 2.4 | 3.9 |
| PSAb | 115 | 103 |
| Fsp3c | 0.3 | 0 |
Calculated partition coefficient (ClogP) values were determined using ACD software.
Polar surface area (PSA) was determined using Pipeline Pilot 2021.
Fraction of sp3 atoms.
The primary optimization objective was to identify potent (<1 μM) cardiac myofibril activators that do not inhibit PDE-3 to enable pharmacological assessment. Improvements in solubility were also needed to ensure accurate PDE-3 inhibition data at higher concentrations and to enable intravenous (IV) dosing. A series of compounds that replaced the biaryl ring with substituents designed to improve solubility by increasing sp3 character and PSA were synthesized (Table 3). Replacement of the internal ring of the biaryl with a 3-pyridyl ring (7) was tolerated but with a significant loss in potency compared to biaryl 3. 2-Pyridyl 9 and 3-pyrimidine 8 were inactive at 40 μM. Several aminopyridine analogs based on azepane 2 were synthesized (10–15). The 3,5-trans-dimethylpiperidine substituent (13) was the most potent aminopyridine but showed PDE-3 inhibitory activity (IC50 = 7 μM). The importance of lipophilic piperidine substituents on biochemical potency is exemplified by inactive piperidine 10 and the >40x eudismic ratio observed with piperidines 13 and 14.
Table 3. Biaryl Optimization.


Cardiac myofibril assay biochemical potency.
PDE-3 inhibition was significantly reduced by replacement of the quinazolinedione ring with a quinazolinone ring (e.g., the aforementioned PDE-3 activity of 13vs16). Comparison of 16 and 13 show that this ring substitution provides a greater than 5-fold reduction in PDE-3 inhibition but also a 10-fold decrease in biochemical potency (Table 4). Substitution at the 7- position of the quinazolinone ring provided analogs 17, 18, and CK-963 with improved biochemical potency. Rat pharmacokinetic assessment of 17, 18, and CK-963 dosed IV in rats showed clearance values < 25% of hepatic blood flow (Qh) and half-lives between 0.6 to 2.3 h. CK-963 possessed the desired IV exposure profile and solubility in a suitable formulation vehicle and was selected for in vivo assessment of cardiac function by echocardiography in rats.
Table 4. Quinazolinedione Replacement.
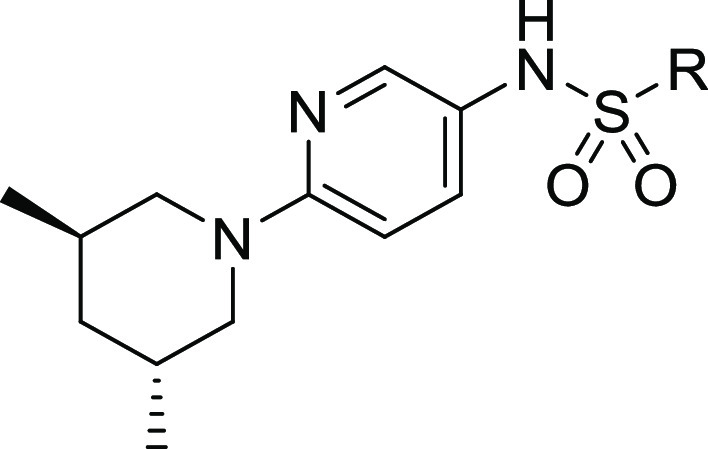
Performed at pH 6.8 using a monopotassium phosphate buffer system.
PK in male Sprague–Dawley rats dosed at 0.5 mg/kg IV using 100% DMSO formulation vehicle.
Rat in vivo clearance.
Rat in vivo half-life.
Rat in vivo volume of distribution.
Plasma protein binding, rat.
NT = Not tested.
CK-963 has a muscle selectivity profile and mechanism of action similar to HTS hits 1 and 2.38 It was selective against fast skeletal myofibrils and smooth muscle myosin and active only in reconstituted sarcomere systems that contained the cardiac thin filament, suggesting that the target is a component of the cardiac troponin regulatory complex. Isothermal calorimetry (ITC) studies confirmed that CK-963 was directly interacting with a cardiac troponin chimera (cNTnC–TnI, 15 kDa) that contains the calcium-sensing and myosin-gating components of troponin. The dissociation constant for this interaction was 11.5 ± 3.2 μM, with changes in enthalpy of −10.3 ± 2.1 kcal/mol and entropy of −12.0 ± 7.5 kcal/(K) (mol). This outcome, along with the result of the sarcomere component swap experiment provides evidence that cardiac troponin is the target of CK-963. The calorimetry result is consistent with previously reported ITC studies using an advanced member of this chemical series that were applied in combination with solution nuclear magnetic resonance to provide the basis for a structural and thermodynamic model for the activation of cardiac troponin.39
A series of experiments in anesthetized Sprague–Dawley rats were performed to evaluate the effect of the selective cardiac troponin activator CK-963 on cardiac function. CK-963 was tested on three separate occasions using continuous or stepwise infusion with cumulative doses as high as 199 mg/kg. Echocardiographic measurements of cardiac contraction were performed throughout the infusion period. Left ventricular fractional shortening (LVFS), the percent change in left ventricular diameter during a contractile cycle, was the primary pharmacodynamic readout with increases in LVFS indicating increased cardiac contractility. The pharmacodynamic response of cardiac function to CK-963 is plotted as percent change in LVFS relative to baseline as a function of total or unbound plasma concentrations (Figure 6). CK-963 increased fractional shortening by about 10% at 9.5 μM total plasma concentration and 0.4 μM unbound plasma concentration (determined by a nonlinear fit of pooled fractional shortening and plasma concentration values using GraphPad software).40 At the highest plasma concentrations measured in these infusion studies (∼100 μM), fractional shortening increased by nearly 100%. The unbound concentration needed to increase fractional shortening by 40% in the echocardiography study was 1.2 μM and is similar to the cardiac myofibril AC40 biochemical potency of 0.7 μM for CK-963.
Figure 6.

Rat echocardiography assessment for CK-963. Data is plotted as fractional shortening as a function of total and unbound plasma concentrations from each recorded time point. The dotted lines indicate a 10 and 40% increase in fractional shortening.
The synthesis of CK-963 is shown in Scheme 1. Acid-catalyzed cyclization of commercially available aminoester 20 provided quinazolinone 21, followed by installation of the sulfonyl chloride in two steps from 21 using a palladium-catalyzed cross-coupling reaction with benzyl mercaptan and then treatment with N-chlorosuccinimide (NCS) and acetic acid.41,42 Sulfonamide 24 was synthesized using a coupling reaction of sulfonyl chloride 23 and amine 30 in the presence of pyridine. Similar coupling conditions using commercially available amines and sulfonyl chlorides were applied to the synthesis of the other sulfonamide analogs in this paper. Dihydroquinazoline CK-963 was synthesized using an SnAr reaction of aryl fluoride 24 and 2-morpholinoethan-1-ol using sodium hydride as the base.
Scheme 1. Synthesis of CK-963.
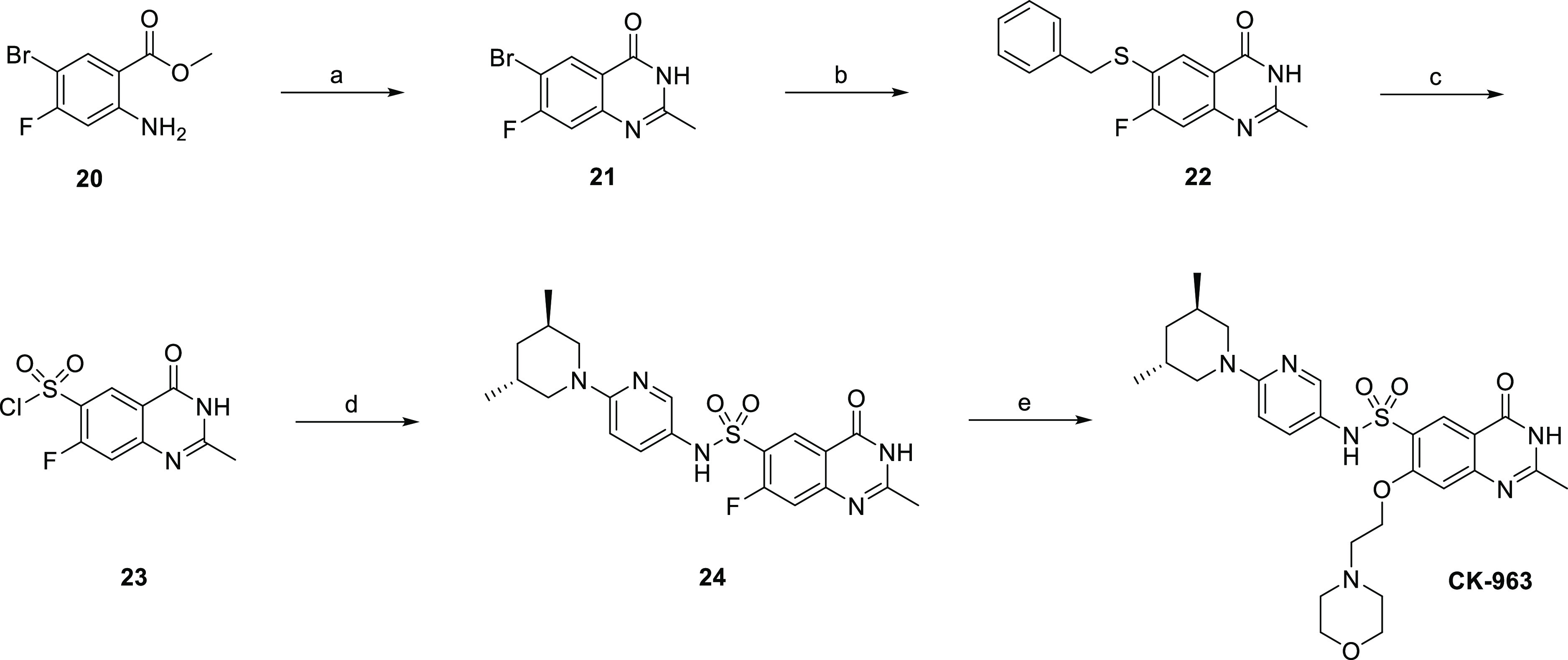
Reagents, conditions, and yields (a) 4 N HCl in dioxanes, CH3CN, 90 °C, 16 h, 93%; (b) benzyl mercaptan, xantphos, Pd2dba3, DIPEA, dioxane, toluene, 90 °C, 6 h, 90%; (c) NCS, acetic acid, water, 0 °C, 5 h, rt, 5 h, 49%; (d) 30, pyridine, CH2Cl2, 1 h, 55%; (e) 2-morpholinoethan-1-ol, NaH, THF, rt, 1 h, 130 °C, 1.5 h, 40%.
Amine 30 was synthesized starting with the separation of an 80% trans/cis mixture of commercially available 3,5-dimethylpiperidine by first benzylating the 3,5-dimethylpiperidine mixture, followed by silica gel column chromatography and then debenzylation using palladium on carbon under a hydrogen atmosphere to give trans 3,5-dimethylpiperidine (27) (Scheme 2). Enantiomerically pure nitropyridine 29 was synthesized using an SnAr reaction of 2-chloro-5-nitropyridine and 27, followed by chiral separation with supercritical fluid chromatography (SFC). Reduction of the nitro group using palladium on carbon under a hydrogen atmosphere provided amine 30.
Scheme 2. Synthesis of 6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-amine.

Reagents, conditions, and yields (a) BnBr, K2CO3, acetone, rt, 4 d, 43%; (b) H2 (25 psi), 20% Pd/C, MeOH, rt, 12 h; (c) 4 N HCl in dioxanes, 100% for steps b and c; (d) 2-chloro-5-nitropyridine, TEA, DMF, 90 °C, 16 h, 29%; (e) chiral resolution using chiral SFC (Chiralcel AD-H, 20% (1:1) isopropanol and MeCN/CO2, 100 bar, 62 mL/min). (f) H2 (50 psi), 10% Pd/C, MeOH, rt, 20 min.
Conclusions
Medicinal chemistry optimization of a cardiac troponin activator series using cardiac myofibril high throughput screening led to CK-963, a compound with sub micromolar biochemical potency, acceptable solubility in a suitable formulation vehicle, selectivity against PDE-3, and adequate exposure in rats to enable pharmacological evaluation of a selective cardiac troponin activator. Rat echocardiography studies using CK-963 showed significant increases in LVFS, and the unbound concentration needed to increase fractional shortening by 10% was about half of the cardiac myofibril biochemical potency. We provide evidence that cardiac troponin is the target of CK-963 based on the direct interaction measured in the ITC study as well as results of the sarcomere component swap experiment that showed activation only occurred in reconstituted sarcomeres containing the cardiac thin filament. Subsequent research activities led to the discovery of a structurally distinct cardiac troponin activator series and a compound that is currently being evaluated in phase I clinical studies, nelutroctiv. This medicinal chemistry discovery story is described in the subsequent article.43 Both novel cardiac troponin activators CK-963 and nelutroctiv could utilized as tool compounds to further understand the mechanisms that regulate cardiac contractile kinetics.44
Experimental Section
General Information
All solvents and reagents were purchased from commercial vendors and used without further purification. 1H NMR were recorded at ambient temperature at 400.13 MHz using a Bruker AVANCE 400 spectrometer. 1H shifts are referenced to the residual protonated solvent signal (δ 2.50 for DMSO-d6, δ 3.31 for MeOH-d4, δ 7.24 for CDCl3). The data are reported as follows: chemical shift in ppm from internal tetramethylsilane on the δ scale, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. Mass spectrometry data were obtained using an Agilent LC/MSD Quad VL system. Normal phase liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on EM Reagents silica gel (SiO2) 60 (230–400 mesh) or using a Biotage Horizon MPLC with Biotage KP-Sil silica gel columns. Reverse phase HPLC purification was performed with an Agilent Series 1100 HPLC equipped with a Phenomenex Gemini C18 Column (5 μm, 150 × 21.2 mm). The typical gradient used for the mobile phase was 20% acetonitrile/water to 90% acetonitrile/water in the presence of 0.1% formic acid over 40 min unless otherwise specified. Unless otherwise noted, the purity for compounds was judged to be >95% as determined by 1H NMR and HPLC at 254 nm. All animal experiments described in this manuscript were performed in compliance with Institutional Animal Care and Use Committee (IACUC) guidelines.
General Procedure for Synthesis of Sulfonamides
To a 20 mL scintillation vial was added amine (1.2 equiv), DMF (0–4 mL/mmol), and pyridine (2.0 equiv), followed by sulfonyl chloride. The reactions were generally stirred for 1–24 h. Some were concentrated and purified using reverse phase HPLC (20–90% CH3CN/H2O over 35 min), and others were worked up using EtOAc and saturated sodium bicarbonate followed by silica gel purification.
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-5-(2-morpholinoethoxy)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (CK-963)
To a 250 mL round-bottom flask was added 2-morpholinoethan-1-ol (16.3 g, 15.0 mL, 124 mmol, 1.7 equiv), followed by sodium hydride (60% dispersion in mineral oil, 1.9 g, 72.0 mmol, 1.0 equiv) in small portions with stirring. The reaction mixture was stirred for 1 h, followed by the addition of N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (3.2 g, 72.0 mmol). The reaction mixture was transferred to a microwave reaction tube, sealed, and heated in a microwave reactor at 130 °C for 90 min. The reaction mixture was then concentrated and purified using reverse phase HPLC (20–90% CH3CN/H2O over 35 min). Ethyl acetate and saturated sodium carbonate solution were used to dissolve the resultant solid, and the organic layer was separated and dried over sodium sulfate to give 1.6 g (40%) of N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-5-(2-morpholinoethoxy)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 9.18 (s, 1H), 8.15 (s, 1H), 7.62 (d, J = 2.6 Hz, 1H), 7.31 (s, 1H), 7.11 (dd, J = 9.1, 2.8 Hz, 1H), 6.65 (d, J = 9.1 Hz, 1H), 4.47 (t, J = 5.3 Hz, 2H), 3.53–3.37 (m, 6H), 3.06 (dd, J = 12.8, 6.8 Hz, 2H), 2.85–2.75 (m, 2H), 2.57–2.50 (m, 4H), 2.34 (s, 3H), 1.83 (pd, J = 6.4, 3.9 Hz, 2H), 1.37 (t, J = 5.8 Hz, 2H), 0.82 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C27H36N6O5S 556.3 Da, measured 557.3 m/z [M + H]+.
trans-3,5-Dimethylpiperidine
To a 85/15 mixture of trans/cis 3,5-dimethylpiperidine (TCI, 320 mL, 2.35 mmol) and K2CO3 (960 g, 6.96 mol) in acetone (8 L) was slowly added benzyl bromide (488 mL, 4.08 mol) while using a water bath to control the reaction temperature below 40 °C. The reaction was stirred at rt for 4 d. The reaction was then filtered, and the filtrate washed with acetone (1 L). The combined filtrates were concentrated and purified using silica gel chromatography (0–5% diethyl ether in hexanes with 0.2% TEA) to give racemic trans-3,5-dimethylpiperidine (200 g, 43%). 1H NMR (400 MHz, Chloroform-d) δ 7.34–7.15 (m, 5H), 3.54–3.28 (m, 2H), 2.37 (d, J = 9.1 Hz, 2H), 2.13–1.97 (m, 2H), 1.90 (ddp, J = 10.0, 6.3, 3.6 Hz, 2H), 1.28 (t, J = 5.8 Hz, 2H), 0.95 (d, J = 6.8 Hz, 6H). LC/MS (APCI) m/z calcd for C14H21N 203.2 Da, measured 204.1 m/z [M + H]+.
trans-3,5-Dimethylpiperidin-1-ium Chloride
To a solution of racemic trans-3,5-dimethylpiperidine (80.0 g, 0.39 mol) in MeOH (500 mL) was added 20% Pd/C (2.1 g, 0.02 mol, Johnson Matthey A402023–20). The reaction was stirred under hydrogen (25 psi) at 45 °C for 12 h and then filtered through Celite. To the filtrate was added HCl (4 N in dioxane, 200 mL) followed by concentration to give trans-3,5-dimethylpiperidin-1-ium chloride (59.0 g, 100%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 3.14 (dd, J = 12.6, 4.0 Hz, 2H), 2.83 (dd, J = 12.5, 7.0 Hz, 2H), 2.20–2.06 (m, 2H), 1.55 (t, J = 5.8 Hz, 2H), 1.07 (d, J = 7.1 Hz, 6H).
2-(trans-3,5-Dimethylpiperidin-1-yl)-5-nitropyridine
To a 2 L round-bottom flask was added trans-3,5-dimethylpiperidin-1-ium chloride (66.7 g, 446 mmol, 1.2 equiv), 2-chloro-5-nitropyridine (60.1 g, 379 mmol, 1.0 equiv), DMF (250 mL), and triethylamine (137 mL, 1000 mmol, 2.6 equiv). The reaction was heated to 90 °C and stirred overnight. The reaction was then diluted with EtOAc (1 L) and washed three times with brine (200 mL each wash). The organic layer was dried over sodium sulfate, filtered, and concentrated. The resultant crude solid was dissolved in a minimum of EtOAc, followed by the addition of 20% EtOAc/hexanes (50 mL). Hexanes were then added until precipitation was observed, and the reaction suspension was stirred at rt for 14 h. The product was filtered, washed with 20% EtOAc/hexanes, and then dried under vacuum to give 2-(trans-3,5-dimethylpiperidin-1-yl)-5-nitropyridine (31.0 g, 29%) as a pale yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.99 (d, J = 2.8 Hz, 1H), 8.12 (ddd, J = 9.6, 2.9, 0.6 Hz, 1H), 6.52 (d, J = 9.6 Hz, 1H), 3.80 (d, J = 12.3 Hz, 2H), 3.36 (dd, J = 13.2, 7.1 Hz, 2H), 2.01 (ddp, J = 10.4, 6.4, 4.0 Hz, 2H), 1.52 (t, J = 5.9 Hz, 2H), 0.94 (d, J = 6.8 Hz, 6H). LC/MS (APCI) m/z calcd for C12H17N3O2 235.1 Da, measured 236.1 m/z [M + H]+.
2-((3S,5S)-3,5-Dimethylpiperidin-1-yl)-5-nitropyridine (Enantiomer 1) and 2-((3R,5R)-3,5-dimethylpiperidin-1-yl)-5-nitropyridine (Enantiomer 2)
2-(trans-3,5-dimethylpiperidin-1-yl)-5-nitropyridine (1.1 g) was resolved using chiral SFC (Chiralcel AD-H, 20% (1:1) isopropanol/MeCN/CO2, 100 bar, 62 mL/min) to give enantiomer 1 (525 mg, [α]20/D = +41.4° (c 0.95, EtOAc)) and enantiomer 2 (520 mg, [α]20/D = −45.0 (c 0.91, EtOAc)). Enantiomers were numbered based on the order of elution, and the absolute stereochemistry of enantiomer 2 was assigned as R,R based on the crystal structure of 19. Enantiomer 1 was therefore assigned as S,S.
Synthesis of 6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-amine
2-((3R,5R)-3,5-Dimethylpiperidin-1-yl)-5-nitropyridine (350 mg, 1.49 mmol, 1 equiv) and Pd/C (80 mg, 0.075 mmol, 0.05 equiv) were suspended in MeOH (35 mL) and stirred under hydrogen (30 psi) for 1 h. The reaction mixture was then filtered through a pad of Celite and concentrated to give 6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-amine (300 mg). LC/MS (APCI) m/z calcd for C12H19N3 205.1 Da, measured 206.1 m/z [M + H]+.
N-([1,1′-Biphenyl]-4-yl)-N-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (4)
A mixture of N-methyl-[1,1′-biphenyl]-4-amine (75 mg, 0.41 mmol, 1 equiv) and pyridine (55 mg, 0.70 mmol, 1.6 equiv) was dissolved in DMF (0.5 mL) and was added to a solution of 2,4-dioxo-1,3-dihydroquinazoline-6-sulfonyl chloride (107 mg, 0.41 mmol, 1 equiv) in DMF (0.5 mL). The reaction mixture was stirred at rt overnight. Methanol (5 mL) was then added to the pink heterogeneous reaction mixture, followed by stirring for 1 h and sonication for 5 min. The resultant solid was filtered and dried to give 87 mg (52%) of pale beige solid. 1H NMR (400 MHz, DMSO-d6) δ 11.66–11.58 (m, 2H), 7.96 (d, J = 2.2 Hz, 1H), 7.72 (dd, J = 8.6, 2.2 Hz, 1H), 7.66 (dt, J = 8.5, 2.9 Hz, 4H), 7.47 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.3 Hz, 1H), 7.30 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.5 Hz, 2H), 3.16 (s, 3H). LRMS (APCI): calcd for C21H17N3O4S 407.1 Da, measured 406.0 m/z [M – H]−.
N-(6-Fluoropyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide
To a 250-round-bottom flask containing 6-fluoropyridin-3-amine (1.22 g, 10.9 mol, 1 equiv), and pyridine (1.78 g, 1.75 mL, 21.8 mmol, 2.0 equiv) at 0 °C was added a suspension of 1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonyl chloride (3.0 g, 10.9 mmol, 1.0 equiv) and CH2Cl2 (50 mL). The reaction was stirred at 0 °C for 1 h and then warmed to rt. The reaction was concentrated, and then methanol (35 mL) was added. The mixture was sonicated and the resultant solids were filtered to give N-(6-fluoropyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (3.8 g, 89%) as a tan solid.
Step 2: 2,4-Dioxo-N-(5-phenylpyridin-2-yl)-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (10)
To a 5 mL microwave reaction vial was added N-(6-fluoropyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (100 mg, 0.29 mmol, 1 equiv) dissolved in NMP (3 mL), triethylamine (0.5 mL), and piperidine (102 mg, 1.2 mmol, 4.0 equiv). The microwave tube was sealed, heated to 220 °C, and stirred for 30 min. The reaction was then cooled, followed by addition of formic acid (1 mL) and purification using reverse phase HPLC (20–90% CH3CN/H2O over 35 min) to give 2,4-dioxo-N-(5-phenylpyridin-2-yl)-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (61 mg, 52%). 1H NMR (400 MHz, Methanol-d4) δ 8.40 (d, J = 2.3 Hz, 1H), 7.99 (dd, J = 8.8, 2.3 Hz, 1H), 7.65 (d, J = 2.8 Hz, 1H), 7.51 (d, J = 8.8 Hz, 1H), 7.30 (dd, J = 9.1, 2.8 Hz, 1H), 6.68 (d, J = 9.1 Hz, 1H), 3.55 (s, 3H), 3.41 (t, J = 5.1 Hz, 4H), 1.61 (q, J = 7.8, 7.3 Hz, 6H). LRMS (APCI): calcd for C19H21N5O4S 415.3 Da, measured 416.1 m/z [M + H]+.
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (13)
To a 20 mL scintillation vial was added 2-((3R,5R)-3,5-dimethylpiperidin-1-yl)-5-nitropyridine (100 mg, 426 μmol, 1 equiv), 10% Pd/C (28 mg), and MeOH (5 mL). The reaction was stirred under a hydrogen atmosphere (50 psi) for 20 min. The reaction mixture was then filtered through a pad of Celite and concentrated to give 6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-amine (87 mg). To a 20 mL scintillation vial was added 6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-amine (43 mg, 0.2 mmol), CH2Cl2 (2 mL) and pyridine (0.2 mL). The reaction was stirred for 1 h, followed by the addition of 20% EtOAc/hexanes (1 mL) and water (5 drops). The resultant solution containing precipitate was sonicated, and the solid then filtered to give N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (29 mg, 31%) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.83 (s, 1H), 9.90 (s, 1H), 8.25 (d, J = 2.3 Hz, 1H), 7.93 (dd, J = 8.8, 2.3 Hz, 1H), 7.64 (d, J = 2.7 Hz, 1H), 7.57 (d, J = 8.9 Hz, 1H), 7.20 (dd, J = 9.2, 2.7 Hz, 1H), 6.79 (d, J = 9.2 Hz, 1H), 3.51 (dd, J = 12.8, 3.8 Hz, 2H), 3.45 (s, 3H), 3.11 (dd, J = 12.9, 6.9 Hz, 2H), 1.87 (dq, J = 12.7, 6.3 Hz, 2H), 1.40 (t, J = 5.8 Hz, 2H), 0.86 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C21H25N5O4S 443.2 Da, measured 444.2 m/z [M + H]+.
N-(6-((3S,5S)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (14)
The exact procedure for the synthesis of 13 was followed to synthesize 14, providing 22 mg (23%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.83 (s, 1H), 9.83 (s, 1H), 8.25 (d, J = 2.3 Hz, 1H), 7.92 (dd, J = 8.8, 2.3 Hz, 1H), 7.65 (d, J = 2.7 Hz, 1H), 7.57 (d, J = 8.9 Hz, 1H), 7.16 (dd, J = 9.1, 2.8 Hz, 1H), 6.72 (d, J = 9.1 Hz, 1H), 3.50 (dd, J = 12.8, 3.7 Hz, 2H), 3.45 (s, 3H), 3.08 (dd, J = 12.8, 6.8 Hz, 2H), 1.86 (dq, J = 10.2, 6.2 Hz, 2H), 1.39 (t, J = 5.8 Hz, 2H), 0.85 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C21H25N5O4S 443.2 Da, measured 444.2 m/z [M + H]+.
2-(3,5-Dimethylpiperidin-1-yl)-5-nitropyridine
To a 50 mL round-bottom flask cooled to 0 °C was added 2-chloro-5-nitropyridine (1.0 g, 6.3 mmol, 1 equiv), 3,5-dimethylpiperidine (1.45 g, 1.70 mL, 12.6 mmol, 2 equiv), triethylamine (0.9 mL, 6.3 mmol, 1 equiv), and THF (20 mL). The reaction was stirred overnight and filtered. The filtrate was washed with brine, dried over sodium sulfate, and concentrated in vacuo to give 1.4 g of crude 2-(3,5-dimethylpiperidin-1-yl)-5-nitropyridine.
6-(3,5-Dimethylpiperidin-1-yl)pyridin-3-amine
To a 100 mL reaction vial was added 2-(3,5-dimethylpiperidin-1-yl)-5-nitropyridine (1.4 g, 6.0 mmol, 1 equiv), methanol (20 mL), ethyl acetate (10 mL), and 10% Pd/C (500 mg). The reaction was stirred under hydrogen (50 psi) for 2 h, followed by filtration, concentration, and purification using silica gel chromatography (0–20% EtOAc/hexanes) to give 6-(3,5-dimethylpiperidin-1-yl)pyridin-3-amine (1.1 g, 90%).
N-(6-((3R,5S)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide
To a 40 mL scintillation vial was added 6-(3,5-dimethylpiperidin-1-yl)pyridin-3-amine (335 mg, 1.22 mmol, 1 equiv), pyridine (2 mL), and 1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonyl chloride (251 mg, 1.22 mmol, 1 equiv). The reaction was stirred for 30 min at 40 °C, concentrated, and purified using silica gel chromatography and then reverse phase HPLC (20–90% CH3CN/H2O over 35 min). The second compound to elute from the column was N-(6-((3R,5S)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazoline-6-sulfonamide (204 mg, 38%). 1H NMR (400 MHz, DMSO-d6) δ 11.83 (s, 1H), 9.84 (s, 1H), 8.24 (d, J = 2.3 Hz, 1H), 7.91 (dd, J = 8.9, 2.4 Hz, 1H), 7.68 (d, J = 2.7 Hz, 1H), 7.57 (d, J = 8.9 Hz, 1H), 7.17 (dd, J = 9.1, 2.8 Hz, 1H), 6.72 (d, J = 9.1 Hz, 1H), 4.15 (dd, J = 12.9, 3.9 Hz, 2H), 3.44 (s, 3H), 2.23–2.13 (m, 2H), 1.73 (d, J = 12.7 Hz, 1H), 1.48 (ddd, J = 22.4, 7.0, 3.6 Hz, 1H), 1.48 (s, 2H), 0.85 (d, J = 6.6 Hz, 6H). LRMS (APCI): calcd for C21H25N5O4S 443.2 Da, measured 442.2 m/z [M – H]−.
6-Bromo-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one
SEMCl (7.5 g. 45 mmol, 1.25 equiv) was added to a stirring solution of 6-bromo-2-methylquinazolin-4(3H)-one (8.0 g, 36 mmol, 1 equiv) and DIPEA (8 mL, 45 mmol, 1.25 equiv) in CH2Cl2 (200 mL). After 14 h, the reaction was concentrated and purified using silica gel column chromatography (10–30% EtOAc/hexanes) to give 6-bromo-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one (8.4 g, 64%) as a yellow oil. 1H NMR (400 MHz, Methanol-d4) δ 8.31 (t, J = 1.9 Hz, 1H), 7.92 (ddd, J = 8.6, 2.1, 1.0 Hz, 1H), 7.53 (dd, J = 8.7, 1.2 Hz, 1H), 5.61 (d, J = 1.3 Hz, 2H), 3.83–3.58 (m, 2H), 2.70 (d, J = 1.3 Hz, 3H), 1.10–0.82 (m, 2H), 0.10 (s, 9H). LRMS (APCI): calcd for C15H21BrN2O2Si 368.1 Da, measured 369.1 m/z [M + H]+.
6-Amino-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one
To a 250 mL round-bottom flask was added 6-bromo-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one (8.4 g, 2.3 mmol, 1.0 equiv), benzophenone imine (4.5, 25 mmol, 1.1 equiv), tris(dibenzylideneacetone)dipalladium (1.05 g, 0.12 mmol, 0.05 equiv), BINAP (2.14 g, 0.35 mmol, 0.15 equiv), sodium tert-butoxide (3.1, 32 mmol, 1.4 equiv), and toluene (100 mL). The reaction mixture was heated to reflux and stirred for 1 h. The reaction was then washed with saturated sodium bicarbonate and the organic layer was separated, dried over sodium sulfate, and concentrated. The resultant crude oil was dissolved in methanol (200 mL), followed by the addition of potassium acetate (6.8 g, 6.9 mmol, 3.0 equiv) and hydroxylamine hydrochloride (4.0 g, 58 mmol, 25.0 equiv). The reaction was stirred at rt for 90 min. The reaction was then washed with saturated sodium bicarbonate, and the organic layer was separated, dried over sodium sulfate, and concentrated. The resultant crude oil was purified using silica gel chromatography to give 6-amino-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one (3.3 g, 47%) as a pale yellow solid.
2-Methyl-4-oxo-3-((2-(trimethylsilyl)ethoxy)methyl)-3,4-dihydroquinazoline-6-sulfonyl Chloride
To a 100 mL round-bottom flask (A) was added copper(II) chloride (0.48 g, 10.8 mmol, 0.3 equiv) and acetic acid (16 mL), and sulfur dioxide was bubbled into the mixture through a gas dispersion tube. In a separate 100 mL round-bottom flask (B) was added 6-amino-2-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)quinazolin-4(3H)-one (3.3 g, 10.8 mmol, 1.0 equiv), acetonitrile (10 mL), and concentrated HCl (5 mL). This mixture (B) was cooled to −5 °C, followed by the addition of sodium nitrite (0.75 g, 10.8 mmol, 1.0 equiv) dissolved in water (5 mL). The reaction mixture (B) was stirred for 5 min at −5 °C. The contents of round-bottom flask B were then poured in round-bottom A, and the combined reaction mixture was stirred for 5 min at −5 °C and then warmed to rt. The reaction was then extracted using EtOAc/water, and the organic layer was separated, washed with saturated sodium bicarbonate solution, dried over sodium sulfate, and concentrated. The resultant crude oil was purified using silica gel chromatography (20–50% EtOAc/hexanes) to give 2-methyl-4-oxo-3-((2-(trimethylsilyl)ethoxy)methyl)-3,4-dihydroquinazoline-6-sulfonyl chloride (2.75 g, 65%) as a pale orange oil. 1H NMR (400 MHz, Chloroform-d) δ 8.91 (d, J = 2.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.4 Hz, 1H), 7.77 (d, J = 8.7 Hz, 1H), 5.57 (s, 2H), 3.73–3.64 (m, 2H), 2.75 (s, 3H), 0.99–0.88 (m, 2H), −0.02 (s, 9H).
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-5-(methylamino)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (16)
2-((3R,5R)-3,5-dimethylpiperidin-1-yl)-5-nitropyridine (100 mg, 0.5 mmol, 1.0 equiv) and Pd/C (28 mg, 10% Pd by mass, 0.026 mmol, 0.05 equiv) were suspended in MeOH (5 mL) and then stirred under a hydrogen atmosphere (50 psi) for 30 min. The reaction was filtered, concentrated, and dried under high vacuum. The resultant solid was dissolved in pyridine (0.08 g, 1.01 mmol, 2 equiv) and CH2Cl2 (2 mL), and 2-methyl-4-oxo-3-((2-(trimethylsilyl)ethoxy)methyl)-3,4-dihydroquinazoline-6-sulfonyl chloride (83 mg, 0.215 mmol, 0.45 equiv) was added. The reaction was stirred for 1 h, filtered through a plug of silica (10 → 30% EtOAc/hexanes), concentrated, and dissolved in MeOH (0.5 mL). HCl (4 N in dioxanes, 3 mL) was then added, and the reaction heated to 90 °C for 10 min. The reaction was cooled to rt, concentrated, and purified using silica gel chromatography (0–10% MeOH/CH2Cl2) to give N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-5-(methylamino)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (16) as a tan solid (19 mg, 20% over 3 steps). 1H NMR (400 MHz, Methanol-d4) δ 8.43 (d, J = 2.1 Hz, 1H), 7.98 (dd, J = 8.6, 2.1 Hz, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 2.7 Hz, 1H), 7.25 (dd, J = 9.2, 2.7 Hz, 1H), 6.67 (d, J = 9.2 Hz, 1H), 3.51 (dd, J = 12.9, 3.7 Hz, 2H), 3.10 (dd, J = 12.9, 6.9 Hz, 2H), 2.46 (d, J = 1.4 Hz, 3H), 1.92 (ddt, J = 13.0, 10.5, 4.3 Hz, 2H), 1.46 (t, J = 5.8 Hz, 2H), 0.91 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C21H25N5O3S 427.2 Da, measured 428.1 m/z [M + H]+.
6-Bromo-7-fluoro-2-methylquinazolin-4(3H)-one
To a 1 L round-bottom flask was added methyl 2-amino-5-bromo-4-fluorobenzoate (25.0 g, 0.1 mol), 4 N HCl in dioxanes (300 mL), and acetonitrile (350 mL). The mixture was heated to 90 °C and stirred overnight. The reaction was then concentrated, and the resultant solid was resuspended in acetonitrile. Aqueous sodium hydroxide (1 N) was used to adjust the pH to 8–9, and the solid was filtered, washed with cold acetonitrile, and dried in vacuo to give 6-bromo-7-fluoro-2-methylquinazolin-4(3H)-one (24.0 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 12.45 (s, 1H), 8.27 (d, J = 7.7 Hz, 1H), 7.54 (d, J = 10.0 Hz, 1H), 3.32 (s, 3H).
6-(Benzylthio)-7-fluoro-2-methylquinazolin-4(3H)-one
6-Bromo-7-fluoro-2-methylquinazolin-4(3H)-one (15.0 g, 58.4 mmol, 1 equiv) was added to a 1 L round-bottom flask and dissolved with dioxane (300 mL) and toluene (300 mL), followed by the addition of diisopropylethylamine (15.0 g, 20.3 mL, 116 mol, 2.0 equiv). The mixture was heated to 90 °C, followed by the addition of phenylmethanethiol (7.6 g, 7.3 mL, 61.3 mmol, 1.05 equiv), xantphos (5.1 g, 8.9 mmol, 0.15 equiv) and tris(dibenzylideneacetone)dipalladium (5.3 g, 5.8 mmol, 0.1 equiv). The reaction mixture was heated at 90 °C for 6 h and then cooled to 0 °C. The resultant solid was filtered, washed with water, and dried in vacuo to give 6-(benzylthio)-7-fluoro-2-methylquinazolin-4(3H)-one (15.7 g, 90%) as a yellow-green solid. 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.26 (s, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.38–7.15 (m, 5H), 4.27 (s, 2H), 2.29 (s, 3H).
7-Fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonyl Chloride
To a 500 mL round-bottom flask was added 6-(benzylthio)-7-fluoro-2-methylquinazolin-4(3H)-one (9.5 g, 31.7 mmol, 1 equiv), acetic acid (200 mL), and water (50 mL). The mixture was cooled with an ice bath to ca. 0 °C, and N-chlorosuccinimide (14.7 g, 110.8 mmol, 3.5 equiv) was added. The reaction mixture was stirred for 5 h at 0 °C, and then at rt for 5 h. The reaction mixture was then extracted using EtOAc (800 mL) and brine (300 mL), and the organic layer was separated, dried over sodium sulfate, and concentrated. The crude product was purified using silica gel chromatography (hexanes/EtOAc) to give 7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonyl chloride (3.9 g, 49%) as a white solid.
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide
To a 1 L round-bottom flask was added 6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-amine (8.0 g, 39 mmol, 1 equiv) and pyridine (200 mL), followed by a suspension of 7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonyl chloride (16.1 g, 58.5 mmol, 1.5 equiv) in methylene chloride (300 mL). The reaction mixture was stirred at rt for 1 h and then concentrated. The crude product was purified using silica gel chromatography (100% Et2O, then 100% ethyl acetate) to give N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (9.5 g, 55%) as a dark purple solid.
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-7-(methylamino)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (17)
To a 20 mL microwave reaction tube was added N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (0.5 g, 1.1 mmol) and methylamine (6 mL, 40% in water). The tube was sealed and the reaction was heated within a microwave reactor at 130 °C for 30 min. The pH of the reaction was then adjusted to 7 followed by extraction with ethyl acetate and brine. The organic layer was dried over sodium sulfate and concentrated. The crude product was purified using reverse phase column chromatography (20–90% CH3CN/H2O over 35 min) to give N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-2-methyl-7-(methylamino)-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (195 mg, 38%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 9.80 (s, 1H), 8.08 (s, 1H), 7.61 (d, J = 2.6 Hz, 1H), 7.07 (dd, J = 9.1, 2.7 Hz, 1H), 6.66 (d, J = 9.2 Hz, 1H), 6.62 (s, 1H), 6.32 (q, J = 4.7 Hz, 1H), 3.48 (dd, J = 12.8, 3.7 Hz, 2H), 3.06 (dd, J = 12.8, 6.8 Hz, 2H), 2.88 (d, J = 4.8 Hz, 3H), 2.27 (s, 3H), 1.81 (dddd, J = 13.4, 9.6, 6.7, 3.8 Hz, 2H), 1.36 (t, J = 5.8 Hz, 2H), 0.81 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C22H28N6O3S 456.2 Da, measured 457.2 m/z [M + H]+.
N-(6-((3R,5R)-3,5-Dimethylpiperidin-1-yl)pyridin-3-yl)-5-methoxy-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (18)
To a microwave reaction tube was added N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-7-fluoro-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide (2.8 g, 6.3 mmol) and sodium methoxide (120 mL of a 25% solution in methanol). The tube was sealed and the reaction was heated to 130 °C for 35 min. The reaction was then concentrated and purified by reverse phase HPLC (20–90% CH3CN/H2O over 35 min) to give a crude solid that was dissolved in ethyl acetate and washed with aqueous sodium bicarbonate and brine. The organic layer was dried over sodium sulfate and concentrated to give N-(6-((3R,5R)-3,5-dimethylpiperidin-1-yl)pyridin-3-yl)-7-methoxy-2-methyl-4-oxo-3,4-dihydroquinazoline-6-sulfonamide 1.61 g (56%) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 9.55 (s, 1H), 8.22 (s, 1H), 7.67 (d, J = 2.6 Hz, 1H), 7.21 (s, 1H), 7.15 (dd, J = 9.1, 2.7 Hz, 1H), 6.65 (d, J = 9.2 Hz, 1H), 4.03 (s, 3H), 3.45 (dd, J = 12.8, 3.7 Hz, 2H), 3.03 (dd, J = 12.8, 6.8 Hz, 2H), 2.33 (s, 3H), 1.82 (pd, J = 6.4, 3.8 Hz, 2H), 1.36 (t, J = 5.8 Hz, 2H), 0.82 (d, J = 6.8 Hz, 6H). LRMS (APCI): calcd for C22H27N5O4S 457.2 Da, measured 458.2 m/z [M + H]+.
Acknowledgments
We thank all members of the Cardiac Muscle Program Team at Cytokinetics and Amgen for their contributions to the data presented in this paper.
Glossary
Abbreviations
- AC40
40% activating concentration
- ATP
adenosine triphosphate
- cTnC
cardiac troponin C
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- HFrEF
heart failure with reduced ejection fraction
- HTS
high throughput screen
- ITC
isothermal calorimetry
- IV
intravenous
- IVIVC
in vitro to in vivo correlation
- LVEF
left ventricular ejection fraction
- LVFS
left ventricular fractional shortening
- NCS
N-chlorosuccinimide
- OM
omecamtiv mecarbil
- PDE
phosphodiesterase
- PK
pharmacokinetics
- PSA
polar surface area
- Qh
hepatic blood flow
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c02412.
Author Contributions
A.M., A.R., C.C., L.A., K.L., S.E.C., and B.P.M. contributed to medicinal chemistry design, planning, and synthesis; J.H. and A.D. planned/executed biological studies; D.T.H. and A.S.M. pharmacology, M.G. DMPK.
The authors declare the following competing financial interest(s): S.E.C, A.R., C.C., J.H., L.A., D.T.H., A.D., M.G., F.I.M., and B.P.M. are currently employed by Cytokinetics.
Supplementary Material
References
- Savarese G.; Becher P. M.; Lund L. H.; Seferovic P.; Rosano G. M. C.; Coats A. J. S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2023, 118 (17), 3272–3287. 10.1093/cvr/cvac013. [DOI] [PubMed] [Google Scholar]
- Braunwald E.Heart Disease: A Textbook of Cardiovascular Medicine, 4th ed.; WB Saunders: Philadelphia, 1992; pp 444–463. [Google Scholar]
- Bui A. L.; Horwich T. B.; Fonarow G. C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8 (1), 30–41. 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam C. S. P.; Docherty K. F.; Ho J. E.; McMurray J. J. V.; Myhre P. L.; Omland T. Recent successes in heart failure treatment. Nature Med. 2023, 29, 2424–2437. 10.1038/s41591-023-02567-2. [DOI] [PubMed] [Google Scholar]
- Heidenreich P. A.; Fonarow G. C.; Opsha Y.; Sandhu A. T.; Sweitzer N. K.; Warraich H. J.; Butler J.; Hsich E.; Pressler S. B.; Shah K.; et al. Economic issues in heart failure in the United States. J. Card. Fail. 2022, 28 (3), 453–466. 10.1016/j.cardfail.2021.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psotka M. A.; Gottlieb S. S.; Francis G. S.; Allen L. A.; Teerlink J. R.; Adams K. F.; Rosano G. M. C.; Lancellotti P. Cardiac calcitropes, myotropes and mitotropes. J. Am. Coll. Card. 2019, 73 (18), 2345–2353. 10.1016/j.jacc.2019.02.051. [DOI] [PubMed] [Google Scholar]
- St. Jean D.; Malinowski J.; Romero A.. Recent Advances Toward New Small Molecule Therapies for Heart Failure. Burger’s Medicinal Chemistry and Drug Discovery; Wiley Online Library, 2021. 28 April. [Google Scholar]
- Kass D. A.; Solaro R. J. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circ. 2006, 113 (2), 305–315. 10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]
- Packer M.; Carver J. R.; Rodeheffer R. J.; Ivanhoe R. J.; DiBianco R.; Zeldis S. M.; Hendrix G. H.; Bommer W. J.; Elkayam U.; Kukin M. L.; Mallis G. I.; Sollano J. A.; Shannon J.; Tandon P. K.; DeMets D. L. Effect of Oral Milrinone on Mortality in Severe Chronic Heart Failure. N. Engl. J. Med. 1991, 325, 1468–1475. 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- Yano M.; Kohno M.; Ohkusa T.; Mochizuki M.; Yamada J.; Kohno M.; Hisaoka T.; Ono K.; Tanigawa T.; Kobayashi S.; Matsuzaki M. Effect of milrinone on left ventricular relaxation and Ca2+ uptake function of cardiac sarcoplasmic reticulum. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1898–H1905. 10.1152/ajpheart.2000.279.4.H1898. [DOI] [PubMed] [Google Scholar]
- Murphy S. P.; Ibrahim N. E.; Januzzi J. L. Heart Failure With Reduced Ejection Fraction: A Review. JAMA 2020, 324 (5), 488–504. 10.1001/jama.2020.10262. [DOI] [PubMed] [Google Scholar]
- Romero A.; Morgan B. P. Cardiac sarcomere activation and its therapeutic application in heart failure. Med. Chem. Rev. 2017, 52, 109–123. 10.29200/acsmedchemrev-v52.ch6. [DOI] [Google Scholar]
- Malik F. I.; Hartman J. J.; Elias K. A.; Morgan B. P.; Rodriguez H.; Brejc K.; Anderson R. L.; Sueoka S. H.; Lee K. H.; Finer J. T.; Sakowicz R.; Baliga R.; Cox D. R.; Garard M.; Godinez G.; Kawas R.; Kraynack E.; Lenzi D.; Lu P.; Muci A.; Niu C.; Qian X.; Pierce D. W.; Pokrovskii M.; Suehiro I.; Sylvester S.; Tochimoto T.; Valdez C.; Wang W.; Katori T.; Kass D. A.; Shen Y.; Vatner S. F.; Morgans D. J. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P. M.; Sykes B. D. Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discovery 2015, 14, 313–328. 10.1038/nrd4554. [DOI] [PubMed] [Google Scholar]
- Morgan B. P.; Muci A.; Lu P.; Qian X.; Tochimoto T.; Smith W. W.; Garard M.; Kraynack E.; Collibee S.; Suehiro I.; Tomasi A.; Valdez S. C.; Wang W.; Jiang H.; Hartman J.; Rodriguez H. M.; Kawas R.; Sylvester S.; Elias K. A.; Godinez G.; Lee K.; Anderson R.; Sueoka S.; Xu D.; Wang Z.; Djordjevic N.; Malik F. I.; Morgans D. J. Jr. Discovery of omecamtiv mecarbil the first, selective, small molecule activator of cardiac myosin. ACS Med. Chem. Lett. 2010, 1, 472–477. 10.1021/ml100138q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planelles-Herrero V. J.; Hartman J. J.; Robert-Paganin J.; Malik F. I.; Houdusse A. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat. Commun. 2017, 8, 190. 10.1038/s41467-017-00176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink J. R.; Diaz R.; Felker G. M.; McMurray J. J. V.; Metra M.; Solomon S. D.; Adams K. F.; Anand I.; Arias-Mendoza A.; Biering-Sørensen T.; Böhm M.; Bonderman D.; Cleland J. G. F.; Corbalan R.; Crespo-Leiro M. G.; Dahlström U.; Echeverria L. E.; Fang J. C.; Filippatos G.; Fonseca C.; Goncalvesova E.; Goudev A. R.; Howlett J. G.; Lanfear D. E.; Li J.; Lund M.; Macdonald P.; Mareev V.; Momomura S.; O’Meara E.; Parkhomenko A.; Ponikowski P.; Ramires F. J. A.; Serpytis P.; Sliwa K.; Spinar J.; Suter T. M.; Tomcsanyi J.; Vandekerckhove H.; Vinereanu D.; Voors A. A.; Yilmaz M. B.; Zannad F.; Sharpsten L.; Legg J. C.; Varin C.; Honarpour N.; Abbasi S. A.; Malik F. I.; Kurtz C. E. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N. Engl. J. Med. 2021, 384, 105–116. 10.1056/NEJMoa2025797. [DOI] [PubMed] [Google Scholar]
- Nieminen M. S.; Fruhwald S.; Heunks L. M. A.; Suominen P. K.; Gordon A. C.; Kivikko M.; Pollesello P. Levosimendan: current data, clinical use and future development. Heart Lung Vessel 2013, 5, 227–245. [PMC free article] [PubMed] [Google Scholar]
- Robertson I. M.; Pineda-Sanabria S. E.; Yan Z.; Kampourakis T.; Sun Y.-B.; Sykes B. D.; Irving M. Reversible covalent binding to cardiac troponin C by the Ca2+-sensitizer levosimendan. Biochem. 2016, 55, 6032–6045. 10.1021/acs.biochem.6b00758. [DOI] [PubMed] [Google Scholar]
- Robertson I. M.; Baryshnikova O. K.; Li M. X.; Sykes B. D. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochem. 2008, 47, 7485–7495. 10.1021/bi800438k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi S.; Pollesello P.; Levijoki J.; Kaheinen P.; Haikala H.; Edes I.; Papp Z. The effects of levosimendan and OR-1896 on isolated hearts, myocyte-sized preparations and phosphodiesterase enzymes of the guinea pig. Eur. J. Pharmacol. 2004, 486, 67–74. 10.1016/j.ejphar.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Kopustinskiene D. M.; Pollesello P.; Saris N. E. Levosimendan is a mitochondrial KATP channel opener. Eur. J. Pharmacol. 2001, 428, 311–314. 10.1016/S0014-2999(01)01350-4. [DOI] [PubMed] [Google Scholar]
- Yokoshiki H.; Katsube Y.; Sunagawa M.; Sperelakis N. Levosimendan, a novel Ca2+ sensitizer, activates the glibenclamide-sensitive K+ channel in rat arterial myocytes. Eur. J. Pharmacol. 1997, 333, 249–259. 10.1016/S0014-2999(97)01108-4. [DOI] [PubMed] [Google Scholar]
- Ørstavik Ø.; Ata S. H.; Riise J.; Dahl C. P.; Andersen G. Ø.; Levy F. O.; Skomedal T.; Osnes J.-B.; Qvigstad E. Inhibition of phosphodiesterase-3 by levosimendan is sufficient to account for its inotropic effect in failing human. Br. J. Pharmacol. 2014, 171, 5169–5181. 10.1111/bph.12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada Y.; Kobayashi M.; Narimatsu A.; Ohizumi Y. Potent stimulation of myofilament force and adenosine triphosphatase activity of canine cardiac muscle through a direct enhancement of troponin C Ca2+ binding by MCI-154, a novel cardiotonic agent. J. Pharmacol. Exp. Ther. 1989, 250, 272–277. [PubMed] [Google Scholar]
- Lues I.; Beier N.; Jonas R.; Klockow M.; Haeusler G. The two mechanisms of action of racemic cardiotonic EMD 53998, calcium sensitization and phosphodiesterase inhibition, reside in different enantiomers. J. Cardiovasc. Pharmacol. 1993, 21, 883–892. 10.1097/00005344-199306000-00006. [DOI] [PubMed] [Google Scholar]
- Strauss J. D.; Zeugner C.; Caspar Rüegg J. The positive inotropic calcium sensitizer EMD 53998 antagonizes phosphate action on cross-bridges in cardiac skinned fibers. Eur. J. Pharmacol. 1992, 227, 437–441. 10.1016/0922-4106(92)90163-p. [DOI] [PubMed] [Google Scholar]
- Herold P.; Herzig J. W.; Wenk P.; Leutert T.; Zbinden P.; Fuhrer W.; Stutz S.; Schenker K.; Meier M.; Rihs G. 5-Methyl-6-phenyl-1-3,5,6-tetrahydro-3,6-methano-1,5-benzodiazocine-2,4-dione (BA 41899): Representative of a novel class of purely calcium-sensitizing agents. J. Med. Chem. 1995, 38, 2946–2954. 10.1021/jm00015a017. [DOI] [PubMed] [Google Scholar]
- Neumann J.; Eschenhagen T.; Grupp I. L.; Haverich A.; Herzig J. W.; Hirt S.; Kalmár P.; Schmitz W.; Scholz H.; Stein B.; Wenzlaff H.; Zimmermann N. Positive inotropic effects of the calcium sensitizer CGP 48506 in failing human myocardium. J. Pharmacol. Exp. Ther. 1996, 277, 1579–1585. [PubMed] [Google Scholar]
- Malik F. I.; Morgan B. P. Cardiac myosin activation part 1: from concept to clinic. J. Mol. Cell. Cardiol. 2011, 51, 454–461. 10.1016/j.yjmcc.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Ebashi S.; Ebashi F.; Kodama A. Troponin as the Ca+2-receptive protein in the contractile system. J. Biochem. 1967, 62 (1), 137–138. 10.1093/oxfordjournals.jbchem.a128628. [DOI] [PubMed] [Google Scholar]
- Spudich J. A.; Watt S. The Regulation of Rabbit Skeletal Muscle Contraction. J. Biol. Chem. 1971, 246 (15), 4866–4871. 10.1016/S0021-9258(18)62016-2. [DOI] [PubMed] [Google Scholar]
- Finer J. T.; Malik F.; Sakowicz R.; Shumate C.; Wood K.. Compositions and assays utilizing ADP or phosphate for detecting protein modulators. U.S. Patent 6,410,254 B1, January 18, 2007.; See Supporting Information and references therein for detailed muscle protein preparation procedures and assay methods.
- Hartman J. J.; Malik F.; Sakowicz R.; Finer J. T.. High throughput sarcomeric assay. U.S. Patent 0,203,835 A1, October 30, 2003.
- Cardiac bovine myofibrils were used for the biochemical assay. There is a high level of similarity for cardiac muscle across species, including humans.
- Li M. X.; Hwang P. M. Structure and function of cardiac troponin C (TNNC1): implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. 10.1016/j.gene.2015.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Spahiu E.; Osten J.; Behrens F.; Grünhagen F.; Scholz T.; Kraft T.; Nayak A.; Amrute-Nayak M. Cardiac ventricular myosin and slow skeletal myosin exhibit dissimilar chemomechanical properties despite bearing the same myosin heavy chain isoform. J. Biol. Chem. 2022, 298 (7), 102070. 10.1016/j.jbc.2022.102070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- All sulfonamide cardiac troponin activators described in this paper increase the ATP hydrolysis rate for both cardiac and slow skeletal myofibrils.
- Poppe L.; Hartman J. J.; Romero A.; Reagan J. D. Structural and thermodynamic model for the activation of cardiac troponin. Biochem. 2022, 61, 741–748. 10.1021/acs.biochem.2c00084. [DOI] [PubMed] [Google Scholar]
- Calculation of FS: (resting length – length at peak contraction)/resting length. Calculation of % change in FS from baseline: ((end diastolic diameter – end systolic diameter)/(end systolic diameter))*100.
- Mispelaere-Canivet C.; Spindler J.-F.; Perrio S.; Beslin P. Pd2(dba)3/Xantphos-catalyzed cross-coupling of thiols and aryl bromides/triflates. Tetrahedron 2005, 61, 5253–5259. 10.1016/j.tet.2005.03.078. [DOI] [Google Scholar]
- Xia M.; Chen S.; Bates D. K. Reactions of benzyl aryl sulfides with excess active halogen reagents. J. Org. Chem. 1996, 61, 9289–9292. 10.1021/jo961565+. [DOI] [Google Scholar]
- Romero A.; Ashcraft L. W.; Chandra A.; DiMassa V.; Cremin P.; Collibee S. E.; Chuang C.; Hartman J.; Hwee D. T.; St. Jean D. J. Jr.; Malinowski J.; DeBenedetto M.; Moebius D. C.; Payette J.; Vargas R.; Yeoman J.; Motani A. S.; Reagan J.; Malik F. I.; Morgan B. P.. Discovery of Nelutroctiv (CK-136), a Selective Cardiac Troponin Activator for the Treatment of Cardiovascular Diseases Associated with Reduced Cardiac Contractility. J. Med. Chem. 2024, 10.1021/acs.jmedchem.3c02413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Martinez D.; Johnston J. R.; Landim-Vieira M.; Ma W.; Antipova O.; Awan O.; Irving T. C.; Bryant Chase P.; Pinto J. R. Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell. Cardiol. 2018, 123, 26–37. 10.1016/j.yjmcc.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.