

Canavan Disease
Canavan disease is a recessively inherited vacuolar leukodystrophy caused by ASPA mutations [1–3]. ASPA encodes aspartoacylase, an oligodendroglial enzyme required for cleavage of the abundant brain amino acid N-acetyl-l-aspartate (NAA) to acetate and l-aspartate [4]. ASPA mutations are relatively common in Ashkenazi Jews, with carrier frequency estimates ranging between 1:40 and 1:60, but also occur, though substantially less often, in many other human populations [3, 5, 6]. The disease classically presents in infancy with ataxia, hypotonia, and failure to acquire normal developmental milestones, often in association with macrocephaly and seizures [3]. In atypical cases in which some aspartoacylase enzymatic activity remains, disease onset is delayed until several years after birth [2, 7, 8]. Neuroimaging shows brain white matter signal abnormalities, and, at later time-points, ventricular enlargement [9, 10]. In vivo proton nuclear magnetic resonance spectroscopy (1H-MRS) documents a 30% or greater elevation in brain NAA concentration ([NAAB]) [10]. Histological studies reveal brain “spongiform” vacuolation, astrogliosis, and dysmyelination [7, 11–13]. These neuropathological abnormalities are most prominent in superficial white matter and neighboring gray matter of the forebrain, cerebellum, and upper brainstem. In more advanced cases, the cerebral ventricles become enlarged, and numbers of brain neurons diminish [7, 12, 13]. No therapies have yet been proven to be effective in preventing or reversing progression of leukodystrophy in Canavan disease.
Aspa Mutant Mice
Aspartoacylase-deficient mice are useful for exploring the pathophysiology of Canavan disease and for preliminary evaluation of new treatment options. The histologically best characterized aspartoacylase-deficient mice are homozygous for an ethyl-N-nitrosourea-induced Aspa nonsense mutation (“Nur7”, Q193X) [14]. These AspaNur7/Nur7 mice, which do not express immunochemically detectable aspartoacylase, and maintain an elevated [NAAB], develop ataxia by postnatal day 21, usually survive into adulthood, but have a diminished median lifespan. Brain astroglial and intramyelinic vacuolation begin between postnatal days 7 and 14 (Fig. 1), followed by cerebral ventricular enlargement and loss of cerebral cortical and cerebellar neurons [14–18].
Fig. 1.
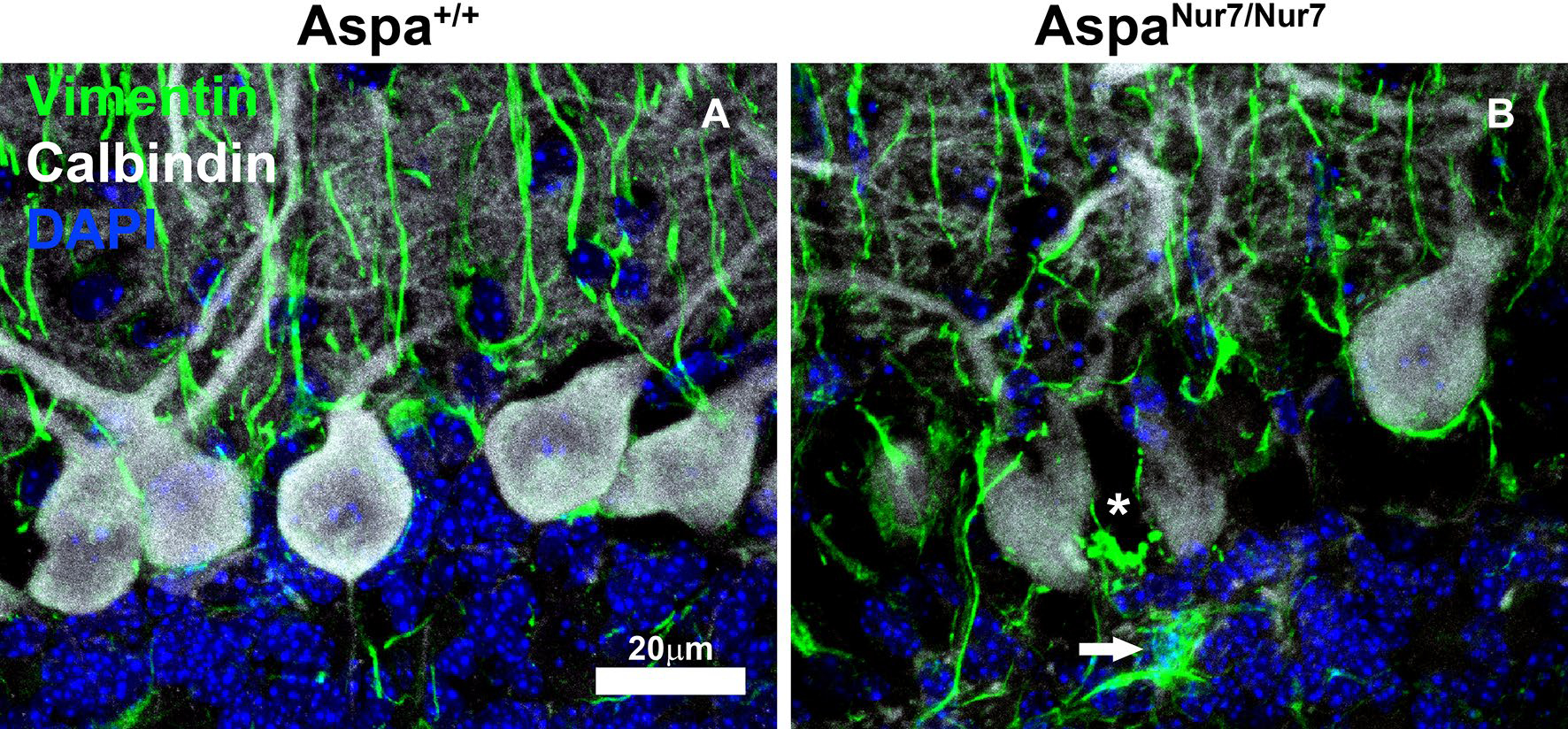
AspaNur7/Nur7 mouse cerebellar vacuolation and astrogliosis. Cryostat sections through cerebellum of a 14 days old wild-type (Aspa+/+) mouse (a) and of a 14 days old aspartoacylase-deficient (AspaNur7/Nur7) mouse (b) were immunostained for calbindin (white) and vimentin (green), then counterstained with DAPI, then viewed by laser scanning confocal microscopy. In the section from the AspaNur7/Nur7 mouse, the Purkinje cell layer is disorganized, and there is a vacuole between two Purkinje cells (asterisk) that is surrounded by vimentin+ fibrils. Also in the AspaNur7/Nur7 section, there is a vimentin+ hypertrophic astrocyte (arrow) in the internal granule cell layer mouse, and Bergmann glial vimentin+ fibrils appear to be fragmented. Scale bar = 20μ m in both panels
Linking Aspartoacylase Deficiency with Vacuolar Leukodystrophy
Two alternative, not mutually exclusive, hypotheses have been advanced to explain how aspartoacylase deficiency might cause vacuolar leukodystrophy in Canavan disease and in aspartoacylase-deficient mice. The “oligodendroglial starvation” hypothesis proposes that dysmyelination results from the inability by aspartoacylase-deficient oligodendroglia to derive acetate from NAA for synthesis of the myelin lipid precursor acetyl-CoA, and perhaps also from an inadequate oligodendroglial supply of NAA-derived l-aspartate to support production of high-energy phosphate compounds [19, 20]. The “NAA toxicity” hypothesis proposes, instead, that astroglial and intramyelinic vacuolation in Canavan disease are caused by impaired brain osmolar homeostasis resulting from elevated [NAAB] [21].
Two sets of data support the oligodendroglial starvation hypothesis. First, in vitro and in vivo isotope studies indicate that carbon atoms derived from NAA are incorporated into CNS myelin lipids [22]. Second, brain acetyl-CoA and ATP concentrations are diminished in aspartoacylase-deficient mice [19, 20]. But the oligodendroglial starvation hypothesis has been somewhat weakened by the demonstration that reducing [NAAB] to undetectably low levels in aspartoacylase-expressing (Aspa+/+) mice by homozygous constitutive deletion of Nat8l, which encodes neuronal N-acetyl transferase 8-like (also referred to as N-acetylaspartate synthetase), an enzyme essential for NAA synthesis [23, 24], does not prevent the mice from achieving full brain myelination [15]. Note, however, that initial central nervous system (CNS) myelination is slowed and CNS myelin composition and structure are altered in Nat8l−/− mice [25, 26].
In support of the “NAA toxicity” hypothesis, homozygous constitutive Nat8l knockout prevents vacuolar leukodystrophy in AspaNur7/Nur7 mice [15, 16] (Fig. 2a–c). Total ablation of NAA synthesis does not ensure a normal lifespan for A spaNur7/Nur7 mice, and blocks their synthesis of the CNS peptide neuromodulator/neurotransmitter NAAG [16, 27]. However, lowering [NAAB] toward the normal range in AspaNur7/Nur7 mice by constitutive knockout of a single Nat8l allele, or by brain delivery of an Nat8l inhibitory short hairpin-RNA via intracerebroventricular administration of an adeno-associated viral vector (AAV), markedly diminishes the severity of vacuolar leukodystrophy (Fig. 2a, b, d), but does not appear to lengthen lifespan [16, 18].
Fig. 2.
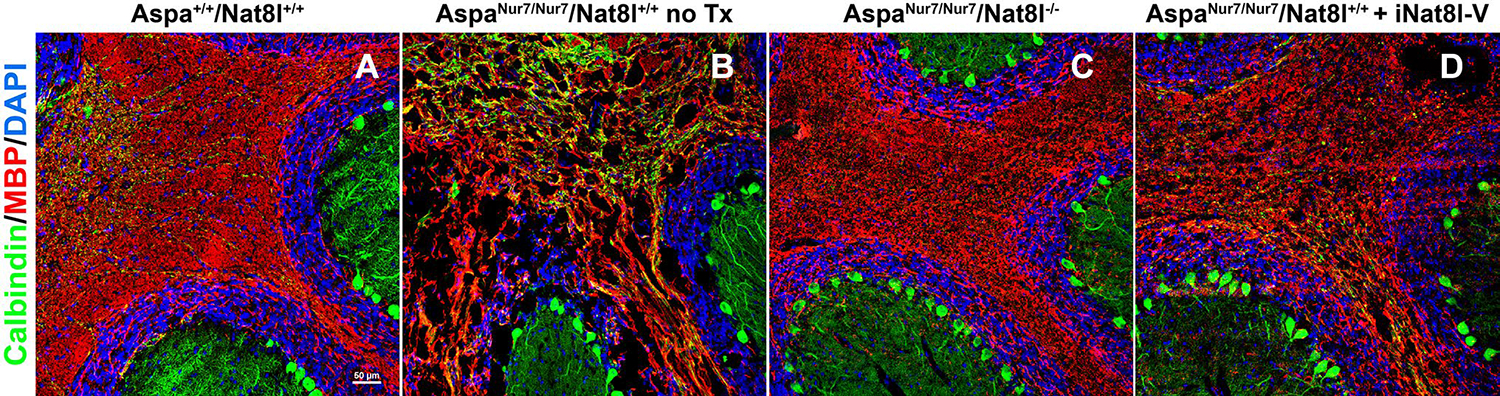
Cerebellar vacuolation and dysmyellnation in AspaNur7/Nur7 mice is prevented by homozygous constitutive Nat8l knockout, and substantially diminished in severity by neonatal brain Nat8l knockdown. Cryostat sections through cerebellum were prepared from brains of 2 months old wild-type (Aspa+/+/Nat8l+/+) (a), a 2 months old untreated A spaNur7/Nur7/Nat8l+/+ mouse (b), a 2 months old Aspa Nur7/Nur7/Nat8l−/− mouse (c), and a 2 months old A spaNur7/Nur7/Nat8l+/+ mouse that had been given an intracerebroventricular AAV carrying an Nat8l short hairpin inhibitory RNA (iNat8l-V) on postnatal day 1 (d). The sections were immunostained for myelin basic protein (MBP) and calbindin, counterstained with DAPI, and viewed by laser scanning confocal microscopy. Cerebellar vacuolation and dysmyelination were prominent in the untreated AspaNur7/Nur7 mouse, but these abnormalities were prevented by homozygous constitutive Nat8l knockout, and diminished in severity by Nat8l knockdown. Scale bar = 50μ m in all panels
The mechanism responsible for the toxicity of elevated [NAAB] has not been established. Cultured neural cells are not harmed by direct exposure to purified NAA [28], and oligodendroglial numbers are not diminished in A spaNur7/Nur7 or Canavan brain white matter [7]. Also, elevating [NAAB] in Aspa+/+ mice to an extent comparable to that in aspartoacylase-deficient mice by feeding NAA methyl ester or by engineering transgenic neuronal overexpression of Nat8l does not elicit clinical or neuropathological abnormalities [29, 30]. Thus, elevated overall [NAAB], is not sufficient to elicit vacuolar leukodystrophy in mice that express aspartoacylase.
Astroglia, but not oligodendroglia, express a sodium-coupled plasma membrane dicarboxylate transporter (NaDC3, encoded by Slc13a3) with sufficient affinity for NAA to maintain normal brain extracellular NAA concentration below 30μM [31, 32]. If astroglia do accumulate NAA from neurons via the action of NaDC3, and then transfer it to oligodendroglia, perhaps via astroglial/oligodendroglial gap junctions, then in the absence of oligodendroglial aspartoacylase, astroglia might over-accumulate NAA and therefore become vacuolated. Providing indirect support for this hypothesis, spontaneous mutations that inactivate ion channel-associated astroglial proteins, or disrupt astroglial/oligodendroglial gap junctions, are sufficient to elicit vacuolar leukodystrophy in A spa+/+ brains [33–35]. This hypothesis could be tested by examining the effects on brain morphology in A spaNur7/Nur7 and A spa+/+ mice of ablating astroglial NaDC3 by constitutive or conditional Slc13a3 knockout.
Future Directions
Advances in in vivo gene editing may ultimately make it possible to correct ASPA mutations in vivo. Until that approach becomes feasible, AAV-mediated brain ASPA transduction is likely to be the most promising avenue to pursue, based on the good results reported with this approach in aspartoacylase-deficient mice. Interestingly, administration of an AAV-ASPA designed to target either oligodendroglia or astroglia has been successful in preventing vacuolar leukodystrophy in neonatal aspartoacylase-deficient mice [36, 37]. It should be noted that the success of ASPA gene therapy in aspartoacylase-deficient mice is compatible with both the oligodendroglial starvation and NAA toxicity hypotheses, since brain aspartoacylase reconstitution normalizes both brain acetyl-CoA content and [NAAB].
An initial attempt at direct brain intraparenchymal AAV2-mediated ASPA gene therapy in a Canavan disease cohort was unsuccessful in preventing progression of clinical neurological deficits and cerebral ventricle enlargement, though it did achieve a slight lowering of [ NAAB] [9]. Among the possible explanations for this disappointing result are that vacuolar leukodystrophy was already far advanced in some patients in the cohort; and that direct brain parenchymal vector administration, while successful in preventing vacuolar leukodystrophy in aspartoacylase-deficient mice, did not elicit sufficiently widespread oligodendroglial and astroglial aspartoacylase expression in the much larger human brains.
Aspartoacylase contributes to acetyl-CoA generation from NAA in various non-neural tissues as well as in oligodendroglia, and aspartoacylase deficiency also causes dysfunction of those tissues. For example, the rate of apoptosis by peritoneal macrophages is increased, and renal distal convoluted tubules are vacuolated, in aspartoacylase-deficient mice. These and other deficits attributable to a lack of extraneural aspartoacylase may contribute to the shortening in median lifespan that has been documented in AspaNur7/Nur7 mice, and would not be expected to be corrected by ASPA gene therapy that is directed solely to the CNS [16, 38–40].
Lowering [NAAB] may provide another effective therapy for Canavan disease, as has been shown in aspartoacylase-deficient mice [15, 16]. Oral lithium citrate administration, a therapy currently advocated for Canavan disease, does lower [NAAB] slightly in children with this leukodystrophy, but this effect, with usually clinically tolerable dosages, was too weak to be more than minimally clinically effective [41]. Instead, it should be possible to lower [NAAB] toward normal in infants and children with Canavan disease by suppressing brain N-acetyltransferase 8-like activity, either via NAT8L knockdown [18] or by development of a druggable N-acetyltransferase 8-like inhibitor [42].
Acknowledgements
This study was supported by Shriners Hospitals Grant 439043; NIH 1R21NS096004-01; and the Dana Foundation.
References
- 1.Matalon R, Michals K, Sebesta D, Deanching M, Gashkoff P, Casanova J (1988) Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet 29:463–471 [DOI] [PubMed] [Google Scholar]
- 2.Mendes MI, Smith DE, Pop A et al. (2017) Clinically distinct phenotypes of Canavan disease correlate with residual aspartoacylase enzyme activity. Hum Mutat 38:524–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoshino H, Kubota M (2014) Canavan disease: clinical features and recent advances in research. Pediatr Int 56:477–485 [DOI] [PubMed] [Google Scholar]
- 4.Madhavarao CM, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM (2004) Immunohistochemical localization of aspartoacylase in the rate central nervous system. J Comp Neurol 472:318–329 [DOI] [PubMed] [Google Scholar]
- 5.Feigenbaum A, Moore R, Clarke J, Hewson S, Chitayat D, Ray PN, Stockley TL (2004) Canavan disease: carrier-frequency determination in the Ashkenazi Jewish population and development of a novel molecular diagnostic assay. Am J Med Genet 124A:142–147 [DOI] [PubMed] [Google Scholar]
- 6.Rivas MA, Avila BE, Koskela J et al. (2018) Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet 14:e1007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jellinger K, Seitelberger F (1969) Juvenile form of spongy degeneration of the CNS. Acta Neuropath (Berl) 13:276–281 [DOI] [PubMed] [Google Scholar]
- 8.Janson CG, Kolodny EH, Zeng B-J et al. (2006) Mild-onset presentation of Canavan’s disease associated with novel G212A point mutation in aspartoacylase gene. Ann Neurol 59:428–431 [DOI] [PubMed] [Google Scholar]
- 9.Leone P, Shera D, McPhee SW et al. (2012) Long-term follow-up after gene therapy for Canavan disase. Science Trans Med 4:165ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janson CG, McPhee SWJ, Francis J et al. (2006) Natural history of Canavan disease revealed by proton magnetic resonance spectroscopy (1J-MRS) and diffusion-weighted MRI. Neuropediatrics 37:209–221 [DOI] [PubMed] [Google Scholar]
- 11.Gambetti P, Mellman WJ, Gonatoas NK (1969) Familial spongy degeneration of the central nervous system (van Bogaert–Bertrand disease). an ultrastructural study. Acta Neuropathol 12:103–115 [DOI] [PubMed] [Google Scholar]
- 12.Adachi M, Schneck L, Cara J, Volk BW (1973) Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan disease). A review. Hum Pathol 4:331–347 [DOI] [PubMed] [Google Scholar]
- 13.Mirimanoff P (1976) La dystrophie spongieuse hereditaire des enfants (Canavan van Bogaert–Bertrand). J Neurol Sci 28:159–185 [DOI] [PubMed] [Google Scholar]
- 14.Traka M, Wollmann RI, Cerda SR, Dugas J, Barres BA, Popko B (2008) Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci 28:11537–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo F, Bannerman P, Mills Ko E, Miers L, Xu J, Burns T, Li S, Freeman E, McDonough JA, Pleasure D (2015) Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann Neurol 77:884–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maier H, Wang-Eckhardt L, Hartmann D, Gieselmann V, Eckhardt M (2015) N-acetylaspartate synthase deficiency corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J Neurosci 35:14501–14516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohn J, Bannerman P, Guo F, Burns T, Miers L, Croteau C, Singhal NK, McDonough JA, Pleasure D (2017) Suppressing N-acetyl-L-aspartate synthesis prevents loss of neurons in amurine model of Canavan leukodystrophy. J Neurosci 37:413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bannerman P, Guo F, Chechneva O, Burns T, Zhu X, Wang Y, Kim B, Singhal NK, McDonough JA, Pleasure D (2018) Brain Nat8l knockdown suppresses spongiform leukodystrophy in an aspartoacylase-deficient Canavan disease mouse model. Mol Ther 26:793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madhavarao CN, Arun P, Moffett JR, Szacs S, Surendram S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W, Namboodiri MA (2005) Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci USA 102:5221–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Francis JS, Wojtas L, Markov V, Gray SJ, McCown TJ, Samulski RJ, Bilaniuk LT, Wang DJ, DeVivo DC, Janson CG, Leone P (2016) N-acetylaspartate supports the energetic demands of developmental myelination via oligodendroglia aspartoacylase. Neurobiol Dis 96:323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baslow MH, Guilfoyle DN (2013) Canavan disease, a rare early-onset human spongiform leukodystrophy: insights into its genesis and possible clinical interventions. Biochimie 95:946–956 [DOI] [PubMed] [Google Scholar]
- 22.Burri R, Steffen C, Herschkowitz N (1991) N-acetyl-L-aspartate is a major source of acetyl groups for lipid synthesis during rat brain development. Dev Neurosci 13:403–411 [DOI] [PubMed] [Google Scholar]
- 23.Wiami E, Tyteca D, Pierrot N et al. (2009) Molecular identification of aspartate N-acetyltransferase and its mutation in hypacetylaspartia. Biochem J 425:127–136 [DOI] [PubMed] [Google Scholar]
- 24.Ariyannur PS, Moffett JR, Manickam P, Pattabiraman N, Arun P, Nitta A, Nabeshima T, Madhavarao CN, Namboodiri AM (2010) Methamphetamine-induced neuronal protein NAT8L is the NAA biosynthetic enzyme: implications for specialized acetyl coenzyme A metabolism in the CNS. Brain Res 1335:1–13 [DOI] [PubMed] [Google Scholar]
- 25.Singhal NK, Huang H, Li S, Clements R, Gadd J, Daniels A, Kooijman EE, Bannerman P, Burns T, Guo F, Pleasure D, Freeman E, Shriver L, McDonough J (2017) The neuronal metabolite NAA regulates histone H3 methylation in oligodendrocytes and myelin lipid composition. Exp Brain Res 235:279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sumi K, Uno K, Noike H, Tomohiro T, Hatanaka Y, Furukawa-Hibi Y, Nabeshima T, Miyamoto Y, Nitta A (2017) Behavioral impairment in SHATI/NAT8L knockout mice via dysfunction of myelination development. Sci Rep 7:16872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neale JH, Olsczewski RT, Zuo D, Jancrura KJ, Profaci CP, Lavin KM, Madore JC, Bzdega T (2011) Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J Neurochem 118:490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolodziejczyk K, Hamilton NB, Wade A, Karadottir R, Attwell D (2009) The effect of N-acetyl-aspartyl-glutamate and N-acetylaspartate on white matter oligodendrocytes. Brain 132:1496–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appu AP, Moffett JR, Arun P, Moran S, Nambiar V, Krishnan JKS, Puthillathu N, Namboodiri AMA (2017) Increasing N-acetylaspartate in the brain during postnatal myelination does not cause the CNS pathologies of Canavan disease. Front Mol Neurosci Jun 2:10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Von Jonquieres G, Spencer ZHT, Rowlands BD et al. (2018) Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol 135:95–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujita T, Katsukawa H, Yodoya E, Wada M, Shimada A, Okada N, Yamamoto A, Ganapathy V (2005) Transport characteristics of N-acetyl-L-aspartate in rat astrocytes: involvement of sodium-coupledhigh-affinity carboxylate transporter NaC3/NaDC3-mediated transport system. J Neurochem 93:706–714 [DOI] [PubMed] [Google Scholar]
- 32.Shannon RJ, van der Heide S, Carter EL, Jalloh I, Menon DK, Hutchinson PJ, Carpenter KLH (2016) Extracellular N-acetylaspartate in human traumatic brain injury. J Neurotrauma 33:319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tress O, Maglione M, May D et al. (2012) Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J Neurosci 32:7499–7518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K (2011) Pathologic and phenotypic alterations in a mouse expressing a Connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet 7:e1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez-Hernandez T, Sirisi S, Capdevila-Nortes X et al. (2011) Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukodystrophy with subcortical cysts. Hum Mol Genet 23:5069–5086 [DOI] [PubMed] [Google Scholar]
- 36.Ahmed SS, Li H, Cao C et al. (2013) A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol Ther 21:2136–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gessler DJ, Li D, Xu H, Su Q, Sanmiguel J, Tuncer S, Moore C, King J, Matalon R, Gao G (2017) Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. JCI Insight 2:e90807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sommer A, Sass JO (2012) Expression of aspartoacylase (ASPA) and Canavan disease. Gene 505:206–210 [DOI] [PubMed] [Google Scholar]
- 39.Gautier EL, Ivanov S, Williams JW et al. (2014) Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med 211:1525–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed SS, Schattgen SA, Frakes AE et al. (2016) rAAV gene therapy in a Canavan’s disease mouse model reveals immune impairments and an extended pathology beyond the central nervous system. Mol Ther 24:1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Assad M, Janson C, Wang DJ, Suri N, Bilaniuk L, Leone P (2010) Lithium citrate reduces excessive intra-cerebral N-acetyl aspartate in Canavan disease. Eur J Paediatr Neruol 14:354–359 [DOI] [PubMed] [Google Scholar]
- 42.Thangavelu B, Mutthamsetty V, Wang Q, Viola RE (2017) Design and optimization of aspartate N-acetyltransferase inhibitors for the potential treatment of Canavan disease. Bioorg Med Chem 25:870–885 [DOI] [PubMed] [Google Scholar]