
Fig. 4 |. Drug combinations with ATR–CHK1–WEE1 inhibitors.
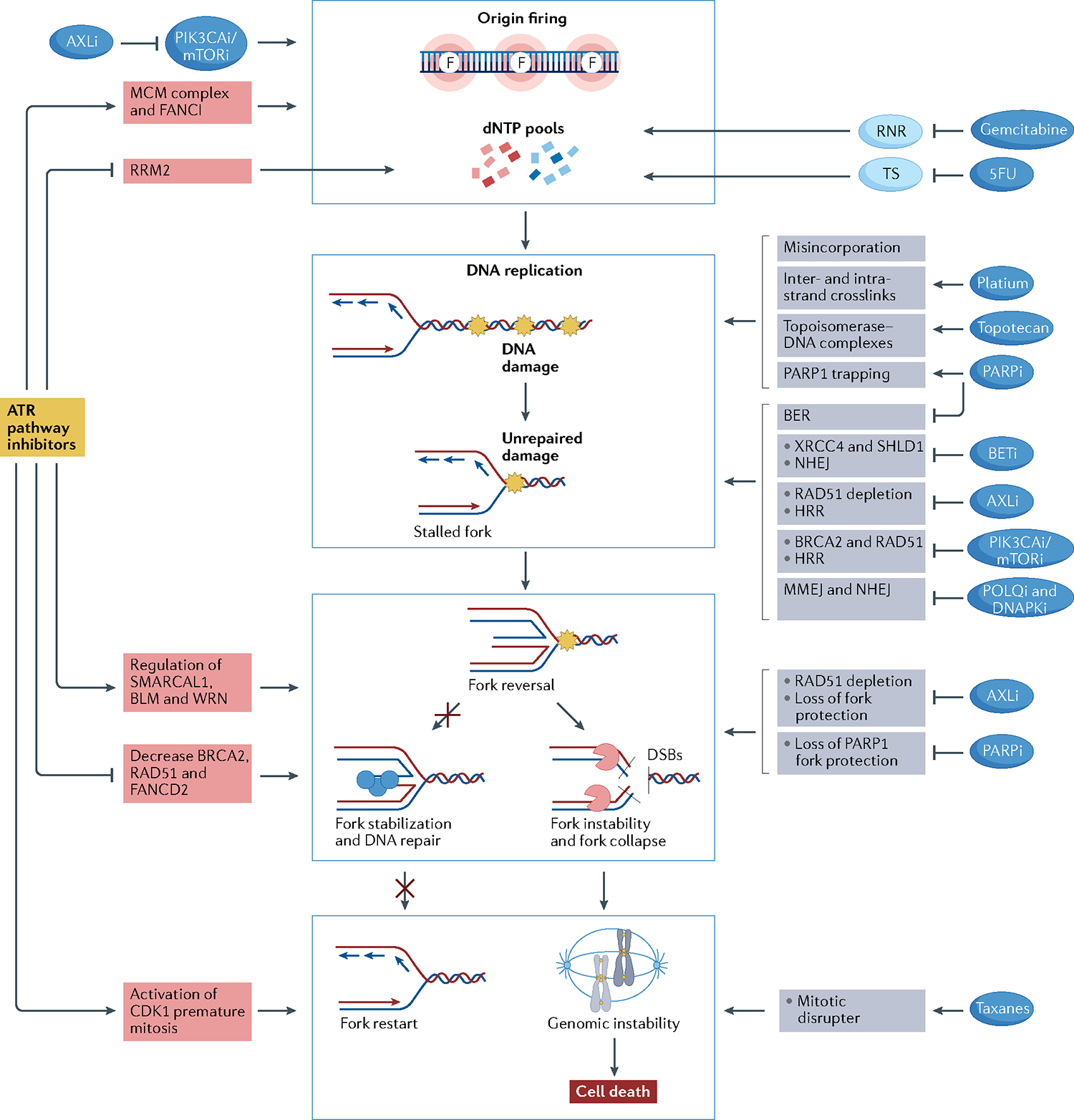
Rationale for drug combinations that promote replication stress, potentiating the effect of ATR–CHK1–WEE1 inhibitors. Red boxes indicate mechanisms of action related to ataxia telangiectasia mutated (ATM) and Rad3-related (ATR) pathway inhibitors; grey boxes indicate mechanisms of action of the other drugs. Central blue squares group the main mechanisms through which drugs may activate replication stress. Drugs may activate replication stress by decreasing deoxynucleotide (dNTP) pools directly or through increasing origin firing (indicated by the letter F, top blue box), increasing DNA damage (second blue box), promoting fork instability (third blue box) and disrupting mitosis (bottom blue box). ATR inhibitors (ATRi), CHK1 inhibitors (CHK1i) and WEE1 inhibitors (WEE1i) decrease dNTP pools by activating origin firing through loss of Fanconi anaemia group I protein (FANCI) phosphorylation and minichromosome maintenance 2–7 complex (MCM2–7) inhibition and by decreasing ribonucleoside-diphosphate reductase subunit M2 (RRM2) expression. PIK3CA/mTOR inhibitors synergize with ATRi, CHK1i or WEE1i by promoting origin firing through increasing cell division cycle 45 (CDC45) expression, which leads to activation of the MCM2–7 complex. Moreover, AXL inhibitors (AXLi) decrease dNTP pools by inhibiting the PI3K–mTOR pathway. Nucleoside analogues reduce intracellular dNTP pools by inhibiting the enzymes responsible for dNTP synthesis. Gemcitabine inhibits ribonucleotide reductase (RNR), and 5-fluorouracil (5FU) inhibits thymidylate synthetase (TS). Drugs may increase DNA damage by directly damaging the DNA or by inhibiting DNA repair mechanisms. Gemcitabine and 5FU lead to DNA damage by their misincorporation into DNA. Platinum salts cause inter- and intra-strand crosslinks, and topotecan generates topoisomerase–DNA complexes, which become obstacles to fork progression. Poly[ADP-ribose]polymerase (PARP) inhibitors (PARPi) create obstacles to fork progression through PARP1 trapping and inhibition of the base excision repair (BER) pathway. Bromodomain and extra-terminal domain (BET) inhibitors (BETi) lead to non-homologous end-joining (NHEJ) inhibition through downregulation of XRCC4 and SHLD1. AXL inhibition results in inhibition of homologous recombination repair (HRR) through RAD51 depletion. PIK3CA/mTOR inhibition leads to HRR inhibition through downregulation of BRCA2 and RAD51. POLQ inhibitors and DNAPK inhibitors directly inhibit microhomology-mediated end-joining (MMEJ) and NHEJ pathways, respectively. ATRi, CHK1i and WEE1i cause fork instability by regulating fork remodellers such as SMARCAL1, Bloom syndrome protein (BLM) and Werner syndrome ATP-dependent helicase (WRN) and by decreasing the activity of fork protectors such as BRCA2, RAD51 and FANCD2. AXLi and PARPi also lead to fork instability by depletion of RAD51 and inhibition of PARP1 fork protection, respectively. Finally, ATRi, CHK1i and WEE1i lead cells to premature mitosis and may synergize with mitotic disrupter agents such as taxanes.