

Abstract
Mantle cell lymphoma (MCL) is recognized as a well‐defined B cell neoplasm characterized by overexpression of cyclin D1 (CCND1), with “classical” and “aggressive” variant subtypes. A small‐cell variant of MCL (small‐MCL), resembling small lymphocytic lymphoma/chronic lymphocytic lymphoma (CLL/SLL), has been added to the World Health Organization classification. However, to the best of our knowledge, there have been no studies focusing on this neoplasm. In the present study, we analyzed 15 cases of CCND1‐positive small‐MCL, including immunohistochemical analysis of Ki‐67 and CCND1 expression, and compared our findings with those of 151 cases of classical MCL. Morphologically, most small‐MCL showed a diffuse growth pattern (76.9%), whereas others featured a very thin mantle zone pattern resembling a reactive follicle (23.1%). Bone marrow involvement and splenomegaly occurred significantly more frequently in small‐MCL than in classical MCL (P < 0.05). Ki‐67 expression in small‐MCL was lower than in classical MCL (mean [±2 SD] 12.5 ± 17.3% and 25.2 ± 25.5%, respectively; P < 0.001), but there was no significant difference in CCND1 expression (P = 0.2445). The 5‐year survival rate in small‐MCL was 83.3%. Although there was no significant difference in outcome between small‐MCL and classical MCL (P = 0.287), only one small‐MCL patient died of the disease. Thus, small‐MCL constitutes a specific subset of indolent lymphoma with distinguishing features, possibly making a major contribution to the accuracy of therapeutic decisions. In addition, clinicians should be aware of the possible presence of small‐MCL to avoid making a misdiagnosis of follicular hyperplasia or CLL/SLL. (Cancer Sci 2011; 102: 1734–1741)
The pathological category of low‐grade B cell non‐Hodgkin’s lymphoma (NHL) basically indicates mature B cell neoplasms with small to medium‐sized cells, such as follicular lymphoma (FL), marginal zone lymphoma (MZL), B cell chronic lymphocytic leukemia/small lymphocytic lymphoma (B‐CLL/SLL), and mantle cell lymphoma (MCL). Although most of these neoplasms usually have an indolent clinical course, MCL is considered to be more aggressive and incurable than the others.( 1 , 2 ) The median overall survival of MCL patients is only 3–5 years with the use of conventional chemotherapies, and adding rituximab has not improved the outcome.( 1 , 2 , 3 ) Accordingly, the use of as intensive chemotherapy as possible is currently considered for any patients diagnosed with MCL.( 4 , 5 ) Conversely, the existence of a specific subset of MCL patients with an indolent clinical course and long survival >5 years has been established beyond any doubt.( 6 , 7 , 8 , 9 , 10 )
Because virtually all cases feature cyclin‐D1 (CCND1) overexpression resulting from t(11;14)(q13;q32), MCL is a relatively well‐defined lymphoma. The classical variant, which usually shows a nodular and/or diffuse growth pattern of atypical medium‐sized B cells, is the most frequent, with the blastoid or pleomorphic (aggressive) variant, which has a median overall survival of <2 years, considered one of the worst types of NHL.( 11 , 12 , 13 , 14 , 15 , 16 )
As reported previously, the immunohistochemically estimated proliferation index (PI) for Ki‐67 is a stronger prognostic factor for MCL than the international prognostic index (IPI).( 17 , 18 ) In addition, we have shown previously that MCL has three morphological evolutionary forms, classical, intermediate, and aggressive, which occur along with increases in the labeling index for CCND1 and Ki‐67.( 19 ) Thus, MCL has a variety of clinical manifestations, with a broad spectrum of PI,( 17 , 18 , 19 ) and recently the importance of morphological variants has received growing recognition.( 13 , 14 , 15 , 16 , 20 )
The World Health Organization (WHO) classification( 11 , 12 ) includes definitions of two additional variants, namely small‐cell and marginal zone‐like MCL. Many studies have included small‐MCL as part of the classical variant, because it reportedly accounts for only 3.6–9.8% of all MCL.( 16 , 19 , 21 ) Rosenwald et al. ( 21 ) reported that patients with small‐MCL had the lowest expression of the proliferation gene signature and the longest survival compared with patients with other variants. These findings suggest that small‐MCL may be an indolent lymphoma. However, to the best of our knowledge, there have been no studies reported that have focused on this neoplasm.
In the present study, combined clinicopathologic and immunophenotypic analyses were performed for a series of 15 cases of small‐MCL. To clarify the clinical and biological features of this disease, an immunohistochemical analysis of Ki‐67 and CCND1 expression was included and compared with expression of these markers in patients with the classical variant of MCL cases. In addition, an immunohistochemical investigation of Sox11 expression was undertaken, which has been reported to be a highly specific marker for both CCND1‐positive and ‐negative MCL.( 22 )
Materials and Methods
Patient population, initial diagnosis, and clinical information. In all, 15 patients with CCND1‐positive small‐MCL, diagnosed between 1981 and 2010 at the Departments of Pathology at Kurume University (Fukuoka, Japan), Nagoya University (Aichi, Japan), and Okayama University (Okayama, Japan), were included in the present study. Informed consent was provided according to the Declaration of Helsinki. For comparison of characteristics in small‐MCL, 151 cases of classical MCL used in a previous study were selected;( 19 ) in addition, to clarify the clinicopathologic characteristics of cases of classical MCL, 44 aggressive MCL cases from the same study were included.( 19 ) The diagnostic materials available for examination comprised six lymph nodes and nine extranodal sites (two from the stomach, one each from the colon, pharynx, spleen, small intestine, tonsil, and bone marrow, and one peripheral blood smear). Tissues were routinely fixed in buffered formalin and embedded in paraffin. To determine the morphologic features of all cases, samples were stained with hematoxylin–eosin (H&E) to assess cytological features and growth patterns. Diagnoses were made independently by three of the authors (K.O., S.N., and T.Y.). If there were differences of opinion among the authors, the cases were discussed until a consensus was reached on a multihead microscope.
After exclusion of cases with classical or aggressive morphology of MCL, the diagnosis of small‐MCL was supported by a combination of cytologic and immunophenotypic features. A small‐MCL has round or oval small nuclei with dense nuclear chromatin and resembles CLL/SLL, implying that it is difficult to distinguish between them without following immunophenotypic studies. Namely, small‐MCL is positive for CD20, CD5, and CCND1, as shown by immunohistochemistry or flow cytometry used to determine the origin of the mantle cells. Basically, in the present study cases with well‐defined nodular components and clear reactive germinal centers were classified as “MZ‐pattern”. Classical MCL consists of medium‐sized atypical lymphoid cells with slightly or markedly irregular nuclear contours, thus closely resembling centrocytes. The term “aggressive MCL” is generally used to designate a combination of two morphological forms, blastoid and pleomorphic. The blastoid form comprises a homogeneous population of cells with a morphology resembling that of lymphoblasts and with scant, indistinct cytoplasm, finely dispersed chromatin, and inconspicuous nucleoli. The pleomorphic form comprises a heterogeneous population of large cells with oval to irregular nuclear contours and frequent prominent nucleoli.
All clinical and laboratory information for the patients was obtained from their medical records and by communicating with the physicians concerned at each of the institutions. Information collated for each patient included the results of physical examination, age, sex, Ann Arbor stage, the International Prognostic Index (IPI; determined by age, clinical stage, performance status, serum lactate dehydrogenase level, and the number of extranodal disease sites),( 23 ) bone marrow involvement, peripheral blood invasion, gut involvement (endoscopic view), hepatomegaly, splenomegaly, and complete white blood counts.
Immunophenotypic studies, Ki‐67 index, and CCND1 index. Flow cytometry and immunohistochemistry were used for the immunophenotypic studies. Flow cytometric immunophenotypic studies were performed with a flow cytometer (FACSCalive; Becton‐Dickinson, Mountain View, CA, USA) and the Cell Quest software program (Becton‐Dickinson) using conventional methods described previously.( 24 ) Cells were stained with FITC‐ or phycoerythrin (PE)‐labeled monoclonal antibodies as follows: CD5 (PE), CD10 (PE), CD23 (PE), and CD20 (FITC). CD5, CD10, and CD23 were obtained from Coulter Clone (Hialeah, FL, USA), whereas CD20 and CD25 were obtained from Becton‐Dickinson. Samples were also evaluated for the expression of markers such as cyclin D1 (NeoMarkers, Fremont, CA, USA), Sox11 (Atlas Antibodies, Stockholm, Sweden), and Ki‐67 (DakoCytomation, Glostrup, Denmark). Formalin‐fixed, paraffin‐embedded tissues were used for all immunohistochemical staining, and antibody dilutions and antigen retrieval procedures were the standardized procedures used in the departments concerned.
For determination of the Ki‐67 and CCND1 indices, sections were examined and scored independently by the same three authors who made the initial diagnosis. Basically, the percentage of tumor cells that stained positive for antibodies against Ki‐67 or CCND1 in the areas of highest expression were calculated and then averaged to yield an immunohistological score of 0–100% for 10 consecutive high‐power fields (×400 magnification).
Molecular analysis of t(11;14)(q13;q32). The t(11;14)(q13;q32) translocation was assessed by conventional cytogenetic studies and/or FISH using previously established protocols.( 25 ) Cells for these studies were taken with the LSI IGH/CCND1 dual‐color, dual‐fusion translocation probe (Vysis, Bergisch‐Gladbach, Germany) from the same samples that were used for the first histopathological analyses.
Statistical analysis. Survival curves were constructed with the Kaplan–Meier method, and the level of significance of differences in survival rates was tested by means of the generalized log‐rank test. Correlations among three groups were examined with the chi‐squared test (Fisher’s exact test). Differences were considered significant if P < 0.05. All data were analyzed with SAS version 9.2 (SAS Institute, Cary, NC, USA).
Results
Clinical features of small‐MCL. The clinical findings for the 15 patients with small‐MCL are summarized in Table 1. Eight patients had systemic lymphadenopathy, whereas the seven patients that did not have confirmed peripheral or internal lymph nodes >1 cm in size are listed separately. Only one case (Case 12) had a lesion of Waldeyer’s throat ring but no lymphadenopathy.
Table 1.
Clinical findings of 15 cases of the small‐cell variant of mantle cell lymphoma
| Case no. | Age/sex | IPI | Lymphadenopathy | WBC (/mm3) | Leukemic change | BM involvement | Hepato/splenomegaly | Endoscopic view | Therapy | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70/M | HI | + | 5370 | + | + | −/+ | – | “Watch and wait” | AWD 5 months |
| 2 | 60/M | H | + | 151 800 | + | + | +/+ | – | R‐Hyper CVAD/MA | AWD 4 months |
| 3 | 60/M | HI | + | 9300 | − | + | −/− | TL of stomach | R‐Hyper CVAD/MA, Allo BMT | ACR 19 months |
| 4 | 75/M | HI | + | 8860 | − | + | −/− | MLP of colon | Modified R‐CHOP | ACR 13 months |
| 5 | 68/M | H | + | 29 200 | + | + | +/+ | ND | CHOP, CHASER, LEED, Auto PBSCT | ACR 19 months |
| 6 | 66/M | LI | + | 3030 | − | + | −/+ | ND | THP‐COP, CHASER | DOD 38 months |
| 7 | 61/M | HI | + | 11 400 | − | + | +/+ | ND | R‐CHOP | AWD 37 months |
| 8 | 61/M | LI | + | 4800 | − | − | −/− | – | Modified CHOP | ACR 47 months |
| 9 | 65/F | L | − | 5680 | − | + | −/+ | TL of stomach | Modified CHOP | AWD 53 months |
| 10 | 63/M | LI | − | 28 200 | + | + | −/+ | – | Endoxan® by oral solution | AWD 29 months |
| 11 | 60/M | HI | −† | 77 100 | + | + | −/+ | TL of stomach, MLP of colon | R‐Hyper CVAD/MA | AWD 2 months |
| 12 | 71/F | H | −‡ | 9300 | + | + | +/+ | MLP of terminal ileum and colon | “Watch and wait” | ACR 66 months |
| R‐CHOP§ | ||||||||||
| 13 | 59/F | HI | −† | 3600 | + | + | −/+ | – | R‐CHOP | ACR 5 months |
| 14 | 63/M | LI | −† | 8900 | − | − | −/+ | MLP of cecum | R‐CHOP, R‐2CDA | AWD 68 months |
| 15 | 65/M | H | − | 5100 | − | − | −/+ | MLP of cecum | R‐CHOP, CHASER | AWD 86 months |
†Confirmed lymph nodes are <1 cm. ‡Case 12 had a lesion of Waldeyer’s throat ring, but no lymphadenopathy. §After a “watch and wait” approach for 64 months, this case was subjected to R‐CHOP therapy (rituximab + cyclophosphamide, adriamycin, vincristine, and prednisone). IPI, International Prognostic Index; WBC, white blood cells; BM, bone marrow; NA, not available; ND, not done; TL, tumorous lesion; MLP, multiple lymphomatous polyposis; AWD, alive with disease; ACR, alive in complete response; DOD, dead of disease; R‐Hyper CVAD/MA, rituximab + fractionated cyclophosphamide, doxorubicin, vincristine, and dexamethasone, alternated with high‐dose methotrexate–cytarabine; Allo BMT, Allogeneic Bone Marrow Transplantation; CHASER, rituximab + dexamethasone, cyclophosphamide, cytarabine, and etposide; LEED, cyclophosphamide, melphalan, etposide, and dexamethasone; Auto PBSCT, autologous peripheral blood stem cell transplantation; THP‐COP, pirarubicin, cyclophosphamide, vincristine, and prednisone; R‐2CDA, rituximab + cladribine.
The initial diagnosis was established for 12 men and three women with a median age of 64 years (range 59–75 years), most of whom had an advanced IPI score (High or High–Intermediate; 66.7%), extranodal involvement (93.3%), and splenomegaly (80%). The most common extranodal site was the bone marrow. Lymphoma cells in the peripheral blood were observed in seven cases, but absolute leukocytosis (>10 000/mm3) with leukemic change was present in only four cases. Gastrointestinal endoscopy identified seven cases with involvement of the gastrointestinal tract. Three of these patients had tumorous lesions (TL) of the stomach and five had multiple lymphomatous polyposis (MLP) of the colon or small intestine (note, one patients showed features of both TL and MLP [Case 11]; Fig. 1a).
Figure 1.
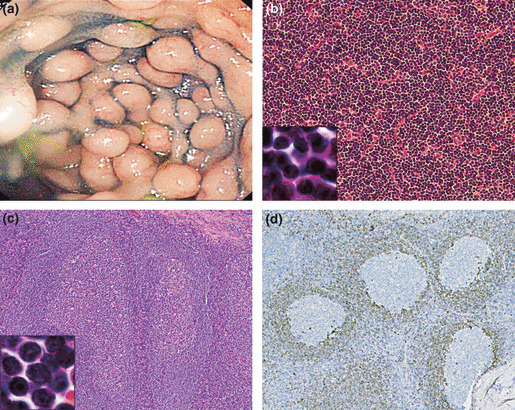
(a) Endoscopy of the colon of Case 11 showed multiple lymphomatoid polyposis involving the entire length of the colon. (b) Histological features of the small‐cell variant of mantle cell lymphoma (small‐MCL) in Case 5 showing a diffuse growth pattern without proliferation centers (original magnification ×200). At higher magnification (original magnification ×2500), small atypical lymphocytes with scant cytoplasm and dense nuclear chromatin are seen. Mitotic features were very rare (H&E). (c) The initial biopsy specimen of a tonsil obtained 4 years earlier from Case 12 shows pronounced follicular hyperplasia (original magnification ×100). The small‐MCL cells (original magnification ×2500) have round or oval small nuclei with dense nuclear chromatin and resemble mature B cells (H&E). (d) In the same specimen shown in (c), CCND1‐positive small lymphoid cells are thinly spread in the mantle zones and very few, if any, tumor cells have spread into interfollicular areas (original magnification ×100).
Following the initial diagnosis, nine of the 15 patients with small‐MCL received combination therapy with cyclophosphamide, adriamycin, vincristine, and prednisone (CHOP) or CHOP‐like chemotherapy regimens (modified CHOP or pirarubicin, cyclophosphamide, vincristine, and prednisone [THP‐COP]), and three who were <60 years of age were treated aggressively with fractionated cyclophosphamide, doxorubicin, vincristine, and dexamethasone, alternated with high‐dose methotrexate–cytarabine (Hyper CVAD/MA). Seven patients received rituximab (R) in addition to these initial chemotherapies. Two cases (Cases 3 and 5) received stem cell transplantation following intensive induction regimens. Conversely, three patients received no systemic chemotherapy after the initial diagnosis because of the clinical behavior of the disease. Two of these cases (Cases 1 and 12) were observed for more than 5 months (“watch and wait”) and one was treated with less aggressive oral chemotherapy using cyclophosphamide (Case 10). Treatment for Case 12 was unexpectedly deferred for 64 months because of misdiagnosis as follicular hyperplasia at the initial examination. Therefore, this patient underwent initial chemotherapy 4 years later, after rebiopsy and a diagnosis of small‐MCL.
Of the 15 patients with small‐MCL, six were alive with complete response at the time of writing and complete response (CR) been induced in two of these patients as a result of stem cell transplantation. Eight patients who relapsed at various times after the first CR were alive with the disease. Only one patient (Case 6) died of the disease, although the response to initial therapy was favorable and immediate, as for the other cases of small‐MCL.
Pathologic features of small‐MCL. The pathologic features of patients with small‐MCL are given in Table 2. We used tissues from 13 cases available for analysis of the pattern of lymphoma cell involvement. Analysis of the tissue samples led to the classification of 10 of the 13 cases as diffuse (Fig. 1b) and three as mantle zone pattern, whereas none showed a nodular pattern. The other morphological characteristics of small‐MCL were that mitotic features were very rare and that there were no proliferation centers or conspicuous histiocytes. In Case 12, a review of a tonsillitis specimen taken 4 years earlier showed pronounced follicular hyperplasia with CCND1‐positive small lymphoid cells spreading in the mantle zones. However, this pattern differed from the conventional mantle zone pattern in that the neoplastic mantle zone was very thin and there was very little, if any, spreading of tumor cells into interfollicular areas (Fig. 1c,d).
Table 2.
Pathological characteristics of the small‐cell variant of mantle cell lymphoma
| Case no. | CD5 | CD10 | CD23 | SOX11 | CCND1 index (%) | Ki67 index (%) | IgH/Bcl1 | Biopsy site | Growth pattern |
|---|---|---|---|---|---|---|---|---|---|
| 1 | + | − | − | Weak + | 63 | 12 | + | LN | Diffuse |
| 2 | + | − | + | + | 30 | 27 | + | LN | Diffuse |
| 3 | + | − | − | − | 56 | 15 | + | LN | Diffuse |
| 4 | + | − | − | − | 61 | 21 | + | LN | Diffuse |
| 5 | + | − | − | + | 47 | 24 | + | LN | Diffuse |
| 6 | + | − | ND | − | 54 | 13 | + | Pharynx | Diffuse |
| 7 | + | − | − | − | 71 | 2 | + | Stomach | Diffuse |
| 8 | + | − | − | ND | 48 | ND | ND | LN | Diffuse |
| 9 | + | − | − | ND | 34 | ND | + | Stomach | Diffuse |
| 10 | + | − | + | ND | 39 | ND | + | PB | NA |
| 11 | + | − | − | + | 63 | 5 | + | BM | NA |
| 12 | + | − | − | − | 82 | 3 | + | Tonsil | Mantle zone |
| 13 | + | + | − | − | 32 | 4 | + | Spleen | Mantle zone |
| 14 | + | − | − | − | 67 | 18 | + | Cecum | Diffuse |
| 15 | + | − | − | − | 49 | 6 | ND | Small intestine | Mantle zone |
LN, lymph node; PB, peripheral blood; BM, bone marrow.
Although all 15 cases could be classified as small‐MCL on the basis of their cytologic features following analysis of H&E‐stained sections, lymphoma cells in the peripheral blood or bone marrow smear, which were available for review in five cases, varied from case to case. Although bone marrow smears of classical MCL show large lymphoma cells with moderately basophilic cytoplasm, reticular chromatin, and slightly abundant cytoplasm (Fig. 2a), small‐MCL lymphoma cells mainly feature a small, round, and slightly irregular nucleus with distinct clumped chromatin and scanty cytoplasm (Fig. 2b) or a small and slightly cleaved nucleus with a high nucleocytoplasmic ratio, dispersed chromatin, and prominent nucleoli (Fig. 2c). In Cases 6 and 11, a high similarity with common CLL was observed (Fig. 2d).
Figure 2.
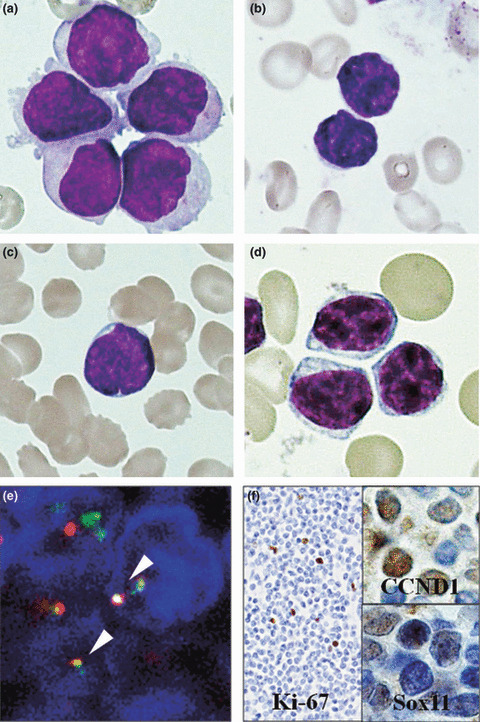
(a) Classical mantle cell lymphoma (MCL) in a bone marrow smear showing large lymphoma cells with moderately basophilic cytoplasm, reticular chromatin, somewhat abundant cytoplasm, and slightly prominent nucleoli. (b) Small‐cell variant of MCL (small‐MCL) cells in the bone marrow aspirate of Case 11 have a small, round, and slightly irregular nucleus with distinct clumping of chromatin and scanty cytoplasm. (c) The bone marrow aspirate of Case 12 shows small lymphoma cells with a high nucleocytoplasmic ratio, slightly cleaved nucleus, dispersed chromatin, and prominent nucleoli. (d) Chronic lymphocytic leukemia in the peripheral blood showing a uniform population of small mature lymphocytes. (a–d) May–Giemsa stain; original magnification ×2500. (e) FISH analysis of Case 1 using the immunoglobulin heavy locus (IGH)/cyclin D1 (CCND1) detection probe, where CCND1 is shown as red, IGH as green, and fusion signals as yellow (white arrows). (f) Neoplastic cells from Case 1 feature a very low Ki‐67‐labeling index (original magnification ×500). The higher magnification views (original magnification ×2000) shown on the upper and lower right from the same case show weak nuclear immunoreactivity for CCND1 and Sox11, respectively.
All 13 cases subjected to cytogenetic and/or FISH studies showed the bcl1 transformation of t(11;14)(q13;q32), as shown in Figure 2(e). Other cytogenetic abnormalities were seen in five cases: deletion of chromosomes 6q and 1p13 in two cases each and the addition of 5q11 and 1p13 in one case each.
All cases showed CD5 expression, one showed CD10 expression and two showed CD23 expression. Expression of Ki‐67, CCND1, and Sox11 was identified immunohistochemically in 12, 15, and 12 cases, respectively. Expression of CCND1 in small‐MCL was detected in all cases and nuclear staining of Sox11 was seen in four of 12 cases. In one of these, nuclear staining of Sox11 was weak. The neoplastic cells in most cases of small‐MCL also exhibited weak nuclear immunoreactivity for CCND1 with a Ki‐67 labeling index of <30% (Fig. 2f).
Clinicopathological features of small‐MCL compared with classical MCL. The clinicopathological features of the 15 cases of small‐MCL were compared with those of classical MCL and the findings are summarized in Table 3. Bone marrow involvement and splenomegaly were observed significantly more frequently in patients with small‐MCL than in patients with classical MCL (P < 0.05). Although an advanced IPI score, non‐nodal clinical features, hepatomegaly, and leukemic changes were identified more frequently in patients with small‐MCL than in patients with classical MCL, the differences did not reach statistical significance. Finally, the expression of CCND1 and Ki‐67 in small‐MCL (mean [±2 SD] 53.1 ± 30.4% and 12.5 ± 17.3%, respectively) were lower than expression in cases of classical MCL (58.1 ± 36.7% and 25.2 ± 25.5%, respectively; P = 0.2445 and P < 0.001, respectively).
Table 3.
Clinicopathological characteristics of the small‐cell variant of mantle cell lymphoma compared with those of classical and aggressive mantle cell lymphoma
| Small‐MCL | Classical MCL | Aggressive MCL | P value (small vs classical) | |
|---|---|---|---|---|
| Total no. patients | 15 | 151 | 44 | |
| Age | 64.47 ± 9.53 | 66.04 ± 20.82 | 68.11 ± 20.28 | 0.564 |
| Sex (n) | 15 | 151 | 44 | 0.7205 |
| Male | 12 | 126 | 32 | |
| Female | 3 | 25 | 12 | |
| IPI score (n) | 15 | 106 | 36 | 0.1045 |
| High/high–intermediate | 10 (67) | 46 (43) | 27 (75) | |
| Lymphadenopathy (n) | 15 | 151 | 44 | 0.2621 |
| No | 7 (47) | 34 (23) | 1 (2) | |
| Hepatomegaly (n) | 15 | 117 | 39 | 0.0563 |
| Yes | 4 (27) | 11 (9) | 11 (28) | |
| Splenomegaly (n) | 15 | 117 | 39 | <0.0001 |
| Yes | 12 (80) | 32 (27) | 20 (51) | |
| BM involvement (n) | 15 | 114 | 34 | 0.0443 |
| Yes | 12 (80) | 53 (47) | 19 (56) | |
| Leukemic change (n) | 5 | 117 | 41 | 0.1245 |
| Yes | 7 (47) | 33 (28) | 17 (42) | |
| Gut involvement (n) | 12 | 99 | 34 | 1 |
| Yes | 7 (47) | 43 (43) | 8 (24) | |
| Growth pattern (n) | 13 | 136 | 42 | 1 |
| Diffuse | 10 (77) | 48 (35) | 38 (91) | |
| Mantle zone | 3 (23) | 18 (13) | 0 (0) | |
| Ki‐67‐index | ||||
| n | 12 | 103 | 30 | <0.0001 |
| Mean ± 2 SD | 12.5 ± 17.3 | 25.2 ± 25.5 | 73.7 ± 28.9 | |
| CCND1‐index | ||||
| n | 15 | 149 | 44 | 0.2445 |
| Mean ± 2 SD | 53.1 ± 30.4 | 58.1 ± 36.7 | 80.1 ± 27.8 | |
Data are shown as the mean ± 2 SD or the number of patients in each group, with percentages in parentheses, as appropriate. MCL, mantle cell lymphoma; IPI, International Prognostic Index; CCND1, cyclin D1.
Clinical follow‐up data were obtained for all 15 patients with small‐MCL. The median follow‐up period for patients with small‐MCL, classical, and aggressive MCL was 38, 66, and 12 months, respectively. The survival time of patients with small‐MCL is shown in Figure 3. Although the overall 5‐year survival rates for small‐MCL were higher (83.3%) than those for classical MCL (65.5%), the difference did not reach statistical significance (P = 0.287). However, univariate analysis showed a clear difference in overall survival time between the three subtypes (P < 0.001), and only one small‐MCL patient (Case 6) died of the disease.
Figure 3.
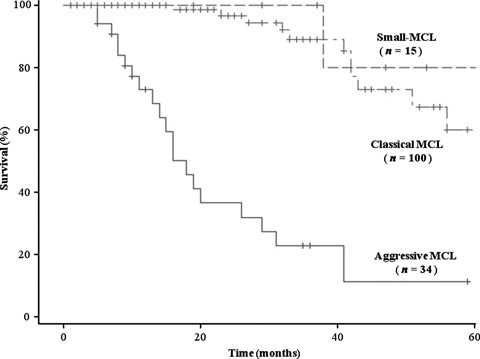
Kaplan–Meier analysis of overall survival of patients with classical mantle cell lymphoma (MCL), aggressive MCL, and the small‐cell variant of MCL (small‐MCL). Although the overall 5‐year survival rate tended to be higher for patients with small‐MCL than classical MCL (83.3 vs 65.6%, respectively), the difference did not reach statistical significance (P = 0.287, log rank test). However, 5‐year survival in both the classical MCL and small‐MCL groups was significantly higher than that in the aggressive MCL group (P < 0.0001, log rank test).
As reported previously,( 19 ) there was no significant difference between the two main MCL subtypes (i.e. classical and aggressive) in terms of non‐nodal disease or bone marrow involvement, although aggressive MCL tended to show a higher IPI score, more advanced hepatomegaly and splenomegaly, more diffuse growth patterns, and higher Ki‐67 and CCND1 indices than classical MCL.
Discussion
There are many overlapping morphologic, immunophenotypic, and sometimes even clinical features between MCL and B‐CLL/SLL. Both are CD5‐positive lymphomas that often show diffuse growth patterns and frequently present with a leukemic picture or splenomegaly.( 26 , 27 ) Thus, the immunohistochemical detection of CCND1 and Sox11 or reactivity for CD23 may prove to be of great help in obtaining an accurate differential diagnosis. However, the differences between the two diseases are most prominently represented by the clinical outcomes of individual patients.( 28 )
Interestingly, recent studies have reported the existence of a specific subset of MCL with an indolent clinical course.( 6 , 7 , 8 , 9 , 10 ) In addition, MCL consists morphologically of a wide variety of CCND1‐positive neoplasms. These facts suggest that the identification of various MCL variants is important for deciding on appropriate therapies or predicting outcomes.
In the present study, we focused on the small‐cell variant of MCL, which features small cells with small, round, or oval nuclei resembling those of CLL/SLL. Our data suggest that this variant is a specific indolent B cell neoplasm with t(11;14)(q13;q32). Briefly, small‐MCL was found to have a high 5‐year survival rate (83.3%) and showed significantly higher bone marrow involvement (P < 0.05) and splenomegaly (P < 0.0001), accompanied by a lower Ki‐67 labeling index (P < 0.001), than cases of classical MCL.
It is of particular interest to examine small‐MCL in terms various aspects of its clinicopathologic characteristics. First, although we confirmed that Sox11 nuclear protein was more frequently detected in cases of classical MCL (20/21), a considerable number of cases of small‐MCL (8/12) lacked Sox11 nuclear expression and the intensity of CCND1 labeling in some of cases was so weak as to make clear detection difficult (data not shown). Second, despite the fact that most cases of small‐MCL showed a predominantly diffuse growth pattern, three cases had hyperplastic germinal centers with a very thin (reactive follicle‐like) mantle zone, which is considered to play an integral part as an initial component. These findings led us to examine the risk of overlooking the disease by diagnosing it as benign lymphadenitis or misdiagnosing it as CLL/SLL.
Sox11 is a highly specific marker for conventional MCL cases.( 22 ) However, whether Sox11 expression could be related to the clinical and biological behavior of MCL remains contentious. Wang et al. ( 29 ) have reported that patients with Sox11‐negative MCL have a shorter survival than patients with MCL with nuclear Sox11 expression; however, our findings and that of another study,( 30 ) provides support for the hypothesis that some indolent MCL lacks Sox11 expression.
It was determined that small‐MCL presents as a systemic disease with a high IPI, which is due, in part, to the high frequency of gastrointestinal tract involvement. Although MLP has been considered to be one of the most common gastrointestinal presentations of MCL, it was reported that the tumorous type is most frequent (85%) and that MLP accounts for only approximately 3.5% of MCL patients with gastrointestinal involvement.( 31 ) In our series, we noted that many patients without lymphadenopathy (non‐nodal small‐MCL) showed gastrointestinal tract involvement with MLP (4/5; 80%). The eight patients with small‐MCL with lymphadenopathy (nodal small‐MCL) had no distinctive clinical features compared with non‐nodal cases.
It has been postulated that the origin of MCL cells is a naïve CD5‐positive pregerminal center B cell because the majority of MCL cases express VH genes without somatic mutations.( 32 ) Other studies have shown that 16–28% of cases of MCL feature hypermutated VH genes,( 33 , 34 ) suggesting that MCL consists of two cell subsets, the majority unmutated and a minority mutated, as seen in CLL/SLL.( 35 , 36 ) Subsequent studies using the same model have led to new discoveries regarding MCL. Some of the patients reported by Orchard et al. ( 7 ) with circulating t(11;14) lymphocytes featured mutated IgVH genes, good prognosis, and non‐nodal disease. Fernandez et al. ( 30 ) also reported that MCL patients who showed an indolent clinical course for more than 2 years without chemotherapy presented with a non‐nodal leukemic disease with predominantly hypermutated IgVH, lack of genomic complexity, and the absence of Sox11 expression. Furthermore, Angelopoulou et al. ( 27 ) found that some MCL patients with an indolent course presented with splenomegaly and a leukemic picture without lymphadenopathy.
As mentioned earlier, we speculated that small‐MCL comprises two different forms, nodal and non‐nodal disease. This notion may provide new insights into the features of indolent MCL. One of these is that non‐nodal small‐MCL may be essentially the same as what has been described as indolent MCL with mutated VH genes or the splenic form of MCL. Moreover, non‐nodal small‐MCL predominantly shows a mantle zone pattern without Sox11 expression, whereas nodal small‐MCL presents a diffuse pattern. However, to put the practical implications of the present study into perspective, it should be emphasized that the pathological characteristic of “small‐cell” as such constitutes a simple predictor.
Although the actual MCL cases with small‐cell features are definitely in a minority, we believe that they are significant for patients with newly diagnosed lymphomas. A small‐MCL constitutes a specific subset of indolent lymphoma characterized by bone marrow involvement, splenomegaly, and a low Ki‐67 labeling index. We should be aware of the possible presence of small‐MCL to avoid making a misdiagnosis of follicular hyperplasia or CLL/SLL. A small‐MCL can be expected to make a major contribution to the accuracy of therapeutic decisions regarding high‐dose or additional chemotherapies.
Disclosure Statement
The authors have no potential conflicts of interest.
Acknowledgments
The authors thank Fumitoh Arima from Shinkoga Hospital, Toshitaka Muto from National Kokura Medical Center, Shiho Fujiwara from NTT Nishinihon Kyushu Hospital, and Osaki Kohichi from the University of Kurume for their help in providing clinical data.
References
- 1. Zucca E, Roggero E, Pinotti G et al. Patterns of survival in mantle cell lymphoma. Ann Oncol 1995; 6: 257–62. [DOI] [PubMed] [Google Scholar]
- 2. Herrmann A, Hoster E, Zwingers T et al. Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol 2009; 27: 511–8. [DOI] [PubMed] [Google Scholar]
- 3. Lenz G, Dreyling M, Hoster E et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long‐term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005; 23: 1984–92. [DOI] [PubMed] [Google Scholar]
- 4. Romaguera JE, Fayad L, Rodriguez MA et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle‐cell lymphoma with rituximab plus hyper‐CVAD alternating with rituximab plus high‐dose methotrexate and cytarabine. J Clin Oncol 2005; 23: 7013–23. [DOI] [PubMed] [Google Scholar]
- 5. Geisler CH, Kolstad A, Laurell A et al. Long‐term progression‐free survival of mantle cell lymphoma after intensive front‐line immunochemotherapy with in vivo‐purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008; 112: 2687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin P, Chadburn A, Christos P et al. Outcome of deferred initial therapy in mantle‐cell lymphoma. J Clin Oncol 2009; 27: 1209–13. [DOI] [PubMed] [Google Scholar]
- 7. Orchard J, Garand R, Davis Z et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood 2003; 101: 4975–81. [DOI] [PubMed] [Google Scholar]
- 8. Bassarova A, Tierens A, Lauritzsen GF, Fossa A, Delabie J. Mantle cell lymphoma with partial involvement of the mantle zone: an early infiltration pattern of mantle cell lymphoma? Virchows Arch 2008; 453: 407–11. [DOI] [PubMed] [Google Scholar]
- 9. Nodit L, Bahler DW, Jacobs SA, Locker J, Swerdlow SH. Indolent mantle cell lymphoma with nodal involvement and mutated immunoglobulin heavy chain genes. Hum Pathol 2003; 34: 1030–4. [DOI] [PubMed] [Google Scholar]
- 10. Espinet B, Sole F, Pedro C et al. Clonal proliferation of cyclin D1‐positive mantle lymphocytes in an asymptomatic patient: an early‐stage event in the development or an indolent form of a mantle cell lymphoma? Hum Pathol 2005; 36: 1232–7. [DOI] [PubMed] [Google Scholar]
- 11. Swerdlow SHB, Isaacson PI, Muller‐Hermelink HK, Nathwani BN, Piris MA, Harris NL (eds). World Health Organization Classification of Tumors. Lyon: IARC Press, 2001. [Google Scholar]
- 12. Swerdlow SH, Campo E, Harris NL, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). World Health Organization Classification of Tumors. Lyon: IARC Press, 2008. [Google Scholar]
- 13. Bernard M, Gressin R, Lefrere F et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001; 15: 1785–91. [DOI] [PubMed] [Google Scholar]
- 14. Weisenburger DD, Vose JM, Greiner TC et al. Mantle cell lymphoma. A clinicopathologic study of 68 cases from the Nebraska Lymphoma Study Group. Am J Hematol 2000; 64: 190–6. [DOI] [PubMed] [Google Scholar]
- 15. Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997; 89: 2067–78. [PubMed] [Google Scholar]
- 16. Tiemann M, Schrader C, Klapper W et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005; 131: 29–38. [DOI] [PubMed] [Google Scholar]
- 17. Raty R, Franssila K, Joensuu H, Teerenhovi L, Elonen E. Ki‐67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002; 69: 11–20. [DOI] [PubMed] [Google Scholar]
- 18. Schaffel R, Hedvat CV, Teruya‐Feldstein J et al. Prognostic impact of proliferative index determined by quantitative image analysis and the International Prognostic Index in patients with mantle cell lymphoma. Ann Oncol 2010; 21: 133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kimura Y, Sato K, Arakawa F et al. Mantle cell lymphoma shows three morphological evolutions of classical, intermediate, and aggressive forms, which occur in parallel with increased labeling index of cyclin D1 and Ki‐67. Cancer Sci 2010; 101: 806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yatabe Y, Suzuki R, Matsuno Y et al. Morphological spectrum of cyclin D1‐positive mantle cell lymphoma: study of 168 cases. Pathol Int 2001; 51: 747–61. [DOI] [PubMed] [Google Scholar]
- 21. Rosenwald A, Wright G, Wiestner A et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003; 3: 185–97. [DOI] [PubMed] [Google Scholar]
- 22. Mozos A, Royo C, Hartmann E et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1‐negative subtype. Haematologica 2009; 94: 1555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. A predictive model for aggressive non‐Hodgkin’s lymphoma. The International Non‐Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med 1993; 329: 987–94. [DOI] [PubMed] [Google Scholar]
- 24. Ohshima KS, Suzumiya J, Kawasaki C, Kanda M, Kikuchi M. Cytoplasmic cytokines in lymphoproliferative disorders: multiple cytokine production in angioimmunoblastic lymphadenopathy with dysproteinemia. Leuk Lymphoma 2000; 38: 541–5. [DOI] [PubMed] [Google Scholar]
- 25. Martin‐Subero JI, Harder L, Gesk S et al. Interphase FISH assays for the detection of translocations with breakpoints in immunoglobulin light chain loci. Int J Cancer 2002; 98: 470–4. [DOI] [PubMed] [Google Scholar]
- 26. Melo JV, Hegde U, Parreira A, Thompson I, Lampert IA, Catovsky D. Splenic B cell lymphoma with circulating villous lymphocytes: differential diagnosis of B cell leukaemias with large spleens. J Clin Pathol 1987; 40: 642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Angelopoulou MK, Siakantariz MP, Vassilakopoulos TP et al. The splenic form of mantle cell lymphoma. Eur J Haematol 2002; 68: 12–21. [DOI] [PubMed] [Google Scholar]
- 28. Asaad NY, Abd El‐Wahed MM, Dawoud MM. Diagnosis and prognosis of B‐cell chronic lymphocytic leukemia/small lymphocytic lymphoma (B‐CLL/SLL) and mantle cell lymphoma (MCL). J Egypt Natl Canc Inst 2005; 17: 279–90. [PubMed] [Google Scholar]
- 29. Wang X, Asplund AC, Porwit A et al. The subcellular Sox11 distribution pattern identifies subsets of mantle cell lymphoma: correlation to overall survival. Br J Haematol 2008; 143: 248–52. [DOI] [PubMed] [Google Scholar]
- 30. Fernandez V, Salamero O, Espinet B et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010; 70: 1408–18. [DOI] [PubMed] [Google Scholar]
- 31. Kohno S, Ohshima K, Yoneda S, Kodama T, Shirakusa T, Kikuchi M. Clinicopathological analysis of 143 primary malignant lymphomas in the small and large intestines based on the new WHO classification. Histopathology 2003; 43: 135–43. [DOI] [PubMed] [Google Scholar]
- 32. Hummel M, Tamaru J, Kalvelage B, Stein H. Mantle cell (previously centrocytic) lymphomas express VH genes with no or very little somatic mutations like the physiologic cells of the follicle mantle. Blood 1994; 84: 403–7. [PubMed] [Google Scholar]
- 33. Walsh SH, Thorselius M, Johnson A et al. Mutated VH genes and preferential VH3–21 use define new subsets of mantle cell lymphoma. Blood 2003; 101: 4047–54. [DOI] [PubMed] [Google Scholar]
- 34. Camacho FI, Algara P, Rodriguez A et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood 2003; 101: 4042–6. [DOI] [PubMed] [Google Scholar]
- 35. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94: 1848–54. [PubMed] [Google Scholar]
- 36. Damle RN, Wasil T, Fais F et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94: 1840–7. [PubMed] [Google Scholar]