

Abstract
Chemoresistance is a major cause of treatment failure in ovarian cancer. Therefore, it is necessary to explore alternative therapeutic methods to overcome drug resistance for ovarian cancer treatment. We previously reported that programmed cell death 4 (PDCD4), a tumor suppressor, significantly suppresses the malignant phenotype of ovarian cancer cells and its lost or low expression in ovarian cancer is associated with unfavorable prognosis of patients. Here we show that PDCD4 improves the sensitivity of ovarian cancer cells to platinum‐based chemotherapy. Overexpression of PDCD4 enhanced chemosensitivity in SKOV3 and CAOV3 cells with low levels of PDCD4, whereas knockdown of PDCD4 reduced chemosensitivity in OVCAR3 cells with high levels of PDCD4. Subsequently, the combination of enforced PDCD4 expression with cisplatin treatment significantly suppressed ovarian tumor growth in a xenograft animal model. The PDCD4 effect appears to be specific for cisplatin and carboplatin, not affecting cyclophosphamide, etoposide, or paclitaxel. Mechanistically, PDCD4 significantly increased cisplatin‐induced cleavage of caspase‐3 and caspase‐8, but had only a slight impact on caspase‐9 cleavage and the expression of Bax and Bcl‐2 in vitro and in vivo. A specific caspase‐8 inhibitor, Z‐ITED‐FMK, attenuated cisplatin‐induced apoptosis in PDCD4‐overexpressing ovarian cancer cells. Taken together, our results indicate that PDCD4 enhances cisplatin‐induced apoptosis by mainly activating the death receptor pathway, and PDCD4 gene transfer in combination with cisplatin therapy may break the resistance of ovarian cancer cells to chemotherapy. (Cancer Sci 2010)
Programmed cell death 4 (PDCD4) was first cloned in mice as MA‐3 and TIS, and its homologues in human (H731, 197/15A), chicken and rat (DUG) were soon identified.( 1 ) The PDCD4 protein has two conserved α‐helical MA‐3 domains which are also present in eukaryotic initiation factor (eIF)4G. Therefore, PDCD4 can suppress protein translation by directly interacting with eIF4A to inhibit the formation of the translation–initiation complex.( 2 ) The crystal structure of the eIF4A and PDCD4 complex has been determined.( 3 , 4 ) In addition, PDCD4 can suppress the transcriptional activity of activator protein (AP)‐1 by inhibiting the expression of MAP4K1 to block the activation of c‐Jun.( 5 , 6 )
Accumulating evidence indicates that PDCD4 is a novel tumor suppressor gene. It has been shown to inhibit tumor promoter‐induced transformation in the mouse epidermal cell line JB6;( 7 ) PDCD4‐deficient mice developed spontaneous tumors of lymphoid origin, and PDCD4 transgenic mice showed significant resistance to tumor induction.( 8 , 9 ) Moreover, loss or reduction of PDCD4 expression was found in several types of human cancer cell lines and primary tumors, such as lung cancer, colorectal cancer, glioma, and ovarian cancer.( 10 , 11 , 12 , 13 , 14 , 15 ) Expression of PDCD4 is regulated by various mechanisms. PDCD4 mRNA was downregulated by increased levels of miR‐21 which can bind to the 3′‐untranslated region of PDCD4 mRNA and promote cell transformation and tumor progression by targeting PDCD4.( 16 , 17 , 18 ) Additionally, methylation of the PDCD4 5′ CpG island blocked PDCD4 expression at the mRNA level in gliomas.( 19 ) At the protein level, S6K‐mediated phosphorylation of PDCD4 triggered its ubiquitinylation by the E3 ubiquitin ligase complex SCF, which may control its abundance in cells.( 20 ) Exposure to TPA regulated PDCD4 degradation through protein kinase C‐mediated activation of the PI3K and MEK signaling pathways.( 21 ) Furthermore, overexpression of PDCD4 suppressed the malignant phenotype in JB6 cells by inhibiting AP‐1 transactivation, inhibited the invasive capacity, and suppressed the malignant phenotype of human ovarian cancer.( 22 , 23 ) Knockdown of PDCD4 significantly promoted invasion, activated β‐catenin/Tcf and AP‐1‐dependent transcription, and stimulated the expression of c‐Myc and u‐PAR.( 24 , 25 )
Recently, two research groups reported that PDCD4 expression enhanced chemosensitivity in renal cancer cell line UO‐31 and prostate cancer cell line PC3.( 10 , 26 ) However, it is unknown whether PDCD4 expression can enhance the chemosensitivity of other tumors, and the underlying molecular mechanism is not clear. In the present study, we provide evidence that PDCD4 enhanced the chemosensitivity of ovarian cancer cells to cisplatin in culture cells and in xenografts. Furthermore, we show that PDCD4 can significantly promote cisplatin‐induced apoptosis by activating the death receptor pathway.
Materials and Methods
Cell culture and transfection. Human ovarian cancer cell lines SKOV3, CAOV3, and NIH‐OVCAR3 were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and the China Center for Type Culture Collection (Wuhan, China). All cancer cells were cultured in recommended media under standard conditions. Transfection of ovarian cells with plasmids pEGFP‐C1 or pEGFP‐C1‐PDCD4 (constructed and provided by Dr. Olubunmi Afonja, New York University, New York, NY, USA) was carried out using Lipofectamine 2000 according to the manufacturer’s protocols (Invitrogen, Carlsbad, CA, USA). The stably expressing PDCD4 cells (SKOV3‐PDCD4) and control cells (SKOV3‐MOCK) were established as described( 23 ) and were cultured in medium with 300 μg/mL G418. Both RT‐PCR and Western blot analysis were used for the detection of PDCD4 expression in transfected cell lines.
RNA isolation and RT‐PCR. Total RNAs were extracted using a modified TRIzol one‐step extraction method (Invitrogen). We carried out RT‐PCR at least three times for each sample according to our previous study.( 23 )
SDS‐PAGE and Western blot. Extraction and detection of protein was done as described.( 23 ) A 60 μg quantity of protein was separated on SDS‐PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). Membranes were then blocked with 1% BSA in TBST containing 0.1% Tween‐20 for 1 h. The filters were then incubated overnight at 4°C with antibodies against PDCD4, Bax, Bcl‐2, cleaved caspase‐3, ‐8, and ‐9 (1:1000; Cell Signaling Technology, Beverly, MA, USA), and β‐actin (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunoblotting was done by incubating the membranes at 4°C overnight followed by secondary antibody (goat anti‐rabbit IgG) conjugated with peroxidase for 1 h at room temperature. After washing, signals were visualized by SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL, USA). Western blot was carried out at least three times for each sample.
Immunohistochemistry. Immunohistochemistry was carried out as previously described.( 23 ) The slides were blocked for endogenous peroxidase activity, preincubated with goat serum, then stained with anti‐PDCD4, anti‐Bax, and Bcl‐2, caspase‐3, ‐8, ‐9 antibodies (1:100) by incubating for 1 h at room temperature. Secondary staining with HRP‐conjugated anti‐mouse/rabbit IgG was carried out using a MaxVision Kit and a DAB Peroxidase Substrate Kit (Maixin, Fuzhou, China). Negative controls for the specificity of immunohistochemical reactions were carried out by replacing the primary antibody with IgG of non‐immunized rabbit/mouse. Immunohistochemistry was carried out twice for each sample.
MTT assay. Cells were seeded into 96‐well plates at 6 × 103 cells/well and allowed to grow overnight, then were treated with different concentrations of cisplatin, carboplatin, cyclophosphamide (CTX), etoposide (VP‐16), and paclitaxel (QiLu Pharmaceutical, Jinan, China). Specific caspase‐8 inhibitor Z‐IETD‐FMK (R&D Systems, Minneapolis, MN, USA) was added 1 h before cisplatin treatment. After 24 h of treatment, 20 μL of 5 mg/mL MTT reagent (Sigma‐Aldrich, St. Louis, MO, USA) was added and incubated in the dark for 4 h. The viability of treated cells was calculated from the average OD570 values compared to that of untreated cells. Each treatment was carried out in triplicate.
Detection of cell cycle and apoptosis. Cells were seeded at a density of 2 × 105 cells in 6‐well plates and were treated with cisplatin. After 24 h of treatment, adherent and floating cells were harvested and cell cycle analysis was carried out according to our previous study.( 23 ) For apoptosis, cells were grown on coverlips at a density of 1.25 × 104 cells per well in 24‐well plates, and were treated with cisplatin. After 24 h of treatment, cells were fixed with 4% paraformaldehyde and nuclei of cells were stained with Hoechst dye (Sigma‐Aldrich), according to the manufacturer’s protocols, and specimens were observed under a fluorescent microscope. Moreover, after treatment with cisplatin, all the treated cells were harvested for TUNEL assay using an In Situ Cell Death Detection Kit, TMR red (Roche, Mannheim, Germany) according to the manufacturer’s recommendation. Apoptosis was assessed by flow cytometer (Beckman Coulter, Fullerton, CA, USA). Each experiment was carried out in triplicate.
Knockdown of PDCD4 expression mediated by siRNA. Two different Silencer Select Pre‐designed siRNA targeting PDCD4 and non‐specific negative control were purchased from Ambion (Austin, TX, USA). For PDCD4‐siRNA‐1: sense, 5′GAGAUGGAAUUUUAUGUAAtt3′, and antisense, 5′UUACAUAAAAUUCCAUCUCca3′; for PDCD4‐siRNA‐2: sense, 5′GG‐CUGGAAUAAUUUCCAAAtt3′, and antisense, 5′UUUGGAAAUUAUUCCAGCCtg3′. The siRNAs were transfected into OVCAR‐3 cells using siPORT NeoFX Transfection Agent (Ambion) according to the manufacturer’s protocol. After 48 h incubation, cells were subjected to RT‐PCR and immunoblotting analysis. The experiment was repeated three times.
In vivo studies using a xenograft animal model. Nude mice, 6–8 weeks old, were inoculated s.c. into the left flank with 5 × 106 SKOV3 cells in 100 μL PBS (n = 33). When tumors reached approximately 100 mm3 in size 1 week later, these mice were randomly divided into six groups: group 1, normal saline (NS, n = 5); group 2, MOCK (n = 5); group 3, PDCD4 (n = 6); group 4, cisplatin (n = 5); group 5, MOCK + cisplatin (n = 6); and group 6, PDCD4 + cisplatin (n = 6). For groups 2 and 5, groups 3 and 6, mice were treated with 20 μg MOCK plasmid and PDCD4 plasmid in a total volume of 50 μL by intra‐tumor injection every 4 days( 27 , 28 ), for a total of four times. At 48 h after transfection, mice in groups 4, 5, and 6 were injected i.p. with 2 mg/kg cisplatin every 4 days, for a total of four injections. Tumors were measured three times a week. Tumor volume was assessed and calculated as follows: length × width × width × 0.4. All mice were killed 1 week after completion of treatment and tumors were removed for weight analysis. Tumors were then fixed in 4% formalin and processed for paraffin embedding for immunohistochemistry. The protocol was approved by the Animal Care and Utilization Committee of Shandong University. This study is also in full compliance with the guidelines for the welfare of animals in experimental neoplasia.
Statistical analysis. Statistical significance was evaluated with data from at least three independent experiments. Statistical analysis was carried out using unpaired the t‐test or one‐way anova (spss 10.0; SPSS, Chicago, IL, USA). Data are presented as the mean ± SEM. For all statistical tests, significance was established at P < 0.05.
Results
Expression of PDCD4 correlates with cytotoxic activity of cisplatin in different ovarian cancer cell lines. To determine the effect of PDCD4 expression on the sensitivity of ovarian cancer cells to cisplatin, we first measured the expression of PDCD4 in three ovarian cancer cell lines by RT‐PCR and Western blot analysis. As shown in Figure 1(a,b), PDCD4 expression was low in SKOV3, moderate in CAOV3, and high in OVCAR3 cells. Results from MTT assays showed that OVCAR3 and CAOV3 cells with relatively high or moderate levels of PDCD4 expression were more sensitive to cisplatin. However, SKOV3 cells with relatively low levels of PDCD4 expression were resistant to cisplatin (Fig. 1c). These results suggest that PDCD4 expression may be associated with high sensitivity to cisplatin in ovarian cancer cell lines.
Figure 1.
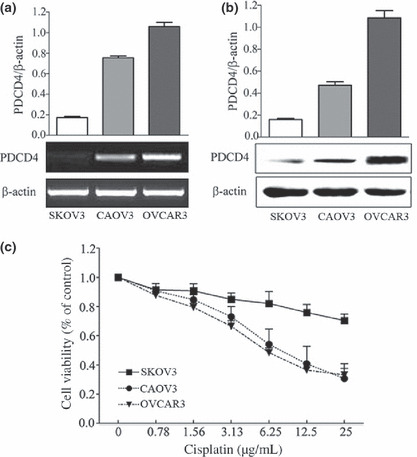
Relation between programmed cell death 4 (PDCD4) expression and the cytotoxic effect of cisplatin on ovarian cancer cell lines. The level of PDCD4 expression in three different ovarian cancer cell lines detected by RT‐PCR (a) and Western blot analysis (b). The bands of interest were further analyzed by densitometer and were normalized to β‐actin. (c) The cytotoxic effect of cisplatin on ovarian cancer cell lines was detected by MTT assay after cells were treated with different concentrations of cisplatin for 24 h. Data are expressed as the mean of triplicate experiments ± SEM.
Overexpression of PDCD4 enhances chemosensitivity of ovarian cancer cells. To investigate the direct impact of PDCD4 on chemosensitivity, we overexpressed PDCD4 in SKOV3 and CAOV3 cells (Fig. 2a,b). As shown in Figure 2(c), there appeared to be high levels of cell death in SKOV3‐PDCD4 cells in the presence of cisplatin (25 μg/mL), whereas there was no observable cell death in SKOV3‐MOCK cells in the presence of the same concentration cisplatin. This result was confirmed by MTT assay, which showed that cell viability of SKOV3‐PDCD4 cells was significantly decreased (P < 0.05, Fig. 2d). Similarly, CAOV3 cells transfected with PDCD4 were more sensitive to cisplatin than control cells (Fig. 2e). Taken together, PDCD4 overexpression appeared to enhance chemosensitivity to cisplatin in ovarian cancer cell lines.
Figure 2.
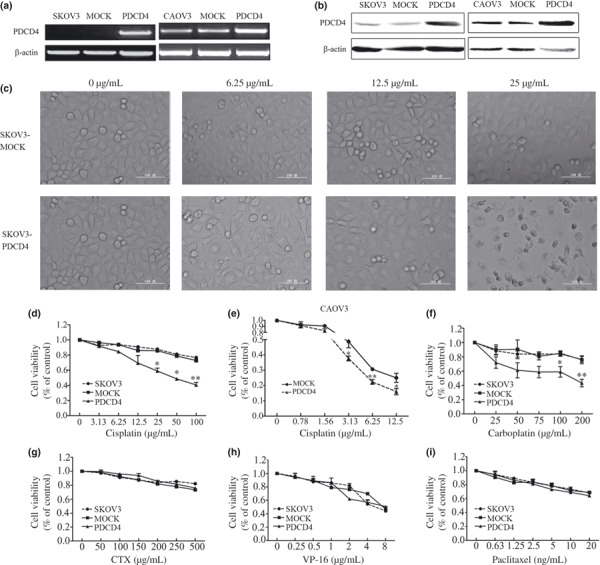
Effect of programmed cell death 4 (PDCD4) on the chemosensitivity of ovarian cancer cells in vitro. (a,b) PDCD4 mRNA and protein expression in SKOV3 and CAOV3 cells were examined by RT‐PCR (a) and Western blot (b) after stable or transient transfection with PDCD4 plasmid (PDCD4) or empty vector (MOCK). Morphological changes (c) showed by phase‐contrast micrography (magnification, ×200) and cell viability (d) detected by MTT assay of SKOV3‐MOCK and SKOV3‐PDCD4 cells after treatment with cisplatin for 24 h. (e) CAOV3 cells were transfected with MOCK and PDCD4 plasmid. Forty‐eight hours after transfection, cells were treated with cisplatin for 24 h and cell viability was detected by MTT assay. (f–i) The cytotoxic effect of carboplatin (f), cyclophosphamide (CTX) (g), etoposide (VP‐16) (h), and paclitaxel (i) on SKOV3, SKOV3‐MOCK, and SKOV3‐PDCD4 cells. Data are expressed as the mean of triplicate experiments ± SEM. *P < 0.05; **P < 0.01.
Moreover, our results showed that the viability of SKOV3‐PDCD4 cells was more decreased than that of SKOV3‐MOCK cells at the same concentration of carboplatin (P < 0.05, Fig. 2f). However, there was no significant difference between the SKOV3‐PDCD4 cells and SKOV3‐MOCK cells when the cells were treated with CTX, VP‐16, or paclitaxel (Fig. 2g–i). These results indicate that PDCD4 expression selectively enhances platinum‐based chemotherapy in ovarian cancer cells.
Knockdown of PDCD4 confers resistance to cisplatin in ovarian cancer cells. To further confirm the effect of PDCD4 on the chemosensitivity of ovarian cancer cells, we knocked down PDCD4 expression by PDCD4‐specific siRNAs in OVCAR3 cells. As shown in Figure 3(a,b), two PDCD4‐specific siRNAs markedly inhibited expression of PDCD4 mRNA by 77% and 76% and PDCD4 protein by 80% and 88%, whereas non‐specific siRNA had no significant effect on PDCD4 expression. Knockdown of PDCD4 with PDCD4‐specific siRNAs significantly increased cell viability when compared with controls treated with non‐specific siRNA (P < 0.05, Fig. 3c). In addition, knockdown of PDCD4 in SKOV3‐PDCD4 cells showed similar results (data not shown). This indicates that downregulation of PDCD4 enhances resistance to cisplatin.
Figure 3.
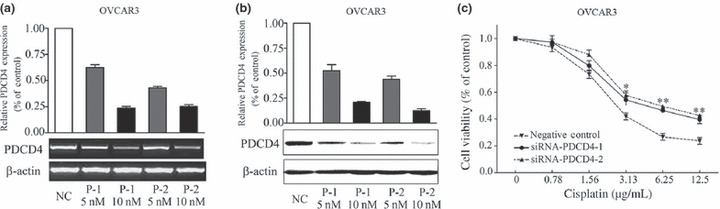
Programmed cell death 4 (PDCD4) knockdown decreased the sensitivity of ovarian cancer cells to cisplatin. (a,b) PDCD4 mRNA and protein expression after transfection with 5 and 10 nM PDCD4 siRNA‐1 (P‐1), siRNA‐2 (P‐2) and negative control siRNA (non‐specific oligonucleotides, NC), as detected by RT‐PCR (a) and Western blot analysis (b). The inhibitory effects of siRNA on PDCD4 expression were further analyzed after normalization to β‐actin. (c) OVCAR3 cells were treated with PDCD4 siRNAs or control siRNA for 48 h prior to treatment with different concentrations of cisplatin. Cell viability was detected by MTT assay after cisplatin treatment for 24 h. Data are expressed as the mean of triplicate experiments ± SEM. *P < 0.05; **P < 0.01.
Combination of PDCD4 and cisplatin significantly inhibits xenograft growth in vivo. To investigate the effect of PDCD4 expression on the chemosensitivity of ovarian cancer in vivo, we established a xenograft animal model as described in “Materials and Methods”. As shown in Figure 4(a), tumors in mice treated with normal saline (NS) or empty vector (MOCK) grew rapidly, whereas xenografts in mice treated with PDCD4 expression plasmid (PDCD4) or cisplatin alone were significantly inhibited. Of note, combined treatment with PDCD4 plasmid plus cisplatin (PDCD4 + cisplatin) inhibited tumor growth to a greater extent than treatment with MOCK plasmid plus cisplatin (MOCK + cisplatin) (n = 6, P < 0.05). After 23 days of treatment, the mice were killed, and tumor size and weight were measured. In line with the growth curve of tumors, tumor volume and weight in the PDCD4 + cisplatin group (0.0375 ± 0.0069 mm3 and 0.0300 ± 0.0044 g, respectively) were markedly lower when compared with that in the cisplatin group (0.1386 ± 0.0185 mm3 and 0.0860 ± 0.0204 g, respectively) or PDCD4 (0.0779 ± 0.0115 mm3 and 0.0533 ± 0.0042 g, respectively) or MOCK + cisplatin (0.1621 ± 0.0090 mm3 and 0.07667 ± 0.0126 g, respectively) (Fig. 4b,c). Furthermore, the tumors injected with PDCD4 plasmids (PDCD4, PDCD4 + cisplatin) expressed higher levels of PDCD4 (Fig. 4d). These data indicate that enforced PDCD4 expression can also enhance the sensitivity of ovarian cancer to cisplatin in vivo.
Figure 4.
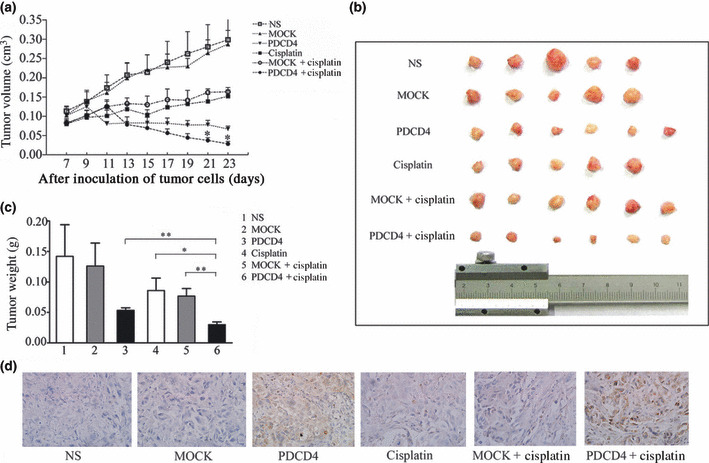
Combination of programmed cell death 4 (PDCD4) with cisplatin inhibited tumor growth in nude mice. SKOV3 ovarian cancer cells were inoculated s.c. When tumors reached approximately 100 mm3, mice were randomly divided into six groups and treated by intratumoral injection with normal saline (NS), empty vector (MOCK), PDCD4 plasmid, cisplatin, MOCK + cisplatin, or PDCD4 + cisplatin. (a) Tumors were measured at different days after treatment and growth curves were drawn. Tumor volume of the PDCD4 + cisplatin group was significantly smaller than those of PDCD4, cisplatin, and MOCK + cisplatin groups (*P < 0.05). (b) Transplanted tumors were excised after different treatments. (c) Tumor weight of six different groups was measured. The tumor weight of the combination treatment group was significantly reduced compared with other groups (*P < 0.05; **P < 0.01). The results are expressed as the mean of each treatment group ± SEM. (d) Expression of PDCD4 in tumor samples detected by immunohistochemistry. Images shown are representative of each group. Magnification, ×400.
PDCD4 expression promotes cisplatin‐induced apoptosis, but not alterations in cell cycle. To determine the mechanism(s) by which PDCD4 enhances sensitivity of ovarian cancer cells to cisplatin, we first analyzed the effect of PDCD4 expression on cisplatin‐induced alterations in the cell cycle. Cisplatin treatment markedly increased cell numbers in S phase, however, PDCD4 overexpression had no significant impact on platinum‐induced alterations in the cell cycle (data not shown). To explore whether PDCD4 expression might alter platinum‐induced apoptosis, we measured apoptosis by Hoechst staining and TUNEL assays. Staining showed that PDCD4 overexpression increased the number of apoptotic cells, characterized by chromatin condensation and nuclear fragmentation after treatment with cisplatin (Fig. 5a). Likewise, results with the TUNEL assay revealed that the combination of PDCD4 expression with cisplatin resulted in a significant increase in apoptosis, whereas control cells treated with cisplatin resulted in minimal apoptosis, especially at the dose of 25 μg/mL (Fig. 5b). Therefore, we can conclude that PDCD4 expression promotes cisplatin‐induced apoptosis.
Figure 5.
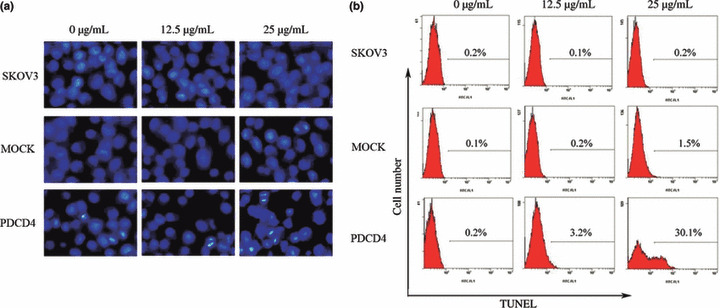
Effect of programmed cell death 4 (PDCD4) expression on cisplatin‐induced apoptosis in ovarian cancer cells. (a) Representative images (magnification, ×400) of cells treated with cisplatin and stained with Hoechst dye. Cells showing nuclear fragmentation and condensation are considered to be undergoing apoptosis. (b) The apoptosis rate of cells treated with cisplatin as determined by TUNEL assay. Data shown are representative of three independent experiments. MOCK, empty vector.
PDCD4 promotes cisplatin‐induced apoptosis by mainly activating death receptor pathways. To further examine the particular apoptotic pathways by which PDCD4 promotes cisplatin‐induced apoptosis, we next measured the expression of several apoptosis‐related proteins. As shown in Figure 6(a), treatment with cisplatin elevated the expression of cleaved caspase‐3 and caspase‐8 in SKOV3‐PDCD4 cells to a greater extent than in SKOV3 or SKOV3‐MOCK cells. However, only a slight increase of cleaved caspase‐9 and Bax was observed in SKOV3‐PDCD4 cells compared with the control cells and there was little, if any, alteration in the expression of Bcl‐2. When Z‐ITED‐FMK was used to inhibit the activity of caspase‐8, cisplatin‐induced apoptosis was attenuated in SKOV3‐PDCD4 cells (Fig. 6b). Furthermore, we detected the expression of these apoptosis‐related proteins in the tumor tissues from tumor‐bearing nude mice. In accordance with the results in cell culture, caspase‐3 and caspase‐8 expression were significantly increased after injected with the PDCD4 plasmid, and were even higher after combined treatment with cisplatin plus PDCD4 plasmid. The level of Bax protein was slightly increased, but there was no significant difference in caspase‐9 or Bcl‐2 expression (Fig. 7). Taken together, these results suggest that the apoptosis of PDCD4‐induced sensitization to cisplatin mainly depends on the activation of the death receptor pathway.
Figure 6.
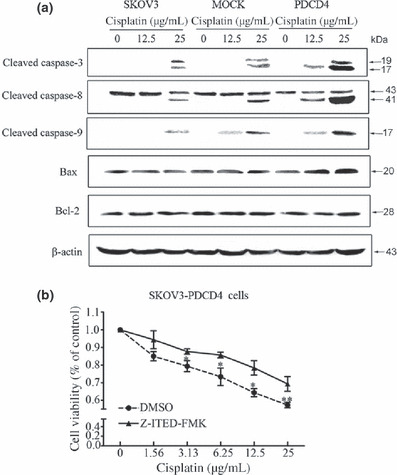
Expression of apoptosis‐related protein in programmed cell death 4 (PDCD4)‐overexpressing SKOV3 ovarian cancer cells after cisplatin treatment. (a) Cultured cells in 6‐well plates were treated with 12.5 or 25 μg/mL cisplatin for 24 h. Expressions of cleaved caspase‐3, ‐8, and ‐9, Bax, and Bcl‐2 in cells were detected by Western blot analysis. Data are representative of three independent experiments. (b) A specific caspase‐8 inhibitor, Z‐IETD‐FMK, was added 1 h before cisplatin treatment and cell viability was determined by MTT assay. Data are expressed as the mean of triplicate experiments ± SEM. *P < 0.05; **P < 0.01.
Figure 7.
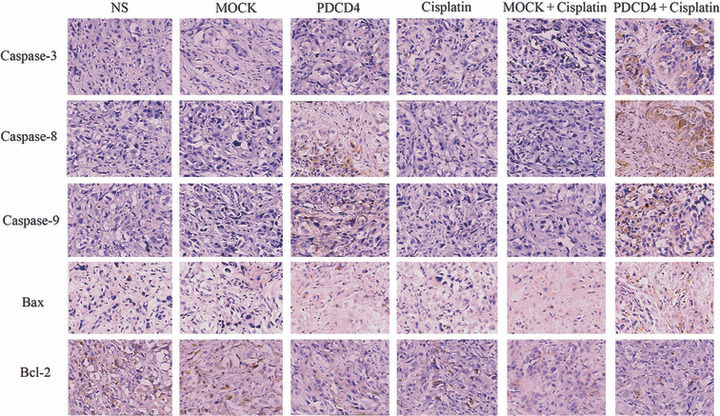
Expression of apoptosis‐related proteins in ovarian cancer tumor samples detected by immunohistochemistry. Xenograft tumors from mice treated with normal saline (NS), empty vector (MOCK), programmed cell death 4 (PDCD4) plasmid, cisplatin, MOCK + cisplatin, and PDCD4 + cisplatin were stained with antibodies against Bax, Bcl‐2, and caspase‐3, ‐8, and ‐9. Images shown are representative of each group. Magnification, ×400.
Discussion
Platinum‐based chemotherapy is the most commonly used method for treatment of advanced ovarian cancer. However, its efficacy is often limited by the existence or development of chemoresistance. Factors that enhance sensitivity of ovarian cancer cells to chemotherapeutic drugs could provide predictive biomarkers or targets for therapy. It has been reported that some tumor suppressor genes, such as PTEN and p53, can increase the chemosensitivity of ovarian cancer cells. Our present data show that PDCD4 can improve the sensitivity of ovarian cancer to platinum drugs in cell lines and in xenografts.
It has been reported that overexpression of PDCD4 increased sensitivity to cisplatin and paclitaxel but not to etoposide or 5FU in PC3 human prostate cancer cells, whereas data from the NCI‐60 human tumor panel suggested that levels of PDCD4 protein had no relation to the sensitivity to cisplatin, VP‐16, and paclitaxel.( 10 , 26 ) Our present results show that PDCD4 could selectively enhance chemosensitivity to cisplatin and carboplatin, but not affect CTX, VP‐16, or paclitaxel chemotherapy. The reasons for the discrepancy may be due to cell‐type specificity of PDCD4 action,( 29 ) which is also found in its other aspects. For example, overexpression of PDCD4 upregulated expression of p21Waf1/Cip1 (a signal molecule associated with cell cycle arrest) in neuroendocrine cells, whereas knockdown of PDCD4 expression caused upregulation of p21Waf1/Cip1 expression in HeLa cells.( 30 , 31 ) In addition, reduced PDCD4 expression did not alter the expression of p21Waf1/Cip1 in acute myeloid leukaemia cells.( 32 ) Therefore, the cell‐type specificity of PDCD4 action may contribute to the different impacts of PDCD4 on drug sensitivity. Moreover, we previously reported that decrease or loss of PDCD4 expression in ovarian cancers was associated with pathological grade and disease‐specific survival of patients, which has been confirmed by a recent report.( 14 , 15 ) We also found that restoration of PDCD4 expression could markedly inhibit the malignant phenotype of ovarian cancer cells in vitro and in vivo.( 23 ) We speculate that loss or reduction of PDCD4 expression may be one reason for chemoresistance in ovarian cancer and that restoration of PDCD4 expression will reverse the resistance of ovarian cancer to chemotherapy and improve the survival of patients with ovarian cancer.
To date, the mechanism by which PDCD4 enhances chemosensitivity remains unclear. It has been reported that PDCD4 expression enhanced geldanamycin‐induced G2‐M arrest accompanied by increased apoptosis.( 10 ) The effect of PDCD4 expression on drug sensitivity was assumed to be due to downregulation of YB‐1 expression, which involves in the resistance against paclitaxel.( 26 ) Here, our results showed that, although cisplatin treatment alone markedly increased cell numbers in S phase and PDCD4 alone could also induce cell cycle arrest in the G2 or S phase as we previously described,( 23 ) action of PDCD4 on the cell cycle was not strong enough to change cisplatin‐induced cell cycle arrest (data not shown). However, PDCD4 overexpression effectively promoted cisplatin‐induced apoptosis. Several reports also indicated that PDCD4 could induce apoptosis of human breast cancer cell line T‐47D in vitro and promote apoptosis of murine lung cancer cells in vivo by significantly increasing pro‐apoptotic proteins such as Bax or decreasing levels of the anti‐apoptotic protein Bcl‐xL.( 33 , 34 ) Apoptosis in hepatocellular carcinoma cells was induced by PDCD4 through mitochondria events and caspase cascade, including increases in cytosolic cytochrome c and mitochondrial Bax and reduction of procaspase‐3, ‐8, ‐9.( 35 ) Our present results showed that the combination of PDCD4 with cisplatin clearly elevated the expression of cleaved caspase‐3 and caspase‐8, but had no obvious effect on activation of caspase‐9 or on expression of mitochondrial‐linked apoptosis‐regulatory proteins Bax and Bcl‐2. Furthermore, when Z‐ITED‐FMK was used to inhibit the activity of caspase‐8, cisplatin‐induced apoptosis was significantly attenuated. Taken together, our data indicated that PDCD4 promoted cisplatin‐induced apoptosis mainly through the death receptor mediated pathway. It has been reported that cisplatin induces apoptosis through primarily activating the death receptor pathway while CTX, VP‐16, and paclitaxel do it mainly through the mitochondrial pathway,( 36 , 37 , 38 , 39 ) which may explain why PDCD4 selectively enhances chemosensitivity to platinum compounds. However, the precise mechanism by which PDCD4 activates the death receptor pathway remains to be further investigated.
In summary, we have found that PDCD4 selectively enhances the sensitivity of ovarian cancer cells to platinum drugs. Mechanistically, PDCD4 promotes cisplatin‐induced cell apoptosis through the death receptor pathway. These results establish that PDCD4 gene transfer in combination with cisplatin therapy may break the resistance of ovarian cancer cells to chemotherapy.
Acknowledgments
This study was supported by funds from the National Natural Science Foundation of China (Grant Nos 30628015 and 30872309), the National “973” Program of China (Grant No. 2011CB503906), the Post‐doctorate Innovation Program of Shandong Province (Grant No. 200802032), the National Natural Science Foundation of Shandong Province (Grant No. ZR2009CM142), and the Graduate Independent Innovation Foundation of Shandong University (Grant No. yzc09084).
References
- 1. Lankat‐Buttgereit B, Göke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell 2009; 101: 309–17. [DOI] [PubMed] [Google Scholar]
- 2. Waters LC, Veverka V, Bohm M et al. Structure of the C‐terminal MA‐3 domain of the tumour suppressor protein Pdcd4 and characterization of its interaction with eIF4A. Oncogene 2007; 26: 4941–50. [DOI] [PubMed] [Google Scholar]
- 3. Loh PG, Yang HS, Walsh MA et al. Structural basis for translational inhibition by the tumour suppressor Pdcd4. EMBO J 2009; 28: 274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang JH, Cho YH, Sohn SY et al. Crystal structure of the eIF4A‐PDCD4 complex. Proc Natl Acad Sci U S A 2009; 106: 3148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang HS, Matthews CP, Clair T et al. Tumorigenesis suppressor Pdcd4 down‐regulates mitogen‐activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol 2006; 26: 1297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bitomsky N, Bohm M, Klempnauer KH. Transformation suppressor protein Pdcd4 interferes with JNK‐mediated phosphorylation of c‐Jun and recruitment of the coactivator p300 by c‐Jun. Oncogene 2004; 23: 7484–93. [DOI] [PubMed] [Google Scholar]
- 7. Cmarik JL, Min H, Hegamyer G et al. Differentially expressed protein Pdcd4 inhibits tumor promoter‐induced neoplastic transformation. Proc Natl Acad Sci U S A 1999; 96: 14037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilliard A, Hilliard B, Zheng SJ et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol 2006; 177: 8095–102. [DOI] [PubMed] [Google Scholar]
- 9. Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res 2005; 65: 6034–41. [DOI] [PubMed] [Google Scholar]
- 10. Jansen AP, Camalier CE, Stark C, Colburn NH. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol Cancer Ther 2004; 3: 103–10. [PubMed] [Google Scholar]
- 11. Chen Y, Knosel T, Kristiansen G et al. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol 2003; 200: 640–6. [DOI] [PubMed] [Google Scholar]
- 12. Mudduluru G, Medved F, Grobholz R et al. Loss of programmed cell death 4 expression marks adenoma‐carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer 2007; 110: 1697–707. [DOI] [PubMed] [Google Scholar]
- 13. Gao F, Zhang P, Zhou C et al. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol Rep 2007; 17: 123–8. [PubMed] [Google Scholar]
- 14. Wang X, Wei Z, Gao F et al. Expression and prognostic significance of PDCD4 in human epithelial ovarian carcinoma. Anticancer Res 2008; 28: 2991–6. [PubMed] [Google Scholar]
- 15. Wei NA, Liu SS, Leung TH et al. Loss of Programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Mol Cancer 2009; 8: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asangani IA, Rasheed SA, Nikolova DA et al. MicroRNA‐21 (miR‐21) post‐transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008; 27: 2128–36. [DOI] [PubMed] [Google Scholar]
- 17. Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR‐21 in breast cancer cells. J Biol Chem 2008; 283(2): 1026–33. [DOI] [PubMed] [Google Scholar]
- 18. Lu Z, Liu M, Stribinskis V et al. MicroRNA‐21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008; 27(31): 4373–9. [DOI] [PubMed] [Google Scholar]
- 19. Gao F, Wang X, Zhu F et al. PDCD4 gene silencing in gliomas is associated with 5’CpG island methylation and unfavorable prognosis. J Cell Mol Med 2009; 13(10): 4257–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1‐ and betaTRCP‐mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006; 314(5798): 467–71. [DOI] [PubMed] [Google Scholar]
- 21. Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res 2008; 68(5): 1254–60. [DOI] [PubMed] [Google Scholar]
- 22. Leupold JH, Yang HS, Colburn NH, Asangani I, Post S, Allgayer H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u‐PAR) gene expression via Sp‐transcription factors. Oncogene 2007; 26(31): 4550–62. [DOI] [PubMed] [Google Scholar]
- 23. Wei ZT, Zhang X, Wang XY et al. PDCD4 inhibits the malignant phenotype of ovarian cancer cells. Cancer Sci 2009; 100(8): 1408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Q, Sun Z, Yang HS. Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both beta‐catenin/Tcf and AP‐1‐dependent transcription in colon carcinoma cells. Oncogene 2008; 27(11): 1527–35. [DOI] [PubMed] [Google Scholar]
- 25. Wang Q, Sun ZX, Allgayer H, Yang HS Downregulation of E‐cadherin is an essential event in activating beta‐catenin/Tcf‐dependent transcription and expression of its target genes in Pdcd4 knockdown cells. Oncogene 2010; 29(1): 128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shiota M, Izumi H, Tanimoto A et al. Programmed cell death protein 4 down‐regulates Y‐box binding protein‐1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res 2009; 69(7): 3148–56. [DOI] [PubMed] [Google Scholar]
- 27. Shi F, Rakhmilevich AL, Heise CP et al. Intratumoral injection of interleukin‐12 plasmid DNA, either naked or in complex with cationic lipid, results in similar tumor regression in a murine model. Mol Cancer Ther 2002; 1(11): 949–57. [PubMed] [Google Scholar]
- 28. Zhou X, Zhang Z, Yang X, Chen W, Zhang P. Inhibition of cyclin D1 expression by cyclin D1 shRNAs in human oral squamous cell carcinoma cells is associated with increased cisplatin chemosensitivity. Int J Cancer 2009; 124(2): 483–9. [DOI] [PubMed] [Google Scholar]
- 29. Lankat‐Buttgereit B, Lenschen B, Schmidt H, Goke R. The action of Pdcd4 may be cell type specific: evidence that reduction of dUTPase levels might contribute to its tumor suppressor activity in Bon‐1 cells. Apoptosis 2008; 13(1): 157–64. [DOI] [PubMed] [Google Scholar]
- 30. Goke R, Barth P, Schmidt A, Samans B, Lankat‐Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1). Am J Physiol Cell Physiol 2004; 287(6): C1541–6. [DOI] [PubMed] [Google Scholar]
- 31. Bitomsky N, Wethkamp N, Marikkannu R, Klempnauer KH. siRNA‐mediated knockdown of Pdcd4 expression causes upregulation of p21(Waf1/Cip1) expression. Oncogene 2008; 27(35): 4820–9. [DOI] [PubMed] [Google Scholar]
- 32. Ozpolat B, Akar U, Steiner M et al. Programmed cell death‐4 tumor suppressor protein contributes to retinoic acid‐induced terminal granulocytic differentiation of human myeloid leukemia cells. Mol Cancer Res 2007; 5(1): 95–108. [DOI] [PubMed] [Google Scholar]
- 33. Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH. Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER‐2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 2004; 23(49): 8135–45. [DOI] [PubMed] [Google Scholar]
- 34. Jin H, Kim TH, Hwang SK et al. Aerosol delivery of urocanic acid‐modified chitosan/programmed cell death 4 complex regulated apoptosis, cell cycle, and angiogenesis in lungs of K‐ras null mice. Mol Cancer Ther 2006; 5(4): 1041–9. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H, Ozaki I, Mizuta T et al. Involvement of programmed cell death 4 in transforming growth factor‐beta1‐induced apoptosis in human hepatocellular carcinoma. Oncogene 2006; 25(45): 6101–12. [DOI] [PubMed] [Google Scholar]
- 36. Seki K, Yoshikawa H, Shiiki K, Hamada Y, Akamatsu N, Tasaka K. Cisplatin (CDDP) specifically induces apoptosis via sequential activation of caspase‐8, ‐3 and ‐6 in osteosarcoma. Cancer Chemother Pharmacol 2000; 45(3): 199–206. [DOI] [PubMed] [Google Scholar]
- 37. Schwartz PS, Waxman DJ. Cyclophosphamide induces caspase 9‐dependent apoptosis in 9L tumor cells. Mol Pharmacol 2001; 60(6): 1268–79. [DOI] [PubMed] [Google Scholar]
- 38. Kaufmann SH. Cell death induced by topoisomerasetargeted drugs: more questions than answers. Biochim Biophys Acta 1998; 1400(1–3): 195–211. [DOI] [PubMed] [Google Scholar]
- 39. Bhalla KN. Microtubule‐targeted anticancer agents and apoptosis. Oncogene 2003; 22(56): 9075–86. [DOI] [PubMed] [Google Scholar]