

Abstract
Human papillomaviruses (HPVs) infect the stratified epithelial organ. The infection induces benign tumors, which occasionally progress into malignant tumors. To elucidate the virus‐induced tumorigenesis, an understanding of the lifecycle of HPV is crucial. In this report, we developed a new system for the analysis of the HPV lifecycle. The new system consists of a novel HPV replicon and an organotypic “raft” culture, by which the HPV‐DNA is maintained stably in normal human keratinocytes for a long period and the viral vegetative replication is reproduced. This system will benefit biochemical and genetic studies on the lifecycle of HPV and tumorigenesis. This system is also valuable in screening for antiviral compounds. We confirmed its usefulness by evaluating the antivirus effect of cytokines. (Cancer Sci 2009 00: 000–000)
The infection of high‐risk type human papillomavirus (HPV) is a major risk factor for cervical cancer.( 1 , 2 , 3 ) The World Health Organization has reported that the cases of HPV‐associated cancers number about half a million, which corresponds to 10% of cancer cases in women.( 4 ) This indicates the importance of the prevention of and urgently developing treatment for the cancer.
In order to control cervical cancer, it is essential to understand the regulatory mechanisms of the HPV infection. The primary target for HPV infection is the epithelial cells (keratinocytes) of the stratified squamous epithelium, and replication of HPV is strictly regulated by the differentiation program of the keratinocytes,( 5 ) making it difficult to analyze the virus lifecycle in standard tissue culture systems. Several tissue culture conditions have been used for studying the differentiation‐dependent lifecycle, such as a suspension culture of keratinocytes using methylcellulose “semisolid” medium,( 6 , 7 ) a calcium‐induced differentiation of keratinocytes,( 8 ) and an organotypic raft culture.( 9 , 10 , 11 ) Among them, the raft culture seems superior for studying the HPV lifecycle, because it is able to reproduce the stratified structure of epithelium, support the production of progeny virions in the differentiated layer, and capture the virus‐induced hyperplasia as in the infected lesions. Although the raft culture has been successfully used for the analysis of the HPV lifecycle in combination with the genomic‐type of HPV‐DNA,( 12 ) the drawbacks to using the culture system are the intricateness in the construction of it and the difficulty in obtaining the cell population maintaining the HPV‐DNA.
In this manuscript, we tried to improve the suitability of the culture system for analysis of the HPV lifecycle. We constructed a new HPV replicon that could be maintained for a long period in comparison with the conventional method utilizing the genomic‐type HPV‐DNA. The raft culture system incorporating the new replicon could reproduce the physiological status of HPV‐infected lesions: virus‐induced hyperplasia accompanied by viral DNA amplification and late gene expression. This new system might accelerate the investigation of the HPV lifecycle. The usefulness of the system was verified by examining the effects of several cytokines on HPV replication and hyperplasia induction.
Materials and Methods
Construction of plasmid DNAs. HPV18 genomic DNA was isolated from a plasmid containing a full‐length HPV18 DNA (GenBank accession no.: X05015). A new HPV18 replicon, p18FLneo, was constructed as illustrated in Figure 1. pEGFP1‐ori(pBR) was constructed by replacing the origin of pEGFP1 (Clontech Laboratories, Mountain View, CA, USA) with pBR322‐ori derived from pPUR (Clontech Laboratories). The long control region (LCR) of HPV18 (nucleotide number 7000 to 100; GenBank no.: X05015) was isolated by PCR, and then cloned into the vector pEGFP1‐ori(pBR); the resultant plasmid was named 18LCR/pEGFP1‐ori(pBR). Full‐length HPV18 genome was cloned into the 18LCR/pEGFP1‐ori(pBR) by using the AflII recognition site. The genomic‐type of HPV18 DNA was obtained by self‐ligation of the full‐length HPV18 DNA by following a method previously described.( 13 )
Figure 1.
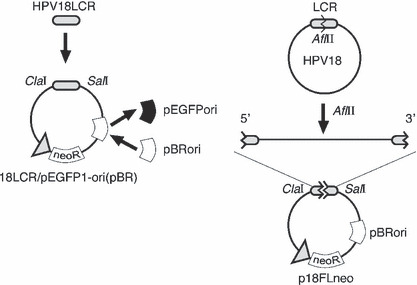
Structure of a new human papillomavirus (HPV) replicon. A new replicon containing the full length of HPV18 genomic DNA constructed with a backbone plasmid, pEGFP1‐ori(pBR), based on pEGFP1 plasmid. The replication origin (ori) element was replaced with that of pBR322. HPV18 long control region (LCR) was cloned into the pEGFP1‐ori(pBR), and then the full‐length HPV18 genome was inserted into the AflII site located in the LCR region. The obtained replicon was named p18FLneo.
Cell culture and transfection. Human foreskin fibroblasts (HFFs) and human foreskin keratinocytes (HFKs) were commercially obtained (Kurabo Industries, Osaka, Japan), and maintained with 10% fetal bovine serum/DMEM and a serum‐free keratinocyte growth medium (KGM) (EpiLife‐KG2; Kurabo Industries), respectively. HFKs were transfected with 2 μg of p18FLneo or 1.5 μg of the genomic‐type HPV18 DNA plus 0.5 μg of pEGFP1 by the nucleofection method (Nucleofector Kit; Amaxa, Cologne, Germany). The HFKs transfected with p18FLneo were cultured under the presence of G418 for more than 4 weeks, then used for the experiments.
Southern hybridization. Total DNA was extracted from the HFKs by following a standard protocol.( 14 ) Five μg of the total DNA was digested with DpnI and BglI, and then the DNA fragments were separated by 0.8% agarose gel electrophoresis, and transferred to a nylon membrane (Hybond N+; Amersham Biosciences UK, Little Chalfont, UK). For the detection of HPV18‐specific DNA, a non‐RI detection system was employed (Digoxigenin [DIG] Wash and Block Buffer Set and anti‐DIG‐alkaline phosphatase [AP]; Roche Diagnostics, Mannheim, Germany). The DIG‐labeled probe for the LCR (7000–100 nt; GenBank no.: X05015) or L1 region (6137–7136 nt; GenBank no.: X05015) of HPV18 was obtained by a PCR‐mediated method (PCR DIG Probe Synthesis Kit; Roche Applied Science, Manheim, Germany). The chemiluminescent signal was visualized with a chemiluminescent image analyzer (LAS‐3000; Fuji Film, Tokyo, Japan).
Organotypic raft culture system. The construction of the organotypic raft culture and the preparation of frozen section have been described previously.( 15 , 16 , 17 ) For BrdU incorporation, 50 g/mL BrdU (Sigma‐Aldrich, St Louis, MO, USA) was added in the medium 6 h before harvest. The thickness of the epidermal layer was measured using image analysis software (AxioVision 3.1; Carl Zeiss Vision, Munich, Germany).
Immunoblot analysis. Total cell lysate of HFKs was obtained with a triple‐detergent buffer (50 mm Tris‐HCl [pH 8.0], 150 mm NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P‐40, 0.5% sodium deoxycholate)( 18 ) supplemented with a protease inhibitor cocktail (0.5 mm PMSF, 0.15 M aprotinin, 1 M E‐64, 1 M leupeptin, 0.5 M EDTA) (Nakarai Tesuque, Kyoto, Japan) and 1 mm dithiothreitol. Equal amounts of cell lysate (5 μg protein) were subjected to SDS–polyacrylamide gel electrophoresis (SDS‐PAGE) and the gel was blotted to a PVDF membrane (Hypond‐P; Amersham Biosciences UK). The equalities of the loaded amounts were confirmed with the anti‐actin immunoblot (1:50000) (clone AC‐15; Sigma‐Aldrich) (data not shown). Antibodies for p53 (1:1000) (Ab‐6; Oncogene Research Products, San Diego, CA, USA) and pRb (1:1000; BD Biosciences Pharmingen, San Diego, CA, USA) were purchased commercially. Horseradish peroxidase (HRP)‐conjugated secondary antibodies (1:3000; Amersham Biosciences UK) and a luminal reagent (Western Blotting Luminol Reagent; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were purchased commercially. The chemiluminescent signal was visualized with a chemiluminescent image analyzer (LAS‐3000; Fuji Film).
Immunohistochemistry (IHC). IHC for the tissue sections on slide glasses was performed as described previously.( 15 , 16 , 17 ) Antibodies for BrdU (1:400) (clone 2B‐1; MBL, Nagoya, Japan) and L1 (1:200) (MAB837; Millipore, Billerica, MA, USA), and p53 (Ab‐6) (Oncogene Research Products), and pRb (BD Biosciences Pharmingen) were purchased commercially.
In situ hybridization (ISH). Detection of HPV18 DNA signals in the tissue sections was performed with the TSA‐biotin system (Perkin‐Elmer, Boston, MA, USA) following the manufacturer’s instructions. The DIG‐labeled DNA for HPV18 LCR region (7000–100 nt; GenBank no.: X05015) (DIG high prime; Roche Diagnostics) was used as the probe. For the detection of DIG‐labeled probe, HRP‐labeled anti‐DIG antibody (Dako, Glostrup, Denmark) was used. After hybridization, biotinyl tyramide working solution, SA‐HRP (streptavidin–HRP), and metal‐enhanced DAB solution (Roche Diagnostics) was used for detection of the signal. All slides were counterstained with hematoxylin.
Cytokine treatment. In monolayer culture, HFKs were incubated with cytokines (interferon [IFN]‐β, 100 units/mL; transforming growth factor [TGF]‐β, 1 ng/mL; tumor necrosis factor [TNF]‐α, 5 ng/mL) for 3 days. IFNβ and TGFβ (Sigma‐Aldrich) and TNFα (Merk Biosciences, San Diego, CA, USA) were purchased from the distributors. In the organotypic raft culture, cytokines were added in the culture medium for 7 days before the sample harvest.
Apoptosis induction by cytokine. 2 × 105 cells were cultured in the growth medium supplemented with the cytokines for 3 days. The treated cells were washed, trypsinized, and fixed with ice‐cold methanol. Apoptotic cells were labeled by M30 antibody (CytoDEATH; Roche Diagnostics) and AlexaFLUOR488‐antimouse antibody (Invitrogen, Carlsbad, CA, USA), and then counted by flow cytometry (BD Biosciences, San Jose, CA, USA).
Results
Construction of new HPV replicon. Genomic‐type HPV‐DNA was used as the replicon to obtain the keratinocytes maintaining HPV‐DNA. The genomic‐type DNA, which was constructed by re‐circularization of full‐length HPV‐DNA, was transfected into the cells with the pSV2neo expressing a neomycin‐resistance gene (neoR).( 13 ) The transfected cells were selected in the culture medium containing G418 for about 1 week, and then the surviving cells were expanded without the drug selection. The raft culture constructed with the cells could reproduce viral vegetative replication as reported previously.( 12 ) By this method, there is no selectable pressure for the maintenance of HPV‐DNA, causing a possible problem in that the cells lose HPV‐DNA. It was also suggested that the transfection of the genomic‐type DNA was inefficient because of its non‐supercoiled structure.( 19 ) To eliminate this potential problem, full‐length HPV18 DNA was inserted into a plasmid containing the neoR expression unit (Fig. 1). The replication of HPVs is regulated by the LCR as the promoter/enhancer for gene expressions at the 5′ region of the coding region and as the transcriptional terminator at the 3′ region. Therefore, the LCR was added at the both sides of the coding region. We named the new replicon p18FLneo (FL is an abbreviation for full length).
We verified the potential of p18FLneo as the replicon by examining whether it could be maintained stably in culture cells. HFKs were transfected with p18FLneo, then cultured under the presence of G418 for 4 weeks. Under the same condition, we found that the mock‐transfected cells were dislodged within a week and that the cells transfected with pEGFP1, which contains the neoR expression unit and is replication‐defective in the HFKs, could not survive for more than 2 weeks. The total DNAs were collected from the cells, and then HPV‐DNA was detected by Southern hybridization analysis. The result indicated that p18FLneo was stably maintained in the HFKs as a HPV18 replicon (Fig. 2A, p18FLneo). In order to validate its potential as the replicon, the genomic‐type of HPV18 DNA was used as the control replicon (Fig. 2A, 18‐ligation). The genomic‐type HPV18 was introduced with the pEGFP1 into the HFKs. The transfected cells were selected under the presence of G418 for 1 week, and then the surviving cells were cultured without the drug‐selection for 3 weeks. The amount of replicated HPV‐DNA was analyzed by Southern blot analysis, and it appeared that the efficiency of the maintenance or the replication of the genomic‐type HPV18 DNA was lower than that of p18FLneo. These results suggested the advantage of p18FLneo as the HPV replicon.
Figure 2.
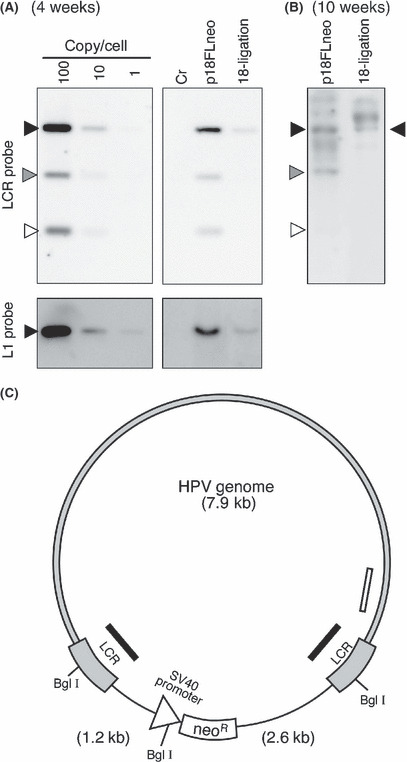
Maintenance of the new human papillomavirus (HPV) replicon in primary keratinocytes. (A) Maintenance of the HPV replicon in the human foreskin keratinocytes (HFKs) 4 weeks after transfection was examined by Southern blot analysis. Total DNA was extracted from the normal HFKs (Cr), the HFKs harboring either the HPV replicon (p18FLneo) or the genomic‐type HPV18 DNA (18‐ligation) and subjected to DpnI + BglI digestion. Each lane was loaded with 5 μg of the total DNA. p18FLneo DNA was used as the control for the copy number per cell. p18FLneo was digested with BglI and applied to the agarose gel at an amount equivalent to 1 copy, 10 copies, or 100 copies per cell. The probe used for the detection of HPV‐DNA was DIG‐labeled 18LCR or 18L1 DNA fragment. Closed triangle indicates the position of the full‐length HPV18 genomic DNA. Gray and open triangles indicate the positions of the fragments containing a portion of long control region (LCR) and the backbone plasmid; those fragments could not be detected with L1 probe. (B) The maintenance of the HPV replicon in HFKs 10 weeks after transfection was examined as described in (A). Closed triangle indicates the position of the full‐length HPV18 genomic DNA. Gray and open triangles indicate the positions of the fragments containing a portion of LCR and the backbone plasmid. The extra signals observed in each lane were considered the integrated form of HPV‐DNA. (C) The scheme of pFL18neo. Gray region indicates HPV18 DNA. Black and white bars indicate the regions targeted by LCR and L1 probes, respectively. The recognition sites for BglI are indicated, and the digestion produced three DNA fragments, 7.9 kb, 2.6 kb, and 1.2 kb.
The HFKs containing p18FLneo could be maintained for more than 10 weeks under the presence of G418. The growth potential of normal HFKs apparently declined after 6 weeks of culturing and the cells acquired senescence status, indicating that the HFKs containing p18FLneo were immortalized by the functions of HPV E6 and E7. The existence of HPV‐DNA in the cells was examined, and it was revealed that the majority of HPV‐DNA was maintained as the episomal status and that some portion of the DNA might be integrated in the host chromosome (Fig. 2B, p18FLneo). In the accompanying experiment, the DNA status in the HFKs containing the genomic‐type HPV‐DNA was examined, and it found that most of the HPV‐DNA was maintained as integrated form (Fig. 2B, 18‐ligation).
Raft culture with HFKs harboring the new HPV replicon. The HFKs or the spontaneously immortalized keratinocytes (normal immortal keratinocytes, NIKS)( 20 ) harboring HPV genomic DNA were used to organize the organotypic raft culture,( 11 , 13 , 21 , 22 ) and they could support HPV replication in a differentiation‐dependent manner. We examined whether the HFKs maintaining p18FLneo (HFKp18FLneo) could also support the HPV lifecycle in the raft culture.
HFKp18FLneo could organize a stratified epithelial structure and it appeared that significant hyperplasia was induced (Fig. 3, H&E). In the epithelial layer of the HFKp18FLneo raft culture, BrdU‐positive cells were detected in the parabasal layer. On the contrary, BrdU‐positive cells were restricted at the basal layer in the raft culture with normal HFKs (Fig. 3, BrdU). This observation suggested that p18FLneo had the potential to disturbe the differentiation program of the epithelial cells and induced hyperproliferation.
Figure 3.
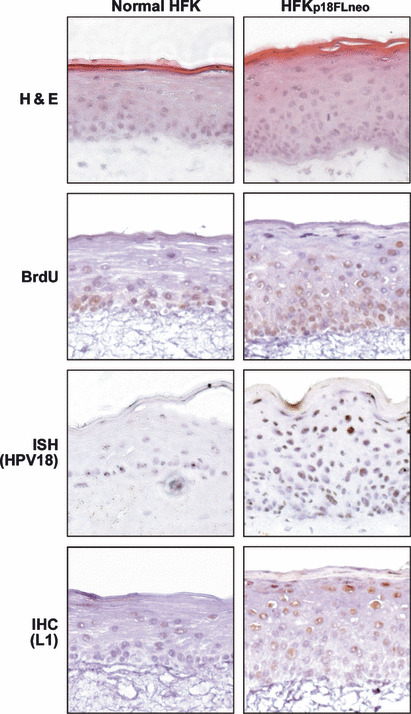
Vegetative replication of the human papillomavirus (HPV) replicon in the raft culture. Thin sections (7 μm) were obtained from the raft culture organized with normal human foreskin keratinocytes (HFKs) and HFKp18FLneo. The DNA synthesis of the cell was monitored by immunohistochemistry for incorporated BrdU. HPV18 DNA was detected by in situ hybridization (ISH) with DIG‐labeled L1 probe. The expression of L1 protein was analyzed by IHC with anti‐L1 antibody.
We next examined whether the late‐phase of the virus lifecycle was reproduced with HFKp18FLneo. By ISH with the HPV18 probe, the cells positive for HPV18 DNA were detected in the suprasurface layer (Fig. 3, ISH), indicating that the copy number of p18FLneo was amplified in the differentiated layer of the epithelium. The expression of the late gene product, L1, was also detected in the differentiated layers by IHC (Fig. 3, IHC). These observations indicate that HFKp18FLneo in combination with the raft culture is an appropriate tool for the analysis of the HPV lifecycle. Although the HFKp18FLneo maintained for long period (10 weeks) was also used for the raft culture, it failed to organize a stratified epithelial layer, indicating it lost the property of normal cellular differentiation.
Application of the new HPV replicon: (1) Effects of cytokines on the latent phase of HPV infection. Cytokine production is a biological response in vivo to virus infection or inflammation. One of the cytokines, type I IFN, is used in chemotherapy for HPV‐positive cervical neoplasia.( 23 ) Other cytokines, TGFβ and TNFα, have been reported to be involved in the response to HPV infection.( 24 , 25 , 26 , 27 , 28 , 29 , 30 ) We examined the effects of these cytokines on HPV replication using the new HPV replicon system.
The monolayer culture of HFKp18FLneo is supposed to represent the status of the latent infection of HPV in basal cells. We first examined the effects of the cytokines on the HFKp18FLneo monolayer culture. We chose doses of cytokines that had minimal effects on the growth of normal HFKs (IFNβ, 100 units/mL; TGFβ, 1 ng/mL; TNFα, 5 ng/mL) in order to observe the specific effects on HPV replication (Fig. 4). These doses of the cytokine treatments had also no significant effects on the growth of HFKp18FLneo. The apoptosis induction by the cytokines was also examined by FACS analysis using an apoptosis‐specific antibody (M30 CytoDEATH; Roche Diagnostics), and it appeared that these cytokine treatments did not induce any apoptotic response (data not shown). Note that the treatment of higher doses of the cytokines induced growth arrest or apoptotic cell death of both normal HFKs and HFKp18FLneo (data not shown).
Figure 4.
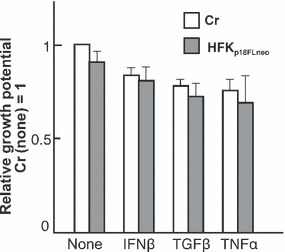
Effects of cytokines on the proliferation of human foreskin keratinocytes (HFKs) harboring p18FLneo. The effects of cytokine treatments (interferon [IFN]‐β, 100 units/mL; transforming growth factor [TGF]‐β, 1 ng/mL; tumor necrosis factor [TNF]‐α, 5 ng/mL) on the proliferation of HFKs were monitored. The growth rate in 72‐h culture of the exponentially growing cells is indicated (growth rate of not treated normal HFKs, 1). The living cells were distinguished by Trypan blue exclusion. The value is the average of at least three independent experiments and the SD is indicated.
Next, we examined the effects of the cytokines on the maintenance of HPV‐DNA in HFKp18FLneo, and the result indicated that IFNβ treatment moderately suppressed HPV‐DNA replication (Fig. 5A). The TGFβ and TNFα treatments did not influence DNA maintenance significantly. In HPV‐infected cells, the expressions of cellular tumor suppressors p53 and pRb are suppressed by the functions of viral oncoproteins E6 and E7, respectively. We analyzed the expressions of p53 and pRb in HFKp18FLneo by immunoblot analysis, and found that they slightly decreased as compared with those in the normal HFKs (Fig. 5B). This weak suppression indicated that the expression levels of the viral oncoproteins were kept low as found in the latent infection of HPV. The weak suppressions of p53 and pRb expressions were not modified significantly by the cytokine treatments.
Figure 5.
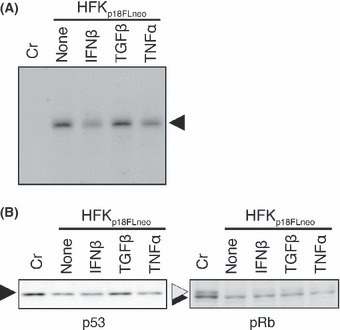
Effects of cytokines on the maintenance of the human papillomavirus (HPV) replicon. (A) The HFKp18FLneo was treated with cytokines for 3 days and total DNA was extracted. The DNA was digested with both BglI and DpnI and 2 μg of it was subjected to Southern blot analysis with the DIG‐labeled L1 probe. Normal human foreskin keratinocytes (HFKs) were used as the control (Cr). (B) Expressions of p53 and pRb in the same cells monitored by immunoblot analysis.
The results indicated that the cytokine treatments used in this report did not affect the growth potential of HFKp18FLneo and the maintenance of HPV‐DNA. Given that HFKp18FLneo is considered to represent basal cells latently infected with HPV, these results suggested that the cytokine treatments had no anti‐HPV effect on the latently infected cells.
Application of the new HPV replicon: (2) Effects of cytokines on the late‐stage of the HPV lifecycle. We next examined the effect of the same set of cytokines on the late‐stage of the HPV lifecycle by using HFKp18FLneo and the raft culture. As described above, the raft culture with HFKp18FLneo showed a moderate hyperplasia at the epithelial layer (Fig. 3), and the hyperplasia could be suppressed by the IFNβ treatment to the level of normal HFKs (Fig. 6). The number of BrdU‐positive cells in the upper layers of epithelium was decreased with IFNβ treatment (Fig. 7, BrdU). As compared with IFNβ, the moderate but significant suppression could be also observed with TGFβ. In contrast, the TNFα could not suppress hyperplasia formation and it activated the invasive potential of the epithelial cells. The number of BrdU‐positive cells was rather increased with TNFα. These results indicated that IFNβ treatment had an apparent suppressive effect on virus‐induced hyperplasia. The results also raised the possibility that TNFα treatment accelerated virus‐induced tumorigenesis.
Figure 6.
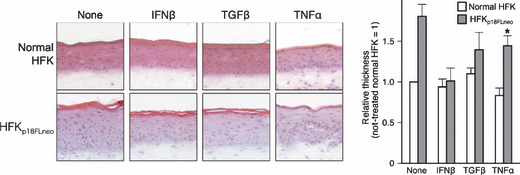
Effects of cytokines on the raft culture constructed with human foreskin keratinocyte (HFK)p18FLneo. Effects of cytokine treatments on the raft culture organized with normal HFKs or HFKp18FLneo were examined. Frozen‐sections of the raft culture were fixed with 4% paraformaldehyde and stained with H&E. The thickness of the epidermis was estimated by microscopic analysis with an image analysis application (AxioVision; Carl Zeiss Vision), and the relative values are indicated as a bar chart (thickness of not treated normal HFK set as 1.0; average value, 95 μm). The value is the average of at least three independent experiments and the SD is indicated. Note that the epidermis of the HFKp18FLneo treated with tumor necrosis factor (TNF)‐α exhibited the invasion phenotype; therefore, the thickness of it could not be measured precisely (indicated with an asterisk).
Figure 7.
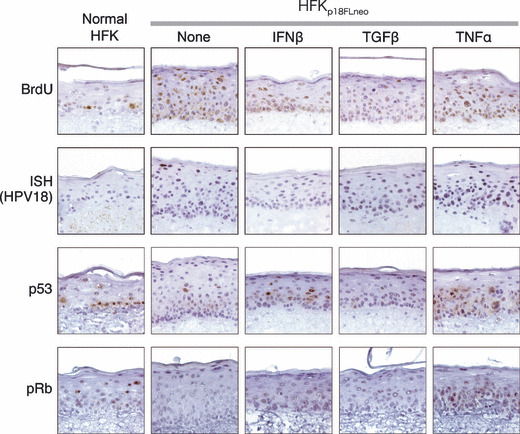
The effects of cytokine treatments on human papillomavirus (HPV) replication. Effects of cytokine treatments on HPV replication and on cellular status were examined by using the raft culture. The DNA synthesis of the cells was monitored by the uptake of BrdU. Incorporated BrdU was detected by immunohistochemistry (IHC) with anti‐BrdU antibody. HPV‐DNA was detected by in situ hybridization (ISH) with DIG‐labeled 18L1 probe (ISH). The expressions of p53 and pRb in the raft culture were monitored by IHC with specific antibodies (p53 & pRb).
Next, the effect of the cytokine treatment on the vegetative viral replication was analyzed. The amplification of viral DNA at the upper layer of epithelium was suppressed by the treatment of IFNβ and TGFβ (Fig. 7, ISH). On the other hand, TNFα treatment activated viral DNA amplification in the broad area of the epithelium. These observations indicated that both IFNβ and TGFβ could suppress the late phase in the HPV lifecycle and TNFα treatment had the opposite effect on it.
It is considered that the viral oncoproteins E6 and E7 are responsible for virus‐induced hyperplasia.( 31 ) We examined the effects of cytokines on the status of p53 and pRb in the raft culture in order to estimate the expression levels of E6 and E7, respectively. The expressions of p53 and pRb were decreased in the epithelial layer of the HFKp18FLneo raft culture as compared to those of normal HFKs (Fig. 7, p53 and pRb). It was supposed that this decrement was caused by the E6 and E7 expressed moderately in HFKp18FLneo. IFNβ treatment recovered p53 expression at the middle layers of epithelium, although TGFβ did not modify the p53 status. TNFα enhanced p53 expression in the epithelium broadly. pRb expression was slightly recovered by all three cytokine treatments. These results indicated that the cytokines affected the expressions of p53 and pRb, although their relation to the antiviral effect remains unclear.
Discussion
A novel HPV replicon. We constructed a new replicon of HPV by integrating several improvements for the stable maintenance of the replicon in the cells, which made it easy to collect cells harboring HPV‐DNA. The major points are summarized below.
-
•
The HPV replicon has a configuration of plasmid DNA, not of genomic DNA. In previous reports, the genomic‐type of DNA was used as a replicon. To obtain the genomic‐structure of DNA, full‐length HPV‐DNA was excised from a plasmid and circularized by self‐ligation.( 13 ) This process is laborious and it is difficult to control the quality of the DNA, which influences the transfection efficiency. The self‐ligated DNA has a non‐supercoiled configuration, and such a form of DNA might not be an appropriate substrate for gene expression and DNA replication. We incorporated, therefore, the HPV‐DNA into a plasmid backbone, which made it easier to control the DNA quality and the constancy of the transfection efficiency was improved.
-
•
In the conventional protocol for obtaining the HFKs harboring HPV‐DNA, HFKs were transfected with both the genomic‐type of DNA and a plasmid expressing a drug‐resistance gene, such as pSV2neo.( 13 ) After a short period of drug selection, the cells were then expanded without drug selection. The HPV replicon used in this report contained a selection marker, an expression unit for a neomycin‐resistance gene, which made it possible to apply the drug‐selection even in the expansion process, ensuring that the obtained cells maintained HPV replicons.
In addition to these points, it is unique that p18FLneo contains the LCR at both sides of the genome (Fig. 1). Although we designed it by taking into account the roles of the LCR as 5′‐ and 3′‐regulatory elements, the artificial structure might have unexpected effects on HPV replication. It is necessary, therefore, to compare carefully the replication potentials between the genomic‐type DNA and p18FLneo. We also need to determine whether the 3′‐LCR unit is required for the maintenance or vegetative replication of HPV‐DNA.
Recently Wang et al. reported a new system for analyzing the HPV18 lifecycle.( 19 ) They used a plasmid containing HPV18 genome into which a neoR expression unit and loxP sites were inserted. The plasmid was co‐transfected into human keratinocytes along with an expression plasmid for a Nuclear Localization Signal (NLS)‐tagged Cre recombinase, resulting in efficient establishment of the cells maintaining almost authentic HPV18 genomic DNA. The most important improvement seemed that they used plasmid DNA instead of the self‐ligated genomic‐type DNA that did not have a supercoiled structure. The system described in this paper employed a plasmid DNA, p18FLneo, as a replicon, suggesting it has the similar advantage. The long‐term selection under the G418 presence is able to be adapted in the system with p18FLneo, but not in the system reported by Wang et al. Although their system could produce very high titer of infectious virus, what made this improvement was unexplained, as commented in a review.( 32 ) It will be necessary to perform side‐by‐side comparison between the systems using p18FLneo and the plasmids used by Wang et al.
Usefulness of the new system supporting efficient HPV replication. The new HPV replicon could be maintained stably in HFKs as shown in Figure 2. When applied in the raft culture, the HFKs maintaining the replicon showed moderate hyperplasia and the HPV‐DNA was amplified at the differentiated layer of epidermis (Fig. 3). The hyperplasia observed with the raft culture was moderate as compared with that induced by high‐risk type E7 expression.( 16 ) The immunoblot analysis of p53 and pRb expressions in the HFKs indicated that the introduction of the HPV replicon suppressed moderately those expressions, suggesting the E6 and E7 expression levels in the HFKs were maintained at low level. It is supposed that the expressions of E6 and E7 are up‐regulated in the course of malignant conversion of the HPV‐infected cells,( 33 ) thus, the raft culture harboring the HPV replicon seems to represent an early stage of the tumor induced by the HPV infection.
Effects of cytokines in protection against virus infection. Living organisms have protection systems for the viral infections. In mammals, a variety of cytokines are produced by either infected cells or periphery cells, which act in the elimination of the infected cells.( 34 ) The system using the new HPV replicon and the raft culture could be used to examine the effect of cytokine treatment on the early stage of HPV‐infected lesions. We selected three cytokines, IFNβ, TNFα, and TGFβ, because there were several reports indicating the relation between these cytokines and HPV infection.( 24 , 25 , 26 , 27 , 28 , 29 , 30 , 35 , 36 )
We first examined the effects of the cytokines on the monolayer culture of the HFKs harboring the HPV replicon, which represented the status of the HPV‐infected basal cells. Under this condition, although IFNβ treatment could suppress HPV‐DNA replication moderately, the treatments of other cytokines, TGFβ and TNFα, had almost no effect on both HPV‐DNA replication and cell proliferation (Figs 4,5). HFKs harboring the 18FLneo under the monolayer culture condition showed little signature of viral infection, and the cytokine treatments examined here might not display their effect on such cells. On the contrary, it has been reported that these cytokines have suppressive effects on the growth of HPV‐positive cell lines.( 24 , 25 , 26 , 27 , 35 , 36 ) The cells used in those reports have malignant or transformed phenotype, which might have cause observations different from ours.
Next, the effects were examined using the raft culture (Fig. 6). Under this condition, IFNβ exhibited strong inhibitory effects on dysplasia formation. It also suppressed the vegetative replication of the virus in the suprabasal layer of epithelium. TGFβ also exhibited a significant inhibitory effect on both dysplasia formation and HPV‐DNA amplification, but the effect was not as drastic as that of IFNβ. In contrast, it appeared that TNFα treatment enhanced HPV replication and induced the invasion of epithelial cells into the dermal layer.
IFNβ has been administered to HPV‐infected lesions including condylomas and early stage cervical intraepithelial neoplasia ( 23 ) By using this model, it was possible to confirm the effectiveness of IFNβ on the treatment of HPV‐related lesions under tissue culture condition, indicating that this system is a useful platform for examination of the detailed action‐mechanisms of IFNβ on HPV replication. Recently, it was reported that p56, a product of ISG56, interacted with viral replication factor E1 and inhibited its function.( 37 ) It will be interesting to examine the induction level of p56 by IFNβ treatment in both the monolayer and raft cultures.
It should be noted that the expressions of E6 and E7 are usually up‐regulated in the advanced stages of cervical neoplasia, and they were reported to interfere with the IFN‐related pathway by interacting with Tyk2,( 38 ) IRF3,( 39 ) IRF1,( 40 ) or IRF9.( 41 ) Although this report described the significant anti‐HPV effects of IFNβ, such effects might be diminished in association with the progression of malignant status.
The observation that TNFα treatment enhanced HPV replication suggests that the inflammatory response accompanied by TNFα production exerts effects opposite to the antiviral response at the early stage of HPV‐infected lesions. In searching for new therapeutic approaches to HPV‐related diseases, it might be important to consider the induction of inflammation, which has the potential to accelerate disease progression.
In this report, we developed a new experimental system that could support the replication of HPV‐DNA for a long period and the differentiation‐dependent lifecycle of the virus. This system will be adapted to screening for other anti‐HPV compounds. It also allows the manipulation of the genetic elements of both host cells and virus; thus, the analysis of regulatory mechanisms of the virus lifecycle and virus‐induced tumorigenesis becomes accessible.
Acknowledgments
We thank the many colleagues for technical assistance and manuscript preparation, K. Sasaki for essential advice and support, Dr. P.M. Howley for providing full‐length clones for HPV18, and Dr. M. Tsunenaga and Dr. T. Kuroki for instructions regarding the organotypic raft culture. This work is supported in part by a Grant‐in‐Aid from the Ministry of Health, Labor and Welfare of Japan for the Third‐Term Comprehensive 10‐year Strategy for Cancer Control. A.S., S.Y., N.K., and H.N. were supported by the 21st Century COE program of the Japan Society for the Promotion of Science (JSPS).
References
- 1. Howley PM, Lowy DR. Papillomavirus and their replication. In: Knipe DM, Howley PM, eds. Fields Virology, 5th edn. Hagerstown: Lippincott Williams & Wilkins,a Wolters Kluwer Business, 2007; 2299–354. [Google Scholar]
- 2. Durst M, Gissmann L, Ikenberg H, Zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A 1983; 80: 3812–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zur Hausen H. Papillomavirus infections–a major cause of human cancers. Biochim Biophys Acta 1996; 1288: F55–78. [DOI] [PubMed] [Google Scholar]
- 4. Castellsagué X, De Sanjosé S, Aguado T et al. HPV and cervical cancer in the 2007 report. Vaccine 2007; 25 (Suppl 3): C1–230. [DOI] [PubMed] [Google Scholar]
- 5. Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 2004; 68: 362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flores ER, Lambert PF. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol 1997; 71: 7167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams JC, Watt FM. Fibronectin inhibits the terminal differentiation of human keratinocytes. Nature 1989; 340: 307–9. [DOI] [PubMed] [Google Scholar]
- 8. Ruesch MN, Laimins LA. Human papillomavirus oncoproteins alter differentiation‐dependent cell cycle exit on suspension in semisolid medium. Virol 1998; 250: 19–29. [DOI] [PubMed] [Google Scholar]
- 9. Meyers C, Frattini MG, Hudson JB, Laimins LA. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992; 257: 971–3. [DOI] [PubMed] [Google Scholar]
- 10. Dollard SC, Wilson JL, Demeter LM et al. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. OFF Genes Dev 1992; 6: 1131–42. [DOI] [PubMed] [Google Scholar]
- 11. Frattini MG, Lim HB, Laimins LA. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation‐dependent late expression. Proc Natl Acad Sci U S A 1996; 93: 3062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moody CA, Fradet‐Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc Natl Acad Sci U S A 2007; 104: 19541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frattini MG, Lim HB, Doorbar J, Laimins LA. Induction of human papillomavirus type 18 late gene expression and genomic amplification in organotypic cultures from transfected DNA templates. J Virol 1997; 71: 7068–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strauss WM. Preparation of genomic DNA from mammalian tissue. In: Ausubel FM, Brent R, Kingston RE et al. , eds. Current Protocols in Molecular Biology. New York: John Wiley & Sons, Inc., 1998; 2.1–2.3. [Google Scholar]
- 15. Yoshida S, Kajitani N, Satsuka A, Nakamura H, Sakai H. Ras modifies proliferation and invasiveness of cells expressing human papillomavirus oncoproteins. J Virol 2008; 82: 8820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ueno T, Sasaki K, Yoshida S et al. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 2006; 25: 4155–64. [DOI] [PubMed] [Google Scholar]
- 17. Tsunenaga M, Kohno Y, Horii I et al. Growth and differentiation properties of normal and transformed human keratinocytes in organotypic culture. Jpn J Cancer Res 1994; 85: 238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sambrool J, Fritsch EF, Maniatis T. Molecular Cloning. New York: Cold Spring Harbor Laboratory Press, 1989; 18.30–3. [Google Scholar]
- 19. Wang HK, Duffy AA, Broker TR, Chow LT. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev 2009; 23: 181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allen‐Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O’Connor SL. Normal growth and differentiation in a spontaneously immortalized near‐diploid human keratinocyte cell line, NIKS. J Invest Dermatol 2000; 114: 444–55. [DOI] [PubMed] [Google Scholar]
- 21. Flores ER, Allen‐Hoffmann BL, Lee D, Sattler CA, Lambert PF. Establishment of the human papillomavirus type 16 (HPV‐16) life cycle in an immortalized human foreskin keratinocyte cell line. Virol 1999; 262: 344–54. [DOI] [PubMed] [Google Scholar]
- 22. Flores ER, Allen‐Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol 2000; 74: 6622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frazer IH, McMillan NAJ. Papillomatosis and condylomata acuminata. In: Penny S‐HaRW, ed. Clinical Applications of the Interferons. London: Chapman and Hall Medical, 1997; 79–91. [Google Scholar]
- 24. Donalisio M, Cornaglia M, Landolfo S, Lembo D. TGF‐beta1 and IL‐4 downregulate human papillomavirus‐16 oncogene expression but have differential effects on the malignant phenotype of cervical carcinoma cells. Virus Res 2008; 132: 253–6. [DOI] [PubMed] [Google Scholar]
- 25. Nindl I, Steenbergen RD, Schurek JO, Meijer CJ, Van der Valk P, Snijders PJ. Assessment of TGF‐beta1‐mediated growth inhibition of HPV‐16‐ and HPV‐18‐transfected foreskin keratinocytes during and following immortalization. Arch Dermatol Res 2003; 295: 297–304. [DOI] [PubMed] [Google Scholar]
- 26. Villa LL, Vieira KB, Pei XF, Schlegel R. Differential effect of tumor necrosis factor on proliferation of primary human keratinocytes and cell lines containing human papillomavirus types 16 and 18. Mol Carcinog 1992; 6: 5–9. [DOI] [PubMed] [Google Scholar]
- 27. Vieira KB, Goldstein DJ, Villa LL. Tumor necrosis factor alpha interferes with the cell cycle of normal and papillomavirus‐immortalized human keratinocytes. Cancer Res 1996; 56: 2452–7. [PubMed] [Google Scholar]
- 28. Woodworth CD, McMullin E, Iglesias M, Plowman GD. Interleukin 1 alpha and tumor necrosis factor alpha stimulate autocrine amphiregulin expression and proliferation of human papillomavirus‐immortalized and carcinoma‐derived cervical epithelial cells. Proc Natl Acad Sci U S A 1995; 92: 2840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kyo S, Inoue M, Hayasaka N et al. Regulation of early gene expression of human papillomavirus type 16 by inflammatory cytokines. Virol 1994; 200: 130–9. [DOI] [PubMed] [Google Scholar]
- 30. Zur Hausen H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin Cancer Biol 1999; 9: 405–11. [DOI] [PubMed] [Google Scholar]
- 31. Dong W, Kloz U, Accardi R et al. Skin hyperproliferation and susceptibility to chemical carcinogenesis in transgenic mice expressing E6 and E7 of human papillomavirus type 38. J Virol 2005; 79: 14899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galloway DA. Human papillomaviruses: a growing field. Genes Dev 2009; 23: 138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res 2002; 89: 213–28. [DOI] [PubMed] [Google Scholar]
- 34. Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev 2007; 220: 214–24. [DOI] [PubMed] [Google Scholar]
- 35. Stanley MA, Pett MR, Coleman N. HPV: from infection to cancer. Biochem Soc Trans 2007; 35: 1456–60. [DOI] [PubMed] [Google Scholar]
- 36. Koromilas AE, Li S, Matlashewski G. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev 2001; 12: 157–70. [DOI] [PubMed] [Google Scholar]
- 37. Terenzi F, Saikia P, Sen GC. Interferon‐inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J 2008; 27: 3311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li S, Labrecque S, Gauzzi MC et al. The human papilloma virus (HPV)‐18 E6 oncoprotein physically associates with Tyk2 and impairs Jak‐STAT activation by interferon‐alpha. Oncogene 1999; 18: 5727–37. [DOI] [PubMed] [Google Scholar]
- 39. Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor‐3 and inhibits its transcriptional activity. Genes Dev 1998; 12: 2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perea SE, Massimi P, Banks L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor‐1. Int J Mol Med 2000; 5: 661–6. [DOI] [PubMed] [Google Scholar]
- 41. Antonsson A, Payne E, Hengst K, McMillan NA. The human papillomavirus type 16 E7 protein binds human interferon regulatory factor‐9 via a novel PEST domain required for transformation. J Interferon Cytokine Res 2006; 26: 455–61. [DOI] [PubMed] [Google Scholar]