

Abstract
We describe novel humanized anti‐CD20 monoclonal antibodies (mAbs) developed for therapeutic use on the basis of their physicochemical properties and cellular cytotoxicity. A distinct correlation between apparent dissociation constants (K d) and apoptotic activity for eight murine anti‐CD20 mAbs (OUBM1–OUBM8) and previously‐developed murine anti‐CD20 mAbs enabled us to categorize anti‐CD20 mAbs into two groups. Group A mAbs had lower K d values and did not induce definite apoptosis, while Group B mAbs had greater K d values and did induce definite apoptosis. A murine version mAb of rituximab, 2B8, belongs to Group B. An epitope analysis showed that the epitope of two murine mAbs, OUBM3 and OUBM6, differed from that of 2B8 or 2F2 (ofatumumab). Two mAbs, OUBM3 from Group A and OUBM6 from Group B, were selected and humanized. As expected, the humanized OUBM3 with the lower K d did not induce apoptosis, while the humanized OUBM6 (hOUBM6) with the greater K d did. Both hOUBM3 and hOUBM6 induced highly‐effective, complement‐dependent cytotoxicity and antibody‐dependent, cell‐mediated cytotoxicity against Burkitt’s and follicular lymphomas. Importantly, hOUBM6 exhibited cellular cytotoxicity against diffuse, large B cells that are less effectively depleted by rituximab and also exhibited effective cytotoxicity against tumor cells from human CD20(+) leukemia and lymphoma patients. These results suggest the potential impact of the further development of our anti‐CD20 mAbs. Our study shows that the selection of mAbs based on their physicochemical parameters, followed by the biological activity assessment for the selected mAbs, is a rational and efficient approach for pharmaceutical mAb development. (Cancer Sci 2009)
Recently, the application of immunotherapy using monoclonal antibodies (mAbs) has increased considerably, and numerous mAb candidates are under development or are being assessed in clinical trials. In the case of mAb development using murine mAbs, the degree of complement‐dependent cytotoxicity (CDC) and antibody‐dependent, cell‐mediated cytotoxicity (ADCC) activity can be measured after humanization, which implies that it is difficult to evaluate the clinical effectiveness of these antibodies (Abs) before humanization. The humanization of large numbers of murine mAbs without data to support future clinical use requires massive investments in terms of time, labor, and funding. Furthermore, the evaluation of bioactivity is often dependent on the assay systems themselves. Therefore, it is highly desirable to select candidates for humanization on the basis of the intrinsic parameters of the mAb itself, such as its dissociation constant and epitope, at an early stage of mAb development. In order to establish an efficient and rational method of developing mAbs, we focused on anti‐CD20 mAbs as the ideal study target for studying the intrinsic properties of mAbs, because data on these Abs is available.
CD20 is a B‐cell‐specific tetra‐transmembrane protein with a molecular weight of 33–37 kDa. It is found both in malignant B cells and in almost all normal B cells other than pro‐B cells and plasma cells. 1 It has been suggested that CD20 plays a role as a store‐operated calcium channel. 2 , 3 , 4 Furthermore, CD20 is known to be bound to Src tyrosine kinases, such as Lyn, Fyn, and Lck, suggesting its involvement in the phosphorylation of intracellular proteins. 5
CD20 has been an effective target for immunotherapies that use mAbs to treat B‐cell‐derived diseases, because it neither internalizes nor dissociates from the plasma membrane upon Ab binding. 6 Rituximab (C2B8, a chimeric version of 2B8) was the first successful mouse/human immunoglobulin G (IgG)1κ chimeric mAb against human CD20 approved by the Food and Drug Administration (FDA) for the treatment of low‐grade, non‐Hodgkin’s lymphoma (NHL). 7 , 8
Three mechanisms have been proposed for the depletion of B cells by rituximab. One mechanism is through the inhibition of cell growth, mainly via the induction of apoptosis by the direct binding of anti‐CD20 Abs to the B cell. 9 , 10 , 11 , 12 The role of anti‐CD20 Abs in the signal transduction pathway that leads to the induction of apoptosis has been studied, although evidence about caspase activation is controversial and the apoptotic pathway induced by anti‐CD20 Abs has not been elucidated. 9 , 11 , 13 , 14 , 15 The two other antilymphoma mechanisms require immunological factors. A second mechanism is CDC that depletes B cells under the presence of complement‐related proteins, and a third mechanism is ADCC that requires effector cells, such as natural killer cells, monocytes, and macrophages. 16 , 17 , 18 , 19 , 20 , 21 , 22 However, the actual mechanism underlying lymphoma depletion in vivo is still unclear. Rituximab has therapeutic effects on CD20‐positive, B‐cell lymphomas. The response rate for rituximab as the single agent for low‐grade, B‐cell lymphomas is reported to be over 50% 23 and is 95% in combination with standard chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone [CHOP]). 24 A phase‐III trial of CHOP with and without rituximab indicated a significant survival benefit when rituximab was used in the treatment of diffuse, large B‐cell lymphomas (DLBCL). 25 Thus, rituximab–CHOP is currently the standard DLBCL therapy. Nevertheless, there is a concern: treatment with rituximab–CHOP is less effective for a significant number of patients during the clinical course of the disease or at the time of relapse. Mechanisms underlying the differences in clinical response have been proposed 26 and have been gradually revealed by recent studies, 27 , 28 , 29 although the means to overcome this resistance have not yet been established.
In the present study, we present a rational approach for the selection and development of therapeutic mAbs based on the physicochemical parameters of Abs: the dissociation constant and epitope. When considered together with the antilymphoma activity of Abs, our rational approach provides novel humanized anti‐CD20 mAbs, which can induce apoptosis and effective ADCC against lymphoma cell lines and has high CDC activity against not only the rituximab‐sensitive lymphoma cell lines, but also lymphoma cell lines that are less effectively depleted by rituximab.
Materials and Methods
Cell cultures. The Burkitt’s lymphoma (BL) cell line, RAJI, and the lymphoblastoid cell line, WiL2‐NS, were obtained from Riken Cell Bank (Ibaraki, Japan) and maintained at the Nanotechnology Center of Osaka University (Osaka, Japan). The follicular lymphoma (FL) cell line, SU‐DHL4, and the DLBCL cell line, RC‐K8, have been described previously. 12 The other DLBCL cell line, Karpas422, was kindly given to us by Dr Karpas (University of Cambridge, Cambridge, UK) as a gift. The cells were maintained in RPMI‐1640 (NACALAI TESQUE, Kyoto, Japan) and supplemented with 10% fetal bovine serum (EQUITECH‐BIO, Kerrville, TX, USA). Humanized anti‐CD20 mAb‐producing Chinese Hamster Ovary (CHO) cells, CD20‐expressing CHO cells, and Human Embryonic Kidney (HEK) 293 T cells were maintained in standard conditions.
Anti‐CD20 mAbs. The extracellular loop of human CD20 produced by using bacterial cells fails to be recognized by the 2B8 mAb because of its unfolded 3‐D structure. This result strongly suggests that producing mAbs by using the unfolded extracellular loop of CD20 is an invalid approach for obtaining anti‐CD20 mAbs that can be bound to the extracellular loop of CD20 on the cell surface. Accordingly, the murine anti‐CD20 mAbs used here and described previously 30 were produced by using CHO cells stably expressing human CD20 as the antigen. Amino acid sequences of the humanized anti‐CD20 mAbs, humanized OUBM6 (hOUBM6) and humanized OUBM3 (hOUBM3), were designed by Dr Eduard Padlan (Kensington, MD, USA) and provided to us through a collaboration between Osaka University and BioMedics (Tokyo, Japan). hOUBM3 and hOUBM6 are the humanized versions of the murine anti‐CD20 mAbs, 1k1782 and 1k1791, respectively, as reported by Nishida et al. 31 Subsequently, mAb‐producing CHO cells were established by using the parental DG44 cell line (Invitrogen, Carlsbad, CA, USA). The molecular weights of the purified mAbs were verified by means of micrOTOF‐Q (Bruker Daltonics, Bremen, Germany). 2B8 was kindly given to us by Zenyaku Kogyo (Tokyo, Japan) as a gift. 2H7 and rituximab were purchased from Medical & Biological Laboratories (Nagoya, Japan) and Roche (Basel, Switzerland), respectively. 2F2, which is also called ofatumumab, 22 was produced using recombinant CHO cells according to the published amino acid sequences. 22
Affinity measurements. RAJI cells were washed with phosphate‐buffered saline (PBS) and resuspended at 5 × 105 cells in 1% bovine serum albumin (BSA) containing PBS, followed by incubation with various concentrations (1.3–20 nM) of anti‐CD20 mAbs at room temperature for 1 h. Unbound Abs were removed by centrifugation. Cells to which anti‐CD20 mAbs were bound on the surface were incubated with an appropriate amount of fluorescein isothiocyanate (FITC)‐conjugated goat antimouse IgG H + L (Chemicon International, Temecula, CA, USA) or FITC‐conjugated goat antihuman IgG Fcγ (Beckman Coulter, Fullerton, CA, USA) for 1 h. After incubation in the dark, the cells were washed and resuspended in PBS. The FITC signal was acquired at 532 nM on a Typhoon 9210 (GE Healthcare, Backinghamshire, UK) and analyzed using Image Quant (GE Healthcare). The fluorescent intensities of different concentrations of FITC‐conjugated secondary Abs in PBS were used to obtain the calibration curve for determining fluorescence intensity versus FITC‐conjugated Ab content. The amounts of bound secondary Abs in each sample were then calculated from the calibration curve. Previously‐reported apparent dissociation constants (K d) of 2B8 and rituximab were employed to estimate the binding ratio between the primary and secondary Abs. 16 The K d was determined by Scatchard plot analysis.
Epitope determination. Recombinant CHO cells stably expressing yellow fluorescent protein (YFP)‐fused wild‐type or mutant CD20 were produced. The variant CD20 contains the amino acid substitutions Ala170Ser/Pro172Ser. The cells were incubated with murine anti‐CD20 mAbs in saturated conditions for 20 min at 4°C. The samples were washed twice and resuspended in 1% BSA containing PBS, followed by further incubation with a spectral red (SPRD)‐conjugated goat antimouse secondary Ab (Beckman Coulter) for 20 min at 4°C. The samples were then washed twice, resuspended in PBS, and plated. YFP and SPRD signals were then detected at 532 nM and 670 nM, respectively, on a Typhoon 9210 and analyzed using Image Quant. The SPRD‐to‐YFP signal ratio was calculated as the binding ratio (SPRD/YFP). Eight kinds of HEK293 T cells that transiently express wild‐type or mutant CD20 were used. The cells were harvested 48 h after transfection and washed with 2 mM EDTA‐containing PBS. Cells were resuspended in 1% BSA containing PBS and incubated with 5 μg/mL (saturated conditions) of rituximab, 2F2, or humanized anti‐CD20 mAbs for 30 min at 4°C. The FITC‐conjugated goat antihuman Fcγ secondary Ab (Beckman Coulter) was used as a secondary Ab. The detection of fluorescent signals was carried out by flow cytometry using EPICS ALTRA (Beckman Coulter).
Characterization of cell lines. The cell surface expression of CD20, CD46, CD55, and CD59 on B‐lymphoma cell lines was analyzed using QIFIKIT (DAKO Cytomation, Glostrup, Denmark), according to the manufacturer’s instructions. The cell surface expression of antigens was calculated as Ab‐binding capacity (sites/cell), according to calibration beads.
Ab‐induced apoptosis. B‐lymphoma cells (2.5 × 105) were washed and resuspended in a culture medium, followed by incubation with a final concentration of 5 μg/mL anti‐CD20 mAbs or control anti‐CD3 mAbs (IMMUNOTECH, Praha, Czech Republic), at 37°C in 5% CO2. After 1.5 h of incubation in the presence or absence of a secondary Ab, unbound Abs were removed by centrifugation. The samples were further incubated for 3 h in the presence of culture medium. Apoptotic activity was then measured with a MEBCYTO apoptosis kit (Medical & Biological Laboratories) according to the manufacturer’s instructions and flow cytometric analysis using EPICS ALTRA.
ADCC. Mononuclear cells separated from the peripheral blood of healthy volunteers by Ficoll–Hypaque centrifugation were used as effector cells. Viable B‐lymphoma cells were labeled with calcein‐AM (Sigma‐Aldrich, St Louis, MO, USA) for 15 min, then washed and resuspended in culture medium. Various concentrations of mAbs were added to obtain different ratios of effector cells to target cells. After 4 h of incubation at 37°C in 5% CO2, released calcein fluorescence was extinguished by FluoroQuench Stain‐Quench Reagent (One Lambda, Canoga Park, CA, USA) and then the preserved calcein fluorescence in the viable cells was detected with FluorImager 595 (GE Healthcare). For maximal and spontaneous lysis, 5% TritonX‐100 and culture medium were used, respectively, instead of the mAbs. Specific ADCC was calculated according to the following equation: ADCC (%) = ([spontaneous lysis − experimental lysis]/[spontaneous lysis − maximal lysis]) × 100.
CDC. Anti‐CD20 mAbs at various concentrations were added to B‐lymphoma cells. As a source of complement, normal human serum prepared from healthy volunteers was added to the samples to 20% v/v. For the negative control, heat‐inactivated (56°C for 30 min) serum was used. After 2 h at 37°C, the samples were stained with propidium iodide (PI) and then specific CDC activity was determined from the percentage of PI‐positive cells by using FACScan analysis (Becton Dickinson, Franklin Lakes, NJ, USA).
CDC assay for tumor cells of human CD20(+) leukemia and lymphoma patients. Tumor cells from one case each of chronic lymphocytic leukemia (CLL), extranodal marginal zone lymphoma mucosa‐associated lymphoid tissues (MALT), and DLBCL were subjected to CDC analysis. Two cases of mantle cell lymphoma (MCL) were also assayed. All tumors were confirmed as CD20(+) by flow cytometry. Frozen cells were thawed, and viable cells were separated by using Ficoll centrifugation at 900g for 3 min. Viable cells were cultured for 2 h at 37°C in RPMI‐1640 supplemented with 10% fetal calf serum in the presence of 10 μg/mL anti‐CD20 mAbs and 20% human serum. The cells were then washed, allowed to react with FITC‐conjugated anti‐CD19 (BD Biosciences, Pharmingen, San Jose, CA, USA) for 15 min, and stained with PI. Through the two‐color flow cytometric analysis using FACScan (Becton Dickinson, USA), the percentage of impaired tumor cells was estimated based on the proportion of PI(+) CD19(+) cells to total PI (±) CD19(+) cells. The CDC activity of each Ab was defined as the difference of the values between the presence or absence of the Ab.
Quantification of C1q. Relative amounts of C1q molecules bound to anti‐CD20 mAbs were determined using FITC‐conjugated polyclonal anti‐C1q Abs, as previously described. 32 For the quantification of relative amounts of anti‐CD20 mAbs bound to cell‐surface CD20 at 5 μg/mL incubation, FITC‐conjugated goat antihuman IgG Fcγ (Beckman Coulter) was used.
Results
K d of murine anti‐CD20 mAbs. In the present study, we developed a novel method for the estimation of K d values of all mAbs by employing fluorescently‐labeled Abs, which enabled us to rapidly obtain precise and quantitative affinity information for mAbs against antigens on the cell surface. The resulting K d values of eight murine anti‐CD20 mAbs show a range of K d values from 0.77 nM to 6.59 nM, while K d values of 2B8 and 2H7 were 3.88 nM and 1.30 nM, respectively (Table 1). The eight murine anti‐CD20 mAbs in this study were named OUBM1 to OUBM8, according to the order of obtained affinity.
Table 1.
Apparent dissociation constant (K d ) values of murine anti‐CD20 monoclonal antibodies (mAbs)
| Clone group | Clone( 30 ) | K d (nM) | Group |
|---|---|---|---|
| 2B8 | 3.88 ± 0.61 | B | |
| 2H7 | 1.30 ± 0.20 | A | |
| OUBM1 | 1k1402 | 0.77 ± 0.21 | A |
| OUBM2 | 1k1228 | 1.15 ± 0.10 | A |
| OUBM3 | 1k1782 | 1.20 ± 0.23 | A |
| OUBM4 | 1k1712 | 1.34 ± 0.16 | A |
| OUBM5 | 1k1736 | 1.46 ± 0.08 | A |
| OUBM6 | 1k1791 | 3.27 ± 0.72 | B |
| OUBM7 | 1k0924 | 3.69 ± 0.63 | B |
| OUBM8 | 1k1422 | 6.59 ± 0.36 | B |
Data represent mean values of three samples in separate two experiments, Anti‐CD20 mAbs in Group A have relatively high affinities with no apoptotic activities, while those in Group B have relatively low affinities and apoptotic activities. Anti‐CD20 mAbs in Group A can induce apoptosis only in the presence of secondary antibodies.
Epitope analysis. We further investigated the epitope specificities of anti‐CD20 mAbs. Alanine 170 and proline 172 in the amino acid sequence of human CD20 are critical to the epitope recognized by anti‐CD20 mAbs, such as rituximab, B1, 2H7, and 1F5. 32 , 33 Murine CD20 has serine at positions 170 and 172 (Fig. 1a). We then examined the binding ability of anti‐CD20 mAbs using CHO cells stably expressing the wild‐type or mutant human CD20, in which amino acids 170 and 172 were both replaced with serine. Since the expression levels of recombinant YFP‐fused CD20 vary among cells, the amount of mAbs bound were evaluated after dividing the signal intensities of the bound mAbs by those of YFP. Figure 1(b) shows the binding ability of each mAb. When the mutant CD20 with Ala170Ser and Pro172Ser was used, the binding ability of all anti‐CD20 mAbs shown, other than OUBM3 and OUBM6, was dramatically reduced. These results indicate that these two mAbs recognize the epitope differently from the other anti‐CD20 mAbs shown, including 2B8 and 2H7. We further verified the responsible amino acids in the epitopes of humanized anti‐CD20 mAbs. The epitope of hOUBM6 was in a CD20 region comprising Glu‐Ser (ES), Arg‐Ala‐His‐Thr (RAHT), and Ile‐Asn‐Ile‐Tyr‐Asn (INIYN), which is different from the epitope of ofatumumab (Fig. 1c).
Figure 1.
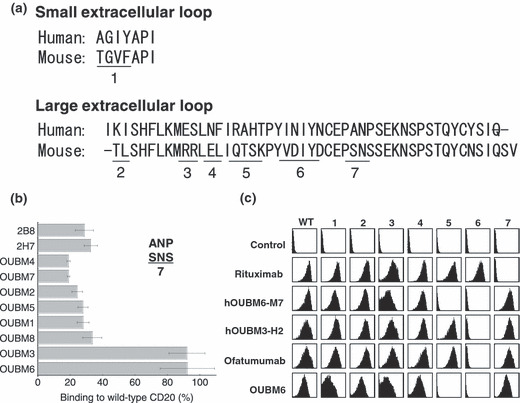
Epitope analysis for murine and humanized anti‐CD20 monoclonal antibodies (mAbs). (a) Sequence comparison between human and murine CD20 extracellular regions. (b) For the first epitope analysis of murine mAbs, Chinese Hamster Ovary (CHO) cells expressing wild‐type or mutant (region 7, A170S/P172S) human CD20 fused with yellow fluorescent protein (YFP) were incubated with murine anti‐CD20 mAbs (5 μg/mL), and the amount of bound mAbs was detected with spectral red (SPRD)‐conjugated secondary antibodies (Abs). SPRD‐to‐YFP signal ratio was taken as the binding ratio, and the percentages of binding ratios for mutant CD20 compared with wild‐type CD20 are shown. Data represent mean values of three separate experiments, and error bars indicate ±SD. (c) For the detailed epitope analysis of humanized mAbs, seven constructs with mutations in the extracellular regions of human CD20 were generated based on the results of the amino acid sequence comparison. HEK293 T cells transiently expressing wild‐type and mutant CD20 were incubated with anti‐CD20 mAbs. Bound anti‐CD20 mAbs were detected using fluorescein isothiocyanate‐conjugated secondary Ab. Data shown here are representative of at least two separate experiments.
Characterization of cell lines. The expression levels of cell surface CD20 of RAJI, WiL2‐NS, SU‐DHL4, and RC‐K8 cells derived from the BL, lymphoblastoid, FL, and DLBCL cell lines, respectively, were quantitatively measured. We employed RC‐K8, because this B‐cell lymphoma with t(4;11)(q21;q23) 34 is a cell line that does not get depleted by rituximab in vitro effectively, 12 although the correspondence of in vitro resistance to rituximab observed to the in vivo resistance has not well clarified. SU‐DHL4 had the highest Ab‐binding capacity with 8 × 105 sites/cell, while WiL2‐NS and RAJI cells exhibited low Ab‐binding capacity with approximately 2 × 105 sites/cell, and RC‐K8 had a value of 6 × 105 sites/cell, which is an intermediate value. Cell surface expression levels of the complement regulatory proteins CD46, CD55, and CD59 were then examined. The most remarkable difference between the four cell lines was the expression level of CD59. RC‐K8 expressed CD59 molecules with 3 × 105 sites/cell, which was more than three times the expression rate of the other cell lines (RAJI: 0.5 × 105 sites/cell; WiL2‐NS: 0.9 × 105 sites/cell; SU‐DHL4: 1.3 × 105 sites/cell).
Apoptotic induction by murine anti‐CD20 mAbs is determined by K d. Early apoptotic events were defined as the FITC‐positive and PI‐negative region, the proportion of which indicates the apoptotic induction ratio, as previously reported. 12 Apoptotic activity was dose dependent and caspase independent (data not shown). The percentages of SU‐DHL4 cells in which apoptosis was induced by the anti‐CD20 mAbs are shown in Figure 2(a,b), according to the K d value of each anti‐CD20 mAb. The relationship between the apoptotic activity and the K d value enabled us to classify the anti‐CD20 mAbs into two groups as follows. Group A was defined as the mAb group with relatively low K d values and no apoptosis as in the negative control, anti‐CD3 mAb. Group A included OUBM1, OUBM2, OUBM3, 2H7, OUBM4, and OUBM5. Group B was defined as the mAb group with greater K d values than mAbs in Group A and with apoptotic induction ratios similar to 2B8. Group B included OUBM6, OUBM7, OUBM8, and 2B8. This classification is further supported by the fact that the mAbs in Group A were able to induce apoptosis only in the presence of a secondary Ab, while the mAbs in group B induced apoptosis themselves and their activity was not enhanced by the addition of a secondary Ab (Fig. 2b). This classification can also be applied to other lymphoma cells, including the Burkitt’s cell line, RAJI, and the DLBCL cell line, RC‐K8, as confirmed in the FL cell line SU‐DHL4 (data not shown). These data suggest that apoptotic induction in B cells upon murine anti‐CD20 mAb binding is most likely to be determined by Ab affinity to CD20.
Figure 2.
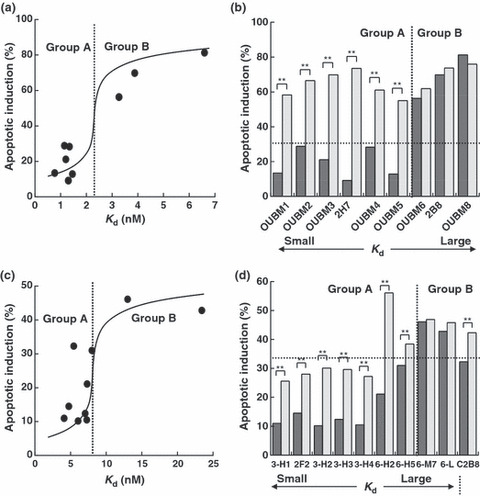
Classification of anti‐CD20 monoclonal antibodies (mAbs) into two groups according to the relationship between apparent dissociation constants (K d) values and apoptotic induction activity. Percentages of early apoptosis (fluorescein isothiocyanate‐positive and propidium iodide‐negative region) are depicted according to K d values. SU‐DHL4 cells were incubated with 5 μg/mL murine anti‐CD20 mAbs or with 5 μg/mL humanized anti‐CD20 mAbs in the absence (black bar) or presence (gray bar) of secondary antibodies (Abs), antimouse Fc, or antihuman Fc. Each value represents the mean value of three samples. Results of murine anti‐CD20 mAbs (a,b) and the results of humanized anti‐CD20 mAbs (c,d) are shown. (d), 3‐H1, 2F2, C2B8, 3‐H2, 3‐H3, 3‐H4, 6‐H2, 6‐H5, 6‐M7, and 6‐L correspond to OUBM3‐H1, ofatumumab, rituximab, OUBM3‐H2, OUBM3‐H3, OUBM3‐H4, OUBM6‐H2, OUBM6‐H5, OUBM6‐M7, and OUBM6‐L, respectively. **P < 0.01.
K d of humanized anti‐CD20 mAbs. Among the murine anti‐CD20 mAbs, OUBM3 and OUBM6 from Group A and Group B, respectively, with an epitope different from that of 2B8 were humanized with a human IgG1κ framework. The K d values of the resulting 16 humanized anti‐CD20 mAbs ranged from 7.08 nM to 23.42 nM in hOUBM6 (Table 2). The K d values of rituximab and ofatumumab were 5.45 nM and 4.76 nM, respectively. Four hOUBM6 (hOUBM6‐H2, hOUBM6‐H5, hOUBM6‐M7, and hOUBM6‐L) were chosen on the basis of their K d values and were subjected to further biological assays. Likewise, four representative hOUBM3 mAbs (hOUBM3‐H1, ‐H2, ‐H3, ‐H4) with K d values ranging from 4.10 nM to 7.27 nM were prepared for biological assays. The selected humanized mAbs had the same type of epitope as the parental murine anti‐CD20 mAbs (Fig. 1c).
Table 2.
Apparent dissociation constant (K d ) values of humanized anti‐CD20 monoclonal antibodies (mAbs)
| mAb | Affinity | Clone | K d (nM) | Group |
|---|---|---|---|---|
| Rituximab | High | 5.45 ± 0.85 | B | |
| hOUBM6 | High | H1 | 7.08 ± 1.67 | A |
| H2 | 7.33 ± 0.52 | A | ||
| H3 | 7.34 ± 0.91 | A | ||
| H4 | 7.92 ± 1.20 | A | ||
| H5 | 8.00 ± 0.91 | A | ||
| H6 | 9.17 ± 0.44 | A | ||
| H7 | 9.56 ± 0.96 | A | ||
| Middle | M1 | 10.09 ± 1.04 | B | |
| M2 | 10.42 ± 1.82 | B | ||
| M3 | 10.46 ± 1.72 | B | ||
| M4 | 11.24 ± 2.09 | B | ||
| M5 | 11.45 ± 1.82 | B | ||
| M6 | 11.76 ± 0.60 | B | ||
| M7 | 13.01 ± 1.65 | B | ||
| M8 | 14.91 ± 1.58 | B | ||
| Low | L | 23.42 ± 1.89 | B | |
| hOUBM3 | High | H1 | 4.10 ± 0.92 | A |
| H2 | 6.06 ± 0.87 | A | ||
| H3 | 7.05 ± 1.16 | A | ||
| H4 | 7.27 ± 1.59 | A | ||
| Ofatumumab | High | 4.76 ± 0.99 | A |
Data represent mean values of three samples in two independent experiments. Anti‐CD20 mAbs in Group A have relatively high affinities with no apoptotic activities, while those in Group B have relatively low affinities and apoptotic activities. Anti‐CD20 mAbs in Group A can induce apoptosis only in the presence of secondary antibodies.
Humanized mAbs exerted effective CDC against lymphomas that are less effectively depleted by rixuximab. CDC activities mediated by the humanized mAbs, hOUBM3 and hOUBM6, which have a different epitope from 2B8, were examined. Unlike the apoptotic induction assay that counts PI‐negative and Annexin V‐positive cells, the CDC assay counts only PI‐positive cells. The number of PI‐positive cells in the complement‐inactivated condition was small (data not shown). However, we subtracted this number from the total number of PI‐positive cells to exclusively estimate the CDC activity mediated by humanized mAbs. Both hOUBM6‐H2 and hOUBM6‐M7 exerted a CDC effect that was comparable to or better than the effect of rituximab against RAJI (Fig. 3a) and SU‐DHL4 (Fig. 3b); the effect was dose dependent. Ofatumumab also showed high CDC activity against RAJI and SU‐DHL4. It is noteworthy that hOUBM6 had highly‐effective CDC, even against RC‐K8 (Fig. 3c) and Karpas422 (not shown), which are DLBCL‐derived cell lines and are less effectively depleted by rituximab. hOUBM3‐H2 also showed slightly higher CDC activity than rituximab against RC‐K8 (Fig. 3c). The CDC activity of other hOUBM3 was similar (data not shown). The resistance of RC‐K8 to rituximab was not attributed to the degree of the CD20 expression level, because RC‐K8 showed more cell surface CD20 expression than RAJI, which is effectively lysed by anti‐CD20 mAbs with a humanized Fc portion. The CDC activities of the humanized anti‐CD20 mAbs, including hOUBM6‐H2, hOUBM6‐M7, and hOUBM3‐H2, were further assessed by using clinical samples of CD20(+) B‐cell lymphoma or leukemia. As shown in Figure 3(d), the in vitro effect of these humanized anti‐CD20 mAbs was better than that of rituximab.
Figure 3.
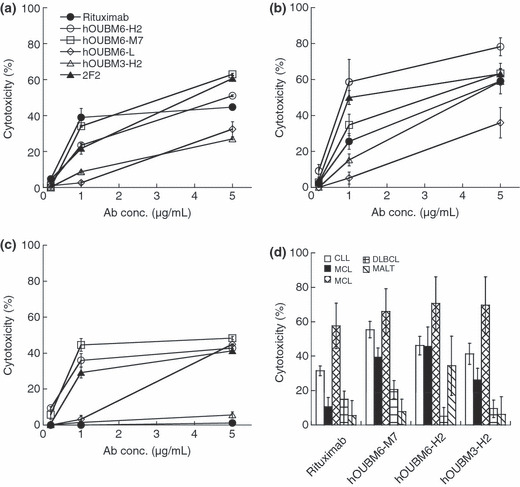
Complement‐dependent cytotoxicity (CDC) induced by humanized anti‐CD20 monoclonal antibodies (mAbs). RAJI (a), SU‐DHL4 (b), RC‐K8 cells (c), and tumor cells of CD20(+) leukemia and lymphoma patients (d) were incubated with normal human serum at 20% v/v for 2 h at 37°C. In the experiments performed using tumor cells of CD20(+) leukemia and lymphoma patients, mAb concentrations were fixed at 10 μg/mL. Lysed cells were detected by propidium iodide (PI) staining (a–c) or by double staining by PI and fluorescein isothiocyanate‐conjugated anti‐CD19. Data points represent the mean values of three samples. CLL, chronic lymphocytic leukemia; MALT, marginal zone lymphoma mucosa‐associated lymphoid tissues; DLBCL, diffuse large B‐cell lymphoma; MCL, mantle cell lymphoma (MCL).
In order to examine marked differences among anti‐CD20 mAbs in mediating CDC against RC‐K8, the capability of each mAb to recruit C1q was quantified. The variation in C1q recruitment had no significant correlation between the amounts of C1q molecules bound to anti‐CD20 mAbs on the cell surface and observed CDC activity (Fig. 4a). These results indicate that the differences observed in C1q levels do not originate from differences in the amount of anti‐CD20 mAb molecules bound to CD20 on the cell surface (Fig. 4b).
Figure 4.
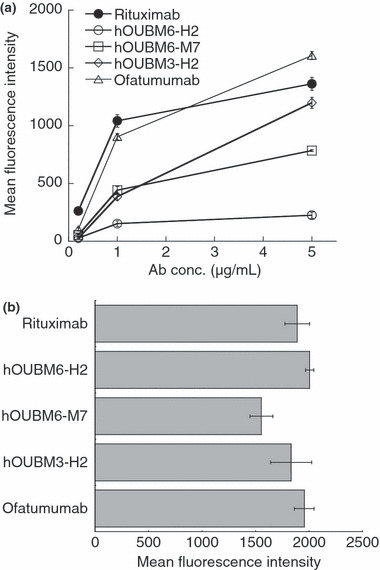
Quantification of C1q molecules bound to immunocomplexes. (a) RC‐K8 cells were incubated with humanized anti‐CD20 monoclonal antibodies (mAbs) at various concentrations for 15 min, followed by the addition of normal human serum to 1% v/v. Relative amounts of bound C1q molecules were determined using fluorescein isothiocyanate (FITC)‐conjugated anti‐C1q polyclonal antibodies (Abs). Data points represent mean values ± SD of three samples. (b) RC‐K8 cells were incubated with 5 μg/mL humanized anti‐CD20 mAbs for 15 min. Relative amounts of bound humanized anti‐CD20 mAbs were determined using FITC‐conjugated secondary Abs. Data points represent mean values ± SD of three samples. Ab, antibody concentration.
Cytotoxicity of humanized mAbs is mediated by effector cells. hOUBM3 and hOUBM6 that mediate effective CDC were subjected to biological assays in the presence of effector cells, such as monocytes and natural killer cells. We confirmed that little activity occurred in the absence of mAbs. Figure 5(a) shows cytotoxic activity with increasing mAb concentrations at the effector‐to‐target (E/T) ratio of 25. Activity increased in a dose‐dependent manner, but became saturated at concentrations at approximately 0.1 μg/mL, which is much lower than what is required for apoptotic induction and CDC activity. These results are consistent with the case of rituximab reported previously. 35 Humanized anti‐CD20 mAbs induced effective ADCC with a strength that was higher than (in the case of hOUBM6) or comparable to (in the case of hOUBM3) rituximab and ofatumumab. Increasing E/T ratios induced enhanced ADCC activity, indicating that the cell lysis we observed was actually mediated by the effector cells (Fig. 5b).
Figure 5.
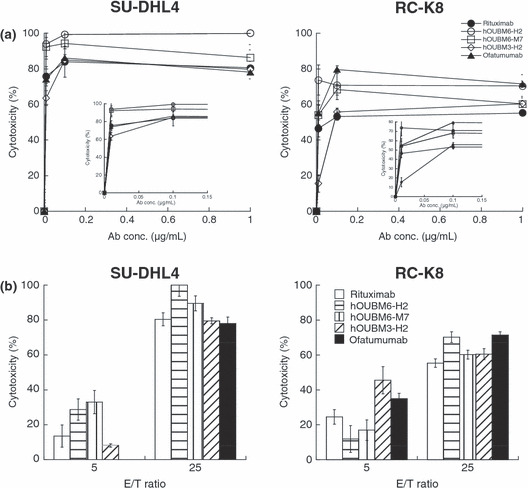
Antibody‐dependent, cell‐mediated cytotoxicity (ADCC) induced by humanized anti‐CD20 monoclonal antibodies (mAbs). Calcein‐labeled cells were incubated with humanized anti‐CD20 mAbs and effector cells for 4 h at 37°C. Preserved calcein fluorescence in viable cells was detected, and cytotoxicity was calculated according to the following equation: ADCC (%) = ([spontaneous lysis − experimental lysis]/[spontaneous lysis − maximal lysis]) × 100. Data points represent the mean values±SD of three samples showing dose‐dependent cytotoxicity (a) and effector‐to‐target (E/T) ratio‐dependent cytotoxity (b) for SU‐DHL4 (left panels) or RC‐K8 cells (right panels). (a,b) Inverted figure is the enlarged version of the concentration region below the 0.15 μg/mL regions. Data points represent the mean values of three samples.
Apoptotic induction by generated humanized mAbs also evaluated by K d . The capability of hOUBM3 and hOUBM6 mAbs to cause apoptotic cell death was investigated. The correlation between K d and apoptotic capability, as observed for murine anti‐CD20 mAbs, was again confirmed in experiments that used SU‐DHL4 (Fig. 3c,d) and the other three cell lines (data not shown). hOUBM6 can also be classified into two groups, A and B, according to their K d values. hOUBM6 had lower K d values and showed no induction of apoptosis (Group A), while hOUBM6 had greater K d values and showed apoptotic activity (Group B). All hOUBM3 had K d values comparable to or lower than the K d values of hOUBM6, which belongs to Group A. As expected, hOUBM3 showed no apoptotic induction and were therefore classified in Group A. The human mAb, ofatumumab, is also classified in Group A. The mAbs in Group A were able to induce apoptosis only in the presence of a secondary Ab (Fig. 3d).
Moreover, for humanized anti‐CD20 mAbs, the relationship between apoptotic induction activity and the K d value held for all tested cell lines that had different CD20 levels on the cell surface. Taking all the experimental facts together, apoptotic induction by anti‐CD20 mAbs appears to be determined simply by Ab‐CD20 affinity, and thus only anti‐CD20 mAbs with an adequate K d can induce apoptosis in B cells.
Discussion
In the current study, we demonstrated a rational strategy for developing effective therapeutic mAbs. A series of murine and humanized anti‐CD20 Abs were characterized based on K d constants, epitopes, and biological activity. The fact that K d clearly correlated with apoptotic activity in both murine and humanized mAbs allowed us to categorize the mAbs into two groups based on K d values (Groups A and B), demonstrating that K d can be employed as an essential parameter to guide mAb development when the K d is adequately and quantitatively measured. However, CDC assays of hOUBM6 and ofatumumab showed that their high CDC activity is effective not only against CD59‐negative cells, but also positive cells like RC‐K8 that are normally less effectively depleted by rituximab. These Abs have epitopes that are different from 2B8 and are thus classified as non‐2B8 types, suggesting that CDC activity depends on epitope type.
Thus, as summarized in Figure 6, anti‐CD20 Abs are classified into four different categories according to two intrinsic parameters: K d and epitope. Anti‐CD20 mAbs with greater K d values (Group B) can induce apoptosis; mAbs with a non‐2B8‐type epitope potentially induce effective CDC, even against CD59‐abundant cell lines. It should be noted that mAbs with K d values much greater than the mAbs examined in this study should be excluded from this classification, because they have limited capability to interact with CD20.
Figure 6.
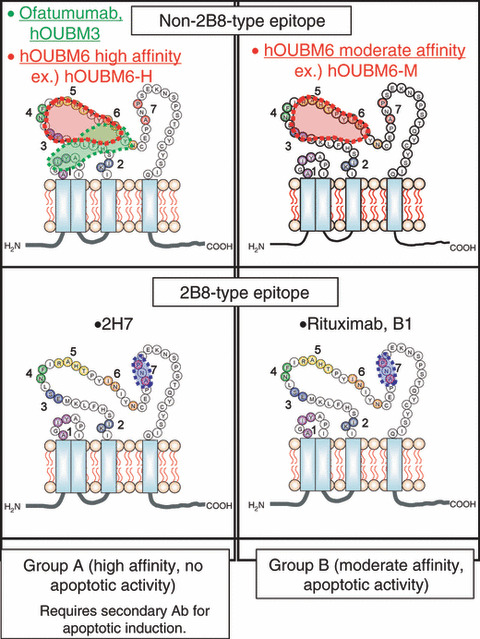
Classification of anti‐CD20 monoclonal antibodies (mAbs). Representative murine anti‐CD20 mAbs and newly‐developed humanized anti‐CD20 mAbs are classified on the basis of affinity and epitope type. Biological activity of the monoclonal antibodies (Abs) vary with region. B1 and 2H7 are murine immunoglobulin G .
It has been generally accepted that Abs with higher affinities exert a greater target cell‐depletion effect. However, our study indicates that both murine and humanized anti‐CD20 mAbs with moderate affinities (those classified in Group B) only induce apoptosis of B cells. Additionally, no apoptotic induction was confirmed in the case of Fab versions for any of the anti‐CD20 mAbs used in the present study (data not shown), and CD20 has been shown to be present as a dimer on the B‐cell surface. 2 Accordingly, it could be proposed that anti‐CD20 mAbs with moderate affinity bind to two CD20 dimers simultaneously, each via one of the two arms, which brings the two CD20 dimers into close proximity of each other, resulting in the triggering of the apoptotic induction signal by undefined factors. Conversely, anti‐CD20 mAbs in Group A (higher affinity) would bind to CD20 via one of the two arms, but not induce apoptosis of B cells. Consistently, cross‐linking by using a secondary Ab with Group A anti‐CD20 mAbs resulted in the induction of apoptosis (Fig. 2a,b). Higher oligomerization of currently‐developed anti‐CD20 mAbs belonging to both groups might be more effective for antitumor activity, as with a rituximab homodimer. 30 , 31 Thus, the regulation of apoptosis by Ab affinity is the first insight into the essential factor for apoptotic induction upon Ab binding.
Regarding the intracellular signaling that occurs upon rituximab binding, several pathways have been considered. Hofmeister et al. 13 demonstrated caspase 3‐dependent apoptosis, while Chan et al. 14 showed a caspase‐independent pathway; Mathas et al. 11 suggested a partly shared pathway with B‐cell receptor‐mediated apoptosis. Recently, Suzuki et al. 36 showed that rituximab inhibits the PI3K–Akt pathway in B‐NHL cell lines. In the present study, we confirmed that the induction of apoptosis by humanized anti‐CD20 mAbs is caspase independent. Furthermore, we observed neither the disappearance of mitochondrial potential nor chromatin digestion after apoptotic induction with the humanized anti‐CD20 that we developed (data not shown).
CDC is considered to be the primary cell‐killing mechanism of rituximab. Under our experimental conditions, hOUBM6 had a more effective CDC than rituximab against RAJI and SU‐DHL4, except for hOUBM6‐L, which was less potent because of its low affinity. Ofatumumab also had highly‐potent CDC, which is consistent with a previous report. 33 Rituximab could not mediate detectable CDC against RC‐K8, in contrast to hOUBM6‐H2, hOUBM6‐M7, and ofatumumab, which showed effective CDC. Furthermore, significant depletion by hOUBM6‐M7, hOUBM6‐H2, and hOUBM3‐H2 was observed in the tumor cells of CD20(+) leukemia and lymphoma patients with DLBCL, MALT, MCL, and CLL, suggesting a potential clinical use for this novel Ab. However, recent studies indicate that the efficacy of Ab therapy might be different for each patient, even for the same disease. 29 , 37 , 38 , 39 Our result shows less‐effective depletion by hOUBM6‐M7 for a DLBCL case and highly‐effective depletion for a CLL case (Fig. 3d) may correspond to these reports. Thus, further investigations into currently‐developed mAbs are required, especially in terms of the effectiveness of these mAbs against not only different types of lymphomas, but also in the same types of lymphoma from different patients.
It has been suggested that the potent CDC of ofatumumab is due to the membrane proximal epitope located around the small extracellular loop of CD20 and the high capacity for C1q recruitment. 40 , 41 However, in our experiments, ofatumumab and rituximab recruited more C1q molecules than hOUBM6‐H2 and hOUBM6‐M7, which indicates that C1q recruitment is not the primary reason for the effective CDC of these compounds. This difference in the number of bound C1q molecules could be attributed to the conformation of each anti‐CD20 mAb on the cell surface. The translocation of CD20 to the detergent‐resistant compartments called lipid rafts after Ab ligation is reported to be necessary for effective CDC. 42 The mobility of CD20 to lipid rafts is considered to be epitope dependent. 43 Furthermore, the mobility of CD20 to lipid rafts after Ab binding is correlated with the distance between the epitope and the plasma membrane. 44 Golay et al. reported a positive relationship between in vitro CDC susceptibility to rituximab and the expression levels of CD20 on the cell surface; 45 however, no such relationship was found in the present study. Golay et al. and other groups have further reported an increase in cytotoxicity by the addition of blocking Abs against the complement regulatory proteins, CD55 and CD59, or siRNA with CD55, implicating the regulation of CDC by CD55 and CD59. 46 , 47 However, Weng et al. 48 suggested that there was no correlation between CDC activity and the expression level of CD20 or the complement regulatory proteins CD46, CD55, and CD59. Our experiments indicate that the rituximab resistance of RC‐K8 to CDC is not due to CD20 levels, but to highly‐expressed CD59 molecules. Our study also indicates the effectiveness of hOUBM6, which have a different epitope than rituximab. Together, these may point to a relationship between epitope and complement regulatory proteins, and in particular, CD59, which is abundant in lipid rafts. A distinct Ab epitope leads to a change in CD20 mobility, which might be attributed to different susceptibility to or mode of interaction with CD59 molecules.
Our classification shown in Figure 6 is based on the physicochemical properties of the mAbs we studied and is different from the classification proposed by Glennie et al., 22 in which anti‐CD20 mAbs are classified into Types I and II, according to their molecular effects (ability of CD20 to translocate to the raft region) or biological effects (apoptotic induction and CDC activity). The relationships between Groups A and B and Types I and II will be clarified in the near future.
Our study indicates that the ability to induce apoptosis could be enhanced simply by controlling Ab affinity. Affinity regulation by structure‐based computer design could be useful for this purpose, 49 and the combination of recent Ab technologies 50 , 51 , 52 will provide highly effective Ab for therapeutic use.
Acknowledgments
This work was initially performed through a collaboration of Osaka University, Tottori University, Osaka City University, and BioMedics, and was supported in part by Special Coordination Funds from MEXT, Japan to KF. This work was supported in part by grants from the Uehara Memorial Foundation to SU. We wish to thank Dr Sadakazu Usuda of the Institute of Immunology for murine antibody production and Mr Michio Nishida for useful discussions in the early stages of our murine antibody research. We also thank Ms Hiroko Miyakoda for technical assistance. We wish to thank Professor Takachika Azuma of Tokyo Science University for critical discussions and insightful comments. We are also grateful to Dr Eduard Padlan for the design of the humanized antibody. We also thank Zenyaku Kogyo for providing Tetsuji Sawada with the 2B8 mAb.
References
- 1. Tedder TF, Engel P. CD20: a regulator of cell‐cycle progression of B lymphocytes. Immunol Today 1994; 15: 450–4. [DOI] [PubMed] [Google Scholar]
- 2. Bubien JK, Zhou LJ, Bell PD, Frizzel RA, Tedder TF. Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2 + conductance found constitutively in B lymphocytes. J Cell Biol 1993; 121: 1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li H, Ayer LM, Lytton J, Deans JP. Store‐operated cation entry mediated by CD20 in membrane rafts. J Biol Chem 2003; 278: 42427–34. [DOI] [PubMed] [Google Scholar]
- 4. Deans JP, Li H, Polyak MJ. CD20‐mediated apoptosis: signalling through lipid rafts. Immunology 2002; 107: 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deans JP, Schieven GL, Shu GL et al. Association of tyrosine and serine kinases with the B cell surface antigen CD20. Induction via CD20 of tyrosine phosphorylation and activation of phospholipase C‐γ1 and PLC phospholipase C‐γ2. J Immunol 1993; 151: 4494–504. [PubMed] [Google Scholar]
- 6. Pulczynski S, Boesen AM, Jensen OM. Modulation and intracellular transport of CD20 and CD21 antigens induced by B1 and B2 monoclonal antibodies in RAJI and JOK‐1 cells‐an immunofluorescence and immunoelectron microscopy study. Leuk Res 1994; 18: 541–52. [DOI] [PubMed] [Google Scholar]
- 7. Maloney DG, Liles TM, Czerwinski DK et al. Phase I clinical trial using escalating single‐dose infusion of chimeric anti‐CD20 monoclonal antibody (IDEC‐C2B8) in patients with recurrent B‐cell lymphoma. Blood 1994; 84: 2457–66. [PubMed] [Google Scholar]
- 8. Maloney DG, Press OW. Newer treatments for non‐Hodgkin’s lymphoma: monoclonal antibodies. Oncology 1998; 10: 63–76. [PubMed] [Google Scholar]
- 9. Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood 1998; 91: 1644–52. [PubMed] [Google Scholar]
- 10. Cardarelli PM, Quinn M, Buckman D et al. Binding to CD20 by anti‐B1 antibody or F(ab’)(2) is sufficient for induction of apoptosis in B‐cell lines. Cancer Immunol Immunother 2002; 51: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mathas S, Rickers A, Bommert K, Dörken B, Mapara MY. Anti‐CD20‐ and B‐cell receptor‐mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer Res 2002; 60: 7170–6. [PubMed] [Google Scholar]
- 12. Taji H, Kagami Y, Okada Y et al. Growth inhibition of CD20‐positive B lymphoma cell lines by IDEC‐C2B8 anti‐CD20 monoclonal antibody. Jpn J Cancer Res 1998; 89: 748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src‐family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3‐dependent apoptosis. Blood Cells Mol Dis 2000; 26: 133–43. [DOI] [PubMed] [Google Scholar]
- 14. Chan HT, Hughes D, French RR et al. CD20‐induced lymphoma cell death is independent of both caspases and its redistribution into triton X‐100 insoluble membrane rafts. Cancer Res 2003; 63: 5480–9. [PubMed] [Google Scholar]
- 15. Alas S, Bonavida B. Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B‐non‐Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down‐regulation of Bcl‐2 and sensitization to cytotoxic drugs. Cancer Res 2001; 61: 5137–44. [PubMed] [Google Scholar]
- 16. Reff ME, Carner K, Chambers KS et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994; 83: 435–45. [PubMed] [Google Scholar]
- 17. Manches O, Lui G, Chaperot L et al. In vitro mechanisms of action of rituximab on primary non‐Hodgkin lymphomas. Blood 2003; 101: 949–54. [DOI] [PubMed] [Google Scholar]
- 18. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 2000; 6: 443–6. [DOI] [PubMed] [Google Scholar]
- 19. Cartron G, Dacheux L, Salles G et al. Therapeutic activity of humanized anti‐CD20 monoclonal antibody and polymorphism in IgG Fc receptor Fcγ RIIIa gene. Blood 2002; 99: 754–8. [DOI] [PubMed] [Google Scholar]
- 20. Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP‐1 monocytes. J Immunol 2006; 176: 2600–9. [DOI] [PubMed] [Google Scholar]
- 21. Anderson DR, Grillo‐López A, Varns C, Chambers KS, Hanna N. Targeted anti‐cancer therapy using rituximab, a chimaeric anti‐CD20 antibody (IDEC‐C2B8) in the treatment of non‐Hodgkin’s B‐cell lymphoma. Biochem Soc Tans 1997; 25: 705–8. [DOI] [PubMed] [Google Scholar]
- 22. Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti‐CD20 monoclonal antibodies. Mol Immunol 2007; 44: 3823–37. [DOI] [PubMed] [Google Scholar]
- 23. McLaughlin P, Grillo‐Lopez AJ, Link BK et al. Rituximab chimeric anti‐CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four‐dose treatment program. J Clin Oncol 1998; 16: 2825–33. [DOI] [PubMed] [Google Scholar]
- 24. Czuczman MS. Treatment of patients with low‐grade B‐cell lymphoma with the combination of chimeric anti‐CD20 monoclonal antibody and CHOP chemotherapy. J Clin Oncol 1999; 17: 268–76. [DOI] [PubMed] [Google Scholar]
- 25. Coiffier B, Lepage E, Briere J et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large‐B‐cell lymphoma. N Engl J Med 2002; 346: 235–42. [DOI] [PubMed] [Google Scholar]
- 26. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 2003; 21: 3940–7. [DOI] [PubMed] [Google Scholar]
- 27. Racila E, Link BK, Weng WK et al. A polymorphism in the complement component C1qA correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin Cancer Res 2008; 14: 6697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Terui Y, Mishima Y, Sugimura N et al. Identification of CD20 C‐terminal deletion mutations associated with loss of CD20 expression in non‐Hodgkin’s lymphoma. Clin Cancer Res 2009; 15: 2523–30. [DOI] [PubMed] [Google Scholar]
- 29. Hiraga J, Tomita A, Sugimoto T et al. Down‐regulation of CD20 expression in B‐cell lymphoma cells after treatment with rituximab‐containing combination chemotherapies: its prevalence and clinical significance. Blood 2009; 113: 4885–93. [DOI] [PubMed] [Google Scholar]
- 30. Nishida M, Usuda S, Okabe M et al. Characterization of novel murine anti‐CD20 monoclonal antibodies and their comparison to 2B8 and c2B8 (rituximab). Int J Oncol 2007; 31: 29–40. [PubMed] [Google Scholar]
- 31. Nishida M, Teshigawara K, Niwa O et al. Novel humanized anti‐CD20 monoclonal antibodies with unique germline VH and VL gene recruitment and potent effector functions. Int J Oncol 2008; 32: 1263–74. [DOI] [PubMed] [Google Scholar]
- 32. Deans JP, Polyak MJ. FMC7 is an epitope of CD20. Blood 2008; 111: 2492. [DOI] [PubMed] [Google Scholar]
- 33. Polyak MJ, Deans JP. Alanine‐170 and proline‐172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood 2002; 99: 3256–62. [DOI] [PubMed] [Google Scholar]
- 34. Akao Y, Seto M, Takahashi T et al. Rearrangements on chromosome 11q23 in hematopoietic tumor‐associated t(11;14) and t(11;19) translocations. Cancer Res 1991; 51: 6708–11. [PubMed] [Google Scholar]
- 35. Hale G, Clark M, Waldmann H. Therapeutic potential of rat monoclonal antibodies: isotype specificity of antibody‐dependent cell‐mediated cytotoxicity with human lymphocytes. J Immunol 1985; 134: 3056–61. [PubMed] [Google Scholar]
- 36. Suzuki E, Umezawa K, Bonavida B. Rituximab inhibits the constitutively activated PI3K‐Akt pathway in B‐NHL cell lines: involvement in chemosensitization to drug‐induced apoptosis. Oncogene 2007; 26: 6184–93. [DOI] [PubMed] [Google Scholar]
- 37. Johnson NA, Savage KJ, Ludkovski O et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 2009; 114: 3533–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Czuczman MS, Olejniczak S, Gowda A et al. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down‐regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 2008; 14: 1561–70. [DOI] [PubMed] [Google Scholar]
- 39. Olejniczak SH, Hernandez‐Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin Cancer Res 2008; 14: 1550–60. [DOI] [PubMed] [Google Scholar]
- 40. Teeling JL, French RR, Cragg MS et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non‐Hodgkin lymphomas. Blood 2004; 104: 1793–800. [DOI] [PubMed] [Google Scholar]
- 41. Teeling JL, Mackus WJ, Wiegman LJ et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 2006; 177: 362–71. [DOI] [PubMed] [Google Scholar]
- 42. Cragg MS, Morgan SM, Chan HT et al. Complement‐mediated lysis by anti‐CD20 mAb correlates with segregation into lipid rafts. Blood 2003; 101: 1045–52. [DOI] [PubMed] [Google Scholar]
- 43. Li H, Ayer LM, Polyak MJ et al. The CD20 calcium channel is localized to microvilli and constitutively associated with membrane rafts: antibody binding increases the affinity of the association through an epitope‐dependent cross‐linking‐independent mechanism. J Biol Chem 2004; 279: 19893–901. [DOI] [PubMed] [Google Scholar]
- 44. Bindon CI, Hale G, Waldmann H. Importance of antigen specificity for complement‐mediated lysis by monoclonal antibodies. Eur J Immunol 1988; 18: 1507–14. [DOI] [PubMed] [Google Scholar]
- 45. Golay J, Lazzari M, Facchinetti V et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B‐cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood 2001; 98: 3383–9. [DOI] [PubMed] [Google Scholar]
- 46. Golay J, Zaffaroni L, Vaccari T et al. Biologic response of B lymphoma cells to anti‐CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement‐mediated cell lysis. Blood 2000; 95: 3900–8. [PubMed] [Google Scholar]
- 47. Terui Y, Sakurai T, Mishima Y et al. Blockade of bulky lymphoma‐associated CD55 expression by RNA interference overcomes resistance to complement‐dependent cytotoxicity with rituximab. Cancer Sci 2006; 97: 72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weng WK, Levy R. Expression of complement inhibitors CD46, CD55, and CD59 on tumor cells does not predict clinical outcome after rituximab treatment in follicular non‐Hodgkin lymphoma. Blood 2001; 98: 1352–7. [DOI] [PubMed] [Google Scholar]
- 49. Lippow SM, Wittrup KD, Tidor B. Computational design of antibody‐affinity improvement beyond in vivo maturation. Nat Biotechnol 2007; 25: 1171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dall’Acqua WF, Cook KE, Damschroder MM, Woods RM, Wu H. Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J Immunol 2006; 177: 1129–38. [DOI] [PubMed] [Google Scholar]
- 51. Niwa R, Natsume A, Uehara A et al. IgG subclass‐independent improvement of antibody‐dependent cellular cytotoxicity by fucose removal from Asn297‐linked oligosaccharides. J Immunol Methods 2005; 306: 151–60. [DOI] [PubMed] [Google Scholar]
- 52. Lazar GA, Dang W, Karki S et al. Engineered antibody Fc mutants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103: 4005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]