

Abstract
We have previously demonstrated that epigenetic silencing of occludin, a tight junction‐associated membrane protein, results in the acquisition of apoptotic resistance to various apoptogenic stimuli, causally contributing to the enhanced tumorigenicity of cancer cells. However, it remains to be examined whether occludin expression in transformed cells has an alternative impact that is important for cancer progression. Here we show that forced expression of occludin induces anoikis and promotes oxidative stress‐induced premature senescence in breast carcinoma cells, which is accompanied by upregulation of negative cell cycle regulators such as p16INK4A, p21Waf1/Cip1 and p27Kip1 but not p53. The senescent phenotype is reversed by specific inhibition of mitogen‐activated protein kinase. Endogenous reexpression of occludin mediated by a synergistic effect with a demethylator and histone deacetylase inhibitor or retinoids that stimulate retinoic acid receptor α is also sufficient for provoking the senescent phenotype. In addition, tumors that developed from occludin‐expressing cells in mice showed a feature of cellular senescence that has not been described as a consequence of occludin signaling. These findings suggest that the loss of occludin expression is at least partially involved in the senescence‐escape program during mammary tumorigenesis. (Cancer Sci 2007; 98: 1027–1034)
Four decades ago, the phenomenon of ‘replicative senescence’ was described as a process leading to irreversible arrest of cell division in which metabolically active cultured human fibroblasts lost the ability to divide upon continuous subculture.( 1 ) Although ‘replicative senescence’ is triggered by progressive telomere shortening, an indistinguishable phenotype, termed ‘premature senescence’, can be acutely produced by a number of cell stresses, including oxidative stress, DNA damage‐inducing agents, and enforced in vitro activated oncogenes.( 2 ) Senescent cells show characteristic changes in cell morphology such as a flattened and vacuole‐rich cytoplasmic shape, and biochemical characteristics such as elevated protein levels of cell cycle inhibitors, including p16 and p21, as well as the upregulation of senescence‐associated β‐galactosidase (SA‐β‐gal) activity and reciprocal negativity for the proliferation marker Ki67.( 3 , 4 , 5 ) Accumulated evidence has revealed that cellular senescence terminates carcinogenesis in a variety of cancer tissues in vitro and in premalignant lesions.( 5 ) However, the physiological relevance of acutely inducible premature senescence has remained controversial for years, in sharp contrast to the confirmed concepts that impaired or decreased responses to various apoptotic signals are associated with oncogenic transformation.( 6 , 7 ) Recent reports have now clarified that cellular senescence is an intrinsic cell stress‐responsive cell cycle exit program, which is regarded as an antioncogenic fail‐safe mechanism to prevent oncogenic activity in aged or potentially damaged cells, and a natural barrier to cancer progression.( 4 , 8 , 9 )
Tight junctions (TJs) are intercellular structures in epithelial and endothelial cells, primarily playing a critical role in cell–cell adhesion.( 10 , 11 ) Occludin was the first identified requisite integral protein for the TJ, but occludin‐deficient cells and animals have fully developed well‐organized TJs structurally.( 12 , 13 , 14 ) Thus it is now well accepted that the claudin family, which has been shown to contain more than 20 members, is the main constituent of the TJ, rather than occludin.( 11 ) We have also demonstrated that the methylator phenotype of occludin exhibits decreased cellular sensitivity to various differently triggered apoptogens, causally enhancing the tumorigenic properties of the transformed cells.( 15 ) In addition, a growing body of evidence has indicated that cancer progression is associated with decreased or impaired TJ phenotypes, and that the loss of TJ‐associated molecules such as occludin is generally correlated with tumor development in carcinogenesis.( 15 , 16 , 17 , 18 ) These findings suggest that occludin is not a simple static component of TJs but has functions in receiving and transmitting cell survival signals, eventually acting as a signal transmitter for the cells at TJs during the carcinogenic scenario.
Based on accumulating evidence concerning occludin, we investigated whether occludin expression in cancer cells had any alternative impact that was important for cancer progression. Here we demonstrate that the expression of occludin in transformed cells induces anoikis in three‐dimensional culture of breast carcinoma cells. In addition, the cellular response in the event of occludin expression shows features of premature senescence, accompanied by upregulation of negative cell cycle regulators such as p16INK4A, p21Waf1/Cip1 and p27Kip1 but not p53. Our observations support the idea that occludin expression in cancer cells is at least partially involved in a senescence‐promoting mechanism that may prevent abnormal cells from entering the carcinogenic program during mammary tumorigenesis.
Materials and Methods
Cell line and transfection. AC2M2 is a metastatic variant of murine breast cancer cell line SP1, and SP1 were isolated from a mammary intraductal adenocarcinoma that arose spontaneously in an 18‐month‐old CBA/J female retired breeder mouse.( 19 ) AC2M2 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Sanko Jyunyaku, Tokyo, Japan), 10 mM HEPES, 100 U/mL penicillin and 100 µg/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere.
Five µg of pcDNA3.1(–)‐occludin, which is a construct comprising full‐length murine occludin cDNA cloned into the response plasmid pcDNA3.1(–) (Invitrogen), was transfected into AC2M2 cells using FuGENE 6 reagent (Roche, Indianapolis, IN, USA). G418 (Sigma, Tokyo, Japan)‐resistant clones were expanded as a monoclonal population. Three weeks later, cell lines were selected to examine the strength of the expression of the introduced gene by quantitative reverse transcription–polymerase chain reaction (RT‐PCR) and western blot analysis. As our preliminary experiment showed that the cell phenotypes observed in the event of occludin overexpression were similar but not the same in at least three different independent clones, these cells were mixed equally to establish stably transfected cell lines and to avoid possible clonal variation. Small interfering RNA (siRNA) targeted to occludin or negative control siRNA (both from Santa Cruz Biotechnologies, Santa Cruz, CA, USA) was also transfected into the cells using FuGENE 6 reagent.
Cell death analyses. Cell death was induced by preventing cells from adhering to the plastic of cell culture dishes.( 15 ) In some experiments, 100 nM all‐trans retinoic acid (atRA) (Sigma) was added and maintained throughout the experiments. Fragmented DNA from dead cells was isolated using a method that preferentially extracts low molecular weight cellular DNA.( 15 ) Gel electrophoresis of the DNA samples was carried out on 2.5% agarose gels and ladder formation due to fragmentation was visualized with ethidium bromide under ultraviolet illumination.
Senescence‐associated β‐galactosidase staining. The cells were exposed to mild oxidative stress under 25 µM H2O2 for up to 5 days, and senescent cells were visualized by SA‐β‐gal staining.( 3 ) The tumors that developed in the mouse subcutaneous tissue were also subjected to this assay. The cells stained positively by the specified procedures were scored using light microscopy by counting the number of cells under low magnification (×100) in 10 separate arbitrarily selected fields in each section. In some experiments, atRA (100 nM), 4(5,6,7,8‐tetrahydro‐5,5,8, 8‐tetramethyl‐2‐naphtamido)benzoic acid (Am580, 10 nM), a mitogen‐activated protein kinase (MAPK) inhibitor (PD98059, 50 µM; Calbiochem‐Novabiochem, San Diego, CA, USA), p38 MAP kinase inhibitor (SB203580, 20 µM; Calbiochem‐Novabiochem) or phosphoinositide‐3‐kinase (PI3K) inhibitor (LY294002, 20 µM; Calbiochem‐Novabiochem) was added and maintained throughout the experiments. To examine the effect of a demethylating agent, the cells were treated with 5′‐aza‐2′deoxycytidine (5′Aza‐dC, 0–5 µM; Sigma) for 48 h, after which a histone deacetylase inhibitor (HDAI) or trichostatin A (TSA, 0–300 nM; Sigma) was added, followed by incubation for an additional 24 h at 37°C.
RT‐PCR and Southern blot analysis. Subsequent to each RT‐PCR analysis, to allow for semiquantitative analysis of signal intensity, we carried out Southern blot analysis using a probe with the full‐length cDNA of each gene.( 15 ) Ribosomal RNA was shown to use the same amounts of RNA for RT reactions. Triplicate independent PCR reactions were carried out to ensure the reproducibility of expression quantification. For the densitometric analysis, signals were quantitated using Scion Image 1.62 (Scion, Frederick, MD, USA). Oligonucleotide sequences of all primers used for the PCR reactions are available on request.
Western blot analysis. Whole‐cell lysates (20 µg) of denatured proteins were electrophoresed on 12–15% sodium dodecylsulfate–polyacrylamide gels and were electroblotted onto nitrocellulose filters. Filters were immunoblotted with antibodies against occludin (Zymed Laboratories, San Francisco, CA, USA), p16INK4A, p21Waf1/Cip1, p27Kip1 (all from Santa Cruz), p53 (Oncogene, San Diego, CA, USA) and β‐actin (Santa Cruz) proteins. The filters were then reacted with corresponding peroxidase‐labeled secondary antibodies. Finally, the immunoreactions were visualized using an enhanced chemiluminescence system (GE Healthcare, Piscataway, NJ, USA).
Terminal deoxynucleotidyl transferase‐meditated dUTP nick‐end labeling assay and immunohistochemistry. Cells cultured on collagen‐coated glass coverslips were subjected to the terminal deoxynucleotidyl transferase‐meditated dUTP nick‐end labeling (TUNEL) assay. Apoptotic cells were visualized using an In Situ Cell Death Detection Kit (Roche). We also carried out a staining procedure other than for TdT as a negative control. For immunohistochemistry, the cells on a coverslip were reacted with the primary antibody against Ki67 (Dako Cytomation, Tokyo, Japan). Cells positively stained by the specified procedures were scored using light microscopy by counting the number of cells under low magnification (×100) in 10 separate arbitrarily selected fields in each section. Staining for actin filaments was done with rhodamine‐phalloidin (1 unit/slide; Molecular Probes, Eugene, OR, USA). All samples were examined with a fluorescence microscope (Zeiss, Tokyo, Japan) or a laser‐scanning confocal microscope (Bio‐Rad, Hercules, CA, USA).
Cell cycle analysis by flow cytometry. Cells were treated with 25 µM H2O2 for 5 days prior to harvesting by centrifugation, and were permeabilized with ice‐cold 70% ethanol for 10 min. After washing with phosphate‐buffered saline (PBS), cells were treated with PBS containing 100 µg/mL DNase free–RNase A at 37°C for 30 min. After centrifugation, cells were suspended in PBS containing 1 µg/mL (4′,6‐diamidino‐2‐phenylindole)‐methanol for 15 min. DNA content was analyzed by FACScan (Becton Dickinson, Tokyo, Japan).
Tumor growth in vivo. We subcutaneously injected cells of AC2M2 and its transfectant (7.5 × 103 cells/mouse) into mammary fat pads of syngenic 6–7‐week‐old female mice (CBA/J; Charles River Japan, Yokohama, Japan).( 15 ) The handling of animals was carried out using protocols approved by the Sapporo Medical University Animal Care Facility.
Statistical analysis. All data represent the mean ± SD of at least three independent experiments, each in triplicate wells. Statistical differences were analyzed using the paired t‐test, and were considered significant when P < 0.05.
Results
Forced expression of occludin promotes anoikis. We established two AC2M2 murine breast carcinoma cell lines constitutively overexpressing wild‐type (wt) occludin, designated AC2M2‐Oc#1 and AC2M2‐Oc#2, as a result of two independent transfections. AC2M2 cells transfected with an empty vector were used as a control (AC2M2‐Vec). Although endogenous expression of occludin was not detectable in AC2M2‐Vec cells by western blot analysis, RT‐PCR and Southern blot analysis demonstrated that AC2M2‐Vec expressed appreciable amounts of endogenous occludin (Fig. 1a). AC2M2‐Oc#1 and AC2M2‐Oc#2 cells expressed occludin mRNA abundantly, and AC2M2‐Oc#2 showed higher expression of occludin than AC2M2‐Oc#1 (Fig. 1a). Our preliminary experiments revealed that occludin overexpression had significant effects both on AC2M2‐Oc#1 and AC2M2‐Oc#2 cells, but AC2M2‐Oc#2 showed more marked changes in each variable than AC2M2‐Oc#1 when compared with the AC2M2‐Vec cells. We thus used AC2M2‐Oc#2 cells, hereafter designated AC2M2‐Oc, in the following studies.
Figure 1.
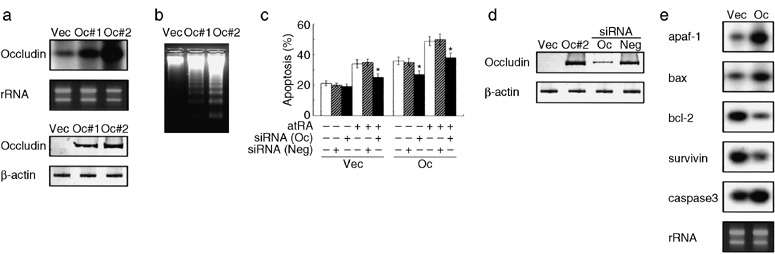
Forced expression of occludin results in enhanced sensitivity to anoikis in AC2M2 cells. (a) Reverse transcription–polymerase chain reaction (RT‐PCR) and Southern blot analysis (upper panel) and western blot analysis (lower panel) to show the expression of occludin in AC2M2‐Vec (Vec), and constitutively overexpressed wild‐type occludin in AC2M2‐Oc#1 (Oc#1) and AC2M2‐Oc#2 (Oc#2) cells. (b,c) Anoikis was induced after adding cells to agarose‐coated dishes to avoid cell attachment. DNA laddering analysis to confirm apoptotic cell death (b). Quantification of anoikis in the presence or absence of 1 µM all‐trans retinoic acid as an apoptotic sensitizer in combination with or without occludin‐specific (Oc) or negative control (Neg) small interfering RNA (siRNA) as determined by terminal deoxynucleotidyl transferase‐meditated dUTP nick‐end labeling assay (c). siRNA (100 nM) was transfected 48 h before anoikis induction. *P < 0.05 versus cells without siRNA transfection. (d) Western blot analysis confirmed the silencing effect of occludin‐specific siRNA (Oc) in AC2M2‐Oc#2 cells (Oc#2), but negative control siRNA (Neg) had no effect on the cells. (e) Occludin expression affects gene expression involving apoptotic machinery. RT‐PCR and Southern blot analysis of indicated apoptosis‐associated genes in AC2M2‐Vec (Vec) and AC2M2‐Oc#2 (Oc) cells.
Breakdown of anoikis (anchorage dependency‐mediated apoptotic cell death) may be a predominant contributor to oncogenic progression.( 6 ) In addition, our previous study has shown that the various cell lines overexpressing occludin gain significant sensitivity to a number of apoptogenic stimuli. We therefore examined whether occludin overexpression promoted anoikis in AC2M2 cells (Fig. 1b). Prior to the apoptotic stimulation, the media used for AC2M2‐Vec and AC2M2‐Oc cells were changed to media without FBS supplementation in the presence or absence of 1 µM atRA as an apoptotic sensitizer.( 20 ) DNA fragmentation analysis demonstrated that forced expression of occludin clearly enhanced the sensitivity to anoikis, and AC2M2 cells that had higher expression of occludin showed increased sensitivity to apoptosis (Fig. 1b). The TUNEL assay also revealed that atRA increased the sensitivity to anoikis both in AC2M2‐Vec and AC2M2‐Oc cells, and that occludin and atRA additively increased anoikis in AC2M2‐Oc cells (Fig. 1c). Based on our observation that occludin‐specific siRNA efficiently suppressed occludin expression in AC2M2‐Oc cells (Fig. 1d), we found that transfection with this siRNA partially but significantly inhibited occludin‐mediated enhancement of cell death in occludin‐expressing cells (Fig. 1c), suggesting a direct mechanistic link between occludin expression and apoptotic sensitization. Importantly, the atRA‐induced increase of apoptotic sensitivity was suppressed by siRNA‐mediated silencing of occludin (Fig. 1c), supporting our previous finding that endogenous occludin induced by atRA could enhance apoptotic sensitivity to apoptogenic stimuli.( 15 )
Because occludin expression affects a wide variety of genes associated with components of the apoptotic machinery, we next examined the alterations in gene expression in the event of occludin overexpression in AC2M2 cells (Fig. 1e). Changes in the pattern of gene expression were consistent with the gene expression profiles determined by an apoptosis pathway‐specific gene array,( 15 ) in which occludin expression downregulated a number of apoptosis‐inhibitory genes such as bcl‐2 and survivin, but conversely upregulated apoptosis‐inducing genes such as apaf‐1 and bax. Genes involved in the apoptosis‐executing pathway, such as caspase 3, were also affected, consistent with the altered phenotype mediated by occludin.
Occludin expression induces premature senescence in AC2M2 cells. Premature senescence is a genetically encoded intrinsic fail‐safe mechanism that has to be overcome by the further acquisition of oncogenic activities during carcinogenesis.( 4 , 8 , 9 ) We next examined the effect of occludin on cellular senescence as assessed by determining the SA‐β‐gal enzymatic activity.( 3 ) When the cells were exposed to a mild oxidative stress that was insufficient to induce apoptotic cell death even in AC2M2‐Oc cells, the occludin‐expressing cells exhibited prominent phenotypic alteration, showing cytoplasmic protrusions and enlarged flat morphology with multiple vacuoles (Fig. 2a). In addition, the percentage of positive reactions to SA‐β‐gal activity was significantly high in AC2M2‐Oc cells (Fig. 2a,b). Interestingly, SA‐β‐gal‐positive AC2M2‐Oc cells were significantly decreased after treatment with occludin‐specific siRNA (Fig. 2b,c), suggesting that occludin was associated with the signaling program to induce premature senescence. We should note here that AC2M2‐Oc cells required 24 h for maximum reduction of occludin after being transfected with occludin‐specific siRNA, and significant suppression of occludin expression was observed for 5 days (Fig. 2c). We could not exclude the possibility that forced expression of occludin might have as yet unidentified effects on the cells. However, the treatment with atRA increased the number of SA‐β‐gal‐positive cells both in AC2M2‐Vec and AC2M2‐Oc, and siRNA targeted to occludin abrogated this effect, indicating that atRA‐mediated upregulation of endogenous occludin was functional in inducing premature senescence in the transformed cells. Observed phenotypes of the cells in the event of occludin expression were not cell‐type specific, because we obtained similar data from three different cell types (data not shown).
Figure 2.
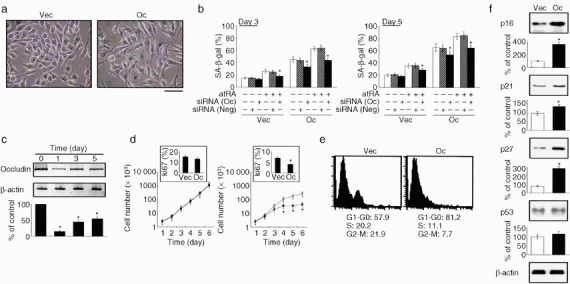
Occludin induces premature senescence in AC2M2 cells under oxidative stress. (a,b) AC2M2‐Vec (Vec) and AC2M2‐Oc (Oc) cells were exposed to 25 µM H2O2 for up to 5 days in the presence or absence of 1 µM all‐trans retinoic acid in combination with or without 100 nM occludin‐specific (Oc) or negative control (Neg) small interfering RNA (siRNA), and were stained for SA‐β‐gal. Note the blue cytoplasmic stain in occludin‐expressing cells after 3‐day exposure to oxidative stress (a), and quantification of senescence‐associated β‐galactosidase (SA‐β‐gal)‐positive cells (b). Scale bar = 40 µm. (c) Western blot analysis confirmed the silencing effect of occludin‐specific siRNA in AC2M2‐Oc cells throughout the experiment. (d) Cell proliferation assay in AC2M2‐Vec (Vec, open symbol) and AC2M2‐Oc (Oc, filled symbol) cells with (right panel) and without (left panel) 25 µM H2O2 for up to 6 days. Insets show the Ki67 labeling indexes at day 6. (e) Flow cytometric analysis of AC2M2‐Vec (Vec) and AC2M2‐Oc (Oc) cells after treatment with 25 µM H2O2 for 3 days. The mean percentage of cells in each cell cycle fraction from independent triplicate experiments is indicated below the histogram. (f) Western blot analysis of cell cycle inhibitors in the presence of 25 µM H2O2 for 3 days. Densitometric analyses for independent triplicate experiments are show below the representative images. *P < 0.05 versus cells without siRNA (b,c) or occludin transfection (d,f).
Occludin expression induces growth arrest under oxidative stress. To study the effect of occludin signaling on proliferation in AC2M2 cells, cell growth was analyzed in a time‐dependent manner. Manual cell counting every 24 h up to day 6 after plating equal numbers of cells showed no significant differences in the growth and DNA synthesis rate, whereas AC2M2‐Oc cells that were exposed to oxidative stress demonstrated significant decreases both in cell growth and DNA synthesis rate (Fig. 2d). Prolonged incubation with H2O2 or incubation with higher concentrations of H2O2 did not result in any further decrease of actively proliferating cells, because AC2M2‐Oc cells underwent apoptotic cell death under these conditions. In addition, cell cycle analyses indicated that occludin expression induced G1 arrest and a concomitant decrease in S‐phase fractions in response to oxidative stress in AC2M2‐Oc cells (Fig. 2e), suggesting that occludin‐induced cellular senescence is involved in the G1/S‐phase transition and might sensitize cells to mitogen‐mediated specific cell cycle checkpoints. Cell cycle inhibitors such as cyclin‐dependent kinase (CDK) inhibitors, including p16INK4A, p21Waf1/Cip1, p27Kip1 and p53, are key mediators of the cellular premature senescence program characterized by cell cycle arrest. We next examined the expression alterations of CDK inhibitors in the event of occludin overexpression in AC2M2 cells under oxidative stress (Fig. 2f). Western blot analysis revealed no significant differences in protein levels between AC2M2‐Vec and AC2M2‐Oc cells without incubation with H2O2, whereas protein increases of p16INK4A, p21Waf1/Cip1 p27Kip1 but not p53, were observed in the presence of H2O2. Withdrawal of the oxidative stress did not result in the release of cell cycle block and the induction of apoptosis. These data provided further support for the possibility that occludin signaling enhanced part of the premature senescence program initiated in response to the cell stressors.
MAPK mediates the induction of senescence in occludin‐expressing cells. The COOH‐terminus of occludin is known to be important in its function, which is responsible for the transmission of various signaling pathways such as MAPK.( 10 , 11 , 15 , 21 ) Consistent with this, deletion mutants with various COOH‐terminal truncations (Fig. 3a) had no effects on increasing SA‐β‐gal‐positive cells in response to oxidative stress (Fig. 3b), suggesting that the 44 amino acids at the COOH‐terminal end of occludin were responsible for the regulation of cellular senescence. We next examined the signal transduction pathways involved in occludin‐mediated premature senescence by using small‐molecule protein kinase inhibitors designed to specifically block signal constituents of protein kinase cascades. A MAPK inhibitor (PD98059) partially but significantly abrogated the enhancement of premature senescence mediated by occludin overexpression, whereas other inhibitors such as a p38 MAPK inhibitor (SB203580) and PI3K inhibitor (LY294002) had no effect (Fig. 3c), suggesting that active engagement of pMAPK signaling molecules was necessary to provoke the cellular senescence program as a result of the activation of occludin signaling.
Figure 3.
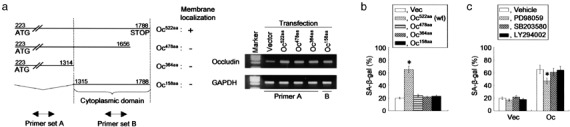
Occludin‐mediated premature senescence is associated with the mitogen‐activated protein kinase signaling pathway. (a) Constructs of deletion mutants used to identify the gene regions responsible for the cellular senescence were as follows: Oc522aa, full‐length murine occludin cDNA (coding region from nucleotides 223–1788 [occludin223–1788], encoding a 522‐amino acid polypeptide; GeneBank accession number, U49185); Oc478aa, occludin construct without 44 amino acids at the COOH‐terminal end; Oc364aa, occludin construct without 158 amino acids at the COOH‐terminal end; Oc158aa, occludin construct with only the cytoplasmic region cloned. Membrane localizations were determined by distribution of enhanced green fluorescent protein (EGFP) signals under fluorescent microscopy after transient transfection with each deletion mutant linked to EGFP (left panel).( 15 ) Transfected genes were examined by quantitative reverse transcription–polymerase chain reaction using primer sets that amplified the non‐cytoplasmic domain (primer set A) or cytoplasmic domain (primer set B) of occludin (right panel), showing that all of the introduced genes were overexpressed in the cells when compared with the vector‐transfected cells. (b) AC2M2 cells transfected with the constructs having various deletions in the COOH‐terminal domain were exposed to 25 µM H2O2 for 5 days, and stained for senescence‐associated β‐galactosidase (SA‐β‐gal). (c) Effects of three kinase inhibitors on cellular senescence in AC2M2‐Vec (Vec) and AC2M2‐Oc (Oc) cells. The cells were treated with PD98059, SB203580 or LY294002 for 24 h prior to stimulation with oxidative stress. Oxidative stress was induced by exposure to 25 µM H2O2 for 5 days and cells were then stained for SA‐β‐gal. *P < 0.05 versus cells without occludin transfection (b) and the treatment (c).
Occludin signaling induces cell spreading. Accumulated evidence has demonstrated that senescent cells not only have different gene expression patterns when compared with reversibly arrested quiescent cells, but also display characteristic morphological changes.( 9 ) To determine the morphological alterations induced in AC2M2 cells by occludin overexpression, we examined the cells with phase‐contrast microscopy at various times (30 min to 6 h) after plating the cells (Fig. 4a,b). Cell shape was indistinguishable between AC2M2‐Vec and AC2M2‐Oc cells, which were regularly round, after 10 min in culture. Cytoplasmic protrusions were observed at 30 min and the cell shape became significantly different after 1 h in occludin‐expressing cells. The size of the AC2M2‐Oc cells increased and they became flattened after 1 h. These differences had gradually increased at 6 h after seeding the cells. After 6 h, a large number of occludin‐expressing cells had actin stress fibers, though these were detected only in a few AC2M2‐Vec cells (Fig. 4c). Occludin‐mediated cell spreading is partially explained by the evidence that occludin directly associates with cytoplasmic scaffolding proteins, submembrane components of TJs, thereby providing a direct linkage to the actin cytoskeleton,( 11 ) and that morphological changes in the cells usually depend on the alterations of actin stress fiber formation.( 22 )
Figure 4.
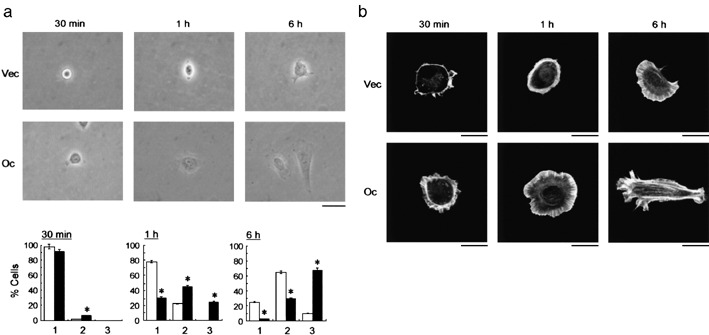
Cell spreading is induced by occludin expression. (a) Morphological changes of AC2M2‐Vec (Vec) and AC2M2‐Oc (Oc) cells examined by phase‐contrast microscopy at indicated time points. Scale bar = 20 µm. (b) Quantification of cell spreading of cultured cells. The number of spreading cells was evaluated by phase‐contrast microscopy in AC2M2‐Vec (open column) and AC2M2‐Oc (filled column) cells, and we categorized cells into three groups: round (a), partially spread (b), and fully spread (c). The results were obtained as the percentage of the cells by counting the cell number under low magnification (×100) in 10 separate arbitrarily selected fields in each section. *P < 0.05 versus cells without occludin transfection. (c) The cells were stained with rhodamine‐phalloidin to detect the actin stress fibers by fluorescent microscopy. Scale bar = 5 µm.
Endogenous occludin is sufficient for provoking cellular senescence. To exclude the possibility that the forced expression of occludin resulted in unidentified artifacts in the cells, we next used a demethylator (5′Aza‐dC) and HDAI (TSA) or retinoids that stimulated retinoic acid receptor (RAR)‐α to induce endogenous occludin expression in the AC2M2 cells (Fig. 5a), in which CpG islands of the occludin promoter were shown to be densely methylated.( 15 ) Although SA‐β‐gal enzymatic activity is a good marker to measure cellular senescence, positive staining using this assay has been observed in cases of cellular stress unrelated to senescence.( 23 ) Thus, senescence was also verified by scoring for the maker Ki67.( 5 , 9 ) Either 5′Aza‐dC or TSA alone or in synergy with these agents, as well as the RAR‐α stimulant was sufficient for increasing the SA‐β‐gal‐positive cells, and conversely for decreasing the Ki67‐positive cells (Fig. 5b,c). Importantly, transfection of occludin‐specific siRNA partially but significantly abrogated the increases of SA‐β‐gal positive cells and the decreases of Ki67 positivity mediated by the treatment with 5′Aza‐dC/TSA and RAR‐α. Our data suggested that the altered phenotypes induced by the treatment with these reagents were, at least in part, responsible for the endogenous reexpression of occludin.
Figure 5.
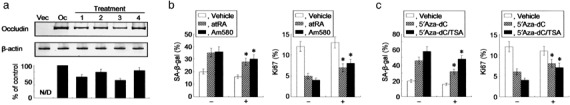
Endogenous occludin is able to promote cellular senescence. (a) Western blot analysis demonstrating that endogenous expression of occludin is induced by the treatment with retinoic acid receptor (RAR)‐α agonists and 5′Aza‐dC/TSA. AC2M2 cells were treated with all‐trans retinoic acid (atRA) (lane 1), Am580 (lane 2), 5′Aza‐dC (lane 3), as well as 5′Aza‐dC and trichostatin A (TSA) (lane 4). Densitometric analyses for independent triplicate experiments are shown below the representative images, and the signal of occludin is defined as 100% in AC2M2‐Oc (Oc) cells without the treatments. N/D, not detectable. (b, c) AC2M2 cells were exposed to 25 µM H2O2 for 5 days with or without 1 µM atRA or 100 nM Am580 (b), and 1 µM 5′Aza‐dC and/or 100 nM TSA (c) in the presence (+) or absence (–) of 100 nM occludin‐specific small interfering RNA (siRNA). Senescence‐associated β‐galactosidase (left panel) and Ki67 (right panel) positive cells were assessed after the treatments. *P < 0.05 versus cells without siRNA transfection.
Occludin expression induces senescent phenotype in vivo. We next investigated whether occludin could induce cellular senescence in vivo. SA‐β‐gal staining revealed diffuse strong positive reactivity in tumors that developed from AC2M2‐Oc cells (Fig. 6a,b). Conversely, cellular proliferation rates assessed by Ki67 labeling indexes were significantly suppressed in occludin‐expressing cells. However, positive staining was not observed in the normal mammary gland or any tumors that developed from AC2M2‐Vec cells, suggesting that occludin signaling was involved in part of the senescence machinery in vivo under physiological cell stress.
Figure 6.
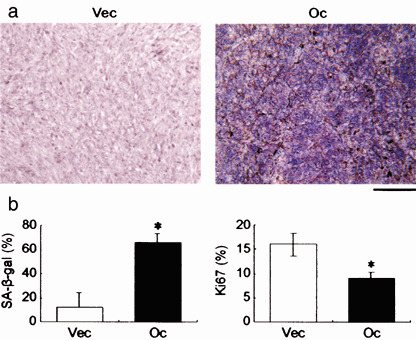
Occludin induces cellular senescence in vivo. Representative results of the senescence‐associated β‐galactosidase (SA‐β‐gal) staining in tumors developed from AC2M2‐Vec (Vec) and AC2M2‐Oc (Oc) cells in mice (a), and quantification of SA‐β‐gal (left panel) and Ki67 (right panel) positive cells in the tissue sections (b). SA‐β‐gal staining was positive in 0 of 24 tumors that developed from AC2M2‐Vec cells (0%) and in 8 of 27 tumors that developed from AC2M2‐Oc cells (29%; P < 0.05). Scale bar = 200 µm. *P < 0.05 versus cells without occludin transfection.
Discussion
Herein we shed light on the novel functions of occludin in cancer cells, which have not hitherto been described as a consequence of occludin signaling, demonstrating that the loss of occludin expression is at least partially involved in the senescence‐escape program during mammary tumorigenesis. Although accumulating studies have shown the downregulation of TJ‐associated proteins such as occludin during carcinogenesis in many cell types,( 15 , 16 , 17 , 18 ) the impact of our findings on breast cancer seems to be limited due to the fact that epigenetic silencing of occludin has not been demonstrated to occur during human breast cancer progression. However, we provided clear evidence that occludin expression in a number of transformed cells showed cellular features of premature senescence that can be triggered when cells are exposed to cell stressors. Although the underlying mechanism explaining how occludin determines the fate of cancer cells (i.e. apoptotic or senescence) in response to cell stressors remains to be clarified, it is possible that the cell fate decision is made depending on the cell type, the type of cell stress, the strength and duration of the stress, and the microenvironment surrounding the cell, as well as on the integrity of tumor suppressor genes.
We found that occludin was associated with a number of apoptosis‐associated genes and components of the lipid signaling pathway such as MAPK. In support of these observations, the Ras/Raf oncogene extracellular signal‐regulated kinase (ERK)/MEK (MAPK/ERK kinase) cascade is of critical importance to show that this protein elicits growth arrest and premature senescence in a variety of cell types.( 2 , 24 , 25 ) We also demonstrated that the COOH‐terminal end of occludin plays an important role in inducing cellular senescence. This is supported by a number of reports showing that the components of lipid signaling interact with the COOH‐terminal region of occludin and mediate cell growth and differentiation.( 26 , 27 , 28 ) It is somewhat surprising that the TJ‐associated gene occludin is able to induce premature senescence in tumor cells. Given the extensive range of physiological activities of occludin at TJs, it is feasible that occludin is not a simple static component of TJs, but has functions in receiving and transmitting cell survival signals, working as a signal transmitter located at the TJ in the cell. This explanation is supported by evidence that occludin directly associates with different types of signaling transduction, and TJ protein directly participates in the control of gene expression via the modulation of transcription machinery in the nucleus.( 11 , 29 ) Recent study has also shown that the second extracellular loop of occludin is a transforming growth factor (TGF)‐β receptor, and occludin regulates TGF‐β type I receptor localization for efficient TGF‐β‐dependent dissolution of TJs during epithelial–mesenchymal transition, which is a cellular transformation crucial for the progression of many epithelial tumors.( 30 ) Consistent with this view, the possible mechanisms underlying premature senescence with high expression of CDK inhibitors caused by forced expression of occludin appear to be explained by the signal transduction from membrane‐anchored occludin to the nucleus.
Non‐cytotoxic cell stress induces premature senescence that is characterized by upregulation of negative cell cycle regulators. Conversely, cell cycle inhibitors are able to provoke the senescent phenotype when, for example, they are overexpressed ectopically in early passage human diploid fibroblasts.( 31 ) Indeed, our observations showed that occludin‐mediated premature senescence was accompanied by upregulation of p16INK4A, p21Waf1/Cip1 and p27Kip1. CDK inhibitors are key mediators of growth arrest in the course of premature senescence, and are implicated in DNA repair to prevent potentially damaged cells from undergoing consequent mitotic catastrophe.( 9 , 24 ) Whether upregulation of CDK inhibitors directly contributes to the senescence‐associated phenotype in occludin‐expressing cells remains to be determined, but it is possible that occludin provokes premature senescence, potentially associated with the enhanced expression of CDK inhibitors. However, we did not observe alteration in the expression of p53 protein in the course of premature senescence, suggesting that the senescence induced by occludin signaling was mediated by p53‐independent mechanisms or mutant p53. The functional requirements for the CDK inhibitors in the senescent state appeared to be heterogeneous in the examined conditions.( 24 , 25 , 31 ) For example, a p53‐independent senescence mechanism was proposed in response to hypermitogenic proto‐oncogene ERBB2 signaling in breast carcinoma cells,( 32 ) though p53 protein is believed to act as an integrator of cell signals for convergence of a broad range of different stressors. Another example was presented by SA‐β‐gal positivity in clinical breast cancer showing that chemotherapy‐induced DNA damage was able to induce senescence in vivo, but that SA‐β‐gal staining was associated with low p53 staining.( 33 )
Recently, senescent IMR90 human fibroblasts have been shown to accumulate a distinct heterochromatin structure, which is designated senescence‐associated heterochromatin foci (SAHF).( 34 ) We did not observe the characteristic features of SAHF during the experiment, and cannot presently explain this observation; however, one possibility is that a lack of intact Rb signaling (and disruption of the p53 pathway) may have limited the ability of occludin‐expressing AC2M2 cells to display SAHF in response to the oxidative stress. Our explanation is supported by the fact that SAHF formation primarily depends on the integrity of the Rb pathway, and eventually involves the combined activities of the Rb/p16INK4a pathway, p53 signaling, and the promyelocytic leukemia protein.( 34 ) In addition, Rb is not essential for all aspects of the senescence program, including the characteristic morphological changes, the accumulation of SA‐β‐gal activity, and the irreversible exit from the cell cycle.( 34 ) We observed that occludin expression in AC2M2 cells promoted oxidative stress‐induced premature senescence, which apparently did not involve SAHF, whereas this was accompanied by upregulation of negative cell cycle regulators and, importantly, withdrawal of the oxidative stress did not result in the release of cell cycle block. These features appear to be sufficient to distinguish senescence from quiescence, a non‐proliferative state that is reversed in response to mitogens.
This study provides significant evidence that occludin signaling is potentially associated with premature senescence machinery. It is now well accepted that the senescence process is an antioncogenic fail‐safe mechanism that may function as a natural brake to tumor development, sharing therapeutic similarities with apoptotic machinery.( 9 , 35 ) Surprisingly, a number of reports have successfully demonstrated that DNA‐damaging agents induce cellular senescence that lacks intact apoptotic responsiveness.( 33 , 36 ) We thus believe that occludin‐mediated induction of cellular senescence can be applied as an anticancer mechanism to blunt oncogenic activity by inducing the cancer cells to exit uncontrolled cellular proliferation before expanding as a clinically relevant tumor mass. While overexpressed protein products or misfolded ones left in the endoplasmic reticulum cause cellular stress, which may consequently have an impact on intracellular signaling, a more important issue was addressed by our observation demonstrating that endogenous reexpression of occludin mediated by a synergistic effect with a demethylator and HDAI or retinoids that stimulated RARa was sufficient to provoke the senescent phenotype in breast carcinoma cells. Given the conceptual similarity to the apoptotic machinery, occludin is thus an appropriate therapeutic target even for cancer cells that do not have detectable expression of endogenous occludin.
Acknowledgments
This study was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Mr Kim Barrymore for help with the manuscript, the Animal Care Facility of Sapporo Medical University School of Medicine, and Dr Hiroyuki Kagechika (School of Biomedical Sciences, Tokyo Medical and Dental University, Tokyo, Japan) for the gift of Am580.
References
- 1. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621. [DOI] [PubMed] [Google Scholar]
- 2. Serrano M, Lin AW, McCurrach ME et al . Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a . Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 3. Dimri GP, Lee X, Basile G et al . A biomarker that identifies senescent human cells in culture and in aging skin in vivo . Proc Natl Acad Sci USA 1995; 92: 9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campisi J. Cellular senescence as a tumor‐suppressor mechanism. Trends Cell Biol 2001; 11: S27–31. [DOI] [PubMed] [Google Scholar]
- 5. Michaloglou C, Vredeveld LC, Soengas MS et al . BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature 2005; 436: 720–4. [DOI] [PubMed] [Google Scholar]
- 6. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001; 411: 342–8. [DOI] [PubMed] [Google Scholar]
- 7. Gozani O, Boyce M, Yoo L et al . Life and death in paradise. Nat Cell Biol 2002; 4: E159–62. [DOI] [PubMed] [Google Scholar]
- 8. Ben‐Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest 2004; 113: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braig M, Schmitt CA. Oncogene‐induced senescence: Putting the brakes on tumor development. Cancer Res 2006; 66: 2881–4. [DOI] [PubMed] [Google Scholar]
- 10. Gumbiner EE. Breaking through the tight junction barrier. J Cell Biol 1993; 123: 1631–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2001; 2: 285–93. [DOI] [PubMed] [Google Scholar]
- 12. Furuse M, Furuse M, Hirase T et al . Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993; 123: 1777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saitou M, Fujimoto K, Doi Y et al . Occludin‐deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol 1998; 141: 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saitou M, Furuse M, Sasaki H et al . Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000; 11: 4131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Osanai M, Murata M, Nishikiori N et al . Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis‐associated genes. Cancer Res 2006; 66: 9125–33. [DOI] [PubMed] [Google Scholar]
- 16. Wang Z, Mandell KJ, Parkos CA et al . The second loop of occludin is required for suppression of Raf‐1‐induced tumor growth. Oncogene 2005; 24: 4412–20. [DOI] [PubMed] [Google Scholar]
- 17. Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res 2005; 65: 9603–6. [DOI] [PubMed] [Google Scholar]
- 18. Tobioka H, Isomura H, Kokai Y et al . Occludin expression decreases with the progression of human endometrial carcinoma. Hum Pathol 2004; 35: 159–64. [DOI] [PubMed] [Google Scholar]
- 19. Elliott BE, Maxwell L, Arnold M et al . Expression of epithelial‐like markers and class‐I major histocompatibility antigens by a murine carcinoma growing in the mammary gland and in metastases: orthotopic site effects. Cancer Res 1988; 48: 7237–45. [PubMed] [Google Scholar]
- 20. Osanai M, Petkovich M. Expression of the retinoic acid‐metabolizing enzyme CYP26A1 limits programmed cell death. Mol Pharamacol 2005; 67: 1808–17. [DOI] [PubMed] [Google Scholar]
- 21. Murata M, Kojima T, Yamamoto T et al . Down‐regulation of survival signaling through MAPK and Akt in occludin‐deficient mouse hepatocytes in vitro . Exp Cell Res 2005; 310: 140–51. [DOI] [PubMed] [Google Scholar]
- 22. Kamada M, Suzuki K, Kato Y et al. von Hippel‐Lindau protein promotes the assembly of actin and vinculin and inhibits cell motility. Cancer Res 2001; 61: 4184–9. [PubMed] [Google Scholar]
- 23. Severino J, Allen RG, Balin S et al . Is beta‐galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res 2000; 257: 162–71. [DOI] [PubMed] [Google Scholar]
- 24. Lin AW, Barradas M, Stone JC et al . Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998; 12: 3008–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J, Woods D, McMahon M et al . Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 1998; 12: 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nusrat A, Chen JA, Foley CS et al . The coiled‐coil domain of occludin can act to organize structural and functional elements of the epithelial tight junction. J Biol Chem 2000; 275: 29 816–22. [DOI] [PubMed] [Google Scholar]
- 27. Sheth P, Basuroy S, Li C et al . Role of phosphatidylinositol 3‐kinase in oxidative‐stress‐induced disruption of tight junctions. J Biol Chem 2003; 278: 49 239–45. [DOI] [PubMed] [Google Scholar]
- 28. Sharma GD, He J, Bazan HE. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross‐talk activation between MAP kinase cascades. J Biol Chem 2003; 278: 21 989–97. [DOI] [PubMed] [Google Scholar]
- 29. Balda MS, Matter K. The tight junction protein ZO‐1 and an interacting transcription factor regulate ErbB‐2 expression. EMBO J 2000; 19: 2024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrios‐Rodiles M, Brown KR, Ozdamar B et al . High‐throughput mapping of a dynamic signaling network in mammalian cells. Science 2005; 307: 1621–5. [DOI] [PubMed] [Google Scholar]
- 31. McConnell BB, Starborg M, Brookes S et al . Inhibitors of cyclin‐dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 1998; 8: 351–4. [DOI] [PubMed] [Google Scholar]
- 32. Trost TM, Lausch EU, Fees SA et al . Premature senescence is a primary fail‐safe mechanism of ERRBB2‐driven tumorigenesis in breast carcinoma cells. Cancer Res 2005; 65: 840–9. [PubMed] [Google Scholar]
- 33. Te Poele RH, Okorokov AL, Jardine L et al . DNA damage is able to induce senescence in tumor cells in vitro and in vivo . Cancer Res 2002; 62: 1876–83. [PubMed] [Google Scholar]
- 34. Narita M, Nuñez S, Heard E et al . Rb‐mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113: 703–16. [DOI] [PubMed] [Google Scholar]
- 35. Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell 2000; 102: 407–10. [DOI] [PubMed] [Google Scholar]
- 36. Chang BD, Broude EV, Dokmanovic M et al . A senescence‐like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 1999; 59: 3761–7. [PubMed] [Google Scholar]