

Abstract
In breast cancer, stromal cells surrounding cancer epithelial cells can influence phenotype by producing paracrine factors. Among many mediators of epithelial–stromal interactions, aromatase activity is perhaps one of the best studied. Clinical data suggest that estrogen‐independent signaling leads to increased proliferation even during therapy with aromatase inhibitors (AIs). Molecular mechanism of crosstalk between the estrogen receptor (ER) and the epidermal growth factor receptor (HER) family have been implicated in resistance to endocrine therapy, but this interaction is unclear. The ability of aromatase to induce estradiol biosynthesis provides a molecular rationale to combine agents that target aromatase activity and the HER pathway. We targeted stromal–epithelial interactions using formestane, which exerts antiaromatase activity, combined with the monoclonal anti‐HER2 antibody herceptin, in a subpopulation of CD44+/CD24low cells sorted from epithelial‐mesenchymal co‐cultures of breast cancer tissues. The growth inhibition was respectively 16% (P < 0.01) in the response to herceptin, 25% to formestane (P < 0.01), and 50% (P < 0.001) with the combination of the two drugs, suggesting that herceptin cooperates with formestane‐induced inhibition of aromatase and this effect could be mediated through HER family receptors. In cells which expressed ERα, formestane/herceptin combination suppressed the mRNA expression of aromatase and HER2 and decreased cyclin D1 expression. These results show that combination therapies involving AIs and anti‐HER2 can be efficacious for the treatment of cancer in experimental models and suggest that subtyping breast tumors gives useful information about response to treatment. (Cancer Sci 2010)
Tumor phenotype is influenced by genetic and epigenetic alterations, tumor stroma, and the systemic environment, which lead to a complex mixture of factors that represent the tumor microenvironment.( 1 ) Estrogens play a critical role in the development of breast cancer. Six enzymes catalyze the conversion of cholesterol to biologically active 17‐β‐estradiol (E2). The aromatase enzyme catalyzes the final and key step of estrogen biosynthesis, that is the conversion of C19 steroids to estrogens. In premenopausal women, most plasma estrogen is synthesized in the ovaries and a small proportion (<10%) derives from peripheral synthesis by aromatase, which is present in subcutaneous fat. As a woman ages, the breast’s epithelial tissue is gradually replaced by adipocytes. In postmenopausal women, estrogens are synthesized by peripheral aromatization from androgens and peripheral aromatase activity increases with age.( 2 ) Most patients with breast cancer are postmenopausal women and 75% of them have estrogen‐dependent tumors defined by estrogen receptor (ER) positivity.( 3 , 4 ) As estrogens synthesized locally or peripherally by aromatase are able to induce mitogenic signals, the ER has long represented the target for endocrine therapies that aim to block the action of estrogen on tumor cells.( 5 , 6 , 7 ) In addition to ER positivity, common features of breast cancers are androgen receptor (AR) positivity( 8 , 9 ) and elevated expression of HER1/HER2.( 10 ) The levels of ERs and of HER1/HER2 in breast tissue distinguish luminal A and B tumors from basal‐like tumors, and are predictive of response to endocrine treatment.( 11 ) Cells purified from breast tissues are categorized according to the cell surface expression of CD44 and CD24, which distinguishes CD24+/CD44− cells (luminal epithelial cells) from CD24−/44+ cells (basal cells),( 12 ) although a considerable heterogeneity in CD44 and CD24 expression was seen both between and within breast tumors.( 13 ) The CD44 cell‐surface antigen identifies tumor‐initiating ability in breast cancer cells, it supports anchorage‐independent growth in vitro and tumor growth in animal models.( 14 ) The tumor‐promoting ability of CD44 results from its association with HER1/HER2.( 15 ) HER2 is overexpressed in 25–30% of breast cancers and stimulates the growth of breast cancer cells.( 10 , 16 ) CD24 expression is known to be lowest in ERα‐positive tumors( 17 ) and CD44 expression is up‐regulated by E2.( 18 ) To examine whether aromatase activity affected HER1/HER2 expression, we sorted and characterized a sub‐population of CD44+/CD24low cells from human breast cancer tissue and evaluated their response to treatment. Our results suggest that sub‐populations of undifferentiated cells could influence the tumor microenvironment and modify the levels of hormones and growth factor receptors in tumor tissue, thereby affecting the expression of prognostic factors and the response to targeted therapy.
Materials and Methods
Materials and control cells. MCF‐7 and MDA‐MB231 were from the American Type Culture Collection (Manassas, VA, USA); human skin fibroblasts (Fibro) are described elsewhere.( 19 ) Fetal bovine serum (FBS) was purchased from Gibco (Invitrogen, Milan, Italy). The final concentrations of the hormones were: estradiol (E2) 10 ng/mL (E2758) and testosterone, 100 nM (T1500), both from Sigma‐Aldrich (Milan, Italy). Trastuzumab (Herceptin) from Genentech (San Francisco, CA, USA), estradiol, and testosterone were dissolved in 70% ethanol. Formestane (Sigma, St. Louis, MO, USA) was dissolved in chloroform at 50 mg/mL. The monoclonal anti‐pancytokeratin and anti‐vimentin antibodies were purchased from Sigma‐Aldrich. Rabbit polyclonal antibodies ERα (HC‐20), cyclin D1 (C‐20), p‐Neu/HER2 (Tyr1248), and mouse monoclonal anti‐actin (sc‐8432) antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Tumor samples. After informed consent was obtained, breast cancer tissues were obtained from patients undergoing surgery for early breast cancer at the University Hospital “Federico II” (Naples, Italy). The institute’s ethics committee approved the study. Patients did not receive neoadjuvant or adjuvant endocrine therapy. Immediately after surgery, proteins were extracted from fragmented aliquots of fresh specimens, and cytokeratins and vimentin were identified by western blot analysis.
Isolation of cells from breast specimens. Tumor specimens were processed as previously described.( 19 ) Briefly, primary cultures were seeded overnight in 24 wells in minimal essential Dulbecco/Ham F12 (1:1) (DMEM/F12 medium) (Sigma‐Aldrich) without phenol red, supplemented with 2 mM glutamine (Sigma‐Aldrich), P+S, 15 mM HEPES (Sigma‐Aldrich), and 5% FBS. Then, the medium was substituted with 0.5% FCS. Cells were cultured at 37°C in a humidified atmosphere of 5% CO2. The medium was renewed twice weekly. Within 3–4 weeks of culture, we obtained sphere‐like aggregates of growing cells. The cells did not form an epithelial‐like flat monolayer, but domes surmounted by aggregates of cells. When the medium was changed to 5% FBS, cells adhered to the dish and reached confluence. We dissociated these cells enzymatically to obtain secondary spheres, leading to amplification of cell numbers. After a gentle trypsinization (2 min at 37°C), we collected only the floating aggregates (300 cells/sphere) to propagate spheres. These were transferred diluted in 0.5% FBS into 96‐well plates. On‐going floating spheres that formed were then transferred to six‐well plates for long‐term experiments. This cell propagating procedure reduced doubling time and generated long‐term cultures (18 months, more than 50 passages). From the co‐cultures, we obtained five different samples of human breast cancer aggregates of cells. All the samples derived from breast tumors classified “luminal A” were histologically classified “ductal” (either invasive or in situ) and all were HER2 negative. HER2 scoring was carried out according to the standard procedure (Dako, Carpintera, CA, USA): HER2‐negative = 0, <10% of the tumor cells stained. Pre‐sorting co‐cultured cells, analysed by western blot for protein expression of phosphorylated‐HER2, were also negative, as previously reported.( 19 ) The experiments reported herein were all performed on sample #2; parallel experiments were carried out on the other samples, and similar results were obtained. For experiments with drugs, the cells were plated, allowed to attach for 48 h, washed with PBS, and incubated without serum for a further 24 h in DMEM/F12. Cells were then treated, as indicated, with testosterone at 100 nM as a substrate for aromatase, the submaximal dose of 70 nM formestane, and 0.7 μg/mL herceptin in 0.5% FCS for 7 days.
Flow cytometry and sorting. Sample and control cells, harvested in 100‐mm dishes, were dissociated by trypsin‐EDTA, counted in a hemocytometer chamber, and 2.5 × 105 cells/sample were incubated for 5 min at RT with 50 μL of FBS. Four separate experiments were performed per sample. All cells were incubated in the dark for 30 min on ice with specific mAb FITC and phycoerythrin (PE) conjugates directed against CD44 and CD24 cell surface antigens. All conjugated antibodies were purchased from Pharmingen (BD Biosciences, Franklin Lakes, NJ, USA). After washing twice with 0.5% PBS–BSA, the suspension was centrifuged and the pellet suspended in 300 μL of 0.5% PBS–BSA. This suspension was filtered with 50‐micron filters to split the cells. Cell analysis and sorting were performed on a triple‐laser cell sorter (MoFlo; DakoCytomation, Fort Collins, CO, USA). FACS analysis was performed using Summit software from DakoCytomation.
Proliferation assay. Growth curves in triplicate were conducted in six‐well plates using 2.5 × 104 cells/well. The number of cells to be plated was the result of preliminary experiments of plating efficiency (data not shown). At the time indicated, after trypsinization, cells were counted in a hemocytometer chamber.
Sphere formation assay and growth in soft agar. To test the ability to form sphere, cells were dissociated and seeded, by serial dilution, in 96‐well plates in DMEM/F12 + 0.5% FCS medium. The final cell dilutions ranged from 1 to 1000 cells/well. The medium was renewed twice weekly. For colony growth in soft agar, spheres were trypsinized, counted, and 104 cells/well were plated in 60‐mm triplicate dishes with 0.3% agar on a 0.5% agar (Type I; Sigma) underlayer DMEM/F12 + 0.5% FBS. The top layer of the corresponding liquid medium was renewed twice weekly. Colonies were counted after 56 days.
Western blot analysis. Western blots were performed on tissue, primary cultures, and cell extracts as indicated in the figures. MCF7 cells, MDA‐MB231, and human skin fibroblasts (Fibro) served as controls. The cells were plated, incubated for 48 h, washed with PBS, incubated for further 24 h in DMEM/F12, and then treated with the supplements indicated for 48 h. Protein preparations from tissues and cells were obtained by lysing samples in 50 mM Tris (pH 7.5), 150 mM NaCl, 1% NonidetP40, 0.1% Triton, 1 mM EDTA, 10 μg/mL aprotinin, and 100 μg/mL phenylmethylsulfonyl‐fluoride. Protein concentration was measured by the Bio‐Rad protein assay (Bio‐Rad, Milan, Italy). Polyacrylamide gels (from 8% to 15%) were prepared as previously described.( 19 ) Prestained molecular weight standards were from Bio‐Rad. Proteins separated on gels were blotted on a nitrocellulose membrane (Hybond‐C pure; Amersham Italia, Milan, Italy). The membrane was stained with Ponceau S (Sigma) to evaluate the success of transfer, and to locate the molecular weight markers. Free protein binding sites were blocked with nonfat dry milk and Tween‐20/TBS solution. The membranes were washed, stained with specific primary antibodies and then with secondary antisera, conjugated with horseradish peroxidase (1:3000; Santa Cruz Biotechnology). The luminescent signal was visualized with the ECL Western blotting detection reagent kit (Amersham Italia) and quantified by scanning with a Discover Pharmacia scanner equipped with a Sun Spark Classic Workstation.
Semi‐quantitative analysis of mRNA by RT‐PCR. RNA was isolated from sample and control cells (MCF7, MDA‐MB241), using TRIzol Reagent (Invitrogen) according to the producer’s instructions. Purity of RNA was checked by measuring the absorbance ratio at 260/280 nm. RNA was stored at −70°C in aliquots of 50 ng/L. Total RNA (1 μg) from the cells was converted to first‐stranded cDNA primed with a random hexamer in a 50 μL reaction volume using a RNA PCR kit (ImProm‐II Reverse Transcription System; Promega, Milan, Italy). The primer pairs, the RT‐PCR conditions used for mRNA amplification, were: ERα 5′‐CCACCAACCAGTGCACCATT 3′‐GGTC‐TTTTCGTATCCCACCTTTC; AR 5′‐CCTGGCTTCCGCAACTTACAC 3′‐GGACTTGTGCATGCGGTACTCA; CYP19 5′‐GAATATTGGAAGGATGCACAGACTC 3′‐GGGTAAAGATCATTTCCAGCATGT; HER1 (EGFR) 5′‐CAACATCTCCGAAAGCCA 3′‐ATAGTCGCCCAAAGTTCCG; and HER2 (c‐erbB2) 5′‐TGCGGCTCGTACACAGGGACTT 3′‐TGCGGGAGAATTCAGACACCAACT. PCR products were analyzed on 2% agarose gels and ethidium bromide staining. Staining intensity was quantified using a Spectroline Transilluminator (model TR365). The expression levels of individual mRNA bands were normalized to that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) 5′‐ACATCATCCCTGCCTCTACTGG and reverse primer: 3′‐AGTGGGTGTCGCTGTTGAAGTC. Reverse transcription–PCR was performed under conditions of linearity to obtain semi‐quantitative amplification responses.
Data analysis. Each experiment was carried out 2–4 times and found to be reproducible.Data of experiments with drugs were analyzed according to Tallarida.( 20 ) We used Prism 3.0 software (GraphPad Software, San Diego, CA, USA) to generate graphs and for statistical analyses. Error bars are presented as SEM. We used anova to identify statistically significant differences among means.
Results
Sorting, isolation, and growth properties of CD44+/CD24− cells. Breast cancers are categorized into luminal‐type, which express luminal keratins, and basal‐type, which express stratified epithelial keratins. CD24+/CD44− expression identifies differentiated luminal epithelial cells, and CD44+/CD24− cells show features of basal cells. We performed flow cytometry and evaluated the expression of CD44 and CD24 of aggregates isolated in our long‐term epithelial‐mesenchymal co‐cultures of breast cancer tissue. To this aim we collected only the floating aggregates detached after trypsin for 2 min at 37°C; these were transferred and maintained in 0.5% FBS in six‐well plates. Control cell lines had different CD44 and CD24 profiles; MCF7 cells consisted mainly of CD24+ expressing cells (luminal‐like), whereas MDA‐MB 231 consisted mainly of CD24− expressing cells (basal/mesenchymal‐like) (Fig. 1a). Human breast cancer cell aggregates consisted of both CD24+ and CD24− populations (mixed luminal and basal cells) (Fig. 1b). Because CD44 supports anchorage‐independent growth in vitro, confers a growth advantage, and is a marker of enrichment of tumor‐initiating cells,( 21 , 22 , 23 ) we sorted the cells with CD44high/CD24low expression (Fig. 1b, R5 square). Cell floating aggregates accounted for 10–15% of the total cell number; thus, the subpopulation of sorted cells represents a small fraction of all the cells in the dissected tissue. To test whether the cell aggregates had luminal or basal features, we immunoblotted for cytokeratins and vimentin in unsorted and sorted cells. We compared samples cells with fibroblasts, MDA‐MB231, and MCF‐7 reference cell lines. Cytokeratins were expressed in MCF7 epithelial cells but not in fibroblasts; vimentin was expressed in fibroblasts, but not in MCF7 epithelial cells. Control MDA‐MB231 cells and sorted S1 and S2 cells expressed both markers. Pre‐ and post‐sorting comparative western blot analysis indicated that sorted cells preserved their original cytoskeletal scaffolds (Fig. 2). Taken together, these data suggest that the sorted CD44+/CD24low cells retained either basal‐like or luminal‐like features.
Figure 1.
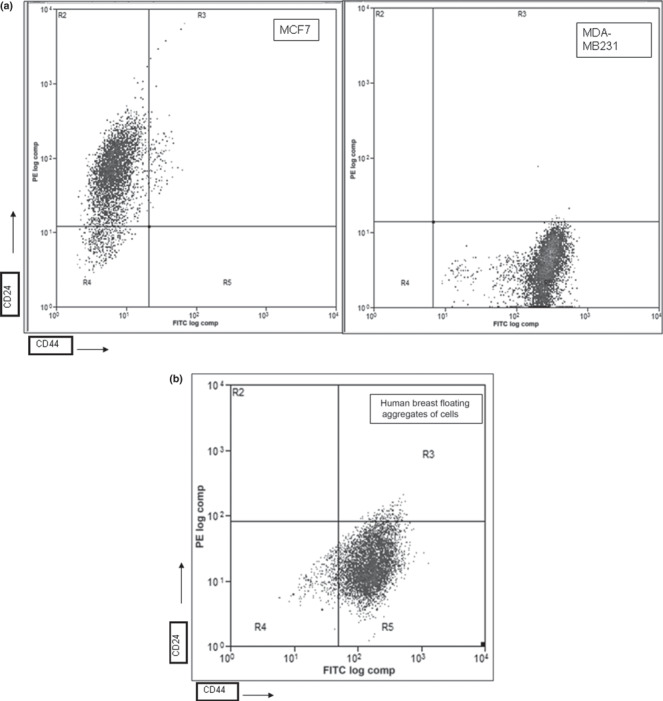
Flow cytometry of CD44/CD24 expression in control MCF7 and MDA‐MB231 cells (a), and human breast floating aggregates of cells (b). Floating aggregates cultured in 0.5% FCS were enzymatically dissociated and 2.5 × 105 cells were analyzed. R5 square indicates CD44+/CD24low sorted cells.
Figure 2.
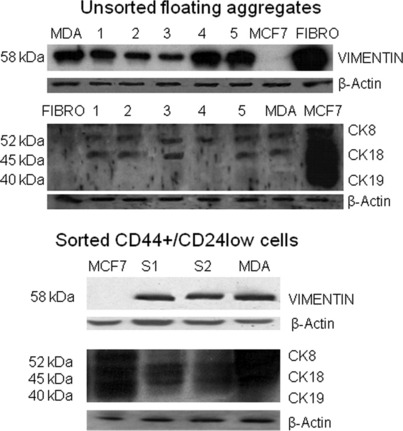
Sorted CD44+/CD24low cells contain populations of cells with either basal‐like or luminal‐like features. Expression of cytokeratins and vimentin in tissue samples and reference cell lines (fibroblasts [Fibro] and MCF‐7), upper panel, and in CD44+/CD24low sorted cells, lower panel. Twenty‐five‐microgram aliquots of five‐tissue extracts (lanes 1–5) and cell lysates (S1, S2), respectively, were electrophoresed through 10% SDS polyacrylamide gels. After transfer onto nitrocellulose membranes, they were subjected to immunoblotting using a pan‐cytokeratin monoclonal antibody that recognizes an epitope common to various cytokeratins (CK8, 52 kDa; CK18, 45 kDa; CK19; 40 kDa), vimentin (58 kDa). Cytokeratins were expressed in MCF7 epithelial cells but not in fibroblasts; vimentin was expressed in fibroblasts but not in MCF7 epithelial cells. Control MDA‐MB231 cells and sorted S1 and S2 cells expressed both markers. β‐Actin served to verify equal loading.
To evaluate the ability of a single cell to form spheres, we cloned, by serial dilution, sorted cells and found that wells of 100 cells and up formed floating aggregates similar to the unsorted cells (Fig. 3). On the cell colonies that expanded after sorting and replating, soft‐agar colony assay was done in quadruplicate; the numbers ± SD of colonies per 10 random fields/dish were S1 = 58 ± 5 and S2 = 43 ± 4, thus corroborating the presence of anchorage‐independent cells.
Figure 3.

Floating aggregates of sorted CD44+/CD24low cells cultured for 3 weeks. Phase‐contrast microscopy of unsorted aggregates (a,b,c) and CD44+/CD24low cells sorted and plated in 0.5% FBS, 4 days after sorting, and 21 days after sorting.
CD44+/CD24low cells express ERα, AR, and phosphorylated HER2 (pNeu/HER2). In subcutaneous fat, aromatase catalyzes the conversion of testosterone to estradiol that acts locally on breast cells through ERα. Since CD44+/CD24− cells proliferate and propagate in low serum (see Materials and Methods), and their growth was not influenced by steroids (Fig. 4a), we analyzed their expression of ERα and AR. The RT‐PCR mRNA expression of ERα in CD44+/CD24low cells untreated and treated with estrogens (E2) and testosterone (T) showed similar levels of transcripts (Fig. 4b). Immunoblotting of cell lysates of CD44+/CD24low treated with E2 and T confirmed the protein levels of ERα (Fig. 4c, upper panel). Since the subpopulation of sorted cells represents a small fraction of all the cells in the dissected tissue, and because these cells expressed ERα and AR whose levels were not influenced by steroids, we explored the alternative signaling route mediated through the receptor tyrosine kinase (RTK) and measured the protein expression of activated HER2 (pNeu/HER2). Figure 4(c), lower panel, shows similar levels of phosphorylated Neu/HER2 in untreated CD44+/CD24low breast cancer cells (c) and treated, respectively, with E2 or T.
Figure 4.
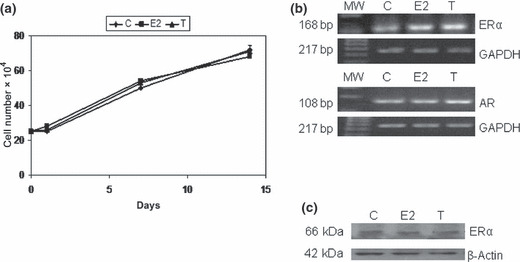
(a) CD44+/CD24low cell growth is not influenced by steroids. Growth curves of triplicate six‐well plates with 2.5 × 104 cells/well cultured with 0.5% FBS (c), or 10 ng/mL estradiol (E2) or 100 nM testosterone (T), for 14 days, and are representative of three different experiments. Error bars indicate SEM. (b) CD44+/24low cells express estrogen receptor (ER)‐α and androgen receptor (AR). Semi‐quantitative RT‐PCR of ERα and AR mRNA of CD44+/CD24low cells cultured in control media 0.5% FBS (c), with 10 ng/mL estradiol (E2), or 100 nM testosterone (T). Standardization by the GAPDH cDNA levels. (c) ERα and pNeu/HER2 protein expression, measured by western blot analysis, of CD44+/CD24low cells in 0.5% FBS (C), with 10 ng/mL estradiol (E2), or 100 nM testosterone (T). β‐Actin to verify equal protein loading.
Cooperative growth inhibitory effect of herceptin and formestane in combination. Aromatase inhibitors (AIs) reduce the progression of breast tumors( 5 ) and clinical trials that combine AIs with HER1/2 inhibitors show benefits rates for ER‐positive patients with metastatic breast cancer and enhanced expression of HER2.( 24 )
First, to determine whether aromatase activity influences the growth of sub‐populations of CD44+/CD24low cells, we constructed growth curves with 100 nM testosterone as a substrate of aromatase and investigated whether this activity affected the response to drugs that inhibit the steroid and RTK pathways. To this aim, we used formestane (70 nM) and herceptin (0.7 μg/mL) alone and in combination for 7 days. As a control for herceptin non‐specific targeting of the ectodomain region of HER2, we also used lapatinib, a reversible tyrosine kinase inhibitor (TKI) that binds intracellularly and inhibits both EGFR and HER2, and obtained similar results (data not shown). The histogram in Figure 5 shows the growth of CD44+/CD24low cells expressed as percentage of viable cells over control. The first bar shows that although testosterone did not enhance cell growth, it caused a 16% decrease in the response to herceptin (three different experiments; P < 0.01) and a 25% decrease in the response to formestane (three different experiments; P < 0.01). The combination of the two drugs exerted a greater inhibitory (50%) (three different experiments; P < 0.001) effect on the growth of CD44+/CD24low cells, suggesting that herceptin cooperates with formestane‐induced inhibition of aromatase and that this effect could be mediated through HER family receptors.
Figure 5.
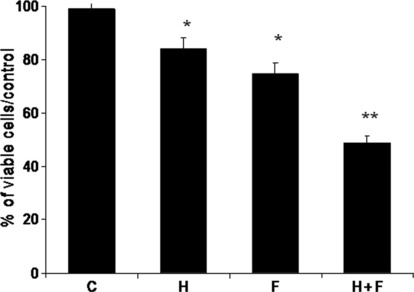
Combined aromatase inhibitor (AI) and herceptin inhibits growth of testosterone‐treated CD44+/CD24low cells. Percentage of viable CD44+/CD24low cells treated as indicated over untreated cells. Data are calculated from growth curves of triplicate six‐well plates with 2.5 × 104 cells/well cultured with 0.5% FBS +100 nM testosterone (C), and 0.7 μg/mL herceptin (H), or 70 nM formestane (F), or both (H+F) for 7 days, and are representative of three different experiments. Error bars indicate SEM. Statistical analysis was done by two‐tailed Student’s t‐test (*P < 0.01; **P < 0.001).
Herceptin and formestane modulate HER1, HER2, and aromatase expression. To address the question of whether HER family receptors mediate the cooperative effect we examined the effect of the drugs on the expression levels of the growth factor receptors HER1 and HER2. We found that herceptin alone, in the presence of testosterone, did not decrease the mRNA levels of HER1/HER2, whereas combined with formestane, it resulted respectively in a 1.5‐ to 3.5‐fold decrease in total HER1 and HER2 mRNA (Fig. 6), which confirms that HER2 can interact with aromatase activity.
Figure 6.
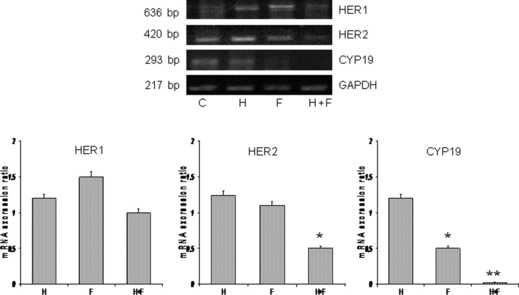
Combined formestane (F) and herceptin (H) inhibit mRNA expression of HER1, HER2, and aromatase (CYP19) in testosterone treated CD44+/CD24low cells (C). The results obtained from RT‐PCR analysis were quantified by densitometry and are displayed in the lower graphs. Statistical differences obtained by comparison with untreated control (C). Error bars indicate SEM of triplicates. Statistical analysis was done by two‐tailed Student’s t‐test (*P < 0.01; **P < 0.001).
Because increased aromatase activity results from increased aromatase gene expression,( 25 , 26 ) we next evaluated the effect of AIs on the levels of mRNA aromatase (CYP19) expression. Formestane alone markedly reduced CYP19 expression (50–60%); the inhibition was greater in the presence of herceptin (85–90%) (Fig. 6). These results suggest that the reduction in aromatase expression could account for the inhibition of growth elicited by the formestane/herceptin combination.
Growth arrest is mediated by cyclin D1. The mitogenic effects of estradiol on breast cancer cells are mediated by cyclin D1.( 27 , 28 ) Stimulation of growth‐arrested cells with members of the EGF family induces D‐type cyclins.( 29 ) Cyclin D1 controls the progression of the cell cycle machinery through the restriction point during the late‐G1 phase when cells commit to DNA synthesis. It is one of the most frequently overexpressed proteins in breast cancer. To investigate whether cyclin D1 plays a role in the growth kinetics of treated CD44+/CD24low cells, we measured protein expression and found that protein levels of cyclin D1 decreased when the formestane/herceptin combination inhibited cell growth (Fig. 7). This result suggests that the ability of CD44+/CD24low cells to proliferate is associated with increased levels of cyclin D1.
Figure 7.
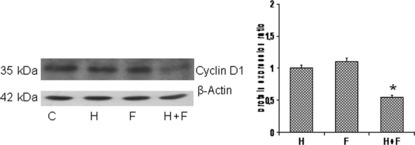
CD44+/CD24low cell growth inhibition is associated with decreased levels of cyclin D1. Lysates of cells treated as reported for growth curves, for 7 days, were immunoblotted for the expression levels of cyclin D1; actin served as loading control. The results obtained from western blotting analysis are quantified by densitometry and displayed in the graph. Statistical differences obtained by comparison with untreated control (C). Error bars indicate SEM of triplicates. Statistical analysis was done by two‐tailed Student’s t‐test (*P < 0.01).
Discussion
The epidermal growth factor receptor (HER) family has been implicated in the development of cancer cell resistance to endocrine therapy.( 29 , 30 ) Clinical data on the benefit of combined treatment with AIs and RTK inhibitors for subgroups of patients with breast cancer are promising, but not in all the patients.( 31 ) The cell surface expression of CD44 and CD24 identifies CD24+/CD44− cells with features of differentiated luminal epithelial cells, and CD44+/CD24− cells with features of basal cells.( 12 ) The levels of ER distinguish luminal A tumors, which are highly ER+, from luminal B and HER2 tumors that have lower ER expression, and co‐express other receptors such as members of the EGF family of growth factor receptors (EGFR/HER1 and erbB2/HER2).( 3 , 4 ) The levels of ER and amplification of HER2 in tumor tissue are respectively predictive of treatment response rates to endocrine therapy and an established prognostic factor in breast cancer.( 4 ) We reported that combined formestane/herceptin treatment of unsorted cells resulted in a percentage of growth inhibition that varied among samples depending on their individual molecular characteristics.( 19 )
In the present study, we investigated the influence of combined formestane/herceptin treatment on a sub‐population of CD44+/CD24low cells from human breast cancer tissue. We show that these cells express cytokeratins and vimentin, possess the features to form sphere‐like aggregates, and contain anchorage‐independent cells. CD44+/CD24low cells express ERα and AR, although E2 and T do not enhance their growth rate. Notably, whereas the breast tumor specimen was classified as HER2‐negative, CD44+/CD24low sorted cells express HER2 phosphorylated.
Co‐targeting of growth factor receptors in ER‐positive breast cancer is a critical issue. In patients with ER‐positive tumors and active growth factor receptor signaling, there is growing evidence that crosstalk between ER and growth factor receptor signaling pathways, especially the HER family, is one of the mechanisms for resistance to endocrine therapy in breast cancer.( 31 , 32 , 33 ) This has created a rationale for using targeted strategies to enhance efficacy of either tamoxifen or estrogen deprivation to overcome endocrine resistance.( 34 ) Several clinical studies now support this idea and have demonstrated a superiority of AIs over tamoxifen in this setting.( 35 , 36 ) In hormone receptor (HR)‐positive, HER2‐positive metastatic breast cancer, two trials demonstrated that the combination either of anastrozole with herceptin( 37 ) or of letrozole and lapatinib( 24 ) significantly prolonged progression‐free survival (PFS) compared with anastrozole or letrozole alone, respectively. In experimental models of hormone‐sensitive ER‐positive breast cancer cells that are initially HER2 negative, HER1 and HER2 pathways may become up‐regulated on development of endocrine resistance over time and a combined growth factor receptor‐ and endocrine‐targeted treatment approach delay acquired resistance.( 38 , 39 , 40 ) In the clinical setting, two randomized phase II trials in HR‐positive metastatic breast cancer patients suggested that the EGFR TKI gefitinib may improve PFS when added to endocrine therapy anastrozole or tamoxifen,( 41 , 42 ) while a double‐blind, placebo‐controlled phase III study( 24 ) that compared letrozole and lapatinib versus letrozole alone showed the inability of lapatinib to delay progression with letrozole in the endocrine‐sensitive, HR‐positive, HER2‐negative population, in contrast to previously reported preclinical and clinical data.( 40 , 41 , 42 ) In this field additional studies are warranted.
Studies analyzing the expression of various genes including estrogen receptor, cytokeratins and HER2 in ductal carcinoma in situ (DCIS) and invasive breast carcinomas demonstrate a high degree of diversity in a subset of tumors.( 43 ) It has been reported that, despite the uniform expression of CD24 or CD44 subsets of tumor cells, these two cell populations are genetically highly heterogeneous and they are likely to display variability for biological and functional traits including tumor‐initiating potential and response to therapeutic agents.( 44 ) In particular, in a study that aimed to evaluate the inter‐ and intra‐tumor expression of stem cell‐related markers at the cellular level in human breast tumors of different subtypes and histologic grade, CD44+/CD24− cells were detected in 69% of all tumors with 100% of the basal‐like and 52% of HER2+ tumors having some of these cells. Within the same tumor, in luminal A tumors, CD44 had higher expression levels in the in situ than in the invasive components, while in HER2+ tumors CD44 expression was higher in the invasive component than in the in situ component.( 45 ) The inter‐ and intra‐tumor heterogeneity of the markers currently used for the categorization of breast tumors into major subtypes (e.g. luminal, HER2+, and basal‐like) reflects the lack of methods for the quantitative assessment of intratumor diversity. Further studies are required to identify features that could be used for establishing the prognosis and predicting the risk of therapeutic resistance.
In our in vitro model system, CD44+/CD24low sorted cells expressed levels of activated HER2. It has been reported that activated HER2 is expressed also in HER2‐negative tumors,( 46 , 47 ) and expression of activated HER2 is associated with poor prognosis in the series of HR‐positive breast cancer patients.( 46 ) This is in agreement with previous reports that show that tumors classified as HER2‐negative express an amount of HER2 protein sufficient to elicit signal transduction upon activation, and this may explain why some patients with HER2‐negative tumors respond to herceptin treatment.( 48 ) Our data are not surprising and confirm other reports that HER2 expression increases the stem/progenitor cell population of both normal and malignant mammary cells, that the effects of HER2 overexpression on mammary tumorigenesis and invasion are due to its effects on the stem cells population, and that these effects are inhibited by herceptin in sensitive cells.( 49 ) When sorted CD44+/CD24low cells were treated with combined submaximal doses of the AIs and herceptin, the drugs cooperated to inhibit growth, which suggests that while the duplication rate cannot be augmented by steroids, the anti‐HER2 drug interferes with the testosterone/aromatase/E2 pathway and inhibits the ability of CD44+/CD24low cells to proliferate. The expression of HER1/HER2 and aromatase (CYP19) partially paralleled the growth inhibition. Although herceptin alone did not decrease the mRNA levels of HER2, its expression levels decreased combined with formestane. Similarly, the inhibition of CYP19 mRNA induced by formestane was greater in the presence of herceptin. Formestane is a type I inhibitor recognized by the active site of aromatase as an alternate substrate; it is converted by aromatase into a reactive intermediate that binds irreversibly and covalently to the binding site of aromatase, permanently inactivating the enzyme.( 6 ) The cross inhibition and the expression of androgen receptor on CD44+/CD24low cells suggest a direct interference with the RTK pathway. Previous studies showed that nonaromatic steroids, precursors or metabolites of endogenous androgens, may serve as ER modulators: they show high binding affinity for human ERα, activate the ERs, and elicit hormonal responses in ERα‐positive cell lines in culture.( 50 ) It is known that the estrogen‐activated ER are translocated into the nucleus and then bind to the specific DNA sequences (ERE) to activate the expression of downstream target genes. In ER‐positive tumors, tamoxifen is one of the most effective therapies, but resistance to tamoxifen is common. Tamoxifen resistant breast tumors are characterized by elevated HER2 levels,( 33 , 51 ) and ER‐positive cell lines overexpressing HER2 acquire resistance to tamoxifen.( 52 ) Clinical features of tamoxifen‐resistant breast cancer are the combined elevation of the amplified in breast cancer‐1 (AIB‐1) and HER2 pathway.( 33 ) An intrinsic transcriptional link between tumors driven by ER and those driven by HER2 has been reported.( 53 ) Hurtado et al. ( 53 ) showed that estrogen‐ER and tamoxifen‐ER complexes directly repress HER2 transcription and implicate paired box gene 2 (PAX 2) as a crucial mediator of ER repression of HER2 by the anticancer drug tamoxifen. PAX2 and the ER co‐activator AIB‐1 compete for binding and regulation of HER2 transcription, the outcome of which determines tamoxifen response in breast cancer cells. The repression of HER2 by ER–PAX2 suggests that aggressive HER2‐positive tumours can originate from ER‐positive luminal tumors by circumventing this repressive mechanism. However, we cannot exclude an in vitro differentiating effect of testosterone that, converted to estradiol by aromatase, could generate ERα‐expressing short‐term transit amplifying cells that give rise to ERα‐positive differentiated cells. Interestingly, in the CD44+/CD24low cells, ERα was expressed in all the conditions tested, and the drug combination failed to completely inhibit growth. This finding warrants further investigation and might explain the correlation between ERα and cyclin D1,( 54 , 55 ) and the correlation between cyclin D1 and anti‐estrogen resistance and disease progression.( 56 ) Cyclin D1 mediates the mitogenic effects of estradiol and controls the EGF stimulated progression of the cell cycle when cells commit to DNA synthesis.( 27 , 28 ) In our study, the level of cyclin D1 was decreased in CD44+/CD24low cells treated with the combination of drugs. This suggests the convergence of mitogenic signals on cyclin D1 and confirms that enhanced cyclin D1 levels are a marker of cell cycle progression.( 57 )
Subtyping breast tumors gives prognostic and predictive information about response to treatment. This study demonstrates that combined AI/herceptin treatment exerts a cooperative inhibition of cell proliferation in a subpopulation of CD44+/CD24low progenitor cells from breast cancer tissue that, in a defined microenvironment, express ERα, HER2, aromatase, and cyclinD1. These features coincide with the clinicopathological characteristics of a large variety of breast tumors( 13 ) and can be modulated by therapeutic agents; the heterogeneity of cells with various CD44/CD24 expressions within individual tumors may be indicative of a cancer stem cell subpopulation giving rise to more differentiated and committed cell populations. This result could lead to a new therapeutic approach that targets subpopulations of cells within the heterogeneous context of tumor tissue. Further studies with the combined use of targeted agent could be useful to understand the mechanism of interaction and to increase the efficiency with which drugs may kill cancer progenitor cells.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by AIRC, Regione Campania, MinisteroUniversità e Ricerca, and Ministero Salute, Italy. We greatly appreciate the help of Drs D. Calabrese and G. Di Martino with the acquisition of human tissue samples. We also are indebted with Jean Ann Gilder for text editing.
References
- 1. Ince TA, Richardson AL, Bell GW et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007; 12: 160–70. [DOI] [PubMed] [Google Scholar]
- 2. Dunning AM, Dowsett M, Healey CS et al. Polymorphisms associated with circulating sex hormone levels in postmenopausal women. J Natl Cancer Inst 2004; 96: 936–45. [DOI] [PubMed] [Google Scholar]
- 3. Sorlie T, Tibshirani R, Parker J et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 2003; 100: 8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sotiriou C, Neo SY, McShane LM et al. Breast cancer classification and prognosis based on gene expression profiles from a population‐based study. Proc Natl Acad Sci U S A 2003; 100: 10393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winer EP. Optimizing endocrine therapy for breast cancer. J Clin Oncol 2005; 8: 1609–10. [DOI] [PubMed] [Google Scholar]
- 6. Santen RJ, Brodie H, Simpson ER et al. History of aromatase: saga of an important biologic mediator and therapeutic target. Endocr Rev 2009; 30: 343–75. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki T, Miki Y, Ohuchi N, Sasano H. Intratumoral estrogen production in breast carcinoma: significance of aromatase. Breast Cancer 2008; 15: 270–7. [DOI] [PubMed] [Google Scholar]
- 8. Isola JJ. Immunohistochemical demonstration of androgen receptor in breast cancer and its relationship to other prognostic factors. J Pathol 1993; 170: 31–5. [DOI] [PubMed] [Google Scholar]
- 9. Suzuki T, Darnel AD, Akahira JI et al. 5alpha‐reductases in human breast carcinoma: possible modulator of in situ androgenic actions. J Clin Endocrinol Metab 2001; 86: 2250–7. [DOI] [PubMed] [Google Scholar]
- 10. Hudelist G, Singer CF, Manavi M et al. Co‐expression of ErbB‐family members in human breast cancer: Her‐2/neu is the preferred dimerization candidate in nodal‐positive tumors. Breast Cancer Res Treat 2003; 80: 353–61. [DOI] [PubMed] [Google Scholar]
- 11. Dowsett M, Houghton J, Iden C et al. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according oestrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann Oncol 2006; 17: 818–26. [DOI] [PubMed] [Google Scholar]
- 12. Shipitsin M, Campbell LL, Argani P et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11: 259–73. [DOI] [PubMed] [Google Scholar]
- 13. Honeth G, Bendahl PO, Ringnér M et al. The CD44+/CD24‐ phenotype is enriched in basal‐like breast tumors. Breast Cancer Res 2008; 10: R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Godar S, Ince TA, Bell GW et al. Growth‐inhibitory and tumor‐ suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008; 134: 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol 2003; 4: 33–45. [DOI] [PubMed] [Google Scholar]
- 16. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 17. Sleeman KE, Kendrick H, Ashworth A et al. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non‐epithelial cells. Breast Cancer Res 2006; 8: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durst B, Sorg RV, Röder G et al. The influence of hormones on CD44 expression in endometrial and breast carcinomas. Oncol Rep 2001; 8: 987–93. [PubMed] [Google Scholar]
- 19. Veneziani BM, Criniti V, Cavaliere C et al. In vitro expansion of human breast cancer epithelial and mesenchymal stromal cells: optimization of a coculture model for personalized therapy approaches. Mol Cancer Ther 2007; 6: 3091–100. [DOI] [PubMed] [Google Scholar]
- 20. Tallarida RJ. Interactions between drugs and occupied receptors. Pharmacol Ther 2007; 113: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al‐Hajj M, Wicha MS, Benito‐Hernandez A et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hurt EM, Kawasaki BT, Klarmann GJ et al. CD44(+)CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer 2008; 98: 756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ponti D, Costa A, Zaffaroni N et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 2005; 65: 5506–11. [DOI] [PubMed] [Google Scholar]
- 24. Johnston S, Pippen J Jr, Pivot X et al. Lapatinib combined with letrozole versus letrozole and placebo as first‐line therapy for postmenopausal hormone receptor‐positive metastatic breast cancer. J Clin Oncol 2009; 27: 5538–46. [DOI] [PubMed] [Google Scholar]
- 25. Salhab M, Reed MJ, Al Sarakbi W et al. The role of aromatase and 17‐beta‐hydroxysteroid dehydrogenase type 1 mRNA expression in predicting the clinical outcome of human breast cancer. Breast Cancer Res Treat 2006; 99: 155–62. [DOI] [PubMed] [Google Scholar]
- 26. Sasano H, Anderson TJ, Silverberg SG et al. The validation of new aromatase monoclonal antibodies for immunohistochemistry – a correlation with biochemical activities in 46 cases of breast cancer. J Steroid Biochem Mol Biol 2005; 95: 35–9. [DOI] [PubMed] [Google Scholar]
- 27. Prall OW, Rogan EM, Musgrove EA et al. c‐Myc or cyclin D1 mimics estrogen effects on cyclin E‐Cdk2 activation and cell cycle reentry. Mol Cell Biol 1998; 18: 4499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altucci L, Addeo R, Cicatiello L et al. 17beta‐Estradiol induces cyclin D1 gene transcription, p36D1‐p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)‐arrested human breast cancer cells. Oncogene 1996; 12: 2315–24. [PubMed] [Google Scholar]
- 29. Arpino G, Wiechmann L, Osborne CK et al. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev 2008; 29: 217–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle 2009; 8: 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicholson RI, McClelland RA, Robertson JF et al. Involvement of steroid hormone and growth factor cross‐talk in endocrine response in breast cancer. Endocr Relat Cancer 1999; 6: 373–87. [DOI] [PubMed] [Google Scholar]
- 32. Shou J, Massarweh S, Osborne CK et al. Mechanisms of tamoxifen resistance: increased estrogen receptor‐HER2/neu cross‐talk in ER/HER2‐positive breast cancer. J Natl Cancer Inst 2004; 96: 926–35. [DOI] [PubMed] [Google Scholar]
- 33. Osborne CK, Bardou V, Hopp TA et al. Role of the estrogen receptor coactivator AIB1(SRC‐3) and HER‐2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003; 95: 353–61. [DOI] [PubMed] [Google Scholar]
- 34. Massarweh S, Osborne CK, Jiang S et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor‐positive, HER‐2/neu‐positive breast cancer. Cancer Res 2006; 66: 8266–73. [DOI] [PubMed] [Google Scholar]
- 35. Banerjee S, Smith IE, Folkerd L et al. Comparative effects of anastrozole, tamoxifen alone and in combination on plasma lipids and bone‐derived resorption during neoadjuvant therapy in the impact trial. Ann Oncol 2005; 16: 1632–8. [DOI] [PubMed] [Google Scholar]
- 36. Ellis MJ, Tao Y, Young O et al. Estrogen‐independent proliferation is present in estrogen‐receptor HER2‐positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol 2006; 24: 3019–25. [DOI] [PubMed] [Google Scholar]
- 37. Kaufman B, Mackey JR, Clemens MR et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2‐positive, hormone receptor‐positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol 2009; 27: 5529–37. [DOI] [PubMed] [Google Scholar]
- 38. Nicholson RI, Hutcheson IR, Knowlden JM et al. Nonendocrine pathways and endocrine resistance: observations with antiestrogens and signal transduction inhibitors in combination. Clin Cancer Res 2004; 10: 346S–54S. [DOI] [PubMed] [Google Scholar]
- 39. Schiff R, Osborne CK. Endocrinology and hormone therapy in breast cancer: new insight into estrogen receptor‐alpha function and its implication for endocrine therapy resistance in breast cancer. Breast Cancer Res 2005; 7: 205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Massarweh S, Osborne CK, Creighton CJ et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 2008; 68: 826–33. [DOI] [PubMed] [Google Scholar]
- 41. Cristofanilli M, Valero V, Mangalik A et al. A phase II multicenter, double‐blind, randomized trial to compare anastrozole plus gefitinib with anastrozole plus placebo in postmenopausal women with hormone receptor‐positive (HR_) metastatic breast cancer (MBC). J Clin Oncol 2008; 26: 44s(suppl; abstr 1012). [Google Scholar]
- 42. Osborne K, Neven P, Dirix L et al. Randomized phase II study of gefitinib (IRESSA) or placebo in combination with tamoxifen in patients with hormone receptor positive metastatic breast cancer. Breast Cancer Res Treat 2007; 106(Suppl 1): S107. [Google Scholar]
- 43. Allred DC, Wu Y, Mao S et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res 2008; 14: 370–8. [DOI] [PubMed] [Google Scholar]
- 44. Park SY, Gönen M, Kim HJ et al. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 2010; 120: 636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park SY, Lee HE, Li H et al. Heterogeneity for stem cell‐related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res 2010; 16: 876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frogne T, Laenkholm AV, Lyng MB et al. Determination of HER2 phosphorylation at tyrosine 1221/1222 improves prediction of poor survival for breast cancer patients with hormone receptor‐positive tumors. Breast Cancer Res 2009; 11: R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singer CF, Gschwantler‐Kaulich D, Fink‐Retter A et al. HER2 overexpression and activation, and tamoxifen efficacy in receptor‐positive early breast cancer. J Cancer Res Clin Oncol 2009; 135: 807–13. [DOI] [PubMed] [Google Scholar]
- 48. Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med 2008; 358(13): 1409–11. [DOI] [PubMed] [Google Scholar]
- 49. Korkaya H, Paulson A, Iovino F et al. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008; 27: 6120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang P, Wen Y, Han GZ et al. Characterization of the oestrogenic activity of non‐aromatic steroids: are there male‐specific endogenous oestrogen receptor modulators? Br J Pharmacol 2009; 158: 1796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Placido S, De Laurentiis M, Carlomagno C et al. Twenty‐year results of the Naples GUN randomized trial: predictive factors of adjuvant tamoxifen efficacy in early breast cancer. Clin Cancer Res 2003; 9: 1039–46. [PubMed] [Google Scholar]
- 52. Benz CC, Scott GK, Sarup JC et al. Estrogen‐dependent, tamoxifen‐resistant tumorigenic growth of MCF‐7 cells transfected with HER2/neu. Breast Cancer Res Treat 1992; 24: 85–95. [DOI] [PubMed] [Google Scholar]
- 53. Hurtado A, Holmes KA, Geistlinger TR et al. Regulation of ERBB2 by oestrogen receptor‐PAX2 determines response to tamoxifen. Nature 2008; 456: 663–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pacilio C, Germano D, Addeo R et al. Constitutive overexpression of cyclin D1 does not prevent inhibition of hormone‐responsive human breast cancer cell growth by antiestrogens. Cancer Res 1998; 58: 871–6. [PubMed] [Google Scholar]
- 55. Bindels EM, Lallemand F, Balkenende A et al. Involvement of G1/S cyclins in estrogen‐independent proliferation of estrogen receptor‐positive breast cancer cells. Oncogene 2002; 21: 8158–65. [DOI] [PubMed] [Google Scholar]
- 56. Reis‐Filho JS, Savage K, Lambros MB et al. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: an immunohistochemical and chromogenic in situ hybridisation analysis. Mod Pathol 2006; 19: 999–1009. [DOI] [PubMed] [Google Scholar]
- 57. Wang Y, Dean JL, Millar EK et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res 2008; 68: 5628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]