

Abstract
Melanoma inhibitory activity (MIA) is an 11‐kDa secretory protein isolated from malignant melanoma cells that is correlated with invasion and metastasis in various human malignancies. We examined MIA expression in 62 oral squamous cell carcinomas (OSCC) by immunohistochemistry. MIA expression was significantly associated with nodal metastasis (P = 0.00018). MIA expression was also associated with expression of high mobility group box‐1 (HMGB1) (P < 0.0001) and lymph vessel density (P < 0.0001). Expression levels of MIA, HMGB1, nuclear factor kB (NFkB) p65 and HMGB1–NFkB p65 binding were significantly higher in a metastatic human OSCC cell line (HSC3) than those in a non‐metastatic OSCC cell line (HSC4). Treatment with receptor for advanced glycation end products (RAGE) antisense or small interfering RNA and human recombinant HMGB1 (hrHMGB1) did not affect MIA expression, whereas HMGB1 antisense or siRNA treatment decreased MIA expression in HSC3 cells. Then HMGB1 enhanced MIA expression as an NFkB cofactor but not as a RAGE ligand. MIA neutralization by MIA antibodies increased extracellular signal‐related kinase 1/2 phosphorylation, but decreased p38 phosphorylation and the expression of vascular epithelial growth factor (VEGF)‐C and ‐D. Treatment with p38 inihibitor decreased VEGF‐C and ‐D expression in HSC3 cells. These results suggest that MIA expression is enhanced by the interaction of intracellular HMGB1 and NFkBp65 and MIA is closely involved in tumor progression and nodal metastasis by the increments of VEGF‐C and VEGF‐D in OSCC. (Cancer Sci 2008; 99: 1806– 1812)
Abbreviation:
- OSCC
oral squamous cell carcinoma
- MIA
melanoma inhibitory activity
- HMGB1
high mobility group box‐1
- RAGE
receptor for advanced glycation end products
- NFkB
nuclear factor kB
Head and neck cancer is the sixth most common malignancy worldwide and the first leading cause of cancer death in South Asia.( 1 ) About 300 000 patients develop OSCC every year in the world.( 2 , 3 ) OSCC has a high potential for nodal metastasis and locoregional invasion,( 4 ) from which over 50% of patients die.( 5 , 6 ) To control lymph‐node metastasis of OSCC, we need to study the molecular aspects of the mechanism of metastasis.
MIA is an 11‐kDa secretory protein isolated from supernatants of HTZ‐19 malignant melanoma cells,( 7 , 8 ) the gene locus of which is mapped to chromosome 19q13.32–13.33.( 9 ) Although previous reports indicated that MIA is correlated with invasion and metastasis in malignant melanoma,( 10 , 11 , 12 ) breast cancer,( 10 ) chondrosarcoma,( 13 ) glioma,( 14 ) and pancreatic cancer,( 15 ) the definite functions of MIA for cancer cells are still unclear.
HMGB1 has a dual role as an extracellular secretory protein and a chromosomal structural protein.( 16 ) HMGB1 works as a cytokine or a growth factor in neural ontogenesis, septic inflammation and neoplasm. HMGB1 is also considered an amphoterin, which is isolated as a motility factor in neurite outgrowth.( 17 ) We previously reported coexpression of HMGB1 and receptor for advanced glycation end products (RAGE), which is a major membrane receptor for HMGB1 and is significantly associated with tumor progression and metastasis( 18 , 19 , 20 , 21 , 22 , 23 , 24 ) and suppression of tumor‐associated macrophages.( 25 , 26 ) As a chromatin structural protein, HMGB1 participates in gene expression, DNA repair and functions of the p53 family.( 27 ) Recently, HMGB1 was revealed to interact with NFkB p65 to accelerate MIA expression.( 28 , 29 ) HMGB1 and NFkB p65 concurrently bind to a 30‐bp region in the promoter region of the MIA gene designated as the highly conserved region (HCR).
MIA is suspected to play an important role as a pro‐metastatic factor in HMGB1‐overexpressing cancers. In the present study, we analyzed the relationship between MIA expression and nodal metastasis and HMGB1 expression in human OSCC.
Materials and Methods
Tumor specimens. Sixty‐two formalin‐fixed, paraffin‐embedded specimens of primary OSCC were randomly selected at Nara Medical University Hospital, Kashihara, Japan. None of the samples was treated using neo‐adjuvant therapy. Medical records and prognostic follow‐up data were obtained from the patient database administered by the hospital. None of the patients was treated before surgery and sample preparation.
Immunohistochemistry. Consecutive 4‐µm sections were cut from each block. Immunohistochemistry was performed as previously described.( 24 ) The sections were subjected to antigen retrieval with pepsin (DAKO, Carpinteria, CA, USA) treatment for 20 min and the immunoperoxidase technique was used in immunostaining. After blockade of endogeneous peroxidase activity by incubation in 3% hydrogen peroxide–methanol for 15 min, the sections were rinsed with phosphate‐buffered saline (PBS) and incubated with diluted primary antibodies: Anti‐MIA antibody,( 10 ) anti‐HMGB1 antibody (Upstate biotechnology, Lake Placid, NY, USA; diluted to 0.5%) and anti‐D2‐40 antibody (a marker for lymph duct endothelial cells recognizing sialo‐glycoprotein type O, Signet Laboratories Inc., Dedham, MA, USA; diluted to 0.5%). After 2 h incubation at room temperature, they were rinsed again with PBS and treated for an hour with the secondary antibody peroxidase‐conjugated antigoat (Medical & Biological Laboratories, Nagoya, Japan) or antirabbit (Medical & Biological Laboratories) diluted to 0.5%. All sections were then rinsed with PBS, color‐developed using diaminobenzidine (DAB) solution (DAKO), washed in water and counterstained with Meyer's hematoxylin (Sigma Chemical, St. Louis, MO, USA). Immunostaining of all samples was performed under the same conditions of antibody reaction and DAB exposure.
Evaluation of immunoreactivity. Immunoreactivity was classified according to Allred's score (AS).( 30 ) We divided the immunoreactivity into four grades by AS: Grade 0, AS is 0; Grade 1, AS is 2–4; Grade 2, AS is 5–6; Grade 3, AS is 7–8. MIA positiveness was exhibited as grades 2 and 3. Labeling index for HMGB1 was calculated as follows: (the number of the HMGB1 positive nucleus/total number of the nucleus) × 100. Anti‐D2‐40 immunostained specimens were observed under 200× magnification microscopy and three maximum lymph vessel density (LVD) fields were selected from around of the tumor cells (the ‘hot spot’). These fields were captured by digital imaging with charge coupled device camera (Olympus, Tokyo, Japan). LVD and mean lymph vessel area (MLA) were measured on the computer‐captured image using NIH Image software (National Institutes of Health, Bethesda, MD, USA).
Cell culture. Human OSCC cell lines, HSC3 and HSC4, were studied. HSC3 cells are tongue squamous cell carcinoma‐derived metastatic cell lines, which provide many sublines with high metastatic potential. In contrast, HSC4 cells show low metastatic potential. No metastatic sublines are derived from HSC4 cells.( 31 ) Controlled cell line was used for U937 (monocytic leukemia cell line, purchased from Dainihon Pharmaceutical, Tokyo, Japan). All cells were maintained in Roswell Park Memorial Institute medium (RPMI)‐1640 (Sigma Chemical) supplemented with 10% fetal bovine serum (Sigma Chemical Co) in 5% CO2 and 95% air at 37°C. Anti‐MIA antibody was used for neutralizing MIA (Santa Cruz Biotechnology) in cultured medium at 2 µL/mL concentration. Rabbit serum (DAKO) was used for control. The cells were also treated with p38 mitogen‐activated protein kinase (MAPK) inhibitor SB239063 (Sigma Chemical) at 5 µM for 12 h.
Short interferent RNA. FlexiTube short interferent RNA (siRNA) for MIA, RAGE and HMGB1 were purchased from Qiagen Genomics (Bothell, WA, USA). AllStars Negative Control siRNA was used for control (Qiagen Genomics). These cells were transfected with 50‐nM siRNA for each gene using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Antisense phosphorothioate(S)‐oligodeoxynucleotide assay. The 18‐mer S‐oligodeoxynucleotide (ODN) for antisense sequence from 6th to 23rd nucleotide of RAGE cDNA (GenBank AB036432) and the 18‐mer S‐ODN for antisense sequence from 1st to 18th nucleotide of HMGB1 cDNA (GenBank X12597) were synthesized and purified by reverse‐phase high performance liquid chromatography (Sigma Genosys, Ishikari, Japan). The sequence of RAGE antisense was 5′‐CTG CTT CCT TCC AGG GTC‐3′; the sequence of HMGB1 antisense was 5′‐AGG ATC TCC TTT GCC CAT‐3′. The sense sequence of the antisense S‐ODN was used for the control S‐ODN. These cells were pretreated with 3 µM of antisense or sense S‐ODN for 6 days with medium exchange and an addition of antisense or sense S‐ODN every 2 days. After pretreatment, the cells were used for further experiments. Cytotoxicity of the antisense S‐ODN were not relevant at the working concentration.( 19 )
Immunoblot analysis. Whole‐cell lysates were prepared as described previously.( 26 ) Fifty‐microgram lysates were subjected to immunoblot analysis in 12.5% sodium dodecyl sulfate–polyacrylamide gels followed by electrotransfer to nitrocellulose filters. The filters were incubated with primary antibody and then with peroxidase‐conjugated immunoglobulin G antibody (Medical and Biological Laboratories, Nagoya, Japan). A γ‐tubulin antibody was used to assess the levels of protein loaded per lane (Oncogene Research Products, Cambridge, MA, USA). The immune complex was visualized using an enhanced chemiluminescence Western‐blot detection system (Amersham, Aylesbury, UK). The primary antibodies used were anti‐MIA, anti‐NFkB p65, anti‐VEGF‐C, anti‐VEGF‐D, anti‐ERK1/2, antiphospho‐ERK1/2, antiphospho‐p38 (Santa Cruz Biotechnology), anti‐integrin α5β1 (Serotec Ltd, Oxford, UK) and anti‐HMGB1 (Upstate Biotechnology).
Immunoprecipitation. For immunoprecipitation, the lysates were precleaned in a lysis buffer containing protein A/G agarose (Santa Cruz Biotechnology) for 1 h at 4°C and subsequently centrifuged. The supernatants were incubated with anti‐HMGB1 antibody (Upstate Biotechnology), or anti NFkB p65 antibody (Santa Cruz Biotechnology) and protein A/G agarose for 3 h at 4°C. The precipitates were collected by centrifugation and washed 5 times with lysis buffer for sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE). For loading control, 5 µL of each preimmunoprecipitated sample (lysate diluted with buffer) was slot‐blotted onto nitrocellulose membrane and stained with Coomassie blue.
Reverse transcriptase–polymerase chain reaction. Total RNA was extracted from cultured cells using the RNeasy Mini Kit (Qiagen Genomics). Total RNA (1 µg) was converted to cDNA with the First‐Strand cDNA Synthesis Kit (Amersham Biosciences, Piscataway, NJ, USA). PCR were performed using Amplitaq Gold Kit (Applied Biosynthesis, Foster City, CA, USA) according to the manufacturer's instructions.
Primer pairs used are listed below: MIA (referred to GenBank NM006533), Sense: 5′‐ACCCTA TCT CCA TGG CTG TG‐3′, Antisense: 5′‐AGG TTT CAG GGT CTG GTC CT‐3′; RAGE (referred to GenBank NM172197), Sense: 5′‐GCT GTC AGC ATC AGC ATC AT‐3′, Antisense: 5′‐ATT CAG TTC TGC ACG CTC CT‐3′; HMGB1 (referred to GenBank NM002128), Sense: 5′‐ATA TGG CAA AAG CGG ACA AG‐3′, Antisense: 5′‐GCA ACA TCA CCA ATG GAC AG‐37; VEGF‐C (referred to GenBank NM005429), Sense: 5′‐GGA AAG AAG TTC CAC CAC CA‐3′, Antisense: 5′‐TTT GTT AGC ATG GAC CCA CA‐3′; VEGF‐D (referred to GenBank NM004469), Sense: 5′‐AGG ACT GGA AGC TGT GGA GA‐3′, Antisense: 5′‐ATC GGA ACA CGT TCA CAC AA‐3′; β‐actin (referred to GenBank NM001101), Sense: 5′‐CAA GAG ATG GCC ACG GCT GCT‐3′, Antisense: 5′‐TCC TTC TGC ATC CTG TCG GCA‐3′. All primers were synthesized by Sigma Genosys (Ishikari, Japan).
Statistical analyses. Statistical differences in MIA expression were tested with the two‐tailed χ2 test. All statistics were calculated using StatView version 4.5 (SAS Institute, Cary, NC, USA) and a P‐value of less than 0.05 was considered statistically significant.
Results
Relationship between MIA expression and clinical parameters. We examined MIA expression in 62 cases of OSCC (Table 1). Expression of MIA was observed in 48.4% of all cases (30/62). Immunoreactivity for MIA was found in the cell membrane and cytoplasm, except in the nuclei of the cancer cells, but was not found in normal epithelium of all cases (Fig. 1).
Table 1.
Relationship between melanoma inhibitory activity (MIA) expression and clinicopathological parameters in oral squamous cell carcinoma (OSCC) patients
| n | MIA expression | P‐value † | ||
|---|---|---|---|---|
| Negative | Positive | |||
| Age | ||||
| –60 | 43 | 25 | 18 | |
| 60– | 19 | 7 | 12 | NS |
| Sex | ||||
| Male | 35 | 17 | 18 | |
| Female | 27 | 15 | 12 | NS |
| Primary site | ||||
| Tongue | 40 | 22 | 18 | |
| Gingiva | 14 | 7 | 7 | |
| Buccal mucosa | 5 | 2 | 3 | |
| Hard palate | 3 | 1 | 2 | NS |
| Histological differentiation | ||||
| Well | 33 | 16 | 17 | |
| Moderately | 26 | 13 | 13 | NS |
| Poorly | 3 | 3 | 0 | |
| T classification (extension of primary tumor)( 47 ) | ||||
| T1, 2 | 21 | 12 | 9 | |
| T3 | 17 | 7 | 10 | |
| T4 | 24 | 13 | 11 | NS |
| Clinical stage( 47 ) | ||||
| I, II | 11 | 9 | 2 | |
| III | 25 | 11 | 14 | |
| IV | 26 | 12 | 14 | NS |
| Histological nodal metastasis( 47 ) | ||||
| n– | 23 | 19 | 4 | |
| n+ | 39 | 13 | 26 | 0.00018 |
| Disease recurrence | ||||
| (–) | 35 | 19 | 16 | |
| (+) | 27 | 13 | 14 | NS |
Calculated χ2 test.
Figure 1.
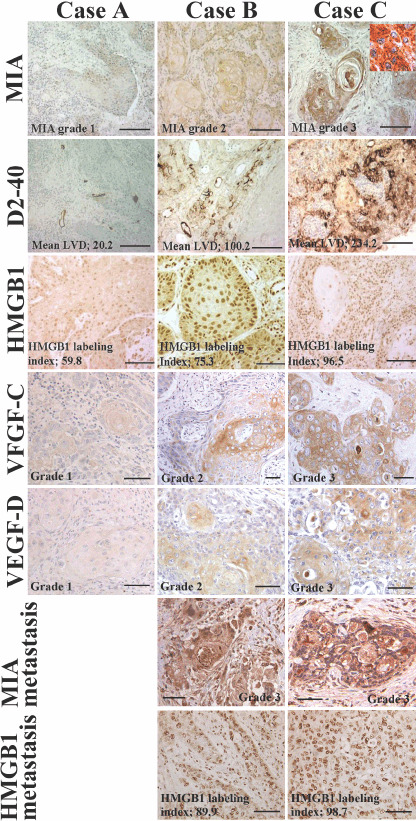
Immunohistochemical staining of melanoma inhibitory activity (MIA), D2‐40, high mobility group box‐1 (HMGB1), vascular endothelial growth factor (VEGF)‐C and VEGF‐D in oral squamous cell carcinomas (OSCC). The expressions of D2‐40, HMGB1, VEGF‐C and VEGF‐D were shown in case A (T2N0M0, stage II, well differentiated OSCC), case B (T2N1M0, stage III, well differentiated OSCC) and case C (T4N2M0, stage IV, well differentiated OSCC). Inset showed MIA localization at the cytoplasm and cytoplasmic membrane. Expressions of MIA and HMGB1 were examined in lymph node metastasis in case B and C. Bar, 100 µm.
A significant association was found between MIA immunoreactivity and histological metastasis of lymph nodes. Four of 23 cases (17.4%) without nodal metastasis (n‐) expressed MIA, whereas MIA expression was found in 26 of 39 (66.7%) cases with nodal metastasis (n+) (P = 0.00018). Representative cases showed significant MIA expression in the nodal metastatic foci (Fig. 1, MIA in metastasis). HMGB1 labeling indices were also high in the nodal metastatic foci (Fig. 1, HMGB1 in metastasis). However, no significant relationship was found between MIA grading and other parameters: age, sex, primary site, histological differentiation, T classification (extension of primary tumor), clinical stage, tumor recurrence and disease‐free survival. To confirm significance of MIA to lymph‐node metastasis in OSCC, we examined LVD and MLA in tumor tissues (Fig. 2a,b). Lymph vessels around the tumor cells were detected by a lymph duct endothelial marker, D2‐40 (Fig. 1). A significant correlation was observed between grading of MIA immunoreactivity and LVD (P < 0.0001) or MLA (P < 0.0001).
Figure 2.
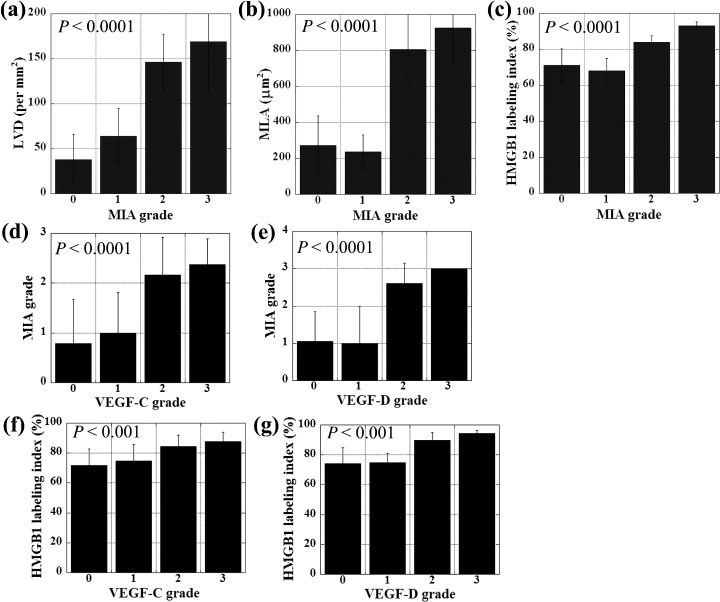
Relationship between MIA expression and lymph vessels, and expressions of HMGB1, VEGF‐C and VEGF‐D. MIA grading was compared with lymph vessel density (LVD) (a), mean lymph vessel area (MLA) (b), HMGB1 labeling index (c), VEGF‐C grading (d),and VEGF‐D grading (e) in OSCC. HMGB1 labeling index was compared with VEGF‐C grading (f), and VEGF‐D grading (g) in OSCC. Expressions of MIA, VEGF‐C, and VEGF‐D were categorized according to Allred's score (AS).( 30 ) Grade 0, AS is 0; Grade1, AS is 2‐4; Grade 2, AS is 5‐6; Grade 3, AS is 7‐8. Error bar, SD.
As a chromatin structural protein, HMGB1 is reported to participate in MIA transcription in association with NFkB.( 28 , 29 ) To examine the role of HMGB1 in MIA expression in OSCC, we compared the expressions of MIA and HMGB1. HMGB1 immunoreactivity was found in cancer cell nuclei (Fig. 1, HMGB1). MIA grading index was significantly correlated with HMGB1 labeling (P < 0.0001) (Fig. 2c).
VEGF‐C and VEGF‐D were examined as the expressions of lymphangiogenesis ‐related growth factors in OSCC cases (Fig. 1). We showed comparison between expressions of VEGF‐C or VEGF‐D and MIA grade or HMGB1 labeling index (Fig. 2d–g). Immunostaining grades of VEGF‐C and VEGF‐D were correlated with MIA grade (P < 0.0001) and HMGB1 labeling index (P < 0.001). All above results suggest that MIA is associated with lymph‐node metastasis of OSCC by up‐regulation of lymphangiogenic factors, VEGF‐C and VEGF‐D.
Relation of MIA expression with HMGB1 or NFkB p65. We next compared the expression level of MIA, HMGB1 and NFkB p65 in metastatic HSC3 and non‐metastatic HSC4 human OSCC cell lines by immunoblotting (Fig. 3). In HSC3 cells, expression levels of MIA, HMGB1 and NFkB p65 were higher than HSC4. U937 monocytic cells, which showed lower HMGB1 and higher NFkBp65 expression levels, expressed MIA at an untraceable level. A physical association between HMGB1 and NFkB p65 was also examined in order to be compared with HMGB1 expression (Fig. 3). The levels of NFkB p65 detected in HMGB1‐immunoprecipitants of HSC3 cells were 4.5‐times higher than that in HSC4 cells. In U937 cells, NFkB p65 binding with HMGB1 is 1/10 of that in HSC4 cells. HMGB1–NFkB p65 binding levels in HSC3, HSC4 and U937 cells were well correlated with those of MIA expression levels.
Figure 3.
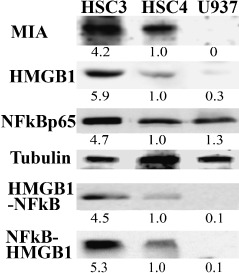
Expression of melanoma inhibitory activity (MIA), high mobility group box‐1 (HMGB1) and nuclear factor kB (NFkB) in human oral squamous cell carcinoma (OSCC) cells. MIA expression was compared with the expressions of HMGB1, NFkBp65 and coprecipitation between NFkBp65 and HMGB1 in HSC3, HSC4 human OSCC cells and U937 monocytic cells. Expressions of MIA, HMGB1 and NFkBp65 were examined by immunoblotting. Tubulin was served as a loading control. Co‐precipitation of HMGB1 and NFkB was examined by immunoprecipitation. A precipitant with anti‐HMGB1 antibody was detected with anti‐NFkBp65 antibody by immunoblotting. Reverse immunoprecipitation was also examined. A precipitant with anti‐NFkBp65 antibody was detected with anti‐HMGB1 antibody.
Role of HMGB1 on MIA expression in HSC3 OSCC cells. HMGB1 has a dual role as a chromatin structural protein and as a cytokine accelerating macrophage‐associated inflammation in cancer growth and invasion. We examined which form of HMGB1 participated in MIA expression (Fig. 4a). Exposure to antisense S‐ODN for HMGB1 receptor RAGE did not affect MIA expression levels. The addition of hrHMGB1 into the culture medium also did not affect MIA expression. In contrast, reduction of intracellular protein by HMGB1 antisense S‐ODN treatment decreased MIA expression. We confirmed the effects of RAGE and HMGB1 on decreasing the MIA expression by using siRNA for HMGB1 (Fig. 4b,c). RAGE siRNA decreased RAGE mRNA expression, but not MIA mRNA, whereas HMGB1 siRNA decreased HMGB1 and MIA mRNA expression in HSC3 cells. These results suggest that HMGB1 is significant in MIA induction as a transcriptional cofactor with NFkBp65 but not as a RAGE ligand.
Figure 4.
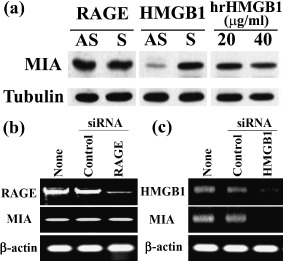
Effects of antisense S‐ODN and siRNA for receptor for advanced glycation end products (RAGE) and high mobility group box‐1 (HMGB1) and hrHMGB1 on melanoma inhibitory activity (MIA) expression in HSC3 human oral squamous cell carcinoma (OSCC) cells. (a) MIA expression was examined by immunoblotting in HSC3 human OSCC cells treated with antisense (AS) or sense (S) S‐ODN for RAGE and HMGB1 and human recombinant HMGB1 (hrHMGB1). Tubulin was served as a loading control. (b, c) MIA expression was examined by reverse transcriptase–polymerase chain reaction in HSC3 cells with or without treatment with siRNA for RAGE and HMGB1 or control siRNA. β‐actin was served as a loading control.
MIA intracellular signals and expressions of VEGF‐C and VEGF‐D. Finally, we examined the MIA effects on HSC3 intracellular signals (Fig. 5). Expressions of VEGF‐C and VEGF‐D were examined by RT‐PCR in HSC3, HSC4 and U937 cells (Fig. 5a). Metastasis‐derived HSC3 cells expressed VEGF‐C and VEGF‐D, whereas original tumor‐derived HSC4 cells expressed only VEGF‐C. U937 monocytes did not express VEGF‐C nor VEGF‐D. As shown in Fig. 5(b), inhibiting the expression of MIA secreted by HSC3 cells with anti‐MIA antibody increased the phosphorylated form of ERK1/2, whereas phosphorylated p38 was decreased. Expressions of pro‐lymphangiogenic growth factors and VEGF‐C and VEGF‐D were inhibited by MIA neutralization in the antibody treatment. To confirm the relationship of p38 phosphorylation level with VEGF‐C or VEGF‐D expression, we examined the effect of p38 inihibitor on expression (Fig. 5c). HSC3 cells treated with p38 inhibitor (SB239063) showed decreased expressions of VEGF‐C and VEGF‐D by RT‐PCR examination. Thus, MIA up‐regulates VEGF‐C and VEGF‐D through p38 activation.
Figure 5.
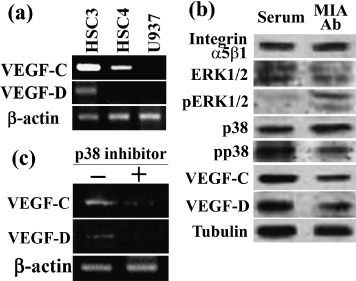
Melanoma inhibitory activity (MIA) intracellular signaling and vascular endothelial growth factor (VEGF)‐C and VEGF‐D expression in HSC3 cells. (a) Expression of VEGF‐C and VEGF‐D was examined by reverse transcriptase–polymerase chain reaction (RT‐PCR) in HSC3, HSC4 and U937 cells. β‐actin was served as a loading control. (b) Effects of anti‐MIA antibody on phosphorylation of extracellular signal‐related kinase (ERK)1/2 and p38 and expressions of integrin α5β1, VEGF‐C and VEGF‐D in HSC3 human oral squamous cell carcinoma (OSCC) cells. Protein levels of MIA, ERK1/2, phosphorylated ERK1/2 (pERK1/2), p38, phosphorylated p38 (pp38), VEGF‐C and VEGF‐D were examined by immunoblotting in HSC3 human OSCC cells treated with MIA antibody. Tubulin was served as a loading control. (c) The effect of inhibition of p38 on expression of VEGF‐C and VEGF‐D. HSC3 cells were treated with p38 inhibitor (SB239063). Expression of VEGF‐C and VEGF‐D was examined by RT‐PCR. β‐actin was served as a loading control.
Discussion
In the present study, we showed immunoreactivity of MIA was significantly correlated with nodal metastasis, LVD and HMGB1 labeling index in OSCC specimens. In in vitro examinations, MIA expression was associated with the expression levels of HMGB1 (not as a cytokine but as an intracellular form) and HMGB1–NFkB p65 binding. In a metastatic OSCC cell line, HSC3 showed higher MIA expression, which was associated with higher expression levels of HMGB1, NFkB p65 and HMGB1–NFkB p65 binding than those in a non‐metastatic OSCC cell line (HSC4). Moreover, MIA expression was associated with VEGF‐C and VEGF‐D expression in HSC3 cells.
HMGB1 is a chromatin structural protein and also acts as a cytokine or growth factor.( 32 , 33 ) Nuclear HMGB1 is recruited in gene replication, repair and transcription, whereas secreted HMGB1 worsens endotoxemic inflammation as a late mediator.( 33 ) Secreted HMGB1 is also known as a pro‐tumoral factor, as is a RAGE ligand.( 34 ) We reported that co‐overexpression of HMGB1 and RAGE is significantly associated with tumor progression and metastasis in gastric cancer,( 24 ) colon cancer,( 21 , 23 ) prostate cancer( 22 ) and malignant transformation of colorectal adenoma.( 35 ) Further, we found that HMGB1 induces apoptosis of macrophages, which is associated with colon cancer metastasis( 25 , 26 ) and enhanced extracellular secretion of HMGB1 in colon cancer.( 36 ) We also reported that a high expression level of RAGE is correlated with tumor progression and recurrence, but not with lymph‐node metastasis in OSCC.( 18 , 19 , 20 ) RAGE activation with HMGB1 as a ligand induced VEGF expression, but not VEGF‐C in HCS3 and HCS4 OSCC cells.( 19 ) In the present study, HMGB1 treatment did not alter MIA expression, whereas HMGB1 antisense S‐ODN treatment decreased MIA expression. HMGB1 is able to bind to HCR located in the promoter region of the MIA gene where HMGB1 interacts with NFkB p65 to enhance MIA expression transcriptionally.( 28 , 29 ) In the present study, we confirmed that physical association between HMGB1 and NFkB p65 regulates induction of MIA in OSCC. From these findings, HMGB1 might induce MIA expression acting as a chromosomal protein.
MIA takes a pivotal role for progression and metastasis in melanoma.( 12 ) MIA promotes cell detachment, migration and invasion and inhibits apoptosis of the cancer cells and infiltration of lymphokine activated killer cells (LAK). MIA binds to fibronectin via SH3 domain‐like structures, which inhibits cell‐to‐stromal attachment.( 37 , 38 ) Further, MIA is able to bind to cell surface integrin α4β1 and α5β1, which suggests MIA might play a role as a ligand for selected integrins.( 39 )
In the present study, we found a significant relationship between MIA expression and LVD in OSCC tumors. Expression of integrin α5β1 in lymph vessel endothelial cells is associated with outgrowth of new lymphatic vessels.( 40 ) MIA might stimulate lymphatic endothelial cells directly to induce lymphangiogenesis. We also found a relationship between MIA expression and VEGF‐C and VEGF‐D expression in HSC‐3 OSCC cells, which expressed integrin α5β1. MAPK activity is reported to be affected by MIA,( 39 ) which we confirmed in the present study. ERK1/2 phosphorylation was recovered by MIA neutralization by the antibody treatment. In contrast, p38 phosphorylation levels were decreased by the antibody treatment. VEGF‐C expression is inhibited by p38 inhibitor but not by ERK1/2 inhibitor.( 41 ) We also confirmed the significance of p38 activation on up‐regulation of VEGF‐C and ‐D in HSC3 cells. The alteration of the signal balance between ERK1/2 and p38 might be associated with up‐regulation of VEGF‐C and ‐D expression by MIA. Further examination will reveal the details of the mechanism of MIA‐dependent VEGF‐C and ‐D induction. VEGF‐C and ‐D are known as strong lymphangiogenic factors in various cancers.( 42 ) Increased VEGF‐C expression is associated with cervical lymph‐node metastasis in head and neck cancer.( 43 ) Although there are still controversies about the role of VEGF‐D in lymph‐node metastasis, VEGF‐D expression is also associated with lymph‐node metastasis in the animal model.( 44 ) Our data suggest that up‐regulation of VEGF‐C and ‐D might explain the relationship between MIA expression and lymph‐node metastasis in OSCC.
HSC3 cells are tongue squamous cell carcinoma‐derived metastatic cell lines, which provide many sublines with high metastatic potential. In contrast, HSC4 cells show low metastatic potential. No metastatic sublines are derived from HSC4 cells.( 45 ) Metastatic HSC3 cells show colony formation in the type‐I collagen matrix, adherence to type‐IV collagen,( 45 ) high heparanase activity( 31 ) and reduced nm23H1 expression and up‐regulated matrix metalloproteinase (MMP)‐2 and ‐9( 46 ) in comparison with HSC4 cells. In the present study, HSC3 cells showed overexpression of MIA, HMGB1, NFkBp65, VEGF‐C and VEGF‐D in comparison with HSC4 cells. Thus the increase of lymphangiogenic capacity might be associated with high potential of lymph‐node metastasis in HSC3. Further examination of the lymphangiogenic capacity might control lymph‐node metastasis in OSCC. In the present study, MIA expression was associated with a high incidence of lymph‐node metastasis, whereas MIA expression did not correlate with recurrence nor with disease‐free survival. Our previous data show that RAGE–HMGB1 coexpression is associated with T classification (extension of primary tumor) but not nodal metastasis in OSCC; however, RAGE expression is closely associated with recurrence and disease‐free survival.( 18 , 19 , 20 ) Many OSCC recurred at the local sites but not from nodal metastasis (data not shown). Local recurrence of OSCC depends on aggressiveness of cancer invasion and anatomical difficulties in the head to obtain sufficient surgical margins. In OSCC, both metastatic potential and local aggressiveness are significant factors to determine the disease outcome.
We showed that MIA was expressed in more than 60% of metastasized OSCC. Considering MIA is a secretory protein, MIA might be a useful marker for metastasis of OSCC, which is detectable in the serum and saliva of OSCC patients. Further examination of MIA might be expected to provide a new target for suppression of lymph‐node metastasis.
Acknowledgments
This work was supported in part by Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, Japan. The authors thank Ms Kaori Isobe for expert assistance with the preparation of this manuscript.
References
- 1. Paterson IC, Eveson JW, Prime SS. Molecular changes in oral cancer may reflect aetiology and ethnic origin. Eur J Cancer B Oral Oncol 1996; 32B: 150–3. [DOI] [PubMed] [Google Scholar]
- 2. Chen YJ, Lin SC, Kao T et al . Genome‐wide profiling of oral squamous cell carcinoma. J Pathol 2004; 204: 326–32. [DOI] [PubMed] [Google Scholar]
- 3. Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer 2005; 5: 127–35. [DOI] [PubMed] [Google Scholar]
- 4. Woolgar JA, Rogers S, West CR, Errington RD, Brown JS, Vaughan ED. Survival and patterns of recurrence in 200 oral cancer patients treated by radical surgery and neck dissection. Oral Oncol 1999; 35: 257–65. [DOI] [PubMed] [Google Scholar]
- 5. Lippman SM, Hong WK. Molecular markers of the risk of oral cancer. N Engl J Med 2001; 344: 1323–6. [DOI] [PubMed] [Google Scholar]
- 6. Hershkovich O, Oliva J, Nagler RM. Lethal synergistic effect of cigarette smoke and saliva in an in vitro model: does saliva have a role in the development of oral cancer? Eur J Cancer 2004; 40: 1760–7. [DOI] [PubMed] [Google Scholar]
- 7. Blesch A, Bosserhoff AK, Apfel R et al . Cloning of a novel malignant melanoma‐derived growth‐regulatory protein, MIA. Cancer Res 1994; 54: 5695–701. [PubMed] [Google Scholar]
- 8. Bosserhoff AK, Hein R, Bogdahn U, Buettner R. Structure and promoter analysis of the gene encoding the human melanoma‐inhibiting protein MIA. J Biol Chem 1996; 271: 490–5. [DOI] [PubMed] [Google Scholar]
- 9. Koehler MR, Bosserhoff A, Von Beust G et al . Assignment of the human melanoma inhibitory activity gene (MIA) to 19q13.32‐q13.33 by fluorescence in situ hybridization (FISH). Genomics 1996; 35: 265–7. [DOI] [PubMed] [Google Scholar]
- 10. Bosserhoff AK, Moser M, Hein R, Landthaler M, Buettner R. In situ expression patterns of melanoma‐inhibiting activity (MIA) in melanomas and breast cancers. J Pathol 1999; 187: 446–54. [DOI] [PubMed] [Google Scholar]
- 11. Poser I, Tatzel J, Kuphal S, Bosserhoff AK. Functional role of MIA in melanocytes and early development of melanoma. Oncogene 2004; 23: 6115–24. [DOI] [PubMed] [Google Scholar]
- 12. Bosserhoff AK, Kaufmann M, Kaluza B et al . Melanoma‐inhibiting activity, a novel serum marker for progression of malignant melanoma. Cancer Res 1997; 57: 3149–53. [PubMed] [Google Scholar]
- 13. Bosserhoff AK, Kondo S, Moser M et al . Mouse CD‐RAP/MIA gene: structure, chromosomal localization, and expression in cartilage and chondrosarcoma. Dev Dyn 1997; 208: 516–25. [DOI] [PubMed] [Google Scholar]
- 14. Hau P, Ruemmele P, Kunz‐Schughart LA et al . Expression levels of melanoma inhibitory activity correlate with time to progression in patients with high‐grade glioma. Oncol Rep 2004; 12: 1355–64. [PubMed] [Google Scholar]
- 15. El Fitori J, Kleeff J, Giese NA et al . Melanoma inhibitory activity (MIA) increases the invasiveness of pancreatic cancer cells. Cancer Cell Int 2005; 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee KL, Pentecost BT, D’Anna JA, Tobey RA, Gurley LR, Dixon GH. Characterization of cDNA sequences corresponding to three distinct HMG‐1 mRNA species in line CHO Chinese hamster cells and cell cycle expression of the HMG‐1 gene. Nucleic Acids Res 1987; 15: 5051–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rauvala H, Pihlaskari R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J Biol Chem 1987; 262: 16625–35. [PubMed] [Google Scholar]
- 18. Sasahira T, Kirita T, Bhawal UK et al . Expression of receptor for advanced glycation end products (RAGE) is associated with angiogenesis in human oral squamous cell carcinoma. Virchows Arch 2007; 450: 287–95. [DOI] [PubMed] [Google Scholar]
- 19. Sasahira T, Kirita T, Bhawal UK, Yamamoto K, Kuniyasu H. Significance of expression of receptor for advanced glycation end products (RAGE) in recurrence of human oral squamous cell carcinoma. Histopathol 2007; 51: 166–72. [DOI] [PubMed] [Google Scholar]
- 20. Bhawal UK, Ozaki Y, Nishimura M et al . Association of expression of receptors for advanced glycation end‐products (RAGE) and invasive and metastatic activity of oral squamous cell carcinoma. Oncology 2005; 69: 246–55. [DOI] [PubMed] [Google Scholar]
- 21. Kuniyasu H, Chihara Y, Kondo H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int J Cancer 2003; 104: 722–7. [DOI] [PubMed] [Google Scholar]
- 22. Kuniyasu H, Chihara Y, Kondo H, Ohmori H, Ukai R. Amphoterin induction in prostatic stromal cells by androgen deprivation is associated with metastatic prostate cancer. Oncol Rep 2003; 10: 1863–8. [PubMed] [Google Scholar]
- 23. Kuniyasu H, Chihara Y, Takahashi T. Co‐expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol Rep 2003; 10: 445–8. [PubMed] [Google Scholar]
- 24. Kuniyasu H, Oue N, Wakikawa A et al . Expression of receptors for advanced glycation end‐products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J Pathol 2002; 196: 163–70. [DOI] [PubMed] [Google Scholar]
- 25. Kuniyasu H, Sasaki T, Sasahira T, Ohmori H, Takahashi T. Depletion of tumor‐infiltrating macrophages is associated with amphoterin expression in colon cancer. Pathobiology 2004; 71: 129–36. [DOI] [PubMed] [Google Scholar]
- 26. Kuniyasu H, Yano S, Sasaki T, Sasahira T, Sone S, Ohmori H. Colon cancer cell‐derived high mobility group 1/amphoterin induces growth inhibition and apoptosis in macrophages. Am J Pathol 2005; 166: 751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stros M, Ozaki T, Bacikova A, Kageyama H, Nakagawara A. HMGB1 and HMGB2 cell‐specifically down‐regulate the p53‐ and p73‐dependent sequence‐specific transactivation from the human Bax gene promoter. J Biol Chem 2002; 277: 7157–64. [DOI] [PubMed] [Google Scholar]
- 28. Poser I, Golob M, Buettner R, Bosserhoff AK. Upregulation of HMG1 leads to melanoma inhibitory activity expression in malignant melanoma cells and contributes to their malignancy phenotype. Mol Cell Biol 2003; 23: 2991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Golob M, Buettner R, Bosserhoff AK. Characterization of a transcription factor binding site, specifically activating MIA transcription in melanoma. J Invest Dermatol 2000; 115: 42–7. [DOI] [PubMed] [Google Scholar]
- 30. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 1998; 11: 155–68. [PubMed] [Google Scholar]
- 31. Ikuta M, Podyma KA, Maruyama K, Enomoto S, Yanagishita M. Expression of heparanase in oral cancer cell lines and oral cancer tissues. Oral Oncol 2001; 37: 177–84. [DOI] [PubMed] [Google Scholar]
- 32. Parkkinen J, Raulo E, Merenmies J et al . Amphoterin, the 30‐kDa protein in a family of HMG1‐type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem 1993; 268: 19726–38. [PubMed] [Google Scholar]
- 33. Czura CJ, Wang H, Tracey KJ. Dual roles for HMGB1: DNA binding and cytokine. J Endotoxin Res 2001; 7: 315–21. [DOI] [PubMed] [Google Scholar]
- 34. Taguchi A, Blood DC, Del Toro G et al . Blockade of RAGE‐amphoterin signalling suppresses tumour growth and metastases. Nature 2000; 405: 354–60. [DOI] [PubMed] [Google Scholar]
- 35. Sasahira T, Akama Y, Fujii K, Kuniyasu H. Expression of receptor for advanced glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch 2005; 446: 411–5. [DOI] [PubMed] [Google Scholar]
- 36. Sasahira T, Sasaki T, Kuniyasu H, Akama Y, Fujii K. Interleukin‐15 and transforming growth factor alpha are associated with depletion of tumor‐associated macrophages in colon cancer. J Exp Clin Cancer Res 2005; 24: 69–74. [PubMed] [Google Scholar]
- 37. Bosserhoff AK, Stoll R, Sleeman JP, Bataille F, Buettner R, Holak TA. Active detachment involves inhibition of cell‐matrix contacts of malignant melanoma cells by secretion of melanoma inhibitory activity. Lab Invest 2003; 83: 1583–94. [DOI] [PubMed] [Google Scholar]
- 38. Jachimczak P, Apfel R, Bosserhoff AK et al . Inhibition of immunosuppressive effects of melanoma‐inhibiting activity (MIA) by antisense techniques. Int J Cancer 2005; 113: 88–92. [DOI] [PubMed] [Google Scholar]
- 39. Bauer R, Humphries M, Faessler R, Winklmeier A, Craig SE, Bosserhoff AK. Regulation of integrin activity by MIA. J Biol Chem 2006; 281: 11669–77. [DOI] [PubMed] [Google Scholar]
- 40. Dietrich T, Onderka J, Bock F et al . Inhibition of inflammatory lymphangiogenesis by integrin alpha5 blockade. Am J Pathol 2007; 171: 361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kobayashi S, Kishimoto T, Kamata S, Otsuka M, Miyazaki M, Ishikura H. Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci 2007; 98: 726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer 2002; 2: 573–83. [DOI] [PubMed] [Google Scholar]
- 43. O‐charoenrat P, Rhys‐Evans P, Eccles SA. Expression of vascular endothelial growth factor family members in head and neck squamous cell carcinoma correlates with lymph node metastasis. Cancer 2001; 92: 556–68. [DOI] [PubMed] [Google Scholar]
- 44. Stacker SA, Caesar C, Baldwin ME et al . VEGF‐D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001; 7: 186–91. [DOI] [PubMed] [Google Scholar]
- 45. Momose F, Araida T, Negishi A, Ichijo H, Shioda S, Sasaki S. Variant sublines with different metastatic potentials selected in nude mice from human oral squamous cell carcinomas. J Oral Pathol Med 1989; 18: 391–5. [DOI] [PubMed] [Google Scholar]
- 46. Khan MH, Yasuda M, Higashino F, Haque S, Kohgo T, Nakamura M, Shindoh M. nm23‐H1 suppresses invasion of oral squamous cell carcinoma‐derived cell lines without modifying matrix metalloproteinase‐2 and matrix metalloproteinase‐9 expression. Am J Pathol 2001; 158: 1785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fleming ID, Cooper JS, Henson DE et al . AJCC Cancer Staging Manual. Lippincott‐Raven, Philadelphia, 1997. [Google Scholar]