

Abstract
In order to study the role of BRCA2 protein in homologous recombination repair and radio‐sensitization, we utilized RNA interference strategy in vitro and in vivo with human tumor cells. HeLa cells transfected with small‐interfering BRCA2 NA (BRCA2 siRNA) (Qiagen) as well as negative‐control siRNA for 48 h were irradiated, and several critical end points were examined. The radiation cell survival level was significantly reduced in HeLa cells with BRCA2 siRNA when compared with mock‐ or negative‐control siRNA transfected cells. DNA double strand break repair as measured by constant field gel‐electrophoresis showed a clear inhibition in cells with BRCA2 siRNA, while little inhibition was observed in cells with negative control siRNA. Our immuno‐staining experiments revealed a significant delay in Rad51 foci formation in cells with BRCA2 siRNA when compared with the control populations. However, none of the non‐homologous end joining proteins nor the phosphorylation of DNA‐dependent protein kinase catalytic subunit was affected in cells transfected with BRCA2 siRNA. In addition, the combined treatment with radiation and BRCA2 siRNA in xenograft model with HeLa cells showed an efficient inhibition of in vivo tumor growth. Our results demonstrate down‐regulation of BRCA2 leads to radio‐sensitization mainly through the inhibition of homologous recombination repair type double‐strand break repair; a possibility of using BRCA2 siRNA as an effective radiosensitizer in tumor radiotherapy may arise. (Cancer Sci 2008; 99: 810–815)
DNA double‐strand breaks (DSBs) are the main cause of cell death induced by ionizing radiation and represent a major threat for the maintenance of genome integrity. These lesions can be produced by ionizing radiation, chemicals and endogenously during replication, V(D)J recombination, and meiosis.( 1 , 2 ) In human cells, DNA DSBs can be repaired by two major processes which are homologous recombination repair (HRR) and non‐homologous end joining (NHEJ). NHEJ is thought to be the main pathway for the repair of ionizing radiation‐induced DSBs in mammalian cells and also plays a role in rearrangement processes during the V(D)J recombination.( 3 ) NHEJ is active in all phases of the cell cycle, but HRR is most effective in late S and G2 phases.( 4 , 5 ) Proteins known to be required for NHEJ thus far include Ku70/Ku80, DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), XRCC4, DNA ligase IV, XLF and Artemis.( 6 , 7 ) HRR pathway employs a large number of proteins including RPA, Rad51, Rad52, Rad54, BRCA1, BRCA2 and MRN complex (Mre11, Rad50 and NBS1).( 8 , 9 ) A recent report indicated that when Hus1 is absent, the effect of cells on ionizing radiation sensitivity is associated with homologous recombination but is independent of NHEJ.( 10 ) It has been shown that the function of BRCA2 in HRR is likely to be mediated through interactions with Rad51, a protein essential for the HRR process. Rad51 is known to bind to the C‐terminus of BRCA2, the BRC repeat region.( 11 , 12 , 13 , 14 )
Women inheriting a defective BRCA2 mutant gene have a significant risk for breast cancer in their life time. Individuals with BRCA2 mutation also show predisposition to ovarian and prostate cancer. BRCA2 mutations have also been found in some pancreatic tumors.( 15 , 16 , 17 , 18 , 19 , 20 ) BRCA2 mutants or defective cells are compromised in their ability to repair DNA DSBs and arrest in the late S/G2 phases of the cell cycle, indicating the cell cycle specific role of HRR pathway.( 21 ) It was also reported that BRCA2 mutated cells displayed radiation hypersensitivity and changes in the expression of DNA repair genes following DNA damage induced by radiation.( 22 , 23 , 24 ) A possible mechanism was suggested by the observed stimulation of single‐strand annealing by loss of wild‐type BRCA2, which in turn lead to an increased lethality from mitomycin C‐induced DNA cross‐links.( 25 )
Very recently, we reported that Hsp90 inhibitor 17‐allylamino‐ 17‐ demethoxygeldanamycin (17‐AAG) inhibited the HRR pathway of radiation induced DSBs, and this was accompanied by the reduced expressions of BRCA2 and Rad51 proteins.( 26 ) In order to further apply the crucial role of BRCA2 in HRR for cancer therapy, we utilized RNA interference (RNAi) strategy in tumor radiosensitization. In this report, we demonstrate that the treatment of BRCA2 siRNA significantly reduces BRCA2 protein and messenger RNA (mRNA) expression, leading to tumor radio‐sensitization in vitro and in vivo mainly through the inhibition of HRR pathway.
Materials and Methods
Antibodies. The following antibodies were used: Mouse monoclonal anti‐Ku80 (Ab‐2 clone 111), mouse monoclonal anti‐Ku70 (Ab‐4 clone N3H10) and mouse monoclonal anti‐DNA‐PKcs (Ab‐4), all purchased from Labvison (Neomarker, USA). Mouse monoclonal anti‐BRCA2 (Ab‐4) was purchased from Calbiochem and Oncogene (Calbiochem, Germany). Rabbit polyclonal anti‐Rad51, rabbit IgG‐HRP and mouse IgG‐HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cy3‐conjugated affinipure goat antirabbit IgG was purchased from Jackson ImmunoResearch. Anti‐DNA‐PKcs‐phosphospecific (2609 Thr) antibody was synthesized by SIGMAGenosys (Japan).
Cell cultures, culture conditions, and irradiation. The human cervix epitheloide carcinoma cell line HeLa was obtained from Cell Resource Center for Biomedical Research, Tohoku University and grown in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics. Irradiation was performed using a Shimadzu Pantak HF‐320 X‐ray machine at a dose rate of 0.93 Gy/min.
siRNA treatment. BRCA2 siRNAs (Product Name: Hs_BRCA2–7_HP Validated siRNA, Qiagen) and negative control siRNA were purchased from Qiagen. 5 × 105 HeLa cells were seeded onto 35 mm plates, and incubated overnight before transfection with 20 nM siRNA using HiperFect transfection reagent (Qiagen) according to the instructions of the manufacturer. Cells were then cultured in normal growth media for 48 h before treatment.
Realtime reverse transcription‐polymerase chain reaction (RT‐PCR). Total RNA was extracted from cells according to the manufacturer instructions using the RNeasy Protect Mini Kit (Qiagen). mRNA silencing was quantified by real‐time PCR using an ABI Prism7500 system (Applied Biosystems, Foster City, CA, USA). The mRNA value for each gene was normalized relative to hGUS (No. 431088E) mRNA levels in RNA sample. The following Assays‐on‐Demand reagents were used in this study: BRCA2 (Hs01037420_m1). All reagents necessary for running a TagMan RT‐PCR assay, including predesigned and optimized Assays were purchased from Applied Biosystems (ABI) and used according to the manufacture's instructions. All the measurements and results were analyzed with an ABI sequence detector software.
Colony‐forming assay. Radiosensitivity was determined by colony forming assay as described previously.( 27 ) Cells in plastic flasks were transfected with siRNA. The plates were irradiated with X‐rays (0−6 Gy). After irradiation, cells were immediately trypsinized, diluted, counted and seeded in 60‐mm dishes at various cell densities. After 2 weeks of incubation, colonies were stained with crystal violet dissolved in methanol. Colonies containing more than 50 cells were counted. The surviving fraction was calculated based on the plating efficiency determined from the negative control siRNA or BRCA2 siRNA transfected cells.
DNA DSB repair assay. The method has been reported previously.( 26 ) To quantify the level of DSBs, cells were irradiated with 20 Gy X‐rays on ice. Immediately after irradiation, the growth medium was aspirated, fresh warm medium was added and HeLa cells were incubated in a CO2 (5%) incubator at 37°C for repair. At each repair point, cells were washed with cold phosphate‐buffered saline (PBS), and trypsinized on ice for 10 min. Cells were washed in cold medium, and the resulting cell pellet was immediately embedded in 1% agarose (InCert agarose, Food Machinery Corporation) at a density of 1.5 × 105 cells/mL (1.0 × 10 cells/plug), and placed on ice. These agarose samples were cut into plugs, and placed in CometAssay Lysis Solution (Trevigen) containing proteinase K for 1 h on ice, and then incubated for 24 h at 50°C. The plugs were equilibrated in TE buffer (Sigma, pH 8.0) for 1 h at room temperature and used for electrophoresis. The plugs were loaded onto 0.6% SeaKem Gold agarose gels (Cambrex) and subjected to electrophoresis at field strength of 0.6 V/cm in 0.5× TBE buffer for 36 h. The gel was stained for 3 h with Ethidium bromide (2 µg/mL), and de‐stained overnight at room temperature in distilled water. The fluorescence intensities ware measured with an UV transilluminator and a digital camera (Kodak, DX290) with an orange filter. National Institute of Health Image software was used for the analysis of DSB damage and the fraction of released DSBs was calculated.
Western blotting. Cells were washed ice‐cold PBS and lyzed in lysis buffer (50 mM Tris‐HCl, 150 mM NaCl, 1 mM EDTA, 2 mM EGTA, 50 mM NaF, 25 mM â‐glycerophospate, 0.5% TritonX‐100, 0.1% SDS, 0.1 mM sodium vanadate, 1 mM DTT, 0.1 mM PMSF, and Complete‐Mini protease inhibitor [Roche Diagnostics]). Protein concentrations were measured using protein assay kit (Pierce) according to the manufacturer's instructions. Total cellular lysates were loaded onto 6 or 12% tris glycine gels (Invitorogen), separated by electrophoresis at a constant voltage (125 V) and electro‐transferred onto nitrocellulose membranes at 42 V. Membranes were blocked for 1 h at room temperature in blocking buffer (25 mM Tris pH 8.0, 125 mM NaCl, 1% Tween 20 [TBS‐T buffer] containing 2% blocking agent) and incubated with primary antibody for 2 h at room temperature. The membranes were washed with TBS‐T buffer three times and incubated with a secondary antibody conjugated to horseradish peroxidase for 1 h. After washing for three times, the blots were visualized by enhanced chemiluminescence method (GE Healthcare) according to the manufacturer's instructions.
Immunofluorescence measurements. Cells were grown on cover‐glasses in 30 mm dish, fixed in 2% paraformaldehyde in PBS for 15 min at room temperature and washed in PBS. Then the cells were permeabilized for 5 min at 4°C in 0.2% Triton X‐100, and blocked in PBS with 1% Bovine Serum Albumin (BSA) for 1 h at 37°C. Cells were then incubated with the primary antibody (rabbit polyclonal anti‐Rad51 (Santa Cruz) for 1 h at 37°C at 1:200 dilutions in PBS with 1% BSA, and washed three times in PBS with 1% BSA for 10 min. The cells were incubated with the secondary antibody (Cy3‐conjugated affinipure goat antirabbit IgG (Jackson ImmunoResearch) for 45 min at 37°C at 1:500 dilutions in PBS with 1% BSA, and washed three times for 10 min in PBS. Cover‐glasses were mounted in 1:1000 dilutions of 4,6‐diamidino‐2‐phenylindole. Fluorescence images were captured using an Olympus DP70 fluorescence microscope for analysis.
Cell cycle analysis. Cells were washed with ice‐cold PBS once and fixed in 70% ethanol. Fixed cells were washed in PBS, then incubated with 1 mg/mL RNase A (Invitorogen) for 20 min at 37°C, washed in PBS and incubated with 0.1 mg/mL propidium iodide (Sigma‐Aldrich) for 20 min on ice. Intensities of fluorescence signals were measured on Becton Dickinson FACS Calibur flow cytometer. At least 10 000 cells were measured for each sample.
In vivo study for xenografted tumor model. A total of 1 × 106 HeLa cells were subcutaneous (s.c.) inoculated in 100 µL of 1 × PBS(–) into the right hind legs of an 8‐week‐old BALB/c‐nu/nµ mice. After 2 weeks when the tumors had reached an average volume of ~50 mm3 the subcutaneous tumors were direct treated with 5 µg of BRCA2 siRNA with atelogene (Koken Co. Ltd, Tokyo, Japan).( 28 , 29 , 30 ) Forty‐eight h after BRCA2 siRNA administration, the mice were placed in a Lucite plate and the tumors were irradiated at 8 Gy by a Cs‐137 gamma‐ray unit with a does rate of 1.6 Gy/min. The size of palpable tumors was measured with calipers every 1–2 days. The tumor volume (V) was estimated by V = Length × Width2/2.
Statistical analysis. Statistical comparison of mean values was performed using the Student's t‐test. Differences with a P‐value of <0.05 were considered statistically significant.
Results
Effect of siRNA on BRCA2 and DNA DSB repair proteins. We examined the effect of BRCA2 siRNA on BRCA2 protein expression. HeLa cells were transfected by the siRNA for 48 h and the protein expression was observed by the Western blot. Compared with the control, BRCA2 protein was reduced as a function of siRNA concentrations (Fig. 1a). The level of BRCA2 protein went down to 15% of the control with 20 nM siRNA transfection. Similarly, BRCA2 mRNA expression measured by real‐time RT‐PCR was reduced and this effect was observed up to 72 h. (data not shown). Judging from these expression data, we decided to use 20 nM as the BRCA2‐siRNA concentration for further studies. We used the negative siRNA to confirm the specificity of our BRCA2 siRNA on BRCA2 protein expression in HeLa cells. The negative siRNA has altered base sequences when compared with the BRCA2 siRNA and gives a slightly lower expression than the mock‐transfected control. We did not find any significant changes in NHEJ associated proteins including DNA‐PKcs, Ku80 and Ku70 in the cells transfected with BRCA2 siRNA (Fig. 1b). BRCA2 mRNA expression measured by real‐time RT‐PCR was reduced to 7% of the control by 20 nM siRNA. (Fig. 1c).
Figure 1.
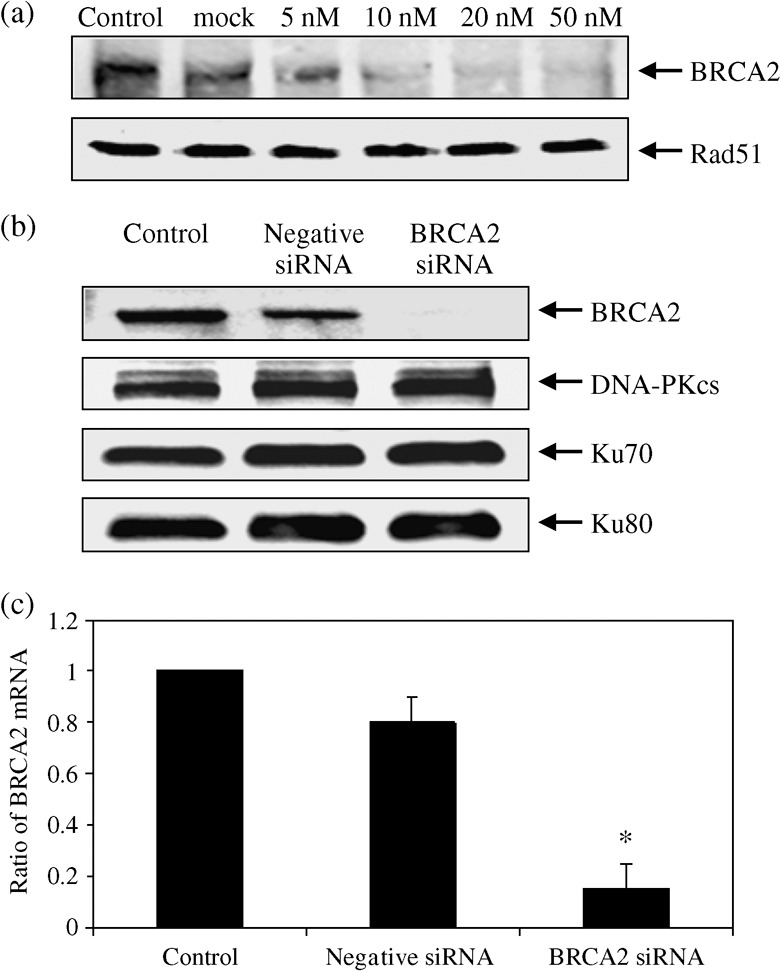
Expression of various DNA double‐strand breaks repair proteins after BRCA2 siRNA transfection. Cells were transfected with various concentrations of BRCA2 siRNA for 48 h and compared with the non‐treated and the mock‐treated control samples. (a) BRCA2 and Rad51 protein expressions were detected by Western blot (upper graph). (b) Cells were transfected with mock‐treated control, negative siRNA or 20 nM BRCA2 siRNA for 48 h, and the levels of BRCA2, DNA‐PKcs, Ku70, Ku80, expressions were determined by Western blots. (c) The expression of BRCA2 messenger RNA was examined by reverse transcription‐polymerase chain reaction. Cells were transfected with 20 nM BRCA2 siRNA for 48 h. Data represent the mean and the standard deviation from three independent experiments. *, P < 0.05 versus control or mock‐transfected cells (Student's t‐test).
BRCA2 siRNA enhances radiosensitivity in human tumor cells. The full‐scale radiation cell survival curves, a critical standard for observing radiation sensitivity,( 31 ) are given in Figure 2. A clear radiosensitizastion can be observed with HeLa cells transfected with BRCA2 siRNA when compared with cells with the mock or the negative control siRNA transfection. This is the first report showing that BRCA2 siRNA induces radiosensitization in human tumor cells.
Figure 2.

Radiation cell survival curves in BRCA2 siRNA‐HeLa cells. Clonogenic survival curves in cells transfected with 20 nM BRCA2 siRNA for 48 h and exposed to 0–6 Gy X‐rays. The cell survival was determined by colony formation assay. Data shown are the mean and the standard deviation of at least three independent experiments.
BRCA2 siRNA impairs rejoining of radiation induced DNA double‐strand breaks. In order to investigate if BRCA2 siRNA affects repair of DNA DSB induced by radiation, we measured rejoining kinetics of DSBs by constant field gel electrophoresis in X‐irradiated HeLa cells.( 32 ) Figure 3a shows typical ethidium bromide stained gel images of irradiated and repaired DNA DSBs. For non‐transfected control cells, the DSB breaks induced at 0 h is mostly rejoined by 3 h after irradiation. For the negative siRNA transfected cells, the kinetics is very similar to the control with a slightly higher background. In contrast, a significantly higher number of remaining breaks was observed in the BRCA2 siRNA transfected samples. The quantified ratios of DSB damage as calculated by the released portion of DNA divided by the total DNA amount are given in Figure 3b. As clearly observed, the remaining DSBs are significantly higher in the BRCA2 siRNA transfected cells than the control samples. There seems to be little change between 3 h and 6 h repair points for the BRCA2 siRNA transfected cells, while the control and the negative siRNA samples showed a good recovery by 6 h. Our data demonstrates that BRCA2 siRNA inhibits rejoining of radiation induced DSBs. These rejoining data seem to reflect the radiation cell survival data presented in Figure 2.
Figure 3.
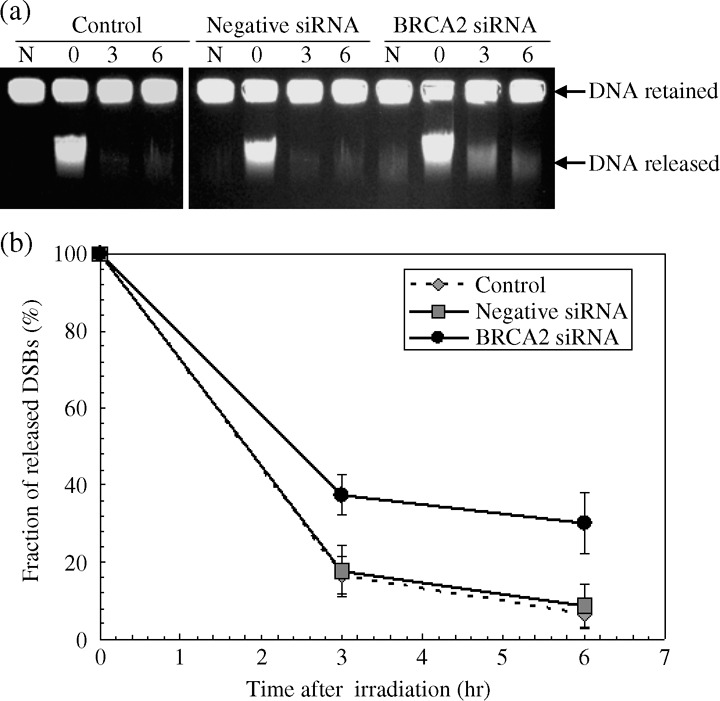
Rejoining kinetics of radiation induced DNA DSBs in HeLa cells. Cells were transfected with mock‐treated (control), negative siRNA or 20 nM BRCA2 siRNA. 48 h after transfection, cells were irradiated with 20 Gy of X‐rays on ice and incubated for various repair periods at 37°C. (a) Typical ethidium bromide stained gel images by constant‐field gel electrophoresis. N designates non‐irradiated control samples. (b) The fraction of migrated broken DSBs obtained by quantifying the intensity of the bands in gels from (a). Data represent the mean and the standard deviation from three separate experiments, P < 0.05 versus control or mock‐transfected cells (Student's t‐test).
HRR type DSB repair is affected by BRCA2 siRNA. To investigate the effect of BRCA2 siRNA in the HRR pathway, we studied the Rad51 foci formation. Rad51 binds to BRCA2 and it forms foci at the sites of DSBs. The numbers of Rad51 foci per nucleus at different time points are shown after cells under various conditions were irradiated with 2 Gy X‐rays. One can observe a distinct difference in Rad51 foci formation between the BRCA2 siRNA transfected cells and the mock (control) or negative siRNA transfected cells (Fig. 4a). The number of cells with more than five Rad51 foci per nucleus was significantly lower (about 16 times) in the BRCA2 siRNA transfected cells than the control or negative siRNA transfected cells at 3 h and 6 h after irradiation (Fig. 4b). The similar tendency was observed up to 72 h after irradiation. We also observed the formation of phosphorylated DNA‐PKcs foci (at 2609 Thr) which are thought to be essential for NHEJ type DSB repair process.( 33 , 34 ) In contrast to Rad51 foci formation, there is little difference observed between the cells with BRCA siRNA transfection and the mock or the negative siRNA transfected cells (Fig. 4c,d).
Figure 4.
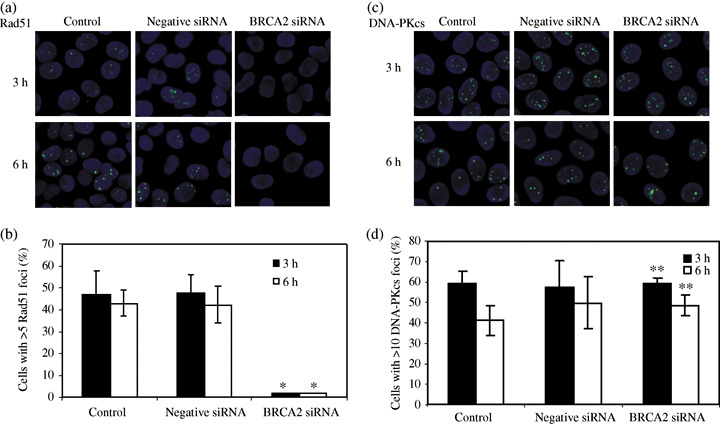
Foci formations of Rad51 and phosphorylated DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) in HeLa cells irradiated with 2 Gy X‐rays. Nuclei were stained with diamidino‐2‐phenylindole. The primary antibody was rabbit polyclonal anti‐Rad51 (a) and antiphosphorylated DNA‐PKcs (2609 Thr) (c). Number of cells containing Rad51. (b) and DNA‐PKcs (d) foci visualized at 3 h (filled columns) and 6 h (open columns) after exposure to 2 Gy X‐rays. Data represent the mean and standard deviation from three separate experiments. *P < 0.05 and **P > 0.05 versus control or mock‐transfected cells (Student's t‐test).
Down regulation of BRCA2 leads to loss of G2/M check point. To determine whether BRCA2 siRNA affect the cell cycle progression, we examined the effect of BRCA2 siRNA on cell cycle distribution. HeLa cells were transfected with BRCA2 siRNA for 48 h, and the data by flow cytometry analysis are given in Table 1. The cell cycle distribution after BRCA2 siRNA transfection (48 h) in non‐irradiated cells was similar to the control or negative siRNA transfected cells. After 6 Gy X‐irradiation, a typical G2 delay was observed for the control and negative siRNA transfected cells. In contrast, there seems no G2 delay detected in the BRCA2 siRNA transfected cells and the profile is similar to non‐irradiated cells. These data may also help explain the hyper radiosensitivity exerted by BRCA2 siRNA as the non‐repaired cells prematurely advance into the next cell cycle phase and collapse. In addition, we did not observe any sub‐G1 population in all of our cell cycle analysis, suggesting that the apoptotic process is not likely to be involved in the radiosensitization process.
Table 1.
Cell cycle distribution of HeLa cells transfected with mock‐treated (control), negative siRNA or 20 nM BRCA2 siRNA. 48 h after transfection, cells were irradiated and cultured for 24 h. Cells were harvested and the cell cycle distribution was analyzed by flow cytometry
| Cell cycle | Sub‐G1 | G1 | S | G2/M |
|---|---|---|---|---|
| No IR | ||||
| Control | 0.9% | 34.4% | 34.9% | 29.9% |
| Negative siRNA | 0.9% | 36.3% | 42.4% | 20.4% |
| BRCA2 siRNA | 1.0% | 42.3% | 32.4% | 24.3% |
| IR: 6 Gy | ||||
| Control | 1.1% | 33.3% | 6.3% | 59.3% |
| Negative siRNA | 1.2% | 27.9% | 12.9% | 61.0% |
| BRCA2 siRNA | 1.4% | 34.9% | 45.1% | 25.7% |
IR, ionizing radiation.
BRCA2 siRNA enhances tumor radio‐response in vivo. We next used a tumor xenograft model to examine the antitumor effect of BRCA2 siRNA with radiation in vivo (Fig. 5). HeLa cells were subcutantaneously injected into the right hind legs of nude mice, and the mice were vehicle‐treated, treated with siRNA alone, radiation alone, or with the combination of the siRNA and radiation. The treatment with BRCA2 siRNA (5 µg/test) alone did not inhibit tumor growth. When mice were irradiated with 8 Gy gamma‐rays, the tumor growth was delayed relative to the non‐irradiated control up to a certain point and then re‐grew. However, the combined treatment significantly inhibited the tumor growth when compared with the radiation treatment alone. The actual tumor samples from the treated mice are given in Figure 5.
Figure 5.
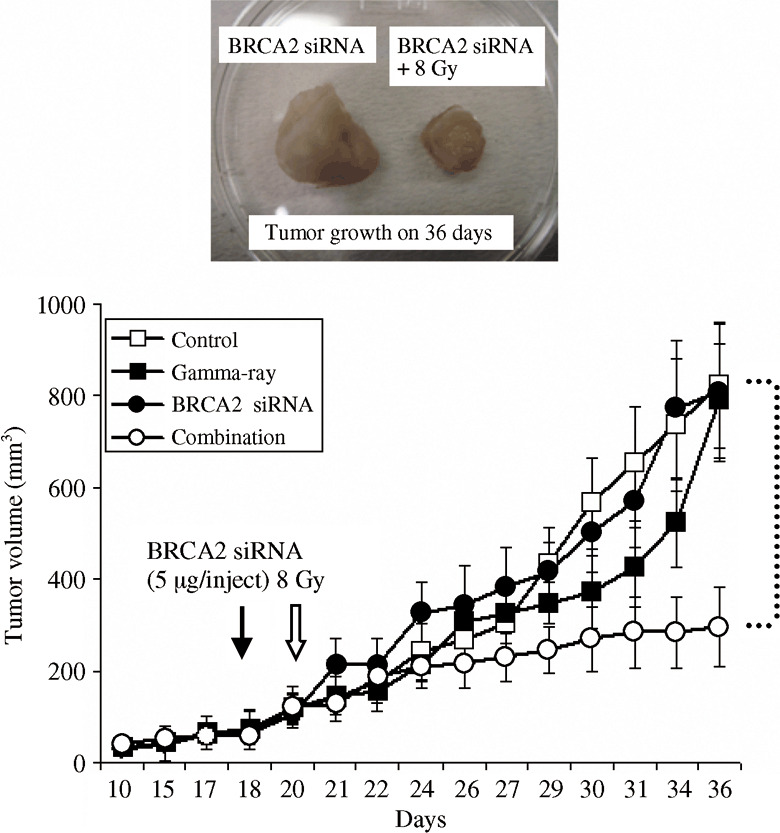
The combined treatment with radiation and BRCA2 siRNA significantly inhibits tumor growth in vivo. BRCA2 siRNA (5 µg/test) were directly injected into HeLa tumor (~50 mm3 on 18 days after inoculation) implanted in BALB/c nude mice. Tumors were irradiated with 8 Gy at 48 h after the injection of BRCA2 siRNA. Closed arrows: BRCA2 siRNA injection; open arrows: radiation. Open square: control; closed square: 8 Gy gamma‐rays; closed circular: BRCA2 siRNA; open circle: combination of BRCA2 siRNA and 8 Gy gamma‐rays. Data represent the mean and standard deviation of tumor size at each measuring point.
Discussion
In this present report, we described the functional roles played by BRCA2 using RNAi strategy in vitro and in vivo using human tumor cells. A significant inhibition of DSB repair was observed in HeLa cells transfected with BRCA2 siRNA, suggesting that BRCA2 is playing an active role in DSB repair process. Although we observed no changes in the expressions of NHEJ proteins and no changes in the phosphorylation status of DNA‐PKcs after BRCA siRNA transfection, the appearance of Rad51 foci was significantly delayed in the BRCA2 siRNA transfected cells. These data seem to demonstrate that BRCA2 is predominantly involved in the HRR pathway of DSB repair. In the past, cells with a truncation mutation of BRCA2 were reported to show defective repair of DNA DSB after exposure to ionizing radiation.( 35 , 36 ) There was one very recent report studying DNA repair responses in the wild type Sp6/HL mouse hybridoma cell line using BRCA2 siRNA, which was inserted separately into a derivative of pSUPER. They showed that the BRCA2‐depleted cells had a significant deficiency in homologous recombination.( 37 )
Our cell survival experiments also showed a clear radio‐sensitization by BRCA2 siRNA in human tumor cells. One report we mentioned above revealed the deregulation of T‐cell apoptosis in T‐cell lineage‐specific Brca2‐deficient mice.( 37 ) In our system, we also assessed the status of cell proliferation in HeLa cells transfected with BRCA2 siRNA. Without radiation, we did not find any significant changes in the cell cycle distribution, but after 6 Gy X‐rays, cells transfected with BRCA2 siRNA did not seem to show a typical G2 block usually associated with radiation and showed the cells arrested at S‐phase. This, in turn, may result in a higher rate of cell death because cells did not have enough time to repair the damage before moving into the next cell cycle phase. Our flow cytometry data also indicate that apoptosis is not the likely cause of enhanced radio‐sensitivity of BRCA2 siRNA transfected cells. BRCA2 is also known to be responsible for DNA replication, the integrity of the genome and the maintenance of chromosomal stability.( 38 )
Finally we studied the tumor growth using nude mice with HeLa xenograft in vivo. In this present study, we used an Atelogene transfect kit to mediate BRCA2 siRNA transfer in vivo. Although radiation alone is not so efficient in controlling the tumor growth, the combination of gamma‐radiation and the BRCA2 siRNA treatment significantly inhibited the tumor growth (Fig. 5). Our results reveal the potential of using siRNA as an efficient radio‐sensitizer for radiation cancer therapy.
In summary, we demonstrated that BRCA2 siRNA significantly enhanced the radio‐sensitivity of human tumor cells in vitro and in vivo. The cause of radio‐sensitization is likely to come from the inhibition DNA DSB repair through the impairment of HRR pathway by the siRNA. Our data suggest that RNAi technology could successfully be applied for an adjuvant to radiotherapy.
Acknowledgments
We thank Dr M. Noguchi, Dr R. Hirayama, Dr K. Suetomi, Dr Y. Ninomiya, Dr T. Kato, Mr Y. Fujii, and Ms. Y. Kamochi for their valuable help in preparation of this manuscript. This work was supported by Japan Society for the Promotion of Science (JSPS) Grant in Aid Scientific Research A 16209036 (R Okayasu).
References
- 1. Van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double‐stranded break connection. Nat Rev Genet 2001; 2: 196–206. [DOI] [PubMed] [Google Scholar]
- 2. Cui X, Yu Y, Gupta S, Cho YM, Lees‐Miller SP, Meek K. Autophosphorylation of DNA‐dependent protein kinase regulates DNA end processing and may also alter double‐strand break repair pathway choice. Mol Cell Biol 2005; 25: 10842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jeggo PA. DNA breakage and repair. Adv Genet 1998; 38: 185–218. [DOI] [PubMed] [Google Scholar]
- 4. Saleh‐Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double‐strand breaks in the S phase of the cell cycle in human cells. Nucl Acids Res 2004; 32: 3683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double‐strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23: 5706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Niewolik D, Pannicke U, Lu H et al . DNA‐PKcs dependence of Artemis endonucleolytic activity, differences between hairpins and 5′ or 3′ overhangs. J Biol Chem 2006; 281: 33900–9. [DOI] [PubMed] [Google Scholar]
- 7. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4‐DNA ligase IV complex to promote DNA nonhomologous end‐joining. Cell 2006; 124: 301–13. [DOI] [PubMed] [Google Scholar]
- 8. D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3: 317–27. [DOI] [PubMed] [Google Scholar]
- 9. Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 2002; 21: 8967–80. [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Hu B, Weiss RS, Wang Y. The effect of Hus1 on ionizing radiation sensitivity is associated with homologous recombination repair but is independent of nonhomologous end‐joining. Oncogene 2006; 25: 1980–3. [DOI] [PubMed] [Google Scholar]
- 11. Mizuta R, LaSalle JM, Cheng HL et al . RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA 1997; 94: 6927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA 1998; 95: 5287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK‐dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434: 598–604. [DOI] [PubMed] [Google Scholar]
- 14. Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH. BRCA2 BRC motifs bind RAD51‐DNA filaments. Proc Natl Acad Sci USA 2005; 102: 8537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wooster R, Bignell G, Lancaster J et al . Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789–92. [DOI] [PubMed] [Google Scholar]
- 16. Goggins M, Schutte M, Lu J et al . Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996; 56: 5360–4. [PubMed] [Google Scholar]
- 17. Miki Y, Swensen J, Shattuck‐Eidens D et al . A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266: 66–71. [DOI] [PubMed] [Google Scholar]
- 18. Tavtigian SV, Simard J, Rommens J et al . The complete BRCA2 gene and mutations in chromosome 13q‐linked kindreds. Nat Genet 1996; 12: 333–7. [DOI] [PubMed] [Google Scholar]
- 19. Teng DH, Bogden R, Mitchell J et al . Low incidence of BRCA2 mutations in breast carcinoma and other cancers. Nat Genet 1996; 13: 241–4. [DOI] [PubMed] [Google Scholar]
- 20. Thorlacius S, Olafsdottir G, Tryggvadottir L et al . A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat Genet 1996; 13: 117–9. [DOI] [PubMed] [Google Scholar]
- 21. Farmer H, McCabe N, Lord CJ et al . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21. [DOI] [PubMed] [Google Scholar]
- 22. Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res 2002; 509: 49–78. [DOI] [PubMed] [Google Scholar]
- 23. Connor F, Bertwistle D, Mee PJ et al . Tumorigenesis and a DNA repair defect in mice with a truncating BRCA2 mutation. Nat Genet 1997; 17: 423–30. [DOI] [PubMed] [Google Scholar]
- 24. Karran P. DNA double‐strand break repair in mammalian cells. Curr Opin Genet Dev 2000; 10: 144–50. [DOI] [PubMed] [Google Scholar]
- 25. Tutt A, Bertwistle D, Valentine J et al . Mutation in BRCA2 stimulates error‐prone homology‐directed repair of DNA double‐strand breaks occurring between repeated sequences. Embo J 2001; 20: 4704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noguchi M, Hirayama D, Yu R et al . Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17‐allylamino‐17‐demethoxygeldanamycin. Biochem Biophys Res Commun 2006; 351: 658–63. [DOI] [PubMed] [Google Scholar]
- 27. Yu, Watanabe D, H, Shibuya H, Miura M. Redundancy of radioresistant signaling pathways originating from insulin‐like growth factor I receptor. J Biol Chem 2003; 278: 6702–9. [DOI] [PubMed] [Google Scholar]
- 28. Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res 2004; 64: 3365–70. [DOI] [PubMed] [Google Scholar]
- 29. Minakuchi Y, Takeshita F, Kosaka N et al . Atelocollagen‐mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res 2004; 32: e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takeshita F, Minakuchi Y, Nagahara S et al . Efficient delivery of small interfering RNA to bone‐metastatic tumors by using EJ. atelocollagen in vivo . Proc Natl Acad Sci USA 2005; 102: 12177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist, 6th edn, Philadelphia, PA: Lippincott Williams & Wilkins, 2006; Section I: 30–46. [Google Scholar]
- 32. Hirayama R, Furusawa Y, Fukawa T, Ando K. Repair kinetics of DNA‐DSB induced by X‐rays or carbon ions under oxic and hypoxic conditions. J Radiat Res (Tokyo) 2005; 46: 325–32. [DOI] [PubMed] [Google Scholar]
- 33. Burma S, Chen DJ. Role of DNA‐PK in the cellular response to DNA double‐strand breaks. DNA Repair (Amst) 2004; 3: 909–18. [DOI] [PubMed] [Google Scholar]
- 34. Chen BP, Uematsu N, Kobayashi J et al . Ataxia Telangiectasia Mutated (ATM) is essential for DNA‐PKcs phosphorylations at the Thr‐2609 cluster upon DNA double‐strand break. J Biol Chem 2007; 282: 6582–7. [DOI] [PubMed] [Google Scholar]
- 35. Patel KJ, Lee VP, Yu H et al . Involvement of BRCA2 in DNA repair. Mol Cell 1998; 1: 347–57. [DOI] [PubMed] [Google Scholar]
- 36. Friedman LS, Thistlethwaite FC, Patel KJ et al . Thymic lymphomas in mice with a truncating mutation in BRCA2. Cancer Res 1998; 58: 1338–43. [PubMed] [Google Scholar]
- 37. Lee SA, Baker MD. Analysis of DNA repair and recombination responses in mouse cells depleted for BRCA2 by SiRNA. DNA Repair (Amst) 2007; 6: 809–17. [DOI] [PubMed] [Google Scholar]
- 38. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108: 171–82. [DOI] [PubMed] [Google Scholar]