

Abstract
Hypoxia‐inducible factors, key transcription factors for hypoxia‐dependent gene expression, play important roles in angiogenesis and tumor growth. The VHL protein binds to the α subunit of (HIF‐α) for its oxygen‐dependent degradation. VHL mutations are found frequently in sporadic RCC. Disruption of VHL results in an abnormal accumulation of HIF‐α, leading to the upregulation of downstream genes such as the vascular endothelial growth factor gene. We constructed a luciferase reporter vector driven by hypoxia‐responsive elements (5HRE/luc) and a therapeutic vector expressing a herpes simplex virus thymidine kinase gene (5HRE/tk). In the transient transfection assay using VHL‐deficient 786‐O cells, constitutive luciferase expression was detected under both aerobic and hypoxic conditions. In contrast, 786‐O cells transfected with a wild‐type VHL showed hypoxia‐inducible luciferase activity. In in vitro MTS assay, 50% of growth inhibition of 786‐O cells stably transfected with 5HRE/tk was achieved with exposure to 0.2 µg/mL of GCV under both aerobic and hypoxic conditions. Xenografts of the stable clone in SCID mice exhibited a marked regression on daily injections of GCV (50 mg/kg) for 10 days. In conclusion, a hypoxia‐responsive vector may have therapeutic potential for RCC with VHL mutations. (Cancer Sci 2005; 96: 288 –294)
Abbreviations:
- CMV
cytomegalovirus
- EPO
erythropoietin
- GCV
ganciclovir
- GLUT‐1
glucose transporter 1
- HA
hemagglutinin
- hCMVmp
human cytomegalovirus minimal promoter
- HIF
hypoxia‐inducible factor
- HRE
hypoxia‐responsive element
- HSV
herpes simplex virus
- HSVtk
herpes simplex virus thymidine kinase gene
- luc
luciferase
- NTR
nitroreductase
- PBS
phosphate‐buffered saline
- RCC
renal cell carcinoma
- RT–PCR
reverse transcription‐polymerase chain reaction
- SDS
sodium dodecylsulfate
- tk
thymidine kinase
- VEGF
vascular endothelial growth factor
- VHL
von Hippel‐Lindau.
Hypoxia‐inducible factors, known as transcription factors, control the expression of genes that play important roles in angiogenesis and tumor growth. 1 , 2 , 3 HIF are composed of a heterodimer of α and β subunits. The α subunit of HIF (HIF‐α) is regulated tightly by oxygen availability, while the β subunit (HIF‐1β) is expressed constitutively. Three HIF isoforms (HIF‐1α, HIF‐2α and HIF‐3α) similar in structure and binding capability to HIF‐1β have been identified. (3) HIF activate transcription by binding to the HRE, which was originally reported in the 3′ flanking region of the human and mouse Epo genes. 4 , 5 Similar HIF binding sites have been found in regulatory regions of other hypoxia‐inducible genes, such as VEGF, EPO and GLUT‐1. 6 , 7 , 8
The molecular mechanisms behind the regulation of HIF have been elucidated by recent studies, and indicate that the VHL protein forms an E3 ligase complex in association with elongin B, elongin C, Cul2 and Rbx1, 9 , 10 , 11 , 12 , 13 which binds to HIF‐α for its oxygen‐dependent degradation via the ubiquitin–proteasome pathway. 14 , 15 , 16 , 17 Furthermore, binding of VHL to HIF requires the hydroxylation of several proline residues within the oxygen‐dependent domain of HIF‐α in the presence of oxygen. 18 , 19 , 20 , 21
Mutations of the VHL tumor suppressing gene are associated with the development of multiple tumors, including hemangioblastomas in the central nervous system, RCC and pheochromocytomas. (22) In sporadic clear cell RCC, which accounts for 75% of RCC, (23) the VHL gene was mutated in 33–57% of cases 24 , 25 , 26 , 27 , 28 and silenced by hypermethylation in an additional 15–19%. 29 , 30 Interestingly, most of the VHL mutations in RCC were located at a particular site within exon 2 encoding the HIF‐binding β domain. (31) Thus, disruption of VHL results in a marked increase in HIF‐1 and/or HIF‐2 activity in non‐hypoxic conditions because of the impaired VHL‐dependent degradation of HIF‐1α or HIF‐2α, leading to the upregulated expression of VEGF, GLUT‐1 and EPO, as demonstrated by studies using VHL‐deficient cell lines, 32 , 33 and clinical samples of RCC. 34 , 35 , 36 , 37 From these findings, we speculated that the dysregulation of HIF‐α caused by VHL mutation in RCC might be exploited as a potential therapeutic target.
Gene therapy has been used in clinical trials for cancer treatment. One of the current problems with cancer gene therapy is the poor targeting selectivity of vectors, leading to a low efficiency of gene transfer to tumor cells and an increased risk of normal tissue toxicity. A tumor‐specific gene therapy targeting aberrant transcriptional control may be a solution because the use of tumor‐specific promoters can regulate the expression of therapeutic genes at a specific site or in a particular tumor. Several published studies have shown vector systems targeting hypoxic regions within solid tumors, utilizing HRE derived from mouse phosphoglycerate kinase‐1, (38) mouse VEGF (39) and EPO. (40) In an earlier study, a vector construct using five copies of HRE derived from the human VEGF gene promoter ligated to a hCMVmp (5HRE/hCMVmp) conferred a marked increase (over 500‐fold) in responsiveness to hypoxia in human fibrosarcoma HT1080 cells. (41) Based on this hypoxia‐inducible promoter system, a therapeutic model targeting tumor hypoxia was established using the gene for Escherichia coli NTR, a prodrug‐activating enzyme. (42) HT1080 cells stably transfected with the 5HRE/hCMVmp‐NTR vector showed hypoxic induction of NTR gene expression in correlation with increased sensitivity to in vitro exposure to the prodrug, and a growth delay was observed in tumor xenografts of the same stable transfectants treated with both intraperitoneal injection of the prodrug and respiration of hypoxic gas. (42) From these findings and results, we expected the hypoxia‐inducible vector to be useful for targeting dysregulation of HIF in VHL‐deficient RCC as well. The purpose of this study was to test the therapeutic potential of the hypoxia‐inducible vector system for RCC harboring VHL mutations.
Materials and Methods
Hypoxia‐inducible vectors. A hypoxia‐inducible vector expressing a firefly luciferase gene with a backbone of pGL3 (Promega, Madison, WI, USA) (5HRE/hCMVmp/luc) was constructed previously. 41 , 43 To generate a 5HRE/tk therapeutic vector, the luciferase gene in the 5HRE/hCMVmp construct with a backbone of pEF/cyto/myc (Invitrogen, Carlsbad, CA, USA) as shown previously, (42) was replaced with a human HSVtk gene (Invivogen, San Diego, CA, USA). Each plasmid construct is shown in Fig. 1.
Figure 1.
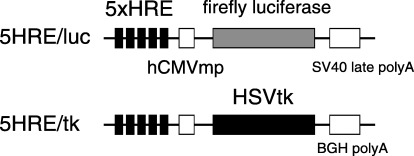
Structure of hypoxia‐inducible plasmids. The constructs of the hypoxia‐inducible reporter plasmid 5HRE/luc and the therapeutic plasmid 5HRE/tk are indicated.
Cell cultures and hypoxic treatment. Human RCC 786‐O cells were purchased from the American Type Culture Collection (Manassas, VA, USA), and 786‐O cells stably transfected with either HA‐tagged wild‐type VHL (786‐O/VHLwt), HA‐tagged truncated VHL 1–115 (786‐O/VHLmt) or an empty vector (786‐O/VHL(–/–)), were provided by Dr Kaelin WG Jr, and are presented as WT8, 115–3, and pRc3, respectively. (44) Untransfected 786‐O cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, while the transfected cells were maintained in the same medium and serum containing 800 µg/mL of G418. Human fibrosarcoma HT1080 cells were maintained in MEMα containing 10% fetal bovine serum. For aerobic incubation, cells were cultured in a well‐humidified incubator with 5% CO2 at 37°C. For hypoxic treatment, cells were cultured in a Bactron II anaerobic environmental chamber (Sheldon Manufacturing, Cornelius, OR, USA) maintained with 90% N2, 5% H2 and 5% CO2.
Transient transfection and luciferase assay. Cells (1 × 105) were seeded in six‐well plates 24 h before transfection. Transfection was carried out with 2 µg of 5HRE/luc, 0.04 µg of the control pRL‐CMV plasmid (Promega) and 6 µL of Superfect Reagent (Qiagen, Hilden, Germany) according to manufacturer's instructions. The medium was replaced with a fresh batch 3 h after transfection. After incubation for 16–20 h, the cells were trypsinized and split. They were then incubated 10–12 h before either hypoxic or aerobic incubation for 18 h. Cell lysates were then prepared with 400 µL of passive lysis buffer using a Dual luciferase assay kit (Promega). Luciferase activity was measured using a Lumat LB 9507 luminometer (Berthold, Bad Wildbad, Germany).
Immunoblotting analysis. Cells (2 × 105) were seeded in a pair of six‐well plates. The next day, one of the plates was kept under hypoxic conditions and the other under aerobic conditions for 18 h. Cells were then collected in 100 µL of 1 × sample buffer (50 mM Tris‐HCl [pH 6.8], 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue and 10% glycerol) and heated at 95°C for 5 min when 20 µL of each sample was immediately loaded on a SDS polyacrylamide gel (10% for VHL detection; 7.5% for HIF‐2α detection) and separated by electrophoresis. To achieve an equal amount of loading between samples, protein volumes were normalized by cell number. Proteins were transferred to a nitrocellulose membrane, blocked with 5% non‐fat milk in Tris‐buffered saline, and incubated with 0.2 µg/mL of anti‐HA antibody (Roche Diagnostics, Indianapolis, IN, USA) for VHL detection or with 0.1 µg/mL of anti‐EPAS1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h for HIF‐2α detection. Detection was carried out with a chemiluminescence‐based method using the ECL Plus Western Blotting Detection System (Amersham Biosciences, Piscataway, NJ, USA).
Stable transfection. To establish stable transfectants of 786‐O/VHL(–/–), 786‐O/VHLmt, 786‐O/VHLwt and HT1080 with the 5HRE/tk vector, 3 × 105 cells were seeded and stably transfected with both 10 µg of 5HRE/tk plasmid and 1 µg of pEF6/Myc‐His‐A plasmid, which expresses a blasticidin‐resistance gene, by a modified calcium‐phosphate method. The cells were then trypsinized 24 h after transfection and cultured in the selection medium containing 5 µg/mL blasticidin for 10 days. After selection, the mixtures of each blasticidin‐resistant cell were used directly for both RT–PCR analysis and in vitro proliferation assay without the isolation as a clone. To establish stable clones of 786‐O cells with 5HRE/tk vector, 3 × 105 of 786‐O cells were plated in a 6 cm dish. The next day, the cells were transfected with 5 µg of plasmid using 15 µL of Superfect Reagent. They were then trypsinized 24 h after transfection and cultured in the selection medium containing 800 µg/mL of G418. The G418‐resistant colonies were isolated and used for in vitro cell proliferation assays and mouse xenograft assays.
In vitro cell proliferation assay. One thousand cells were seeded in each well of two 96‐well plates and allowed to attach overnight. Cells were treated with medium in the absence or presence of GCV (Invivogen) at various concentrations for 24 h in either hypoxic conditions for 18 h and aerobic conditions for a subsequent 6 h, or else in aerobic conditions for 24 h. The medium was then replaced with fresh medium without the GCV, and subsequent aerobic incubation was carried out for an additional 72 h. Growth inhibition was determined by colorimetric quantification using a Celltiter 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega). Briefly, 10 µL of MTS tetrazonium solution (3‐[4,5‐dimethylthiazol‐2‐yl]‐5‐[3‐carboxymethoxyphenyl]‐2‐[4‐sulfophenyl]‐2H‐tetrazolium, inner salt) was added to each well. After incubation for 2 h, absorbance at 490 nm was measured using a Microplate Reader (Bio‐Rad, Hercules, CA, USA). Cell viability was calculated as the ratio of the absorbance value at each condition against that incubated in medium without GCV under continuous aerobic conditions.
Semiquantitative RT–PCR analysis. Blasticidin‐resistant stable transfectants of 786‐O/VHL(–/–), 786‐O/VHLmt, 786‐O/VHLwt and HT1080 with 5HRE/tk vector were cultured under aerobic and hypoxic condition for 18 h, and total RNA was extracted using an RNA extraction kit (Qiagen). Complementary DNA was synthesized from 2.5 µg total RNA using an oligo dT‐Adaptor Primer (Takara Biomedicals, Tokyo, Japan). Primers used for PCR were HSV/tk‐forward: 5′‐ATA TCG TCT ACG TAC CCG AG‐3′; HSV/tk‐reverse: 5′‐CGC ACC GTA TTG GCA AGC AG‐3′; GAPDH‐forward: 5′‐ACC ACA GTC CAT GCC ATC AC‐3′; and GAPDH‐reverse: 5′‐TCC ACC ACC CTG TTG CTG TA‐3′. The PCR was carried out to amplify the HSV/tk and GAPDH genes for 25 and 20 cycles, respectively. The PCR products were separated by agarose gel electrophoresis and stained with ethidium bromide.
Mouse xenograft assay. Three million cells were suspended in 100 µL PBS and inoculated in the right flank of male 6–8‐week‐old C.B‐17/lcr‐scid Jcl mice (Clea Japan, Tokyo, Japan). When the tumor volume had reached approximately 200 mm3, mice were treated daily with 50 mg/kg GCV or a comparable volume of PBS by intraperitoneal injection for 10 days. Tumors were measured using a caliper and tumor volume was calculated according to the following equation: volume = 0.5 × a × b2 (a, larger diameter; b, the smaller diameter). The study was approved by the ethical committee of the Kyoto University Institute of Laboratory Animals.
Results
Constitutive luciferase expression of a hypoxia‐inducible vector in VHL‐deficient and VHL‐mutated RCC cells. To test the activity of a hypoxia‐inducible vector in VHL‐deficient 786‐O RCC cells, luciferase activity was examined following transient transfection with a 5HRE/luc vector (Fig. 1). In 786‐O/VHL(–/–) cells, strong luciferase expression was detected under both aerobic and hypoxic conditions. This observation was completely different from that for HT1080 cells, as shown in a previous report. (41) We also tested 786‐O cells transfected with the wild‐type VHLcDNA (786‐O/VHLwt), and those transfected with the truncated VHL 1–115 (786‐O/VHLmt), which is a C‐terminal truncation mutant lacking a region frequently altered in sporadic and VHL‐related RCC. (44) 786‐O/VHLwt showed an inducible luciferase activity in a hypoxia‐dependent manner, while 786‐O/VHLmt showed a marked expression under both aerobic and hypoxic conditions (Fig. 2A). In addition, we examined the expression of VHL and HIF‐2α in an immunoblotting analysis. A protein with the predicted size of the VHL protein was detected in each of 786‐O/VHLwt and 786‐O/VHLmt. HIF‐2α protein was detected under both aerobic and hypoxic conditions in 786‐O/VHL(–/–) and 786‐O/VHLmt, while hypoxia‐dependent HIF‐2α expression was detected in 786‐O/VHLwt (Fig. 2B), supporting the result of the luciferase assay. Thus, the expression pattern of a hypoxia‐inducible vector became constitutive via mutation of the VHL gene.
Figure 2.
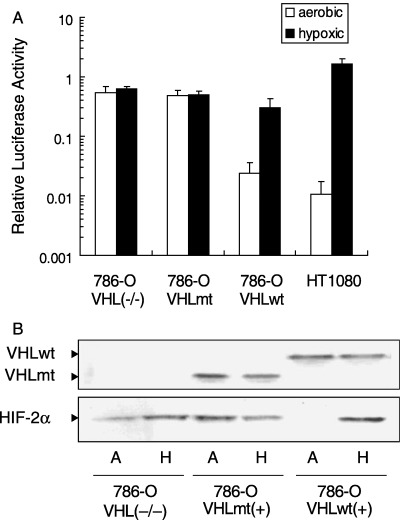
Constitutive gene expression from the hypoxia‐inducible promoter in VHL‐deficient and VHL‐mutated 786‐O cells. (A) Dual luciferase assay was carried out using 786‐O/VHL(–/–), 786‐O/VHLmt, 786‐O/VHLwt and HT1080. The cells were transiently transfected with both 5HRE/luc vector and pRL‐CMV, and cultured under aerobic (open bar) or hypoxic (closed bar) conditions. To normalize the firefly luciferase activity from the 5HRE/luc vector, renilla luciferase activity from pRL‐CMV was used as an internal control. The normalized luciferase activity under hypoxic conditions was divided by the one under aerobic conditions to calculate the relative luciferase activity. Results are the mean of three independent experiments ± SD. (B) Immunoblots against VHL (upper) and HIF‐2α (lower) were carried out using 786‐O/VHL(–/–), 786‐O/VHLmt and 786‐O/VHLwt cells under aerobic (A) and hypoxic (H) conditions.
In vitro cytotoxicity of 5HRE/tk influenced by different VHL statuses. To test the therapeutic efficacy, we constructed a plasmid expressing a HSVtk gene based on the same hypoxia‐inducible system (Fig. 1). The 5HRE/tk vector was introduced into 786‐O/VHL(–/–), 786‐O/VHLmt, 786‐O/VHLwt and HT1080 cells, and stable transfectants were treated with various concentrations of GCV for 24 h under either 18 h of hypoxic followed by 6 h of aerobic conditions, or continuous aerobic conditions. The growth inhibitory effects were determined by MTS assay 96 h after the start of treatment. In 786‐O/VHL(–/–) and 786‐O/VHLmt, a growth inhibition rate of 50% was achieved with exposure to less than 0.2 µg/mL GCV under both aerobic and hypoxic conditions. On the other hand, the growth inhibition in both 786‐O/VHLwt and HT1080 were observed only under hypoxic conditions, while no significant growth inhibition was observed with exposure up to 10 µg/mL of GCV under aerobic conditions (Fig. 3A). HSVtk transcription levels were examined using semiquantitative RT–PCR analysis (Fig. 3B). High levels of HSVtk transcripts were detected under both aerobic and hypoxic conditions in 786‐O/VHL(–/–) and 786‐O/VHLmt, but only under hypoxic conditions in 786‐O/VHLwt and HT1080 cells, indicating HSVtk transcription levels correlated with sensitivity to GCV. From these results, in vitro antitumor effects of 5HRE/tk and GCV gave rise to RCC cells with mutations in the VHL gene both under aerobic and hypoxic conditions as well as to hypoxic cells, while cells with wild‐type VHL were spared under aerobic conditions.
Figure 3.
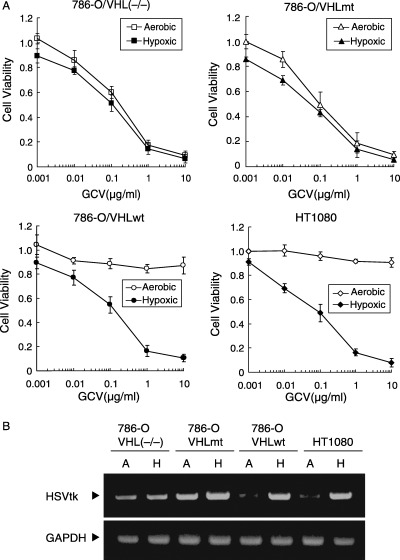
Cytotoxicity of 5HRE/tk and GCV in VHL(–/–) and VHLmt cells. (A) 786‐O/VHL(–/–), 786‐O/VHLmt, 786‐O/VHLwt and HT1080 were exposed to GCV under aerobic (open) or hypoxic (closed) conditions. Cell viability was quantified using the MTS assay at 72 h after the end of the GCV treatment, and calculated as the ratio of the absorbance value at each condition against that incubated in medium without GCV under continuous aerobic conditions. Results are the mean of three independent experiments ± SD. (B) Cells were cultured under aerobic (A) or hypoxic (H) conditions, and expression of HSVtk mRNA was assessed using semiquantitative RT–PCR using a specific primer set. GAPDH mRNA was also analyzed as an internal control.
In vivo antitumor effects in SCID mouse xenografts of 786‐O cells transfected with 5HRE/tk. To prepare the mouse xenograft model, selected clones of 786‐O transfected with 5HRE/tk were established and screened by MTS assay. Among the transfected clones, clone 9 was used for further analysis, indicating a growth inhibition rate of 50% with exposure to less than 0.1 µg/mL GCV under both aerobic and hypoxic conditions (Fig. 4A). On the other hand, the growth of untransfected 786‐O cells was not inhibited by up to 10 µg/mL of GCV regardless of hypoxic treatment (Fig. 4A). To confirm in vivo therapeutic efficacy, a growth delay assay was carried out. Tumor‐bearing mice were treated with daily injections of GCV (50 mg/kg) or PBS for 10 days. In mice with xenografts of clone 9, marked tumor regression was observed during GCV treatment, while tumors treated with PBS continued to grow (Fig. 4B). Among the six mice treated with GCV, three mice showed tumor regrowth after cessation of GCV, while three mice showed stable tumor size (Fig. 4C). On the other hand, the tumor xenografts of untransfected 786‐O cells grew regardless of GCV treatment (Fig. 4B).
Figure 4.
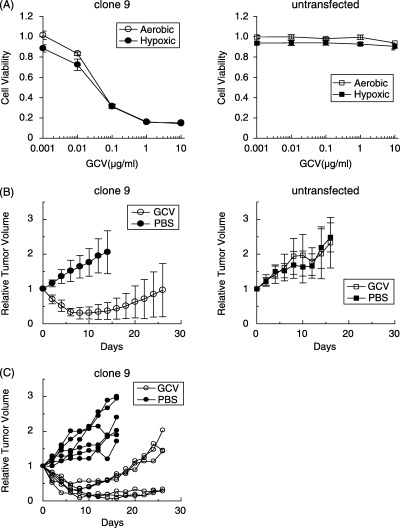
In vivo antitumor efficacy in tumor xenografts consisting of 786‐O clone stably transfected with 5HRE/tk. (A) MTS assay was carried out with the stable 786‐O clone 9 transfected with 5HRE/tk (left) and untransfected 786‐O cells (right), indicating hypersensitivity to GCV in clone 9 under both aerobic (open) and hypoxic (close) conditions. (B) A growth delay assay was carried out using xenografts derived from clone 9 (left) and untransfected 786‐O cells (right). Tumor‐bearing SCID mice were treated by daily intraperitoneal injection of either 50 mg/kg GCV (open) or a comparable volume of PBS (closed) for 10 days. Relative tumor volume as a function of days from the start of treatment is indicated. Each point and error bar is the mean (n = 4–6) and SD. (C) Individual tumor growth derived from clone 9 is indicated.
Discussion
The most common type of human kidney tumors, clear cell RCC, often have mutations of the VHL gene, resulting in an abnormal accumulation of HIF‐α and upregulation of hypoxia‐dependent gene expression driven by HRE regardless of oxygen status. We have developed a hypoxia‐inducible vector using HRE derived from human VEGF to target hypoxic cells existing in solid tumors and to overcome the resistance of hypoxic cells to chemotherapy and radiotherapy. Here, we demonstrated a therapeutic model for VHL‐deficient RCC using the hypoxia‐inducible vector system.
First, we confirmed that HIF transcriptional activity is dysregulated by VHL mutations. Luciferase activity was remarkably increased in response to hypoxia in HT1080 and 786‐O/VHLwt cells transfected with the 5HRE/luc vector. In 786‐O/VHL(–/–) and 786‐O/VHLmt, however, strong luciferase expression was detected under both aerobic and hypoxic conditions (Fig. 2A). Figure 2B shows that such dysregulation in 786‐O cells is presumably mediated by constitutive HIF‐2α expression, because 786‐O cells lack HIF‐1α. (45) These results are consistent with reports by Maxwell et al. and Hu et al. 45 , 46
HSVtk is a common prodrug‐activating gene used in preclinical and clinical trials. GCV is phosphorylated specifically by HSVtk to its monophosphate, which is subsequently converted to the di‐ and tri‐phosphate forms by guanylate kinase and other cellular kinases. GCV‐triphosphate can be incorporated into elongating DNA, causing inhibition of DNA replication and single strand breaks. (47) In this study, we constructed HSVtk in conjunction with the 5HRE/hCMVmp promoter as a therapeutic vector. HT1080 and 786‐O/VHLwt stably transfected with 5HRE/tk showed that hypoxia‐inducible transcription of HSVtk correlated with increased sensitivity to GCV, as had been demonstrated by Shibata et al. using the same promoter system. (42) Here, 786‐O/VHL(–/–) and 786‐O/VHLmt with the 5HRE/tk vector showed hypersensitivity to GCV, together with constitutive HSVtk transcription under both aerobic and hypoxic conditions. From these results, our hypoxia‐inducible vector has a selective therapeutic effect not only on hypoxic cells but also on RCC with VHL mutations.
According to the mechanism in action described above, the HSVtk–GCV system is particularly suitable for eradication of rapidly dividing tumor cells. On the other hand, because activated GCV is an S‐phase‐specific cytotoxin, it is necessary that target cells must be actively dividing in S‐phase at the time of exposure. (47) As shown in Fig. 4B, xenografts of 786‐O cells transfected with 5HRE/tk showed a marked response to GCV and reduction in size during GCV treatment. However, half of them showed regrowth after cessation of GCV (Fig. 4C). This may be because elimination of activated GCV reactivated division of surviving cells that were not in S‐phase during GCV treatment, and because administration dose and/or duration of GCV treatment might have been insufficient to eradicate tumors. In this experimental setting, we did not plan to use 786‐O/VHLwt as a control because Iliopoulos et al. had already shown that 786‐O subclones transfected with the wild‐type VHL gene suppressed tumor formation in the nude mouse xenograft model. (44)
As shown in Fig. 3A, 786‐O/VHLwt and HT1080 transfected with 5HRE/tk exhibited clear differences in sensitivity to GCV under aerobic and hypoxic conditions. These transfectants have no significant growth inhibition with exposure up to 10 µg/mL of GCV under aerobic conditions, suggesting the possibility of the use of the 5HRE promoter to reduce toxicity to normal tissues where hypoxic area dose not usually exist. It would be certain that the use of constitutive promoters such as CMV instead of HRE drive high expression of HSVtk in normal cells that have wild‐type VHL even under aerobic conditions, which may damage normal cells. In vivo toxicity, however, remains to be evaluated with systemic administration of CMV or HRE vectors using a clinically relevant gene delivery system such as viral vectors. Binley et al. reported that use of the OBHRE promoter reduced hepatotoxicity with systemic administration of adenoviral vectors. (48)
In this study, we demonstrated the proof‐of‐principle for a therapeutic model exploiting dysregulation of the VHL/HIF pathway in RCC, providing for the potential application of a hypoxia‐inducible vector system to the novel therapeutic treatment of RCC. For clinical application, however, further experiments should be conducted using gene delivery systems such as adenoviral or retroviral vectors, bacteria and macrophages. Of note, a report published during the preparation of this paper demonstrated a therapeutic effect using an oncolytic wild‐type adenovirus with HRE from human VEGF gene promote on VHL‐deficient RCC. (49)
In present clinical practice, patients with RCC are treated mainly with surgical resection for primary lesions. Metastatic RCC are treated with immunotherapy using interferon‐α or interleukin‐2, but are still difficult problems. Radiotherapy and chemotherapy are often ineffective. A tumor‐specific gene therapy using the hypoxia‐responsible vector system may be an option for the treatment of RCC in addition to these therapeutic modalities. Recently, several new therapeutic approaches for RCC have been tested in clinical trials using a radiolabeled chimeric monoclonal antibody targeting CAIX, (50) or using a neutralizing antibody to VEGF. (51)
In conclusion, the hypoxia‐inducible vector system may have therapeutic potential for RCC with VHL mutations. Further study using delivery systems such as viral vectors, bacteria and macrophages should be conducted for clinical application.
Acknowledgments
The work was supported in part by Grants‐in‐Aid for Scientific Research in Priority Areas (12217066) and for Exploratory Research (14657213) from the Japanese Ministry of Education, Culture, Sports, Science and Technology. We thank Dr Kaelin WG Jr for kindly providing the 786‐O cell lines stably transfected with different versions of the VHL gene. We also thank Akiyo Morinibu for her excellent technical assistance.
References
- 1. Semenza GL. HIF‐1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002; 8: S62–7. [DOI] [PubMed] [Google Scholar]
- 2. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9: 677–84. [DOI] [PubMed] [Google Scholar]
- 3. Safran M, Kaelin WG, Jr . HIF hydroxylation and the mammalian oxygen‐sensing pathway. J Clin Invest 2003; 111: 779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia‐inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA 1991; 88: 5680–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madan A, Curtin PT. A 24‐base‐pair sequence 3′ to the human erythropoietin gene contains a hypoxia‐responsive transcriptional enhancer. Proc Natl Acad Sci USA 1993; 90: 3928–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldberg MA, Schneider TJ. Similarities between the oxygen‐sensing mechanisms regulating the expression of vascular endothelial growth factor and erythropoietin. J Biol Chem 1994; 269: 4355–9. [PubMed] [Google Scholar]
- 7. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia‐inducible factor 1. J Biol Chem 1994; 269: 23 757–63. [PubMed] [Google Scholar]
- 8. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia‐inducible factor 1. J Biol Chem 1996; 271: 32 529–37. [DOI] [PubMed] [Google Scholar]
- 9. Duan DR, Humphrey JS, Chen DY, Weng Y, Sukegawa J, Lee S, Gnarra JR, Linehan WM, Klausner RD. Characterization of the VHL tumor suppressor gene product: localization, complex formation, and the effect of natural inactivating mutations. Proc Natl Acad Sci USA 1995; 92: 6459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG, Jr . Binding of the von Hippel‐Lindau tumor suppressor protein to elongin B and C. Science 1995; 269: 1444–6. [DOI] [PubMed] [Google Scholar]
- 11. Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD. The von Hippel‐Lindau tumor‐suppressor gene product forms a stable complex with human CUL‐2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA 1997; 94: 2156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG, Jr . Regulation of hypoxia‐inducible mRNAs by the von Hippel‐Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol 1998; 18: 732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG Jr, Elledge SJ, Conaway RC, Harper JW, Conaway JW. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999; 284: 657–61. [DOI] [PubMed] [Google Scholar]
- 14. Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor‐alpha binding and ubiquitylation by the von Hippel‐Lindau tumor suppressor protein. J Biol Chem 2000; 275: 25 733–41. [DOI] [PubMed] [Google Scholar]
- 15. Kamura T, Sato S, Iwai K, Czyzyk‐Krzeska M, Conaway RC, Conaway JW. Activation of HIF1α ubiquitination by a reconstituted von Hippel‐Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA 2000; 97: 10 430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia‐inducible factor requires direct binding to the beta‐domain of the von Hippel‐Lindau protein. Nat Cell Biol 2000; 2: 423–7. [DOI] [PubMed] [Google Scholar]
- 17. Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia‐inducible factor‐1 alpha by the von Hippel‐Lindau tumor suppressor protein. Embo J 2000; 19: 4298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in HIF‐1 alpha by pVHL. Nature 2002; 417: 975–8. [DOI] [PubMed] [Google Scholar]
- 19. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG, Jr . HIFα targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464–8. [DOI] [PubMed] [Google Scholar]
- 20. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF‐α to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 2001; 292: 468–72. [DOI] [PubMed] [Google Scholar]
- 21. Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP. Structure of an HIF‐1α–pVHL complex: hydroxyproline recognition in signaling. Science 2002; 296: 1886–9. [DOI] [PubMed] [Google Scholar]
- 22. Kim W, Kaelin WG, Jr . The von Hippel‐Lindau tumor suppressor protein: new insights into oxygen sensing and cancer. Curr Opin Genet Dev 2003; 13: 55–60. [DOI] [PubMed] [Google Scholar]
- 23. Linehan WM. Molecular targeting of VHL gene pathway in clear cell kidney cancer. J Urol 2003; 170: 593–4. [DOI] [PubMed] [Google Scholar]
- 24. Bailly M, Bain C, Favrot MC, Ozturk M. Somatic mutations of the von Hippel‐Lindau (VHL) tumor‐suppressor gene in European kidney cancers. Int J Cancer 1995; 63: 660–4. [DOI] [PubMed] [Google Scholar]
- 25. Foster K, Prowse A, Van Den Berg A, Fleming S, Hulsbeek MM, Crossey PA, Richards FM, Cairns P, Affara NA, Ferguson‐Smith MA, et al . Somatic mutations of the von Hippel‐Lindau disease tumour suppressor gene in non‐familial clear cell renal carcinoma. Hum Mol Genet 1994; 3: 2169–73. [DOI] [PubMed] [Google Scholar]
- 26. Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, et al . Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7: 85–90. [DOI] [PubMed] [Google Scholar]
- 27. Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, Nagashima Y, Kitamura H, Latif F, Zbar B, et al . Frequent somatic mutations and loss of heterozygosity of the von Hippel‐Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res 1994; 54: 2852–5. [PubMed] [Google Scholar]
- 28. Whaley JM, Naglich J, Gelbert L, Hsia YE, Lamiell JM, Green JS, Collins D, Neumann HP, Laidlaw J, Li FP, et al . Germ‐line mutations in the von Hippel‐Lindau tumor‐suppressor gene are similar to somatic von Hippel‐Lindau aberrations in sporadic renal cell carcinoma. Am J Hum Genet 1994; 55: 1092–102. [PMC free article] [PubMed] [Google Scholar]
- 29. Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER. Inactivation of the von Hippel‐Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL‐independent pathway in clear cell renal tumourigenesis. Genes Chrom Cancer 1998; 22: 200–9. [DOI] [PubMed] [Google Scholar]
- 30. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al . Silencing of the VHL tumor‐suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994; 91: 9700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brauch H, Weirich G, Brieger J, Glavac D, Rodl H, Eichinger M, Feurer M, Weidt E, Puranakanitstha C, Neuhaus C, Pomer S, Brenner W, Schirmacher P, Storkel S, Rotter M, Masera A, Gugeler N, Decker HJ. VHL alterations in human clear cell renal cell carcinoma. association with advanced tumor stage and a novel hot spot mutation. Cancer Res 2000; 60: 1942–8. [PubMed] [Google Scholar]
- 32. Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up‐regulation of hypoxia‐inducible factors HIF‐1α and HIF‐2α under normoxic conditions in renal carcinoma cells by von Hippel‐Lindau tumor suppressor gene loss of function. Oncogene 2000; 19: 5435–43. [DOI] [PubMed] [Google Scholar]
- 33. Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny‐Baron G, Marme D. Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel‐Lindau tumor suppressor protein. Cancer Res 1996; 56: 2299–301. [PubMed] [Google Scholar]
- 34. Turner KJ, Moore JW, Jones A, Taylor CF, Cuthbert‐Heavens D, Han C, Leek RD, Gatter KC, Maxwell PH, Ratcliffe PJ, Cranston D, Harris AL. Expression of hypoxia‐inducible factors in human renal cancer. relationship to angiogenesis and to the von Hippel‐Lindau gene mutation. Cancer Res 2002; 62: 2957–61. [PubMed] [Google Scholar]
- 35. Wiesener MS, Munchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A, Jurgensen JS, Gruber G, Maxwell PH, Loning SA, Frei U, Maher ER, Grone HJ, Eckardt KU. Constitutive activation of hypoxia‐inducible genes related to overexpression of hypoxia‐inducible factor‐1α in clear cell renal carcinomas. Cancer Res 2001; 61: 5215–22. [PubMed] [Google Scholar]
- 36. Igarashi H, Esumi M, Ishida H, Okada K. Vascular endothelial growth factor overexpression is correlated with von Hippel‐Lindau tumor suppressor gene inactivation in patients with sporadic renal cell carcinoma. Cancer 2002; 95: 47–53. [DOI] [PubMed] [Google Scholar]
- 37. Na X, Wu G, Ryan CK, Schoen SR, Di'Santagnese PA, Messing EM. Overproduction of vascular endothelial growth factor related to von Hippel‐Lindau tumor suppressor gene mutations and hypoxia‐inducible factor‐1 alpha expression in renal cell carcinomas. J Urol 2003; 170: 588–92. [DOI] [PubMed] [Google Scholar]
- 38. Dachs GU, Patterson AV, Firth JD, Ratcliffe PJ, Townsend KM, Stratford IJ, Harris AL. Targeting gene expression to hypoxic tumor cells. Nat Med 1997; 3: 515–20. [DOI] [PubMed] [Google Scholar]
- 39. Koshikawa N, Takenaga K, Tagawa M, Sakiyama S. Therapeutic efficacy of the suicide gene driven by the promoter of vascular endothelial growth factor gene against hypoxic tumor cells. Cancer Res 2000; 60: 2936–41. [PubMed] [Google Scholar]
- 40. Ruan H, Su H, Hu L, Lamborn KR, Kan YW, Deen DF. A hypoxia‐regulated adeno‐associated virus vector for cancer‐specific gene therapy. Neoplasia 2001; 3: 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shibata T, Giaccia AJ, Brown JM. Development of a hypoxia‐responsive vector for tumor‐specific gene therapy. Gene Ther 2000; 7: 493–8. [DOI] [PubMed] [Google Scholar]
- 42. Shibata T, Giaccia AJ, Brown JM. Hypoxia‐inducible regulation of a prodrug‐activating enzyme for tumor‐specific gene therapy. Neoplasia 2002; 4: 40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shibata T, Akiyama N, Noda M, Sasai K, Hiraoka M. Enhancement of gene expression under hypoxic conditions using fragments of the human vascular endothelial growth factor and the erythropoietin genes. Int J Radiat Oncol Biol Phys 1998; 42: 913–16. [DOI] [PubMed] [Google Scholar]
- 44. Iliopoulos O, Kibel A, Gray S, Kaelin WG, Jr . Tumour suppression by the human von Hippel‐Lindau gene product. Nat Med 1995; 1: 822–6. [DOI] [PubMed] [Google Scholar]
- 45. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 1999; 399: 271–5. [DOI] [PubMed] [Google Scholar]
- 46. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia‐inducible factor 1α (HIF‐1α) and HIF‐2α in hypoxic gene regulation. Mol Cell Biol 2003; 23: 9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol 2001; 187: 22–36. [DOI] [PubMed] [Google Scholar]
- 48. Binley K, Askham Z, Martin L, Spearman H, Day D, Kingsman S, Naylor S. Hypoxia‐mediated tumour targeting. Gene Ther 2003; 10: 540–9. [DOI] [PubMed] [Google Scholar]
- 49. Cuevas Y, Hernandez‐Alcoceba R, Aragones J, Naranjo‐Suarez S, Castellanos MC, Esteban MA, Martin‐Puig S, Landazuri MO, Del Peso L. Specific oncolytic effect of a new hypoxia‐inducible factor‐dependent replicative adenovirus on von Hippel‐Lindau‐defective renal cell carcinomas. Cancer Res 2003; 63: 6877–84. [PubMed] [Google Scholar]
- 50. Divgi CR, Bander NH, Scott AM, O'Donoghue JA, Sgouros G, Welt S, Finn RD, Morrissey F, Capitelli P, Williams JM, Deland D, Nakhre A, Oosterwijk E, Gulec S, Graham MC, Larson SM, Old LJ. Phase I/II radioimmunotherapy trial with iodine‐131‐labeled monoclonal antibody G250 in metastatic renal cell carcinoma. Clin Cancer Res 1998; 4: 2729–39. [PubMed] [Google Scholar]
- 51. Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA. A randomized trial of bevacizumab, an anti‐vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003; 349: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]